

Abstract
The autosomal dominant hyper-IgE syndrome (HIES, ‘Job's syndrome’) is characterized by recurrent and often severe pulmonary infections, pneumatoceles, eczema, staphylococcal abscesses, mucocutaneous candidiasis, and abnormalities of bone and connective tissue1,2. Mutations presumed to underlie HIES have recently been identified in stat3, the gene encoding STAT3 (signal transducer and activator of transcription 3) (refs 3, 4). Although impaired production of interferon-γ and tumour-necrosis factor by T cells5, diminished memory T-cell populations, decreased delayed-type-hypersensitivity responses and decreased in vitro lymphoproliferation in response to specific antigens6 have variably been described, specific immunological abnormalities that can explain the unique susceptibility to particular infections seen in HIES have not yet been defined. Here we show that interleukin (IL)-17 production by T cells is absent in HIES individuals. We observed that ex vivo T cells from subjects with HIES failed to produce IL-17, but not IL-2, tumour-necrosis factor or interferon-γ, on mitogenic stimulation with staphylococcal enterotoxin B or on antigenic stimulation with Candida albicans or streptokinase. Purified naive T cells were unable to differentiate into IL-17-producing (TH17) T helper cells in vitro and had lower expression of retinoid-related orphan receptor (ROR)-γt, which is consistent with a crucial role for STAT3 signalling in the generation of TH17 cells7–14. TH17 cells have emerged as an important subset of helper T cells15 that are believed to be critical in the clearance of fungal16 and extracellular bacterial17 infections. Thus, our data suggest that the inability to produce TH17 cells is a mechanism underlying the susceptibility to the recurrent infections commonly seen in HIES.
We studied three groups of subjects: healthy controls with no apparent immunological defects, HIES individuals with defined mutations in stat3, and individuals (termed ‘HIES-like’) with some combination of elevated IgE, atopic dermatitis, skeletal abnormalities and susceptibility to infection, but without recurrent staphylococcal abscesses or candidiasis or stat3 mutations (Table 1).
Table 1. Summary of patient characteristics.
| Patient ID | Age (yr) | Sex | stat3 mutation | HIES clinical score | Serum IgE* (IU ml−1) | Skeletal abnormalities | Recurrent candidiasis/staphylococcal abscess or lung infection | Atopic dermatitis |
|---|---|---|---|---|---|---|---|---|
| 1† (J112) | 6 | M | 1970 A→G (SH2) | 31 | 4,190 | + | + | + |
| 2 (J088) | 7 | M | 1145 G→T (DNA) | 70 | 18,000 | + | + | + |
| 3 (J083) | 10 | F | 1909 G→A (SH2) | 78 | 5,070 | + | + | + |
| 4 (J005) | 13 | F | 1144 C→T (DNA) | 60 | 18,600 | + | + | + |
| 5 (J100) | 22 | F | 1909 G→A (SH2) | 76 | 1,020 | + | + | + |
| 6 (J017) | 23 | M | 1145 G→A (DNA) | 82 | 20,500 | + | + | + |
| 7‡ (J002) | 24 | F | 1865 C→T (SH2) | 85 | 8,550 | + | + | + |
| 8† (J112) | 51 | M | 1970 A→G (SH2) | 79 | 6,380 | + | + | + |
| 9 (J054) | 40 | F | 1393 T→G (DNA) | 78 | 239 (47,338) | + | + | + |
| 10‡ (J002) | 56 | M | 1865 C→T (SH2) | 90 | 26 (2,392) | + | + | + |
| 11 | 48 | M | 1268 G→A (DNA) | 65 | 1,340 (20,700) | + | + | + |
| 12 | 37 | F | 1909 G→A | 94 | 495 (25,058) | + | + | + |
| 13 (J015) | 36 | F | 1861 T→G | 96 | + | + | + | |
| 14§ | 16 | M | None | 27 | 11,100 | − | −(other recurrent infections) | − |
| 15§ | 13 | F | None | 30 | 14,000 | − | −(other recurrent infections) | − |
| 16§ | 18 | F | None | n.a. | 136 | − | −(other recurrent infections) | − |
| 17 | 15 | M | None | 49 | 6,880 | + | − | + |
| 18 | 15 | M | None | 55 | 160 (69,280) | + | − | + |
| 19 | 4 | F | None | n.d. | >30,000 | + | − | + |
n.a., not available; n.d., not determined.
