

Abstract
We present an experimental analysis of the plasmonic scattering properties of gold nanoparticles controllably placed nanometers away from a gold metal film. We show that the spectral response of this system results from the interplay between the localized plasmon resonance of the nanoparticle and the surface plasmon polaritons of the gold film, as previously predicted by theoretical studies. In addition, we report that the metal film induces a polarization to the single nanoparticle light scattering, resulting in a doughnut-shaped point spread function when imaged in the far-field. Both the spectral response and the polarization effects are highly sensitive to the nanoparticle–film separation distance. Such a system shows promise in potential biometrology and diagnostic devices.
Surface plasmons are highly versatile electromagnetic modes that occur at optical wavelengths at the surface of metallic structures and that exhibit unique characteristics due to the participation of the conduction electrons within the metal. Because of their enormous potential as sensors and miniaturized photonic components, a wide range of plasmonic systems have been studied in great depth and have been implemented or are being actively considered for biomedical and photonic applications. A particularly interesting plasmonic system that has received somewhat less attention is that of a metal nanoparticle interacting with a metal film.1–10 This system has been predicted to display a wealth of interesting optical phenomena caused by the complex interaction of the localized surface plasmon of the nanoparticle with the delocalized thin film surface plasmon polariton, which is especially strong when the nanoparticle is within a distance of ∼50 nm to the film surface. Here, by measuring the light scattered from individual gold nanoparticles positioned controllably above a gold film using polyelectrolyte layers, we are able to probe the coupling effects and relate them to prior numerical studies. We find that the nanoparticle–thin film coupled system produces remarkably consistent and highly sensitive wavelength shifts and polarization effects in the scattered light, suggesting that the system might be useful for metrology or diagnostic applications.
Surface plasmons are strongly dependent on the shape, size, and composition of the metallic surface on which they reside11–22 and on the dielectric value of the proximate medium.23–31 Surface plasmons can be delocalized and propagating, as on the surface of thin films, or can be strongly confined to subwavelength regions, as on metallic nanoparticles (NPs). When a NP has dimensions much smaller than the wavelength of excitation, a resonance condition can occur at which the elastic scattering cross section of the particle increases dramatically and the local electromagnetic fields are strongly enhanced. Although they have dimensions smaller than the diffraction limit of light, plasmon resonant NPs can, nevertheless, be readily observed individually in a microscope under appropriate illumination conditions where background scattering is minimized.32,33 Since the plasmon resonance of a NP is strongly affected by its interaction with the local environment, the far-field scattering from the nanoparticle provides a simple and convenient means to study the near-field coupling effects of interest here.
While several authors have recently presented numerical results for the coupling of a nanoparticle to a metallic film, the system considered by Leveque and Martin8,9 is the closest to the geometry considered in our experiments, and our observations agree qualitatively with their numerical results. Leveque and Martin performed full-wave numerical scattering simulations based on iteratively constructing the Green's tensor for a system consisting of a nanosized gold cube supporting a localized surface plasmon (LSP) above a gold film which supports propagating surface plasmon polaritons (SPPs). Experimentally, both of these plasmons can be excited by a variety of optical methods. One common method, used in this work and simulated by Leveque and Martin, is to introduce a prism beneath the film. White light is brought into the prism in such a manner that it undergoes total internal reflection (TIR) and produces an evanescent field that decays exponentially away from the prism surface (Figure 1a). The evanescent field selectively excites the SPP mode of the thin film at one wavelength that has the same in-plane wave vector and energy as the incoming light (45° incidence), which in turn interacts with the NPs situated on or above the film. Although the SPPs are confined modes, the coupling to the NPs allows the light to scatter into the far-field. The evanescent field also directly excites the LSP of the NPs; thus, this illumination drives both resonances of the system simultaneously, and both are scattered by the NP into the far-field.
Figure 1.
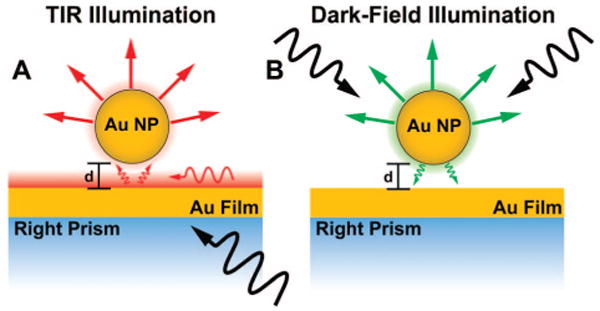
Simplified representation of the different illumination schemes used to excite single nanoparticles (NPs) coupled to the gold film (NP–film). In both cases, the far-field scattered light from the NP can be collected and analyzed by the microscope objective. (A) White light total internal reflectance (TIR) illumination at 45° excites a single surface plasmon polariton (SPP) mode of the film and the localized surface plasmon (LSP) of the NP, with an evanescent field which decays exponentially with increasing distance (d) from the surface. (B) White light dark-field illumination (using a DF microscope objective) excites the LSP of the NP, which can then radiate into either the far-field (measured by microscope) or a nonresonant continuum of 2-D gold film SPP modes.
