

Abstract
DNA methylation is a critical epigenetic mark that is essential for mammalian development and aberrant in many diseases including cancer. Over the past decade multiple methods have been developed and applied to characterize its genome-wide distribution. Of these, Reduced Representation Bisulfite Sequencing (RRBS) generates nucleotide resolution Illumina-based libraries that enrich for CpG-dense regions by methylation-insensitive restriction digestion. Here we provide an extensive, optimized protocol for generating RRBS libraries and discuss the power of this strategy for methylome profiling. We include information on sequence analysis and the relative coverage over genomic regions of interest for a representative mouse MspI generated RRBS library. Contemporary sequencing and array-based technologies are compared against sample throughput and coverage, highlighting the variety of options available to investigate methylation on the genome-scale.
Introduction
DNA methylation is the only known covalent modification to the eukaryotic genome and serves multiple functions in plants and higher animal phyla. In mammals it is primarily observed at cytosine residues within the symmetrical CpG dinucleotide, which provides a mechanism for stable marker inheritance through enzymatic recognition of newly synthesized, hemi-methylated DNA [1]. Moreover, this heritability is maintained independently of any underlying nucleotide sequence, making cytosine methylation a truly "epigenetic" mark [2,3].
In mammals, CpG methylation is essential for development and is generally regarded as a terminal silencer of expression, though it also plays roles in more nuanced programs such as maintenance of parental allele-specific imprinting and dosage compensation by X inactivation in females [2,3]. Moreover, the contributing paternal genome within a fertilized zygote demonstrates an active global demethylation that provides strong evidence towards CpG methylation’s dynamic potential as an epigenetic modifier, not exclusively as a terminal silencer [4–6]. Substantial alterations of DNA methylation have been observed in multiple cancers, characterized by localized hypermethylation at target gene promoters and a global loss of methylation [7,8].
Experimental analysis of DNA methylation states is complicated by the fact that it does not alter base pairing and is lost during PCR amplification. Traditionally, measurement of methylated cytosine has instead relied on a chemical reaction using high temperature, low pH, and treatment with sodium bisulfite, a protocol that specifically deaminates unmethylated cytosines and converts them into uracils, while leaving methylated cytosines unchanged [9]. Subsequent PCR amplification of bisulfite converted DNA replaces uracil by thymine, giving rise to a methylation-specific single nucleotide polymorphism that is detectable by conventional sequencing and alignment against the reference sequence.
Recent technical advances such as tiling microarrays and high-throughput sequencing have dramatically increased the scale at which DNA methylation can be analyzed [3,10]. Some array-based strategies utilize bisulfite converted DNA to probe defined regions by bimodal hybridization of either unconverted or converted sequences. Others enrich directly for methylated DNA by co-precipitation using either methyl-binding proteins or antibodies that target the 5-methyl-CpG hapten. Methylated DNA immunoprecipitation (MeDIP) has provided preliminary methylome profiles of mammalian promoters and the entire Arabidopsis genome [11–13], but even the most advanced array technologies are only capable of screening pre-selected genomic regions. High-throughput sequencing strategies address this latter issue and achieve greater coverage (MeDIP-Seq) [14]. While MeDIP-seq can identify immunoprecipitable 5-methyl-CpG-containing fragments, it cannot determine the methylation status of individual CpGs within the fragment. Recently, a bisulfite conversion protocol compatible with ultra-high throughput sequencing (BS-seq) was used to assess the Arabidopsis methylome at single-CpG resolution with approx. 20-fold sequencing coverage [15,16]. However, the larger genome-size (3 Gb in human vs 120 Mb in Arabidopsis) and a more uneven CpG distribution renders a comparable study extremely costly in mammalian models.
Reduced Representation Bisulfite Sequencing (RRBS) utilizes the same high-throughput sequencing strategy as BS-seq, but enriches its libraries by digesting genomic DNA with restriction endonucleases that are specific for CpG containing motifs (Figure 1A and 1B) [17–19]. RRBS therefore provides an enhanced coverage for the CpG dinucleotide and yields single base pair resolution data within multiple regions of interest, including CpG islands, promoters and enhancer elements (Figure 1C). CpG fragment enrichment substantially reduces the sequencing depth required for whole genome coverage by under-representing CpG poor, constitutively methylated, intergenic regions. Instead, a small but reproducible subset of CpG-rich restriction fragments in the genome is sequenced at sufficient depth. Because RRBS operates by fragmenting DNA at specific restriction sites and sequencing coverage relative to the reduced representation genome is high, the vast majority of fragments are sequenced in all RRBS analyses for a given species, increasing the method’s utility for comparative DNA methylation profiling [10,18,19].
Figure 1. Reduced Representation Bisulfite Sequencing.