Peak values in parentheses.
Parent–child pairs.
Siblings with similar phenotypes.
We observed that IL-17-producing T cells were barely detectable among peripheral blood mononuclear cells (PBMCs) from subjects with HIES on stimulation with staphylococcal enterotoxin B (SEB) (Fig. 1a). The frequency of SEB-induced interferon (IFN)-γ-producing CD4 T cells from PBMCs of subjects with HIES was similar to that of healthy controls, whereas the frequency of cells producing IL-2 and/or tumour-necrosis factor (TNF) was slightly reduced. Fewer of the IFN-γ-producing CD4 T cells from subjects with HIES also produced TNF and/or IL-2 compared with healthy controls (Fig. 1b). IL-17-producing T cells were present in PBMCs from the HIES-like cohort with no mutations in stat3, suggesting that the lack of IL-17 production in HIES has a critical function in susceptibility to the specific infections seen in HIES, and also that elevated serum IgE, atopic dermatitis or low frequency of memory T cells (data not shown) are not independently associated with severe defects in the TH17 axis (Fig. 1a). The production of IL-21 and IL-22, which have been described as both TH1 and TH17 cytokines9,10,18, was not significantly lower in subjects with HIES than in healthy controls. Additionally, among subjects with HIES, IL-21 was derived predominantly from TH1 cells, suggesting that the induction of IL-21 and IL-22 is not uniquely dependent on normal STAT3 signalling in humans (Supplementary Fig. 1a–c). Although the HIES-like subjects had significantly fewer IFN-γ-producing cells than healthy controls did (Fig. 1a), they are not susceptible to the specific infections seen in HIES, namely recurrent candidiasis and staphylococcal abscesses.
Figure 1. Lack of IL-17 production in SEB-stimulated PBMCs from HIES patients.

a, PBMCs from cohorts of HIES patients with stat3 mutations (red circles), HIES-like patients (blue circles) and healthy individuals (green circles) were stimulated overnight with SEB in the presence of brefeldin A and stained as described in Methods. The frequency of memory CD4 T cells (gated as described in Methods) that produced IFN-γ, IL-2, TNF or IL-17 was then assessed. b, Memory CD4 T cells that produced IFN-γ were analysed for their ability to co-produce IL-2 and TNF by using SPICE (Simplified Presentation of Incredibly Complex Evaluations) as described in Methods. Statistical significance was determined with the Mann–Whitney test. Significant P values are shown in red, and median values with horizontal bars.