The theoretical study by Leveque of the scattering properties of the single NP–film system showed that the SPP resonance wavelength is not affected by the presence of the single NP, which would be expected given that the NP represents an insignificant perturbation to the delocalized SPP supported by the (much larger) infinite thin film. However, their calculations revealed that the LSP resonance undergoes a blue shift as the distance between the particle and the film is increased. This result is consistent with previous experimental work that showed a very similar behavior for LSPs excited on nanoisland films in close proximity to a metal slab.34 As pointed out by Holland and Hall, the LSP frequency blue shift can be explained by treating the NP as a dipole placed above a conducting plane, for example, by using the theory of Chance, Prock, and Silbey.35
Here, we confirm and complete these previous results by experimentally characterizing the scattering of single NPs under two illumination schemes. In addition to 45° white light TIR illumination, we apply dark-field (DF) illumination (Figure 1b), which provides decoupled information on the interactions of the NP–film system due to the distinct excitation conditions. Dark-field illumination is particularly useful for imaging small scattering objects by exciting the sample with a hollow cone of light with numerical aperture (NA) greater than the collection N.A. In our experiments, the light cone is produced by a microscope objective that illuminates the sample from the air side. In this configuration, light can directly excite the LSP resonance of the NP, but it cannot directly couple to the SPPs of the gold film because of the inherent mismatch between the in-plane wavectors' kspp of these modes and those of the photons traveling in the air region. However, the NP provides an indirect way to excite the SPPs of the gold film at all wavelengths; due to its subwavelength nature, the NP generates a broad range of wavevectors that include the kspp subset when it scatters white light. In other words, the NP does not preferentially couple to any specific SPP but rather interacts with the whole continuum of SPP modes which span the visible spectrum. For this reason, the LSP does not lose its identity because it does not interact with a discrete mode of the film. The resonant LSP scattering of the nanoparticle radiates both into the far-field (as detected by the microscope) and into the nonresonant continuum of SPP modes within the gold film.
We complement the spectral measurements of the plasmons as outlined above with characterization of dramatic polarization effects induced by the film interaction with the NP, which can be accounted for by the simple dipole–metal surface model. Conveniently, we are able to measure these polarization effects from images of the far-field scattering of single NP resonators, similar to what has been previously observed by researchers using far-field fluorescence imaging to characterize the three-dimensional orientation of single-molecule dipole emitters.36–40
To create an experimental version of the NP–film system, gold NPs (60 nm diameter) were immobilized either directly on gold films (45 nm thickness) or on polyelectrolyte (PE) spacers deposited on the gold films. The concentration of NPs deposited on the film surface was intentionally kept low (<0.05% surface area coverage) in order to facilitate single NP characterization and to minimize both interparticle LSP coupling and shifts to the gold film SPPs, which can be induced by high concentrations of any surface defect. The NPs were imaged and their spectra taken using a Nikon dark-field (DF) microscope with a 100× DF (0.9 NA) objective. The sample slides were index-matched with oil to the top surface of a right prism that was mounted on a microscope stage and were illuminated with a 100 W Xenon lamp either through the DF objective (unpolarized) or through an optical fiber in TIR alignment. For the TIR configuration, a multimode fiber (1 mm) was directed normal to the prism sidewall for illumination. The angle of incidence at the metal film was 45°, and the illumination was P-polarized for the TIR (in air) data presented here.
In both illumination schemes, the background scattering was minimized, and the spot size spanned an area larger than the microscope's total field-of-view, ensuring uniform excitation of all of the particles in the image area. The microscope was fitted with an adjustable pinhole aperture and mirror located at the image plane, which was used to select an individual NP scatterer, remove as much background light as possible, and relay the signal to the spectrometer (Acton 2300SPI, with CoolSnap HQ detector). A secondary port on the column enabled switching between spectral acquisition and imaging with either a grayscale CCD (CoolSnap ES) or a color digital camera (Nikon D70). At least five gold NPs were characterized at each PE layer thickness using both types of illumination. All single NP spectra were background corrected by subtracting the spectrum from an apertured region of the substrate with no NPs and normalized to correct for the wavelength response of the apparatus by dividing with the scattered spectrum from a white scattering standard (Labsphere).
A general comparison between the scattering properties of 60 nm gold NPs in a quasi-uniform dielectric (glass/water interface) and 60 nm gold NPs electrostatically immobilized on a 45 nm gold film is shown in Figure 2. The LSP resonance and hence the scattering profile of a gold NP are significantly altered when placed directly on the gold film. Assuming the NP scatters as a polarizable dipole, an image dipole is induced in the film in response to the presence of a NP. The in-plane (parallel to the film) dipole moments of the image are opposite to those of the NP, so that the net scattering from the NP vanishes when light is polarized such that the electric field is parallel to the film (Figure 2a and c). The NP and its image can then only be polarized normal to the surface (Figure 2b and d). The point spread function (PSF) intensity distribution at the image plane for the radiation emanating from a dipole moment with its axis oriented parallel to the microscope optical axis (normal to the film surface) has a characteristic doughnut shape (Figure 2d and f), which is the result of spatial filtering of the collected scattered light.41 In addition, the doughnut shaped PSF is significantly broader than the PSF for a typical un-polarized scatterer.
Figure 2.