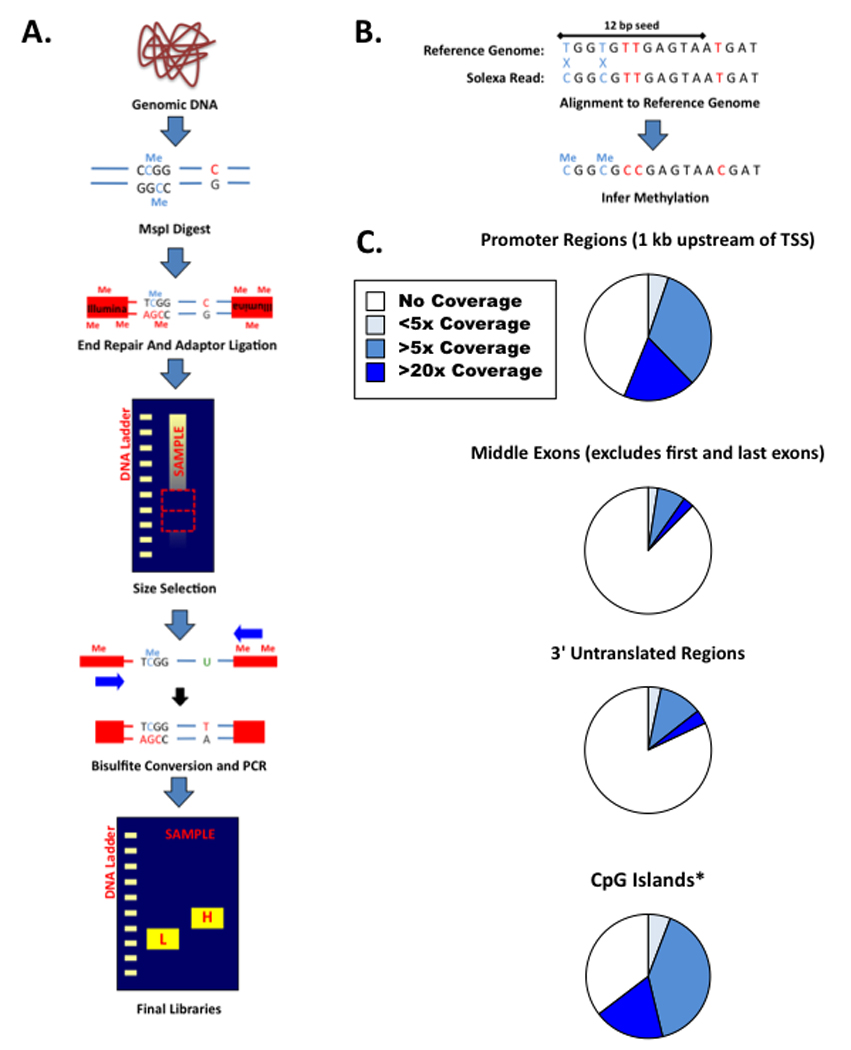
- General outline of RRBS library construction protocol including all key steps
- Overview of RRBS read alignment
-
Sequencing coverage at selected regions of interest within the mouse genome as generated from a representative V6.5 mouse Embryonic Stem (ES) cell library.*CpG Islands are as described in Ref. 30 and extend beyond 700 bp in length.
Here we provide an extended protocol for generating RRBS libraries, emphasizing critical steps, checkpoints for quality control, and portions that can be customized to meet the needs of more specialized studies (Figure 1). To date, the majority of RRBS libraries generated within our lab have been in Mus musculus [18], and the protocol is largely adapted to provide robust coverage of this genome, but all of the general principles and most of the specific steps extend to other organisms such as human. This review concludes with a discussion on the fundamental distinctions and attributes of other screening protocols, as well as on future directions for genome-scale DNA methylation mapping.
DNA Isolation
The method for isolation of genomic DNA can critically affect the quality of an RRBS library. Cultured cells are first washed with standard PBS solution, trypsinized and pelleted at 1000 rpm for 5 minutes. Cell pellets are then resuspended in a prepared lysis buffer consisting of 100mM Tris HCl (pH 8.5), 5 mM EDTA, 0.2% SDS, 200 mM NaCl. Proteinase K (Invitrogen) is added fresh to a final concentration of 300 µg/ml [20]. Traditionally, 1 ml of Proteinase K / lysis buffer solution is used per million cells and this proportion can be scaled accordingly [21]. Isolated tissue samples are prepared similarly, but are finely minced with a scalpel or passed through a syringe within the lysis buffer before Proteinase K is added. Samples are incubated overnight at 55° C to ensure that genomic DNA is completely dissociated from any contaminating nucleosomal or DNA binding proteins. The length of incubation and amount of Proteinase K corresponds directly to the quality of genome scale libraries [18,21]. Nucleosomal contamination can alter the properties of DNA denaturation and prevent suitable cytosine-to-uracyl conversion, as the bisulfite reaction requires single stranded DNA molecules. Unconverted, unmethylated CpGs will be misread as artifactual methylation events; additionally, retained cytosine bases within a non-CpG sequence context might be discarded by automated alignment platforms when using short read-length platforms. RNase A (Roche) can be optionally included during the lysis step, but may only be necessary if a very precise quantification of starting template is necessary.
