

Abstract
Glioblastoma multiforme (GBM) is the most common primary brain cancer in adults. Despite significant advances in treatment and intensive research, the prognosis for patients with GBM remains poor. Therapeutic challenges for GBM include its invasive nature, the proximity of the tumor to vital brain structures often preventing total resection, and the resistance of recurrent GBM to conventional radiotherapy and chemotherapy. Gene therapy has been proposed as a useful adjuvant for GBM, to be used in conjunction with current treatment. Work from our laboratory has shown that combination of conditional cytotoxic with immunotherapeutic approaches for the treatment of GBM elicits regression of large intracranial tumor masses and anti-tumor immunological memory in syngeneic rodent models of GBM. In this review we examined the currently available animal models for GBM, including rodent transplantable models, endogenous rodent tumor models and spontaneous GBM in dogs. We discuss non-invasive surrogate end points to assess tumor progression and therapeutic efficacy, such as behavioral tests and circulating biomarkers. Growing preclinical and clinical data contradict the old dogma that cytotoxic anti-cancer therapy would lead to an immune-suppression that would impair the ability of the immune system to mount an anti-tumor response. The implications of the findings reviewed indicate that combination of cytotoxic therapy with immunotherapy will lead to synergistic antitumor efficacy with reduced neurotoxicity and supports the clinical implementation of combined cytotoxic-immunotherapeutic strategies for the treatment of patients with GBM.
Keywords: Immunotherapy, apoptosis, cancer models, HMGB1, Flt3L, HSV1-TK
BRAIN CANCER: A CHALLENGING THERAPEUTIC TARGET
Glioblastoma multiforme is the most common primary brain cancer in adults, affecting ~18,000 patients every year in the USA (www.cbtrus.org). In children, brain tumors account for 21% of childhood cancers and constitute the leading cause of solid tumor cancer death (www.cbtrus.org). The standard treatment of care consists in surgical resection followed by radiotherapy and chemotherapy [1, 2]. Despite significant advances in current therapeutic approaches, the prognosis of GBM remains dismal [1, 3-5]. Due to the diffuse nature of GBM and the proximity of the tumor to vital brain structures, complete tumor resection is practically impossible and the tumor often recurs in an area close to the original resection cavity [3]. Another therapeutic challenge of this disease is the intrinsic resistance of glioblastoma cells to radiotherapy and chemotherapy [5, 6]. The limit of radiation that normal brain can tolerate, up to 60 Gy, appears to be below the levels required to induce GBM cell death [7]. On the other hand, invading GBM cells, which give rise to recurrences, have been reported to be resistant to cytotoxic therapies due to the constitutive activation of antiapoptotic signaling pathways [3]. Also, expression of DNA repair enzyme O6-methylguanine-DNA methyltransferase (MGMT) in GBM cells gives them resistance to temozolomide and other alkylating agents [4]. In 40-50% of GBM patients methylation of MGMT promoter leads to inhibition of its expression, conferring sensitivity to temozolomide and increasing the median survival of surgery+radiation+temozolomide treated patients from 12 to 27 months [5]. Nevertheless, although traditional therapies allow an increase in the survival and quality of life of GBM patients, they are not curative and long-term survival is very rare [1-5]. Thus, novel therapeutic approaches and adjuvants to be employed in combination with standard therapeutic strategies are sorely needed for these patients. Here we review the evidence that support the combination of cytotoxic and immunotherapeutic approaches for the treatment of GBM.