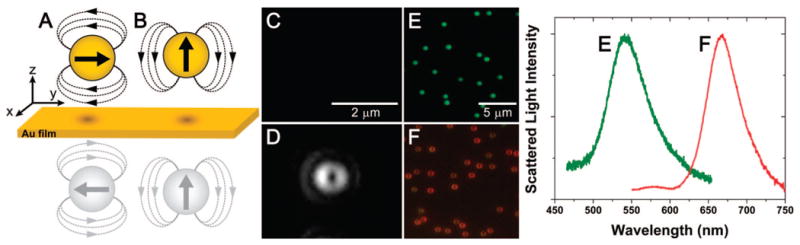
Optical effects observed from a gold nanoparticle (NP) placed on a gold film. (A) Simplified representation of a plasmonic NP interaction with a gold film. The dipole parallel to the gold film is canceled out by the induced image dipole. (B) The dipole perpendicular to the gold film resonantly couples to the induced image dipole, is red shifted, and scatters asymmetrically. (C) Experimentally observed far-field scattering from a 60 nm gold NP on a 45 nm gold film illuminated with polarization parallel to the film (scattering is damped and is virtually undetectable). (D) The same NP as that in (C) but with a component of the illumination polarized perpendicular to the film, which couples to the vertically oriented dipole and is scattered into the far-field. (E) Color image and typical spectrum of 60 nm gold NPs in a quasi-uniform dielectric (at a glass–H2O interface) and (F) on the surface of the 45 nm thick planar gold film (under dark-field illumination). Spectra amplitudes are normalized.
The resonant scattering of a 60 nm gold NP in a uniform dielectric of air or water has a peak in the green region of the optical spectrum; in contrast, when this same NP is placed on a gold film and illuminated with dark-field white light, the resonance is dramatically red shifted (Figure 2e and f). This spectral shift is again consistent with the interaction between two closely spaced NPs illuminated such that the electric field is along the symmetry axis. It should be noted that the scattering spectrum of the NP in contact with the gold film contains one predominant peak, corresponding to the LSP resonance, because dark-field illumination is not capable of preferentially exciting any SPP mode of the gold film.
To probe the scattering properties of the NP–film system between the two extremes shown in Figure 2–NP in a uniform dielectric and NP directly on the films–a controllable separation between the NPs and the underlying gold films was introduced by a layer-by-layer (LBL) deposition of polyelectrolyte (PE) layers of thicknesses from 0.64 to 22.3 nm (Figure 3a). Ellipsometry measurements revealed that the thickness of the PE thin film on each gold-coated slide increased linearly with the number of bilayers (∼4.1 nm) deposited (Figure 3b), consistent with previous studies.42,43
Figure 3.
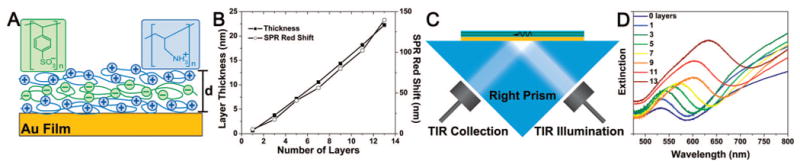
Layer-by-layer polyelectrolyte (PE) assembly on gold film with no nanoparticles present. (A) Alternating PE layers always terminated in a positively charged top surface to enable chemisorption of gold NPs. (B) Ellipsometric measurement suggest a ∼4 nm bilayer thickness. (C) Experimental configuration for exciting and measuring the surface plasmon polariton (SPP) of the gold film with TIR evanescent white light at 45° incidence. (D) Measurement of SPP extinction from the gold films exhibits the expected red shift as the dielectric thickness increases, which is also summarized in (B), where the SPP extinction minimum is plotted versus the number of deposited PE layers. Note that by changing the angle of illumination, the wavelength of the SPP mode would shift so it was kept at 45° throughout all “in-air” measurements presented in this article.
The dielectric value of the PE layers changes the electromagnetic environment of the nanoparticles and the film. While the SPP of the gold film that is resonantly excited by 45° TIR white light illumination is significantly red shifted by the PE layers, they likely produce a small effect on the localized plasmon resonance of the NP relative to the coupling interaction between the nanoparticles and film. In order to deconvolve the complex scattering of the NP–film under this TIR illumination predicted by Leveque and Martin, we first characterized the resonant extinction of the gold film SPP for each PE layer thickness. For these surface plasmon resonance (SPR) measurements, the reflected light from TIR off of the gold–PE films was collected by a second multimode (1 mm diameter) optical fiber and directed to the spectrometer. These SPR measurements (Kretschmann configuration, Figure 3b, c, d) showed that the preferentially excited SPP mode of the gold–PE film red shifted with increasing dielectric layer thickness, as expected. We did not attach nanoparticles on the surface of the gold films for these measurements; thus, we independently characterized the gold film SPP at this angle of incidence.
In a first set of single NP scattering experiments, we investigated the polarization properties of scattering from isolated NPs as a function of their distance from the gold film. The distance measurements which we report in this paper all refer to the spacing between the bottom surface of the 60 nm NP and the top surface of the film, which is set by the PE layer. All of the effects that we observe occur for distances much smaller than the radius of the NP. Figure 4a and b shows that the well-defined doughnut shape of NPs illuminated using TIR quickly evolves toward a more typical airy pattern when the distance from the metal film increases from 0 to 22.3 nm. This behavior suggests that the horizontal dipole moment, which disappears when the NP is touching the metal surface, quickly recovers when the NP is slightly moved away from the film. The transition from a doughnut shape is even more spectacular when the system is excited by DF illumination, as illustrated in Figure 4c and d for the same nanoparticles. Interestingly, Figure 4a–d shows that at each separation distance, the particle-to-particle aspect and color variations are small for the respective illumination conditions, indicating a very uniform and reproducible effect of the gold film on the NPs and a very uniform polymer layer.
Figure 4.