After digestion, genomic DNA is extracted using a standard phenol:chloroform protocol. A 1:1 volume ratio of phenol:chloroform:isoamyl alcohol (25:24:1, Invitrogen) is added to each sample, vortexed, and centrifuged for 5 minutes in a tabletop microcentrifuge at maximum speed. The aqueous fraction is removed and precipitated with 250 mM NaCl and a 2.5 : 1 ratio of ethanol, followed by overnight incubation at −20 °C. DNA is subsequently pelleted by centrifugation, washed with 70% ethanol, and resuspended in DNAse-free water. For samples where total genomic yield is expected to be below 100 ng, 10 µg of Glycogen (Roche) serves as a visible, high recovery co-pellet during centrifugation steps. Final yield may be quantified by spectrophotometer, fluorometer, or qPCR based methods, depending on the amount or preciousness of the sample. If protein contamination is observed during quantification, column-based DNA purification systems are suitable for secondary cleanup.
Methylation-Insensitive Digestion
While genome scale libraries constructed from sonicated DNA provide an unbiased selection of available DNA fragments, digestion with CpG containing restriction enzymes addresses the fundamental CpG asymmetry present within vertebrate genomes and enriches for regions of known functional interest, such as CpG islands, promoter regions, and enhancer elements [18]. The vast majority of CpG dinucleotides occur infrequently, such that a randomly selected sonication fragment frequently contains not even a single CpG dinucleotide for DNA methylation analysis at a current read length of 75 bp, given an average density of approximately one CpG per 100 base pairs in mammalian genomes. Restriction-based enrichment ensures that every sequenced read will contain information regarding at least one CpG purely because one is included within the target sequence. MspI, for instance, recognizes and cleaves the sequence CCGG and MspI derived libraries provide information regarding the methylation state of nearly 90% of CpG islands within the mouse genome. The relative representation for regions of interest covered by a standard MspI-digested mouse library is summarized in Figure 1C.
Genomic DNA is fragmented in a 30 µl reaction containing 10 U of MspI (NEB) per 1 µg DNA and supplier provided enzyme buffer. For libraries constructed using > 100 ng of starting template, complete digestion is ensured by overnight incubation at 37 °C. After incubation, add 1 µl of 0.5 M EDTA to chelate essential cofactors and arrest the reaction. Complete digestion is confirmed by running a small aliquot of the complete reaction on a 4–20% – Criterion precast TBE gel (Bio-Rad) and staining with SYBR green. If digestion is complete, distinct, redundant microsatellite bands whose size can be predicted from in silico digests of the genome (Figure 2), should be visible; incomplete digestion is noted by a more prevalent, high molecular weight smear at the top of the gel. Complete digestion is essential to ensure an unbiased coverage of available MspI fragments. Libraries derived from incompletely digested samples will be enriched for redundant microsatellite regions. If digestion is complete, samples are purified using Phenol:chloroform as described above. Purified, digested DNA is eluted into 15 µl 10 mM Tris-HCl (pH8).
Figure 2. MspI Generated RRBS Libraries from Mouse Genomic DNA.
- Overlap of sequenced/aligned CpGs with specific regions of interest in the mouse genome for typical low (40–120bp) and high (120–220bp) fragment libraries (single CpGs can simultaneous overlap with more than one region class)
- Size distribution of in silico digested fragments compared to experimentally generated RRBS libraries (low and high are combined). Only unique sequences are aligned. Asterisks (*) denote redundant microsatellite fragments.
- Diagnostic gel of high (H) and low (L) fragment libraries generated from V6.5 mouse Embryonic Stem (ES) cells. Asterisks (*) correspond to microsatellite markers as predicted in silico.
End Repair and Adapter Ligation of DNA Fragments
In more widely used chromatin immunoprecipitation protocols, sequencing adapters are added as a terminal step, after the enriched DNA has been fractioned or isolated by target binding [22]. However, within these systems, genomic DNA fragments are never altered and remain in an unconverted, double-stranded state. RRBS and BS-seq protocols alternatively rely on bisulfite conversion, which will change nucleotide sequence and retain converted fragments as single stranded DNA that will be incompatible with adapter ligation [18]. To prepare samples for the addition of Illumina/Solexa adapters, the 5’ CG overhangs generated by MspI digestion must first be end repaired using Klenow DNA polymerase lacking 3’–5’ exonuclease activity (NEB). Digested samples are repaired in 50 µl reactions containing 10 U Klenow (3’ 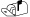 5’ exo−) and supplied 1x enzyme buffer. This step utilizes modified dNTP concentrations containing 1 nM dGTP, 1 nM 5’ methylated dCTP, and an excess of 10 nM dATP to provide sufficient 5’ adenosine overhangs that are essential for ligation to the Illumina/Solexa adapters which contain a 3’-T overhang. Reactions are incubated at 30 °C for 20 minutes followed by a second 37 °C incubation for an additional 20 minutes. The reaction is stopped by the addition of 1 µl 0.5 M EDTA and purified by reaction cleanup columns or phenol : chloroform. Samples are eluted in 10 µl 10 mM Tris-HCl (pH 8).