We next tested whether TH17 cells could be generated from CD4 T cells of subjects with HIES. Naive CD4 T cells (CD4+CD8−CD45RO−CD11adullCD27+CD31+) from healthy controls and subjects with HIES were sorted by flow cytometry and then cultured in TH17 conditions7,10 with anti-CD2/CD3/CD28 beads in the presence of anti-IFN-γ alone, or with the addition of either IL-6 and IL-1β, or IL-23 and IL-1β, for 12 days. TH17 cell differentiation was observed in all CD4 T-cell cultures from healthy controls. However, CD4 T cells from the majority of subjects with HIES showed variable survival when cultured with either IL-6 or IL-23 in the presence of IL-1β, but not when cultured without additional cytokines. Surviving cells did not make any IL-17, but IFN-γ-producing CD4 T cells were present, although at a lower frequency than among CD4 T cells from controls (Fig. 2a–e). ROR-γt mRNA expression in HIES T cells after 48 h under TH17-polarizing conditions was significantly lower in T cells from subjects with HIES than in healthy controls (Fig. 2f). Introduction of mutant stat3 from subjects with HIES, with either SH2 domain or DNA-binding mutations, into normal human naive CD4 T cells also resulted in a decrease in ROR-γt mRNA expression under the same TH17-polarizing conditions (25% decrease for the SH2 domain mutation, and 65% for the DNA-binding mutation), showing that mutant STAT3 suppresses ROR-γt expression in a dominant-negative fashion (Supplementary Fig. 1d). Given the variable cell survival in the TH17 differentiation experiments, we directly tested the effects ofIL-1β onT-cell survival by culturing PBMCs from subjects with HIES and from healthy individuals overnight, and measuring subsequent IL-1β-induced apoptosis by staining with 7-amino-actinomycin D (7-AAD) and 3,3′-dihexyloxacarbocyanine iodide (DiOC6). Apoptosis was significantly increased among CD4 T cells from subjects with HIES in comparison with healthy individuals or HIES-like individuals in the presence of IL-1β (Supplementary Fig. 2a). We observed no differences between controls and subjects with HIES in the percentage of FoxP3+ cells in sorted naive CD4 T cells or among total CD4 T cells (data not shown). Furthermore, IL-4 production was not enhanced in stimulated purified naive CD4 T cells from subjects with HIES; even after one round of priming under TH2-polarizing conditions, IL-4 production was undetectable in either subjects with HIES or controls. In addition, one round of priming in TH1-polarizing conditions or conditions with no added cytokines led to similar frequencies of IFN-γ-producing cells in subjects with HIES and in controls (Supplementary Fig. 3).
Figure 2. Failure of TH17 generation from naive cells of patients with HIES.
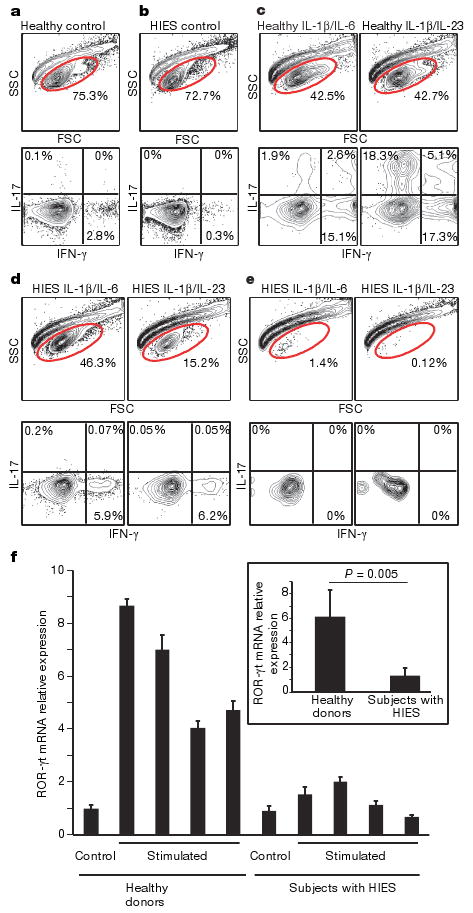
A total of 50,000 naive T cells from healthy controls and patients with HIES were stimulated with anti-CD2/CD3/CD28 microbeads and the indicated cytokines as described in Methods. a–e, Dot plots of intracellular cytokine staining from healthy control (a) and HIES (b) cells stimulated without any cytokines, and from control subjects (c) and subjects with HIES (d, e) stimulated in the presence of IL-6 and IL-1β, or with IL-23 and IL-1β. e, Representative forward and side scatter plots of HIES cells after stimulation for 12 days show the poor recovery after stimulation with either IL-6 and IL-1β, or IL-23 and IL-1β. d, The one subject sample that had good cellular recovery. f, Naive CD4 T cells from four healthy donors and four subjects with HIES were freshly lysed or stimulated with anti-CD3 and anti-CD28 in the presence of IL-23 for 48h. ROR-γt mRNA expression was detected by quantitative RT–PCR. Values shown are relative expression levels of triplicate samples (means and s.d.). Inset: average expression levels for the four healthy donors compared with those for the four subjects with HIES. Statistical significance was determined with a two-tailed unpaired Student t-test.