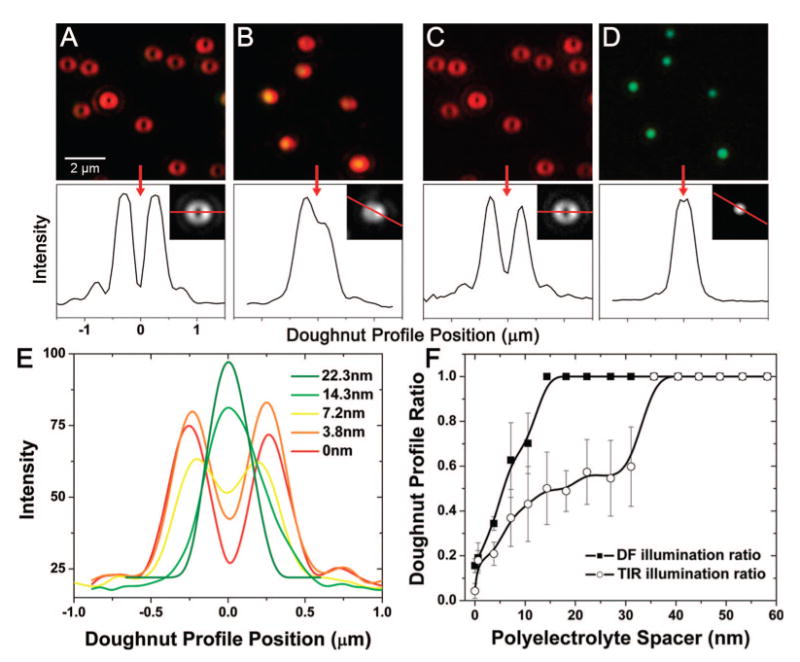
Microscope images (color and grayscale CCD) of scattering from 60 nm gold nanoparticles (NPs) on a 45 nm thick gold film. Line plots through the grayscale CCD images demonstrate the far-field image pattern resulting from polarized dipole radiation. (A) NPs spaced 0 nm from the gold film, excited with TIR white light. (B) NPs spaced 22.3 nm from the gold film by a PE layer and excited with TIR white light. (C) NPs spaced 0 nm from gold film and excited with dark-field white light. (D) NPs spaced 22.3 nm from gold film, excited with dark-field white light. Note: the same NPs are shown in (A) and (C) and in (B) and (D). The doughnut-shaped profile can be used as a metric to track the distance of the NP from the surface of the gold film. (E) Image line plot through single gold NP scatterers at incrementally increasing distances above the gold film. (F) Accumulated analysis of many NP scatterers (10 at each PE layer, standard deviation error bars included), plotting the ratio of the central minimum intensity to the maximum intensity of the PSF as a function of the distance above the surface of the gold film.
To gain more insight into the transition from a vertically polarized dipole to an unpolarized scattering, we have plotted the ratio of the PSF central intensity to the PSF maximum intensity as a function of NP spacing over the gold film. This ratio was obtained from single intensity versus position line plots through the doughnut image, as those shown in Figure 4e. It should be noted that the ratio that we use to quantify the doughnut is only relevant when there exists a minimum intensity at the center of the point spread function. For example, the 14.3 and 22 nm curves of Figure 4e lead to the same ratio of 1, although the PSF is still evolving. A more accurate analysis of the NP–film scattering polarization would require a fit of the two-dimensional doughnut-shaped image to a combination of the calculated PSFs of the horizontal and vertical dipoles.41 Incorporating such a peak-fitting routine into a full-field image process could potentially provide an accurate and rapid topological map of NP–film spacing.
Figure 4f reveals that the polarization transition occurs within the first 15 nm for the DF illumination but that it is much slower for the curve corresponding to the TIR excitation. In other words, the differences between the two curves suggest that the dipoles excited in each case are different. This observation is further supported by the fact that these dipoles do not radiate at the same wavelength; for example, in Figure 4b and d, we observe that the scattering peak of a NP spaced 22.3 nm above the gold film is in the red when illuminated by TIR and in the green when illuminated by DF. As will be discussed in the next paragraphs, these spectral differences occur because the TIR illumination can lead to NP scattering of the SPP mode that is resonantly excited on the gold film, whereas the DF illumination only leads to scattering of the LSP resonance of the NP.
We now analyze the spectral signature of the NP–film system in greater detail. The scattering spectra of NPs at various distances above a gold film have distinctive trends that are dependent on whether the TIR (SPP–LSP scattering) or DF (predominant LSP scattering) illumination is used for excitation. Figure 5 shows a sequence of color images and the scattering spectra from single NPs on the gold film at increasing PE spacer thicknesses. In Figure 5a, the NP–film system is excited by DF illumination, and a blue shift of the scattering is observed as the PE thickness is increased and the particle is spaced at larger distances away from the metal film. This shift corresponds to what is predicted in refs 8, 9, 34, and 35 for the LSP of a nanoparticle as it is moved away from a metal film. The observed spectra are consistent with our interpretation of dark-field coupling directly with the LSP of the NP, which can scatter both into the far-field and, with a multitude of wave vectors, into the continuum of SPP modes that are supported by the gold film. Dark-field illumination does not preferentially excite any of the SPP modes, and therefore, the LSP resonance dominates the scattering spectra.
Figure 5.