5’ exo−) and supplied 1x enzyme buffer. This step utilizes modified dNTP concentrations containing 1 nM dGTP, 1 nM 5’ methylated dCTP, and an excess of 10 nM dATP to provide sufficient 5’ adenosine overhangs that are essential for ligation to the Illumina/Solexa adapters which contain a 3’-T overhang. Reactions are incubated at 30 °C for 20 minutes followed by a second 37 °C incubation for an additional 20 minutes. The reaction is stopped by the addition of 1 µl 0.5 M EDTA and purified by reaction cleanup columns or phenol : chloroform. Samples are eluted in 10 µl 10 mM Tris-HCl (pH 8).
Illumina Genome Analyzer sequencing requires that cytosines within the adapter sequences be maintained. We use Illumina adapters that contain 5-meC instead of C to prevent deamination during the bisulfite reaction [18]. Adapter oligonucleotides are annealed at a concentration of 15 µM in 10 mM Tris-HCl, pH 8, 0.1 mM EDTA and 10 mM NaCl by heating to 98 °C for 5 minutes in a heat block followed by a gradual return to room temperature. Klenow treated DNA fragments are reacted in 50 µl reactions containing 2.5 µl of 15 µM annealed Illumina adapters, 1 µl of 400 U/µl T4 DNA Ligase (NEB), and reaction buffer. For sample containing less than 1 µg of DNA, we use 1 µl 2000 U/ul concentrated T4 DNA ligase (NEB) in a 20 µl reaction to maximize the ligation efficiency. The reaction is incubated overnight at 16°C, stopped by 1 µl 0.5 M EDTA and purified by phenol:chloroform extraction. Samples are eluted in 10 µl Tris-HCl (pH=8.5).
Size Selection of Adapter-Ligated Fragments
Size selection of MspI-digested, Illumina adapter-ligated samples provides the second enrichment step through which large fragments, which are generally CpG poor, and small fragments, which are too frequently redundant and difficult to align, can be eliminated from the final libraries. Samples are run on a 3% NuSieve 3:1 agarose 0.5x TBE gel, flanked by one or more lanes containing 50 bp or Low Molecular Weight DNA ladders (NEB) at ~90 – 100 V until the bromophenol blue within the loading dye has run 4–5 cm. Lanes containing the ladders are removed and stained with SYBR green for 30 min, then the molecular weights of interest are carefully marked using a fine pipette tip or toothpick such that small dots that signify the boundary of each region to be extracted can be easily seen. Gel excision is performed by aligning these markers on either side of the unstained gel and precisely cutting straight lines using a clean razor for each cut. Slicing through multiple sample lanes with one razor can lead to contamination that may deter comparative analysis between methylated and unmethylated regions, especially if the starting template is low. Gel slices containing the regions of desired DNA are purified using QIAGEN gel purification columns. Samples are eluted into 20 µl 10 mM Tris-HCl (pH 8). If more than one size window is excised per sample, they are kept separate from the extraction step forward.
Size selection depends upon the regions of interest for a given study and the desired trade-off between breadth and depth of coverage. To obtain ~90% of the annotated CpG islands within the M. musculus genome with reasonably low enrichment for redundant intergenic elements, two gel bands are cut from each sample ranging between 40–120 and 120–220 bp (Table 1 and Figure 2). Generating two smaller, rather than one 40–220bp library was introduced to reduce possible size-related amplification and/or sequencing biases. If the size selection step is performed before adapter ligation, the molecular weight of each DNA marker band should correspond directly to the location of sample fragments. However, in this adapted protocol, compensating for the ~50 bp length of each adapter is not directly additive because the forked adapters cause the fragments to run somewhat slower than expected. To generate RRBS fragments of 40–120 and 120–220 bp MspI fragments we excise adapter-ligated fragments that run at 160–240 and ~240–340 bp. If additional coverage is desired or a different organism used, PCR amplification using genomic PCR primers LPX 1.1 and 2.1 from Illumina for each 100 bp span of sample will identify the running properties of adapted samples. This primer pair can also be used after gel extraction to perform an analytical PCR to gauge ligation efficiency and quality. 10 µl PCR reactions are set up for each gel purified high and low molecular weight fractions and tested over a range of cycle numbers. At this step, any polymerase is sufficient for amplification as the DNA template still exists in a native, double stranded state, but it is important to note that amplification of the final bisulfite converted library can be affected by the chosen enzyme as discussed below. Reactions are performed under the following thermocycler conditions: 94 °C for 5 min, n × (94°C for 20 sec, 65 °C for 30 sec, 72 °C for 30 sec), 72 °C for 7 min. Sample amplification can be best assessed using 4–20% PAGE gels run against 50 or 20 bp DNA ladders and stained with SYBR green. When starting with 1 ug of total genomic DNA, PCR products can be visible after as few as 8–10 cycles. Quality of the library can be predicted by demonstrated scaling over increasing cycle number, the presence of clear, concise microsatellite bands (Figure 2), and, when constructing multiple libraries, consistency of the selected size. The presence of contaminant microsatellite bands or other aberrant amplification outside of the size-selected range can affect the overall quality of the library and are a sign of imprecise gel extraction.