The failure of HIES T cells to differentiate into TH17 cells is likely to be due to mutated stat3; however, aberrant STAT3 DNA binding in primary T cells from HIES patients has not previously been reported. We therefore performed an electrophoretic mobility-shift assay with purified HIES T cells treated with the potent stat3-activating and TH17-promoting cytokine IL-21. We found that, even in the presence of normal levels of phosphorylated STAT3, binding of STAT3 to DNA was undetectable, whereas robust binding was found in similarly treated purified T cells from healthy donors (Supplementary Fig. 4). Therefore, although IL-21 production seems normal in HIES, its signalling is not.
Although cells capable of producing IL-2, TNF and IFN-γ were present in the memory CD4 T-cell population from subjects with HIES (Fig. 1), it remained possible that the lack of TH17 cells in the affected individuals was due to a more global failure of T-cell maturation and differentiation. Indeed, consistent with previous studies6 was our observation that the frequency of memory CD4 and CD8 T cells, defined by the expression of CD45RO, was significantly decreased in subjects with HIES, and in many of the HIES-like subjects (data not shown). PBMCs were stimulated with streptokinase (a streptococcal antigen), Candida albicans (CA) or Staphylococcus aureus (SA), all antigens from microorganisms commonly associated with the infections that characterize HIES. The percentage of memory CD4 T cells producing IL-2, TNF and IFN-γ in response to CA or streptokinase was not decreased in HIES-like subjects or in subjects with HIES in comparison with healthy controls. However, HIES T cells showed little or no antigen-specific IL-17 production (Fig. 3a–c), whereas both healthy controls and subjects with HIES were able to mount antigen-specific IL-17 responses. There was significant lymphocyte cell death in the cultures from patients with HIES that had been incubated with SA (Supplementary Fig. 2b). Although SEB stimulation did not cause cell death, other staphylococcal T-cell mitogens or innate immune ligands might be responsible. In a previous study19 a microarray analysis of purified T cells from subjects with HIES stimulated with SA showed diminished IL-17 mRNA expression, providing further evidence that pathogen-induced IL-17 production is impaired in subjects with HIES.
Figure 3. Lack of IL-17 production despite antigen-specific IFN-γ, IL-2 and TNF production from subjects with HIES.

PBMCs from cohorts of healthy subjects (green circles) HIES-like subjects (blue circles) and individuals with HIES (red circles) were stimulated overnight with Candida albicans (a), streptococcal kinase (b) or Staphylococcus aureus (c) in the presence of brefeldin A, then stained as described in Methods. The production of IFN-γ, IL-2, TNF or IL-17 by memory CD4 T cells (gated as described in Methods) in response to individual antigen stimulation is shown. Statistical significance was determined with the Mann–Whitney test. Significant P values are shown in red, and median values with horizontal bars.
HIES is a multifaceted disease, a major component of which is immunodeficiency. Herein we describe a potential mechanism for the susceptibility to particular fungal and extracellular bacterial infections, namely the inability to generate TH17 cells. Mice with a targeted mutation in IL-17 are susceptible to C. albicans and the extracellular bacterium Klebsiella pneumoniae16,17, presumably as a result of abnormal antimicrobial peptide expression20 and neutrophil recruitment21, mobilization and influx16. This suggests that it is the absence of TH17 cells themselves, and not other STAT3-dependent mechanisms, that render subjects with HIES susceptible to these infections. The cell death we have observed in vitro in response to S. aureus may be responsible for the lack of cytokine responses to SA ex vivo (Fig. 3c), and this may have implications for the control of S. aureus infection in vivo. Further examination of mouse models of the dominant-negative HIES stat3 mutations should help test the role of IL-17 in anti-staphylococcal responses more directly. IL-1β, which also induced cell death in subjects with HIES, is not only crucial for the generation of TH17 cells, it is also critical to the clearance of S. aureus in mice through neutrophil recruitment22.