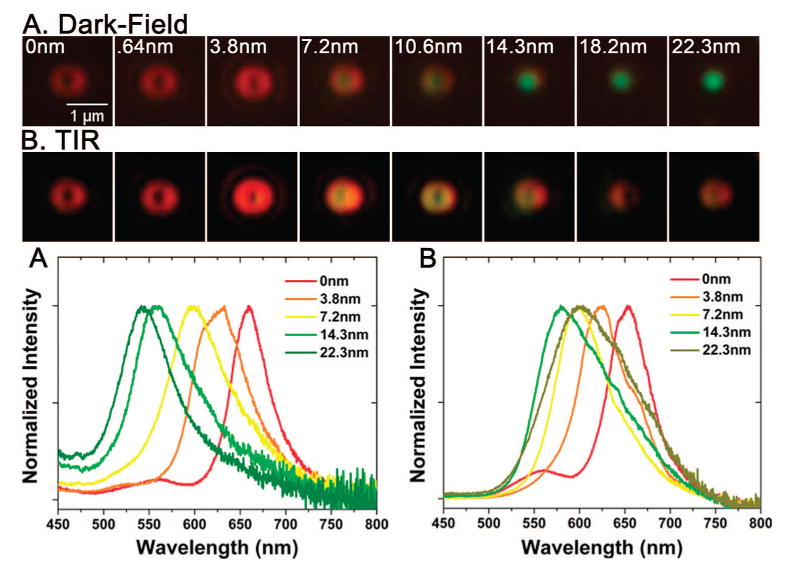
Resonant scattering as NPs are spaced with increasing distance (0-22.3 nm via polyelectrolyte spacers) from the gold film. (A) Single NP scattering (top, color images; bottom, normalized spectra) when NP-film is illuminated with dark-field illumination shows a blue shift with increasing distance of the NP from the gold film, indicative of the LSP resonance. (B) Single NP scattering (from the same NPs as those in (A)) when NP-film is illuminated with an evanescent field from a 45° incidence TIR shows a more complex spectral shift, which reflects the convoluted SPP-LSP resonances. Spectra amplitudes are normalized.
In Figure 5b, the NP–film is illuminated by 45° white light TIR, and in this case, the NP scattering spectrum has the features of a two-peak system. The NP scattering spectra at each distance are influenced by the red-shifting SPP mode, which is resonantly excited on the film as the PE layer increases, and by the blue-shifting LSP of the NP as the NP moves away from the gold film. The shifting spectral trend with distance is clearly more complex than the trend seen with DF illumination. It is important to note, however, that in both illumination schemes, the NP scattering spectra are very sensitive to the spacing between the NP and the film.
A summary of the measured resonances of our NP–film system is presented in Figure 6, which shows the peak positions of the SPR extinction measured from the bulk gold film with no NPs present using TIR illumination (red triangles, data from Figure 2c), the LSP measured from single NP scatterers when excited by DF illumination (blue squares, data from Figure 5a), and the LSP–SPP scattering measured from single NPs when excited by TIR illumination (green circles, data from Figure 5b). The error bars in these plots represent the standard deviation between five distinct NP spectra taken for each condition and demonstrate the uniformity of NP scattering at each dielectric layer. Noteworthy here is the high sensitivity of the NP LSP scattering to spacing between the NP and the film from 0 to 22.3 nm (blue squares). This LSP resonance peak shift is 5.6 nm per 1 nm change in spacing.
Figure 6.
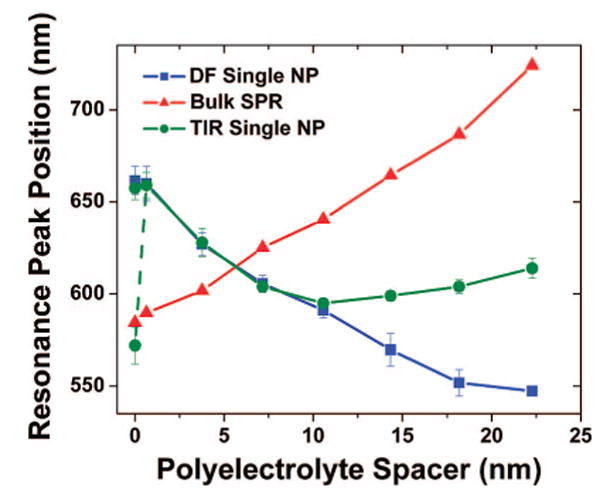
Peak positions of the measured plasmon resonances as a function of the polyelectrolyte (PE) spacer thickness. The SPR extinction (red triangles) is collected from the gold film measurements using 45° TIR illumination (as shown in Figure 2) and red shifts with increasing PE thickness. Dark-field (DF)-illuminated single NPs (blue squares, five at each layer) exhibit a uniform blue shift (LSP scattering), and 45° incidence TIR evanescent field-illuminated single NP–film scatterers (green circles, five at each layer) exhibit a resonance that tracks the LSP trend from a 0 to ∼10 nm spacer distance and then the SPP resonance (SPR, which exists at this illumination angle) for distances >10 nm. The single NP scatterers include standard deviation bars to indicate NP-to-NP spectral variation. The dashed line and additional data point for the NP–film scatterers at 0 nm indicates a second peak existing in the spectrum at that spacer distance.