Table 1.
Coverage of MspI digest fragments based on read length in mouse and human.
| Species | Enzyme | Cut | Read length |
# Fragments |
Bases | CpGs | of all | CpG islands (>= 10) |
of all |
|---|---|---|---|---|---|---|---|---|---|
| Human | MspI | 40–220 | 51 | 647,902 | 5.5 Mb | 2,801,861 | 10% | 23,324 | 83% |
| Human | MspI | 40–220 | 75 | 647,902 | 6.7 Mb | 3,238,402 | 12% | 23,649 | 84% |
| Human | MspI | 40–220 | 100 | 647,902 | 7.2 Mb | 3,401,082 | 12% | 23,835 | 84% |
| Mouse | MspI | 40–220 | 51 | 333,104 | 2.8 Mb | 1,275,597 | 6% | 13,633 | 85% |
| Mouse | MspI | 40–220 | 75 | 333,104 | 3.5 Mb | 1,445,830 | 7% | 13,800 | 86% |
| Mouse | MspI | 40–220 | 100 | 333,104 | 3.8 Mb | 1,506,712 | 7% | 13,883 | 87% |
Bisulfite Conversion and Scaling of the Final Library
Recently, multiple commercial kits for bisufite sequencing have become available. In general, these kits are superior to the classical conversion protocols based on generic ingredients, providing greater consistency and lower overall degradation during the reaction. We use the EpiTect system from Qiagen as our standard and follow the provided protocol as described with some exceptions. If analytical PCR demonstrated successful ligation, the entire 20 µl of eluted library may be converted. While the stock thermocycler settings provided by Qiagen provide sufficient precision for classic, single gene methylation analysis such as Combined Bisulfite Restriction Analysis (COBRA) or bisulfite conversion followed by TOPO Cloning/Sequencing, optimization experiments conducted within our lab have concluded that these settings do not provide adequate conversion on the genome scale. Others have introduced similar modifications to the manufacturer’s protocol. To ensure highest possible quality of sequenced reads, overall conversion efficiency > 97% for unmethylated cytosines is required. Conversion efficiency can be improved by an extended 14 hour thermocycler protocol with the following phases: 99 °C for 5 min, 60 °C for 25 min, 99 °C for 5 min, 60 °C for 85 min, 99 °C for 5 min, 60 °C for 175 min, 6 x (95 °C for 5 min, 60 °C for 90 min), 20 °C indefinitely. This protocol has been empirically optimized to provide the greatest possible conversion efficiency while minimizing overconversion of methylated cytosines and sample degradation. However, it should be noted that this protocol is predominantly conducted on murine samples and that sequence dependent effects on overall conversion efficiency is not understood. It may therefore be necessary to optimize several additional conditions or even additional commercial products for RRBS in other systems. Bisulfite cleanup is performed as described; the final elution step conducted twice per column for a final volume of ~40 µl.
Before final scaling of converted libraries, it is essential to perform a semi-quantitative PCR set across a reasonable cycle range; excessive cycle number can introduce unwanted point mutations, fragment size biases, and biases against methylated CpG rich regions of interest or underrepresented fragments. PCR is performed as described above and the range post-conversion for visible product can be expected to be ~3–7 cycles higher than observed when assessing ligation efficiency. The optimal cycle number should be determined for each independent sample individually, as variation introduced over this lengthy protocol is expected to result in a range of several cycles even when starting templates between samples are equivalent. In PCR following bisulfite conversion, the choice of polymerase is essential and must be uracyl insensitive to avoid so-called uracyl stalling or “poisoning” that would otherwise drastically reduce quality and yield. Pfu Turbo Cx Hotstart (Stratagene) is an attractive option as it provides high-rate read-through of uracyl bases but retains additional proofreading activity to reduce mutation. Samples are again run on a 4%–20% Criterion precast TBE gel and stained for 30 min with SYBR green.