Lack of IL-17 may also function in the eosinophilia of HIES, because neutralization of IL-17 on allergenic challenge of sensitized mice leads to increased eosinophilic disease23, whereas the elevated IgE levels in subjects with HIES are likely to have a complex basis. IL-21, which can signal through STAT1 and STAT5, as well as STAT3, could be involved, because one study of IL-21R−/− mice reported elevated IgE levels24, although another did not25. Because IL-21 is critical for immunoglobulin production26, and because non-IgE immunoglobulin levels are normal in HIES, one could speculate that the normal IL-21 production we have observed in the face of aberrant, but not absent, IL-21 signalling might lead to the preferential expansion of IgE-producing B-cells, the phenotype seen in HIES27. Thus, although the inability to generate TH17 cells may explain the susceptibility to the bacterial and fungal infections that characterize HIES, it may not be the underlying mechanism for the other features of the syndrome.
Thus, we have demonstrated a human genetic disease that results in the failure to generate TH17 cells in vitro and their absence in vivo. Studies of individuals with HIES may help to explain the role of TH17 cells in infection and in other forms of dysregulated immunity, and may help in understanding the role of STAT3 in T-helper differentiation in humans. Furthermore, our findings suggest that therapeutic approaches to HIES may focus on therapies aimed at improving IL-17 production28.
Methods Summary
Both study and control subjects were evaluated under an Institutional Review Board-approved natural history of HIES protocol at the Clinical Center at the National Institutes of Health (NIH). Stat3 sequences were confirmed in all patients with HIES and HIES-like patients. PBMCs were prepared by Ficoll centrifugation of venous blood from healthy volunteers and patients. Stimulations with mitogens and antigens and intracellular cytokine staining were performed overnight as described29. In vitro cytokine differentiation was performed as described with some modifications10, and ROR-γt was induced with IL-23 and measured by quantitative reverse-transcriptase-mediated polymerase chain reaction (RT–PCR).
Methods
Subjects
Both study and control subjects were evaluated under an Institutional Review Board-approved natural history of HIES protocol at the Clinical Center at the National Institutes of Health (NIH). Study subjects were diagnosed with HIES by experienced clinicians, assisted by a diagnostic scoring system comprising immunological and non-immunological features, in which a score of more than 40 is consistent with HIES, a score of 20–40 is indeterminate, and a score of less than 20 is not suggestive of HIES30. The diagnosis was confirmed by the identification of stat3 mutations. HIES-like subjects were diagnosed with an illness typically lacking one of the major characteristic features of HIES (abscesses, high serum IgE, recurrent staphylococcal pulmonary infections, pneumatoceles or candidiasis, eczema, and/or connective/skeletal tissue abnormalities) and in whom no stat3 mutations were identified. All of the patients were in their usual state of health at the time that blood was drawn. PBMCs were prepared from venous blood by density-gradient centrifugation.
Flow cytometry
Eighteen-parameter flow cytometric analysis was performed with a FACSAria (Becton Dickinson). CD11a or IFN-γ (BD) fluorescein isothiocyanate (FITC), IL-17 (eBioscience) or CD31 (BD) phycoerythrin (PE), TNF (BD) Cy7PE, CD4 (Invitrogen, Carlsbad, CA) Cy5.5PE, IL-2 (BD) or IL-21 (eBioscience) allophycocyanin (APC), CD3 (BD) Cy7APC, CD45RO (Beckman Coulter) Texas Red PE (TRPE), violet amine reactive dye (Vivid) (Invitrogen), and CD8 (Invitrogen) Quantum-dot 705 (QD705) were used as the fluorophores.At least 300,000 live lymphocytes were collected. The list-mode data files were analysed with FlowJo (Tree Star). Functional capacity was determined after boolean gating and subsequent analysis was performed with Simplified Presentation of Incredibly Complex Evaluations (SPICE, version 2.9; a gift from M. Roederer). All values used for analysing proportionate representation of responses are background-subtracted. Naive T cells for priming conditions were sorted as Vivid−CD4+CD8−CD27+CD45RO−CD11adullCD31+. Memory CD4 Tcells were gated as CD3+Vivid−CD4+CD8−CD27+CD45RO+. Sort purities were consistently greater than 99%.