Figure 6 shows clearly that the light scattered from the NP when using specific-angle TIR illumination can be dominated by either the LSP resonance or the SPP modes resonantly excited on the gold film (green circles). While the coupling of the SPR to the NP scattering is complicated by the fact that both of the resonances are shifting as the PE spacer layer increases, we can qualitatively describe the NP scattering behavior as a consequence of the unique evanescent fields present on the surface of the gold film that consist of both broad-band white light and SPP waves. When the NP is very close to the film (at d < 10 nm), it is apparent that the LSP of the nanoparticle is predominantly scattered because the spectral shift matches the trend of the DF-illuminated NP–film (blue squares). This result suggests that the spectrum of the excitation field present in this regime is comparable to the DF white light illumination and that the SPP mode also excited by the evanescent field does not efficiently couple to the NP scatterer principally as a result of the mismatched resonances. However, as the NP is spaced away from the surface, the evanescent fields contribute less and less to the NP scattering because their amplitude decays rapidly in the air region. In contrast, the SPP waves decay more slowly than the evanescent waves. The SPP mode effectively becomes the only wave that is measurably scattered by the NP, which explains why the scattering data exhibit an upturn at d > 10 nm and then a red shift similar to the curve obtained from the resonant SPP extinction spectra (red triangles). There remains a wavelength offset between the measured resonant SPP extinction (red triangles) and the scattering of the NP in TIR illumination at d > 10 nm (green circles). We attribute this shift to a combination of several effects. Primarily, there is an inherent spectral shift between extinction and scattering measurements. In addition, the scattering properties of a NP strongly vary with the wavelength and incident field distribution; therefore, the SPR spectral shape as measured from our extinction experiments is inevitably distorted when it is measured through the scattering of the NP.
The extreme sensitivity and remarkable uniformity of the NP–film system suggests its potential use for biosensing or nanometrology, such as for the detection of conformational changes in biomacromolecules or binding events between biological molecules. We consider many biosensing applications utilizing the interaction between coupled nanoparticles, such as the plasmonic “molecular ruler” reported in a recent publication by Sonnichsen et al.,44 as adaptable to the NP–film system. Because such applications would likely require aqueous environments, we characterized the NP–film interactions for the case where water rather than air was the surrounding medium.
A dove prism was used to direct incident illumination at 73° from the normal of the glass slide, an angle greater than the critical angle of the glass-water interface. This ensures TIR at the glass-water interface, which results in an evanescent field capable of exciting both a single SPP mode of the gold film and the resonant LSP of the gold NPs coupled to the film. Imaging of the NP scatterers in solution was accomplished by placing 10 μL of water and a coverslip on the sample and approaching with an oil immersion objective (Nikon 100×, 1.3 NA). The resonant scatterers appear as bright, doughnut-shaped point sources in a very dark background (Figure 7a), similar to the way they appeared in our experiments in air. Additional airy rings were visible when imaging in water, which is a result of the increased aberrations inherent to imaging through the dielectric mismatch at the interface between the glass coverslip and the liquid. In this configuration, we were not using a DF objective, and thus, specific excitation and detection of only the LSP, as done in previous experiments, was not possible. However, the insights provided by the previous DF illumination measurements can be applied to the observations made of the NP-film-coupled LSP-SPP resonance excited by the specific-angle TIR illumination in the aqueous environment.
Figure 7.
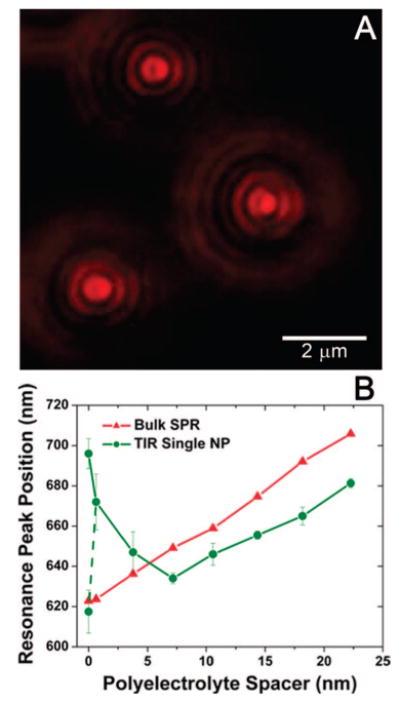
Plasmon resonant NPs spaced above a gold film with the increasing polyelectrolyte (PE) spacer distance measured in water. (A) Color image of the NP scatterers spaced 3.8 nm from the gold film. (B) Peak positions of the resonant SPP extinction (SPR) using 73° TIR illumination for the gold film with no NPs present (red triangles) and for the single NP scatterers under the same illumination conditions (green circles) at various PE spacer thicknesses. The dashed line and additional data point for the NP scatterers at 0 nm indicates a second peak arising in the resonance spectrum.