ANIMAL MODELS FOR PRECLINICAL TESTING
Transplantable Tumor Models: Syngeneic GBM in Immunecompetent Rodents and Human GBM in Nude Mice
As a prelude to the implementation of gene therapy clinical trials for glioblastoma multiforme, it is critical to test potential novel therapies in relevant animal models of this disease. The ideal brain tumor model should exhibit predictable and reproducible intracranial growth patterns, have histopathological and biochemical resemblance to human GBMs and be non-immunogenic. There are several models available in which it is feasible to study the efficacy and toxicity of different therapeutic approaches for this disease, i.e., anti-angiogenic agents, proapoptotic molecules, immunotherapy, etc. Implantation of rodent glioma cells has proven an excellent intracranial brain tumor model due to their efficient tumorigenesis, reproducible and fast growth rates and accurate knowledge of the tumor location [8]. Syngeneic murine models, i.e. GL26 cells in C57BL6 mice [9, 10], SMA-560 cells in VMDK mice [11], CNS-1 cells in Lewis rats [9, 12], F98 and RG-2 cells in Fisher rats [13-15], are non-immunogenic, constituting an excellent tool for studying the response of brain tumors to immunotherapy [9, 15-17]. These models display some of the histopathological features of human GBM, including infiltration of tumor cells throughout the surrounding brain parenchyma (Fig. 1), areas of necrosis, micro-vascular hyperplasia, hemorrhages, presence of palisades; they are technically relatively easy to develop and highly reproducible [10, 12, 16-19], constituting a good model to test therapeutic efficacy in vivo. The fact that these animal models have an intact immune system, makes them a valuable tool to test immunotherapeutic approaches [16, 17, 20-23].
Fig. (1). Infitration of rat GBM cells into the adjacent brain parenchyma.
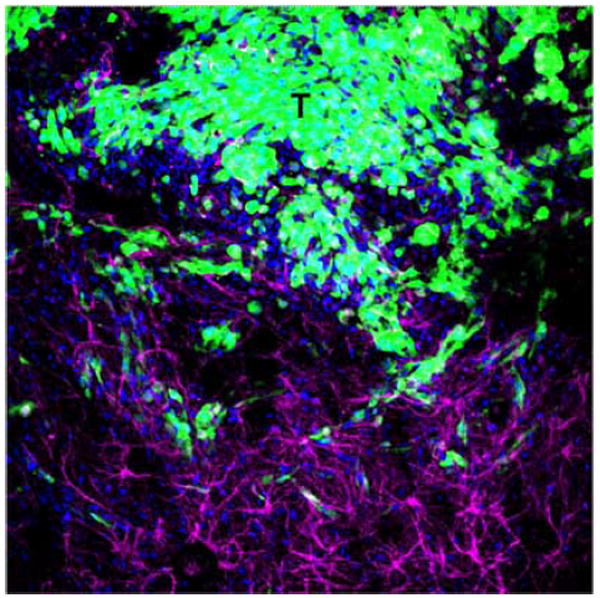
Representative confocal microphotograph shows rat GBM CNS-1 cells expressing Green Fluorescent Protein (GFP) infiltrating the adjacent brain parenchyma. Astrocytes (magenta) were stained using an anti-GFAP antibody. Nuclei were stained with DAPI (blue). T: tumor area. Width of illustrated field: 2mm.
Human glioma xenografts implanted in immunocom-promised mice have been extensively employed in preclinical brain cancer research. Although their xenogeneic nature impairs the study of immune-mediated anti-tumor strategies, they allow assessing the efficacy of therapeutic approaches in human GBM cells in the context of normal brain tissue. In fact, human xenografts exhibit histopatological features that resemble the human GBM and retain gene amplifications detected in the in situ tumors [24, 25]. Also, these models are very useful for the initial evaluation of novel imaging techniques [26] as well as new therapies for GBM, including antiangiogenic therapy [27-29], chemotherapy [30], radiotherapy [31], targeted toxins [32], cytotoxic [33] or conditionally replicative oncolytic viruses [34-38]. In summary, in view of their reproducibility and availability, transplantable rodent models of GBM constitute excellent preclinical models to test the efficacy and toxicity of novel gene therapy approaches for GBM.