Intracellular cytokine T-cell assay
Stimulation was performed on fresh or frozen lymphocytes as described elsewhere29. Freshly isolated or freshly thawed lymphocytes were resuspended at 106ml−1 in RPMI medium supplemented with 10% heat-inactivated fetal calf serum (R10; Invitrogen) and with 1μg ml−1 anti-CD28 andanti-CD49d(BD) antibodies. Staphylococcus enterotoxin B (Sigma) was used to stimulate T cells mitogenically. Streptokinase (Varidase, Wyeth), Candida albicans (Greer), and heat-killed Staphylococcusaureus (Calbiochem) were used to stimulate antigen-specific cells. All stimulations were performed in the presence of brefeldin A (1μg ml−1; Sigma) for16 h at 37 °C. All cells were surface-stained for phenotypic markers of interest and stained intracellularly for cytokines (intracellular cytokine staining). The frequencies of memory CD4 Tcells (CD3+Vivid−CD4+CD8−CD45RO+CD27+) responding to stimuli are presented.
In vitro differentiation of naive CD4 T cells
A total of 50,000 sorted, Vivid−CD3+CD4+CD31+CD27+CD11adullCD45RO− naive T cells from patients and controls were placed in 96-well round-bottomed plates with 25,000 CD2/CD3/CD28 human T-cell stimulation beads (Miltenyi Biotec) in the presence of 10μg ml−1 anti-IFN-γ alone or with either 20 ngml−1 IL-6 and 20 ngml−1 IL-2, or 20ngml−1 IL-23 and 20ng ml−1 IL-1β. Cells were then cultured for four or five days, after which the culture was split and cells were placed in 20U ml−1 IL-2 for a further seven days. Cells were then stimulated with 12-O-tetradecanoylphorbol-13-acetate and ionomycin in the presence of brefeldin A for 4 h, fixed with Fix/Perm buffer (BD), permeabilized with saponin buffer, and stained for intracellular cytokines.
ROR-γt mRNA quantitative RT–PCR
NaiveCD4+CD45RA+CD45RO−T cells were purified from donor peripheral blood by magnetic cell sorting with a human naive CD4+ T-cell isolation kit (Miltenyi Biotec). Purified cells were freshly lysed in RLT lysis buffer (Qiagen) or activated with plate-bound anti-CD3, soluble anti-CD28 (BD) and IL-23 (10ngml−1; R&D Systems) for 48h. Total RNA was isolated with an RNeasy kit (Qiagen). Complementary DNA was synthesized with TaqMan Reverse Transcription reagents (Applied Biosystems), using random hexamers as primers in accordance with the manufacturer's instructions. Hypoxanthine guanine phosphoribosyltransferase (HPRT) was used as an endogenous control. TaqMan primers and probes for human ROR-γt and HPRT were purchased from Applied Biosystems. All samples were analysed in duplicate or triplicate with the ABI Prism 7500 Fast Real-Time PCR System (Applied Biosystems). Relative expression levels of the genes of interest were calculated with the 2ΔΔCt method, with expression levels in unstimulated healthy patient cells assigned an arbitrary value of 1.
Statistical analysis
Mann–Whitney tests and t-tests were performed with Prism 4.0 software (Prism).
Supplementary Material
Acknowledgments
This work was supported by the intramural program of the National Institutes of Health.