The final library is synthesized by large-scale amplification of the remaining bisulfite converted sample. Eight reactions are established to include 2–4 µl of bisulfite converted template, 1.25 U Pfu Turbo Cx Hotstart Polymerase, supplied buffer, 150 nM 1.1/2.1 Illumina Primers, and 1 mM dNTPs. Thermocycler conditions are as described above for the analytical PCR protocol and the reactions are combined after completion and can be cleaned using commercially available reaction clean-up kits. The cycle number for this final step is empirically determined to be the lowest cycle number in which a band of the expected range is observed after 30 minutes of SYBR green staining in the semi-quantitative step; while this amount may appear exceedingly low, the final library yield is usually sufficient for sequencing and can be increased by providing additional template within the final reactions. Final libraries are obtained after a second electrophoresis step using a 3% 3:1 NuSieve Agarose 0.5x TBE gel. The libraries are best detected by SYBR green staining and extracted using commercially available kits; the final volume of each library should exceed the requirements for sequencing by 5–10 µl such that some residual sample may be subjected to secondary validation protocols before sequencing. Concentration is expected to be relatively low, in the single ng/µl range, so the use of spectrophotometry or high sensitivity fluorometry is recommended; concentrations are confirmed by running 3 µl of the final libraries on Criterion Precast TBE gels and the samples are normalized to the requirements of the sequencing facility used.
Validation of Libraries
Sequencing by Illumina Genome Analyzers remains the most costly step in generating genome-wide methylation profiles, so some care should be taken to ensure that the generated libraries are of the highest possible quality before submitting them for high-throughput sequencing. This post-synthesis analysis can most easily be confirmed by visual inspection of the final diagnostic gel (Figure 2). Individual molecular weight bands for single samples are not expected to dramatically overlap and all microsatellite MspI digested repeats occurring within selected size ranges are expected to be present at a greater intensity, though if they are exclusively seen, libraries were most likely generated from incompletely digested template. The absence of predicted bands within any size range indicates degradation. Moreover, if libraries generated from multiple samples are synthesized together, they should be highly similar in size ranges and quality to provide the highest possible co-occurring fragment representation to facilitate comparison. Generally, validation of samples by visual inspection provides an accurate gauge of quality and ensures reasonable coverage within the selected range.
Sequencing and Analysis
Bisulfite treated, amplified libraries are sequenced using Illumina Genome Analyzer technology capable of producing at least 75 nucleotide reads from the standard sequencing primer. Each alignable read yields at least 1 CpG of interest as an inherent characteristic of RRBS in which the template genome is fractionated via the MspI target sequence. 5 to 10 million reads are generated per lane on an 8-lane flow cell, which is generally sufficient for an approx. 8-fold median coverage of aligned sequences. Sequences are called using standard Illumina software, but require a unique bioinformatic pipeline to successfully align non-complex, bisulfite converted DNA reads to the in silico fragmented genome. The stringency with which bisulfite converted sequence reads are aligned depends on the organism and experiment in question. Here, we provide a brief summary of the steps in which high quality Illumina data is generated for murine tissues using MspI digestion. This pipeline is similar to efforts described previously to align Shotgun Bisulfite Sequencing (BS-seq) data to Mus musculus and Arabadopsis genomes.
Two sets of reference sequences of MspI fragments between 25 and 250 bp from the organism of interest are constructed in silico, one in which a normal nucleotide context is maintained and one in which all Cs are converted to Ts; the converted reference assumes a complete alignment of a given read only when conversion efficiency of cytosine to thymine is 100%. Residual Cs within each read are also converted to Ts and aligned against the converted reference sequence of all predicted MspI generated fragments. Reads are subsequently matched to the reference genome by identifying all perfect 12 bp alignments and extending from either end of the established seed without gaps. Mismatches are identified and counted, but ignore cases where a T in the read is matched to a C in the unconverted reference. Instead, C-to-T mismatches are marked for downstream methylation analysis. The best alignment is kept only in cases where the next best alignment has > 2 more mismatches and reads that do not meet this stringency criterion are discarded as non-unique. For any given mammalian high-throughput bisulfite library, the number of discarded reads can be expected to greatly exceed those that are unique. However, the vast majority of remaining reads is high quality and contains relevant CpG methylation data, as each read will at least provide information for the 5’ CpG of the MspI overhang. Conventional Bisulfite-Seq libraries rely on a third reference library, in which genomic G nucleotides are converted to A to compensate for the conversion of cytosines on the G-poor complementary strand synthesized during the PCR. However, due to the directionality of the Solexa sequencing process, this additional alignment step is unnecessary as sequencing proceeds exclusively from the amplified copy of the MspI-mitigated 5’CGG containing strand. Bioinformatic approaches to defining the methylated state of a given region continue to improve in their statistical power and various methods exist for downstream analysis of RRBS-derived data. However, even relatively rudimentary definitions and tools can be used to reasonably interpret overall methylation for any given site or region. The methylation level of an individual cytosine base is calculated as the number of aligned reads containing a C at any C-T mismatched position divided by the number of reads containing either a C or a T (which is the total number high-quality reads containing that position). The overall DNA methylated state of a given region can be estimated as the mean methylation across all of its CpGs with subsequent sequence coverage. The general precision this measurement is determined by the coverage of high quality reads. As a convention, sequence coverage > 5 × for any given CpG dinucleotide is sufficient to yield reasonable insight towards its methylated status. To pursue more nuanced analysis of generated methylation profiles, genome browsers provide additional genomic and epigenomic contexts and databases exist that provide computationally defined annotation of regional CpG densities [22].