Optical fiber collection of the light reflected from the 73° TIR white light illumination was used to measure the extinction of the gold films with PE layers in water. Because the PE layers have a higher refractive index than water (1.58 compared to 1.33 for water, Supporting Information), increasing the number of PE layers causes a measurable red shift of the SPR mode that we excite at this angle of illumination (Figure 7b, red triangles). Similar to the behavior observed for the NP-film in air using TIR illumination, the NP scatters either predominantly the LSP or the resonant SPP mode of the gold film, depending on the spacer distance. The peak position of the far-field scattering spectra of the NP-film blue shifts from spacer distances of 0 to 7.2 nm and then red shifts as the distance between the NP and gold film further increases (Figure 7b, green circles). Note that the PE layer thicknesses reported here and in Figure 7 are underestimated since they were characterized in a dry state via ellipsometry. It has been shown before that PE layers swell as they are rehydrated with water. See Supporting Information for discussion as well as single NP scattering spectra from NPs at each spacer height. Once again, we plot the peak positions from five single NP scatterers at each PE layer and include the standard deviation indicating NP-to-NP variation. The red-shifted NP LSP resonance at d < 7 nm is excited by the white light evanescent field and does not effectively scatter the gold film SPP mode to the far-field. However, as the NP is moved away from the surface, the LSP blue shifts, the SPP mode also excited by the evanescent field red shifts, and the evanescent field intensity rapidly decays, resulting in a transition at ∼7 nm distance (for aqueous) from the LSP-dominated far-field scattering of the NP to SPP-dominated far-field scattering. We observe a similar offset between the spectral peak positions of the NP SPP-dominated scattering for d > 7 nm (red triangles) and the gold film SPR extinction measurements (green circles) to the offset observed in air (Figure 6). For the range between 0 and ∼7 nm where the NP resonance is LSP-dominated, the NP scattering has a high sensitivity to changes in spacer distance, with a spectral peak shift of 7.8 nm for every 1 nm change in distance. In this range, we might expect to measure spectral shifts of the LSP resonance caused by conformational changes to biological molecules acting as linkers between the NP and the film. For separation greater than ∼7 nm, the spectral shift reflects only the red-shifting SPR of the gold film caused by the increasing dielectric thickness. In the hypothetical case of a small number of molecules modulating the spacing of a single NP from the film, the measured SPR of the gold film would not shift, effectively flattening the green curve in Figure 7 for spacing >7 nm, implying an insensitivity of the NP scattering to separation from the film in this regime. In such a case, where measuring the distance between the NP and the film in an aqueous environment (TIR illumination) is the goal, utilizing a metal film or specific angle of illumination which does not result in a SPP mode in the same wavelength range as the nanoparticle LSP would reduce the contribution of the SPP to the NP scattering and perhaps increase the NP-film spacing range over which the LSP can be measured. In future work, we intend to measure NP-film systems consisting of other combinations of metals, NP shapes, and angles of TIR excitation, allowing us to specifically tailor the positions of the LSP resonance and the excited SPP modes, which may improve the suitability of the aqueous NP-film system for specific applications.
We have presented a detailed experimental characterization of the distance-dependent resonant scattering of 60 nm gold nanoparticles over a 45 nm gold film which corroborates former studies of NP-film interaction and unveils additional interesting scattering polarization phenomena. We have shown that when the NP is in close proximity to the metal film, damping of the horizontal polarization dipole results in vertically polarized NP scattering and a doughnut-shaped far-field image. In addition, the plasmonic resonance of the NP scattering is significantly red shifted. We have also shown that both of these effects are responsive to nanometer-scale changes in the distance between the NP and the gold film. Furthermore, the localized surface plasmon (LSP) of the NP and the surface plasmon polariton (SPP) mode excited on the gold film by TIR illumination which couples to the NP have been independently characterized at incrementally increasing NP-film separation distances in water and in air. The spectral shift of the LSP driven by the proximity of the NP to the gold film is on the order of 6 nm for every 1 nm change in distance. Assuming that the plasmon resonance peaks can be resolved at 1 nm wavelength separation; this sensitivity should enable measurement of Ångstrom-scale distances, which are comparable to molecular bond lengths.
Combining simultaneous characterization of the distance-dependent NP scattering polarization (doughnut-shaped image) with plasmonic spectra analysis of the NP-film system in an aqueous environment could lead to the development of an ultrasensitive biometric detector for sensing subtle environmental changes, such as temperature, pH, or molecular conformation that induce changes in the separation between plasmonic NPs and a metal film.
Supplementary Material
Acknowledgments