Footnotes
Author Contributions J.D.M., J.M.B., A.L., B.J.H., K.M.E., Y.K., A.H., H.Z.E., M.L.P. and A.I.A. performed experiments and analysed data. A.F.F., C.S., J.D. and S.M.H. evaluated patients and obtained samples. J.D.M., J.M.B., A.L., J.O'S., S.M.H., W.E.P. and D.C.D. conceived the study, designed experiments, interpreted data and wrote the manuscript.
Supplementary Information is linked to the online version of the paper at www.nature.com/nature.
Full Methods and any associated references are available in the online version of the paper at www.nature.com/nature.
References
- 1.Buckley RH, Wray BB, Belmaker EZ. Extreme hyperimmunoglobulinemia E and undue susceptibility to infection. Pediatrics. 1972;49:59–70. [PubMed] [Google Scholar]
- 2.Grimbacher B, et al. Hyper-IgE syndrome with recurrent infections—an autosomal dominant multisystem disorder. N Engl J Med. 1999;340:692–702. doi: 10.1056/NEJM199903043400904. [DOI] [PubMed] [Google Scholar]
- 3.Minegishi Y, et al. Dominant-negative mutations in the DNA-binding domain of STAT3 cause hyper-IgE syndrome. Nature. 2007;448:1058–1062. doi: 10.1038/nature06096. [DOI] [PubMed] [Google Scholar]
- 4.Holland SM, et al. STAT3 mutations in the Hyper-IgE syndrome. N Engl J Med. 2007;357:1608–1619. doi: 10.1056/NEJMoa073687. [DOI] [PubMed] [Google Scholar]
- 5.Del Prete G, et al. Defective in vitro production of γ-interferon and tumor necrosis factor-α by circulating T cells from patients with the hyper-immunoglobulin E syndrome. J Clin Invest. 1989;84:1830–1835. doi: 10.1172/JCI114368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Buckley RH. The hyper-IgE syndrome. Clin Rev Allergy Immunol. 2001;20:139–154. doi: 10.1385/CRIAI:20:1:139. [DOI] [PubMed] [Google Scholar]
- 7.Acosta-Rodriguez EV, Napolitani G, Lanzavecchia A, Sallusto F. Interleukins 1β and 6 but not transforming growth factor-β are essential for the differentiation of interleukin 17-producing human T helper cells. Nature Immunol. 2007;8:942–949. doi: 10.1038/ni1496. [DOI] [PubMed] [Google Scholar]
- 8.Korn T, et al. IL-21 initiates an alternative pathway to induce proinflammatory TH17 cells. Nature. 2007;448:484–487. doi: 10.1038/nature05970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nurieva R, et al. Essential autocrine regulation by IL-21 in the generation of inflammatory T cells. Nature. 2007;448:480–483. doi: 10.1038/nature05969. [DOI] [PubMed] [Google Scholar]
- 10.Wilson NJ, et al. Development, cytokine profile and function of human interleukin 17-producing helper T cells. Nature Immunol. 2007;8:950–957. doi: 10.1038/ni1497. [DOI] [PubMed] [Google Scholar]
- 11.Yang XO, et al. STAT3 regulates cytokine-mediated generation of inflammatory helper T cells. J Biol Chem. 2007;282:9358–9363. doi: 10.1074/jbc.C600321200. [DOI] [PubMed] [Google Scholar]
- 12.Chen Z, et al. Selective regulatory function of Socs3 in the formation of IL-17-secreting T cells. Proc Natl Acad Sci USA. 2006;103:8137–8142. doi: 10.1073/pnas.0600666103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Laurence A, et al. Interleukin-2 signaling via STAT5 constrains T helper 17 cell generation. Immunity. 2007;26:371–381. doi: 10.1016/j.immuni.2007.02.009. [DOI] [PubMed] [Google Scholar]
- 14.Mathur AN, et al. Stat3 and Stat4 direct development of IL-17-secreting Th cells. J Immunol. 2007;178:4901–4907. doi: 10.4049/jimmunol.178.8.4901. [DOI] [PubMed] [Google Scholar]