Conclusions and Future Directions
Reduced Representation Bisulfite Sequencing is one of several technologies currently used to profile DNA methylation on a genome-wide scale [10]. Previously described libraries sequenced using the Illumina Genome Analyzer platform have demonstrated reasonable coverage of nearly 1 million distinct CpG dinucleotides utilizing short 36 bp reads [18]. As technology improves, new platforms will provide greater sequencing length and depth for lower costs, such that enrichment based bisulfite sequencing methods can be adapted to gradually approach total CpG coverage and global methylation maps. Shotgun-Bisulfite sequencing already benefits from extended read lengths, and its sole detriment remains the excessive cost necessary to reach the depth required for accurate DNA methylation mapping. While the dramatically reducing cost of sequencing ensures BS-seq mapping will eventually become the preferred method for generating reference methylomes, many experimental and diagnostic needs will require more specific, high throughput methods to profile the methylated states of specific genomic subsets. Moreover, future endeavors to conduct epiallelic association studies or to optimize clinical diagnostics will require the rapid analysis of multiple samples. Strategies for nucleotide level methylome profiling include RRBS, and this technique is a method of choice to assay multiple genomic features based upon intrinsic CpG context
Several complementary and/or alternative methods for DNA methylation profiling at a specific subset of CpG dinucleotides exist. When probing many samples over a known CpG subset, array-based technologies may provide sufficient information. Illumina Infinium® BeadChip arrays can analyze 12 samples simultaneously and provide specific methylated states of nearly 28,000 CpGs per sample. For even greater sample numbers, the Illumina GoldenGate® Assay provides limited CpG information, but can run up to 96 samples per array. The respective coverage for these systems are low, 1.9 CpGs per gene (~15,000 genes) for Infinium® and 4.1 CpGs per gene for GoldenGate® (371 genes), but these representative sequences are only suitable for profiling the DNA methylation state of preselected promoter regions. While originally designed for human cancer screening, these technologies can be adapted to include cytosine bases within regions specific for the sample and experiment at hand. Other array based systems, such as HpaII tiny fragment Enrichment by Ligation-mediated PCR (HELP) and Comprehensive High-Throughput Array-Based (CHARM) analysis measure DNA methylation by comparative hybridization of selectable/enriched methylated and unmethylated regions [23,24].
Recently, hybrid capture approaches have used advanced Illumina sequencing technologies to successfully cover 22,000 specific genomic regions by selective hybridization and pulldown with biotinylated RNA probes [25]. While this study intended to gain deep sequencing information of the entire human exome, the method can be extended to select target regions outside of exons and to include additional steps for high-throughput bisulfite sequencing. Given a reasonable probe set, hybrid capture strategies could provide additional means for enrichment that could yield comprehensive, nucleotide-resolution maps. Efforts to use “padlock capture” strategies to analyze human CpG island methylation demonstrate the applicability of other enrichment methods to measure mammalian methylomes [26,27]. However, unlike RRBS, the regions of interest within these methods must be pre-selected, introducing sequence independent coverage biases.