A.C. and S.Z. acknowledge support by the NSF through Grant CMS-0609265 (NIRT). We thank Olivier Martin for helpful discussions and F. Nelson Nunalee for contributions to experiments leading up to those presented here.
Footnotes
Supporting Information Available: A detailed description of the experimental protocol for sample preparation and characterization. Additional single particle spectra and a plot of the amplitude of the NP scattering with increasing distance above the gold film. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Tokareva I, Minko S, Fendler JH, Hutter E. J Am Chem Soc. 2004;126:15950–15951. doi: 10.1021/ja044575y. [DOI] [PubMed] [Google Scholar]
- 2.He L, Smith EA, Natan MJ, Keating CD. J Phys Chem B. 2004;108:10973–10980. [Google Scholar]
- 3.Papanikolaou N. Phys Rev B. 2007;75:235426. [Google Scholar]
- 4.Nordlander P, Le F. Appl Phys B. 2006;84:35–41. [Google Scholar]
- 5.Le F, Lwin NZ, Steele JM, Kall M, Halas NJ, Nordlander P. Nano Lett. 2005;5:2009–2013. doi: 10.1021/nl0515100. [DOI] [PubMed] [Google Scholar]
- 6.Nedyalkov N, Sakai T, Miyanishi T, Obara M. Appl Phys Lett. 2007;90:123106. [Google Scholar]
- 7.Nedyalkov N, Sakai T, Miyanishi T, Obara M. J Phys D. 2006;39:5037–5042. [Google Scholar]
- 8.Leveque G, Martin OJF. Opt Express. 2006;14:9971–9981. doi: 10.1364/oe.14.009971. [DOI] [PubMed] [Google Scholar]
- 9.Leveque G, Martin OJF. Opt Lett. 2006;31:2750–2752. doi: 10.1364/ol.31.002750. [DOI] [PubMed] [Google Scholar]
- 10.Eah SK, Jaeger HM, Scherer NF, Wiederrecht GP, Lin XM. J Phys Chem B. 2005;109:11858–11861. doi: 10.1021/jp0511395. [DOI] [PubMed] [Google Scholar]
- 11.Orendorff CJ, Sau TK, Murphy CJ. Small. 2006;2:636–639. doi: 10.1002/smll.200500299. [DOI] [PubMed] [Google Scholar]
- 12.Dahmen C, von Plessen G. Aust J Chem. 2007;60:447–456. [Google Scholar]
- 13.Sonnichsen C, Franzl T, Wilk T, von Plessen G, Feldmann J. New J Phys. 2002;4:93. doi: 10.1103/PhysRevLett.88.077402. [DOI] [PubMed] [Google Scholar]
- 14.Wang X, Zhang ZY, Hartland GV. J Phys Chem B. 2005;109:20324–20330. doi: 10.1021/jp054233u. [DOI] [PubMed] [Google Scholar]
- 15.Nehl CL, Liao HW, Hafner JH. Nano Lett. 2006;6:683–688. doi: 10.1021/nl052409y. [DOI] [PubMed] [Google Scholar]
- 16.Murphy CJ, Gole AM, Hunyadi SE, Orendorff CJ. Inorg Chem. 2006;45:7544–7554. doi: 10.1021/ic0519382. [DOI] [PubMed] [Google Scholar]
- 17.Mock JJ, Barbic M, Smith DR, Schultz DA, Schultz S. J Chem Phys. 2002;116:6755–6759. [Google Scholar]
- 18.Gao Y, et al. J Cryst Growth. 2005;276:606–612. [Google Scholar]
- 19.Sherry LJ, Chang SH, Schatz GC, Van Duyne RP, Wiley BJ, Xia YN. Nano Lett. 2005;5:2034–2038. doi: 10.1021/nl0515753. [DOI] [PubMed] [Google Scholar]
- 20.Kottmann JP, Martin OJF, Smith DR, Schultz S. Phys Rev B. 2001;64:235402/235401–235402/235410. [Google Scholar]
- 21.Murphy CJ, San TK, Gole AM, Orendorff CJ, Gao JX, Gao L, Hunyadi SE, Li T. J Phys Chem B. 2005;109:13857–13870. doi: 10.1021/jp0516846. [DOI] [PubMed] [Google Scholar]
- 22.Jin RC, Cao YW, Mirkin CA, Kelly KL, Schatz GC, Zheng JG. Science. 2001;294:1901–1903. doi: 10.1126/science.1066541. [DOI] [PubMed] [Google Scholar]
- 23.Mock JJ, Smith DR, Schultz S. Nano Lett. 2003;3:485–491. [Google Scholar]
- 24.Miller MM, Lazarides AA. J Phys Chem B. 2005;109:21556–21565. doi: 10.1021/jp054227y. [DOI] [PubMed] [Google Scholar]
- 25.McFarland AD, Van Duyne RP. Nano Lett. 2003;3:1057–1062. [Google Scholar]
- 26.Raschke G, et al. Nano Lett. 2004;4:1853–1857. [Google Scholar]
- 27.Alivisatos P. Nat Biotechnol. 2004;22:47–52. doi: 10.1038/nbt927. [DOI] [PubMed] [Google Scholar]
- 28.Khlebtsov NG, Trachuk LA, Mel'nikov AG. Opt Spectrosc. 2005;98:77–83. [Google Scholar]
- 29.Marinakos SM, Chen SH, Chilkoti A. Anal Chem. 2007;79:5278–5283. doi: 10.1021/ac0706527. [DOI] [PubMed] [Google Scholar]
- 30.Nath N, Chilkoti A. Anal Chem. 2002;74:504–509. doi: 10.1021/ac015657x. [DOI] [PubMed] [Google Scholar]
- 31.Nusz GJ, Marinakos SM, Curry AC, Dahlin A, Hoeoek F, Wax A, Chilkoti A. Anal Chem. 2008;80:984–989. doi: 10.1021/ac7017348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schultz S, Smith DR, Mock JJ, Schultz DA. Proc Natl Acad. 2000;97:996–1001. doi: 10.1073/pnas.97.3.996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Raschke G, Kowarik S, Franzl T, Sonnichsen C, Klar TA, Feldmann J, Nichtl A, Kurzinger K. Nano Lett. 2003;3:935–938. [Google Scholar]
- 34.Holland WR, Hall DG. Phys Rev Lett. 1984;52:1041. doi: 10.1103/PhysRevLett.56.2838. [DOI] [PubMed] [Google Scholar]
- 35.Chance RR, Prock A, Silbey R. Phys Rev A. 1975;12:1448. [Google Scholar]
- 36.Bartko AP, Dickson RM. J Phys Chem B. 1999;103:11237–11241. [Google Scholar]
- 37.Bohmer M, Enderlein J. J Opt Soc Am B. 2003;20:554–559. [Google Scholar]
- 38.Patra D, Gregor I, Enderlein J. J Phys Chem A. 2004;108:6836–6841. [Google Scholar]
- 39.Bartko AP, Dickson RM. J Phys Chem B. 1999;103:3053–3056. [Google Scholar]
- 40.Moerland RJ, Taminiau TH, Novotny L, van Hulst NF, Kuipers L. Nano Lett. 2008;9:606. doi: 10.1021/nl073026y. [DOI] [PubMed] [Google Scholar]
- 41.Novotny L, Hecht B. Principles of Nano-Optics. Cambridge University Press; Cambridge, U.K.: 2006. [Google Scholar]
- 42.Arys X, Jonas AM, Laschewsky A, Legras R, Mallwitz F. In: Supramolecular Polymers. Ciferri A, editor. Taylor & Francis; Boca Raton, FL: 2005. pp. 651–710. [Google Scholar]
- 43.Buscher K, Graf K, Ahrens H, Helm CA. Langmuir. 2002;18:3585–3591. [Google Scholar]
- 44.Sonnichsen C, Reinhard BM, Liphardt J, Alivisatos AP. Nat Biotechnol. 2005;23:741–745. doi: 10.1038/nbt1100. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.