- 15.Steinman L. A brief history of T(H)17, the first major revision in the TH1/TH2 hypothesis of T cell-mediated tissue damage. Nature Med. 2007;13:139–145. doi: 10.1038/nm1551. [DOI] [PubMed] [Google Scholar]
- 16.Huang W, Na L, Fidel PL, Schwarzenberger P. Requirement of interleukin-17A for systemic anti-Candida albicans host defense in mice. J Infect Dis. 2004;190:624–631. doi: 10.1086/422329. [DOI] [PubMed] [Google Scholar]
- 17.Happel KI, et al. Divergent roles of IL-23 and IL-12 in host defense against Klebsiella pneumoniae. J Exp Med. 2005;202:761–769. doi: 10.1084/jem.20050193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wolk K, Sabat R. Interleukin-22: a novel T- and NK-cell derived cytokine that regulates the biology of tissue cells. Cytokine Growth Factor Rev. 2006;17:367–380. doi: 10.1016/j.cytogfr.2006.09.001. [DOI] [PubMed] [Google Scholar]
- 19.Tanaka T, et al. Distinct gene expression patterns of peripheral blood cells in hyper-IgE syndrome. Clin Exp Immunol. 2005;140:524–531. doi: 10.1111/j.1365-2249.2005.02805.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kao CY, et al. IL-17 markedly up-regulates β-defensin-2 expression in human airway epithelium via JAK and NF-κB signaling pathways. J Immunol. 2004;173:3482–3491. doi: 10.4049/jimmunol.173.5.3482. [DOI] [PubMed] [Google Scholar]
- 21.Ye P, et al. Requirement of interleukin 17 receptor signaling for lung CXC chemokine and granulocyte colony-stimulating factor expression, neutrophil recruitment, and host defense. J Exp Med. 2001;194:519–527. doi: 10.1084/jem.194.4.519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Miller LS, et al. MyD88 mediates neutrophil recruitment initiated by IL-1R but not TLR2 activation in immunity against Staphylococcus aureus. Immunity. 2006;24:79–91. doi: 10.1016/j.immuni.2005.11.011. [DOI] [PubMed] [Google Scholar]
- 23.Schnyder-Candrian S, et al. Interleukin-17 is a negative regulator of established allergic asthma. J Exp Med. 2006;203:2715–2725. doi: 10.1084/jem.20061401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ozaki K, et al. A critical role for IL-21 in regulating immunoglobulin production. Science. 2002;298:1630–1634. doi: 10.1126/science.1077002. [DOI] [PubMed] [Google Scholar]
- 25.Pesce J, et al. The IL-21 receptor augments Th2 effector function and alternative macrophage activation. J Clin Invest. 2006;116:2044–2055. doi: 10.1172/JCI27727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kuchen S, et al. Essential role of IL-21 in B cell activation, expansion, and plasma cell generation during CD4+ T cell–B cell collaboration. J Immunol. 2007;179:5886–5896. doi: 10.4049/jimmunol.179.9.5886. [DOI] [PubMed] [Google Scholar]
- 27.King CL, et al. Frequency analysis of IgE-secreting B lymphocytes in persons with normal or elevated serum IgE levels. J Immunol. 1991;146:1478–1483. [PubMed] [Google Scholar]
- 28.Mucida D, et al. Reciprocal TH17 and regulatory T cell differentiation mediated by retinoic acid. Science. 2007;317:256–260. doi: 10.1126/science.1145697. [DOI] [PubMed] [Google Scholar]
- 29.Pitcher CJ, et al. HIV-1-specific CD4+ T cells are detectable in most individuals with active HIV-1 infection, but decline with prolonged viral suppression. Nature Med. 1999;5:518–525. doi: 10.1038/8400. [DOI] [PubMed] [Google Scholar]
- 30.Grimbacher B, et al. Genetic linkage of hyper-IgE syndrome to chromosome 4. Am J Hum Genet. 1999;65:735–744. doi: 10.1086/302547. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.