Reduced representation methylation profiling provides an enrichment strategy that eliminates non-CpG containing regions and in silico generated, genome-wide restriction maps predict selectable size ranges that increase the frequency in which functional genomic contexts are represented. Size selection of CpG containing fragments dramatically reduces the sequencing depth that is required to provide adequate coverage for regions of interest. Adapter bar-coding methods would conceivably allow multiple libraries to be sequenced simultaneously and indexed by sample during alignment, which even given contemporary costs would bring RRBS within a comparable range to most array-based platforms at an improved CpG coverage [28]. The development of higher efficiency sequencing platforms promise to bring nucleotide resolution methylation mapping to a full genome scale [10]. Moreover, new approaches such as nanopore sequencing offer an affordable future technology that could provide high-resolution 5’ methylcytosine discriminatory mapping without chemical conversion or amplification of the genomic template [29]. Sequencing technologies and library construction protocols are improving in lockstep with recent discoveries that illuminate the functional dynamics of CpG methylation in mammalian development and human disease. The generation of genome-wide methylation maps will provide an invaluable new dimension to dissect the epigenetic basis for cellular identity.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Bestor TH. Hum. Mol. Genet. 2000;9:2395–2402. doi: 10.1093/hmg/9.16.2395. [DOI] [PubMed] [Google Scholar]
- 2.Bird A. Genes Dev. 2002;16:6–21. doi: 10.1101/gad.947102. [DOI] [PubMed] [Google Scholar]
- 3.Bernstein BE, Meissner A, Lander ES. Cell. 2007;128:669–681. doi: 10.1016/j.cell.2007.01.033. [DOI] [PubMed] [Google Scholar]
- 4.Santos F, Hendrich B, Reik W, et al. Dev. Biol. 2002;241:172–182. doi: 10.1006/dbio.2001.0501. [DOI] [PubMed] [Google Scholar]
- 5.Reik W, Dean W, Walter J. Science. 2001;293:1089–1093. doi: 10.1126/science.1063443. [DOI] [PubMed] [Google Scholar]
- 6.Mayer W, Niveleau A, Walter J, et al. Nature. 2000;403:501–502. doi: 10.1038/35000656. [DOI] [PubMed] [Google Scholar]
- 7.Jones PA, Baylin SB. Nat. Rev. Genet. 2002;3:415–428. doi: 10.1038/nrg816. [DOI] [PubMed] [Google Scholar]
- 8.Jones PA, Baylin SB. Cell. 2007;128:683–692. doi: 10.1016/j.cell.2007.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Frommer M, McDonald LE, Millar DS, et al. Proc. Natl. Acad. Sci. USA. 1992;89:1827–1831. doi: 10.1073/pnas.89.5.1827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Beck S, Rakyan VK. Trends Genet. 2008;24:231–237. doi: 10.1016/j.tig.2008.01.006. [DOI] [PubMed] [Google Scholar]
- 11.Weber M, Davies JJ, Wittig D, et al. Nat. Genet. 2005;37:853–862. doi: 10.1038/ng1598. [DOI] [PubMed] [Google Scholar]
- 12.Weber M, Hellmann I, Stadler MB, et al. Nat. Genet. 2007;39:457–466. doi: 10.1038/ng1990. [DOI] [PubMed] [Google Scholar]
- 13.Zhang X, Yazaki J, Sundaresan A, et al. Cell. 2006;126:1189–1201. doi: 10.1016/j.cell.2006.08.003. [DOI] [PubMed] [Google Scholar]
- 14.Down TA, Rakyan VK, Turner DJ, et al. Nat. Biotechnol. 2008;26:779–785. doi: 10.1038/nbt1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cokus SJ, Feng S, Zhang X, et al. Nature. 2008;452:215–219. doi: 10.1038/nature06745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lister R, O’Malley RC, Tonti-Filippini J, et al. Cell. 2008;133:523–536. doi: 10.1016/j.cell.2008.03.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Meissner A, Gnirke A, Bell GW, et al. Nucleic Acids Res. 2005;33:5868–5877. doi: 10.1093/nar/gki901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Meissner A, Mikkelsen TS, Gu H, et al. Nature. 2008;454:766–770. doi: 10.1038/nature07107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jeddeloh JA, Greally JM, Rando OJ. Genome Biol. 2008;9:231. doi: 10.1186/gb-2008-9-8-231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Meissner A, Eminli S, Jaenisch R. Methods Mol. Biol. 2009;482:3–19. doi: 10.1007/978-1-59745-060-7_1. [DOI] [PubMed] [Google Scholar]
- 21.Warnecke PM, Stirzaker C, Song J, et al. Methods. 2002;27:101–107. doi: 10.1016/s1046-2023(02)00060-9. [DOI] [PubMed] [Google Scholar]
- 22.Mikkelsen TS, Ku M, Jaffe DB, et al. Nature. 2007;448:553–560. doi: 10.1038/nature06008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Irizarry RA, Ladd-Acosta C, Wen B, et al. Nat. Genet. 2009;41:178–186. doi: 10.1038/ng.298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Khulan B, Thompson RF, Ye K, et al. Genome Res. 2006;16:1046–1055. doi: 10.1101/gr.5273806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gnirke A, Melnikov A, Maguire J, et al. Nat. Biotechnol. 2009;27:182–189. doi: 10.1038/nbt.1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Deng J, Shoemaker R, Yie B, et al. Nat. Biotechnol. 2009;27:353–360. doi: 10.1038/nbt.1530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ball MP, Li JB, Gao Y, et al. Nat. Biotechnol. 2009;27:361–368. doi: 10.1038/nbt.1533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Craig DW, Pearson JV, Szelinger S, et al. Nat. Methods. 2008;5:887–893. doi: 10.1038/nmeth.1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Clarke J, Wu HC, Jayasinghe L, et al. Nat. Nanotechnol. 2009;4:265–270. doi: 10.1038/nnano.2009.12. [DOI] [PubMed] [Google Scholar]
- 30.Bock C, Walter J, Paulsen M, et al. PLoS Comput. Biol. 2007;3:e110. doi: 10.1371/journal.pcbi.0030110. [DOI] [PMC free article] [PubMed] [Google Scholar]