Endogenous Brain Cancer Models
Over the past two decades, scientists have developed a greater understanding of the molecular and genetic basis of brain tumorigenesis [39]. Evidence of the downregulation of tumor suppressor genes such as p53 and PTEN as well as elevated expression of growth factors, and their cognate tyrosine kinase receptors, such as PDGF and EGFR are found in a high percentage of human GBM tumors [39-42]. Researchers have exploited the role of these molecular pathways in brain tumor development to induce endogenous brain tumors in rodents. Thus, genetic engineering of mouse genes or intracranial delivery of oncogenic transgenes in adult mice and rats have been attempted in order to trigger the development of endogenous brain tumor in rodents. Germline deletion of the tumor suppressor genes p53 and NF1 increased the susceptibility of mice to develop astrocytomas [43]. These mice exhibit a wide range of astrocytoma stages, with tumor growth detected in 50-70% of the mice and median survival times of 6-8 months [44]. This model is a valuable tool to study the development of secondary glioblastoma upon loss of p53. Germline deletion of other tumor suppressor genes, such as PTEN and Rb has also been attempted [45]. However, deletion of certain genes can lead to embryonic lethality or to the generation of tumors in other organs, limiting the utility of these models [45]. Transgenic mice that display cell type-specific overexpression of oncogenes have been employed to study genetic abnormalities in astrocytes and neural progenitors. This has proven useful to establish the role of oncogenes in the tumorgenesis and progression of GBM [46, 47]. Overexpression of the transcription factor E2F1 under the transcriptional control of the GFAP promoter led to the formation of astrocytomas in p53 KO mice, suggesting a role for E2F1 as an oncogene in the formation of brain tumors [44]. Considering that cell type-specific expression of certain genes is lethal during early development [45], oncogene overexpression has also been approached by delivery of gene therapy vectors into the brain of pre-natal or adult rodents, leading to the formation of endogenous brain tumors. These tumors harbor the genetic abnormalities found in human GBM, as well as the histopathological hallmarks of human GBM, including the aggressive invasive behavior. The use of viral or plasmid based vectors to introduce genetic aberrations permits the tight anatomical restriction of tumor-forming genetic events to specific areas of the brain. Furthermore, viral and plasmid vectors allow for the delivery of multiple tumorigenic genes in any combination, thereby reducing the amount of time required to generate germline transgenic mouse models. Thus, endogenous rodent GBM models constitute a very promising and stringent animal model of GBM which recapitulates the most salient histopathological features, molecular attributes, and heterogeneity of human GBM in a syngeneic rodent background. However, the applicability of the endogenous brain tumor models to assess the pre-clinical efficacy of experimental therapeutics is still limited due to the long latency and the variable reproducibility of these models.
Extensive evidence from across this developing field suggests that formation of endogenous brain tumors using viral vectors or plasmid systems to deliver oncogenes is somewhat variable. The degree of penetrance, tumor latency, and histopathological characteristics are dependant on the species and age of animals, the identity of specific genetic alterations and the vector system used to deliver them, and the anatomical location of genetic alterations. Retroviral-mediated delivery of PDGF into the adult rat white matter leads to formation of brain tumors with histopathological features that resemble human GBM; 100% of the animals succumb due to tumor burden 14-20 days after injection [48]. However, when retro-PDGF is delivered into the brain of newborn mice brain tumor formation only occurred in ~40% of the animals within 14-29 weeks [49]. The incidence and grade of brain tumor formation in mice has been suggested to be dependant on the levels of expression of PDGF [50]. Newborn mice were administered with retroviral vectors encoding a PDGF gene that lacks its regulatory sequences, which leads to higher levels of PDGF expression. Within 4-12 weeks, 100% of these mice developed invasive glioblastoma that exhibited neo-vascularization and tumor cell infiltration throughout the brain parenchyma [50].
In order to mimic the multiple genetic lesions encountered in human GBM, retroviral vectors that encode growth factors and a cycline-dependent kinase (cdk) were injected in the brain of neo-natal mice harboring additional mutations in tumor suppressor genes. Delivery of a constitutively active form of epidermal growth factor receptor gene (EGFR) in combination with basic fibroblast growth factor (bFGF) or ckd4 into the brain of neo-natal mice that are deficient in INK4a–ARF or p53 tumor suppressor genes led to formation of GBM in ~50% of the animals, while single mutations were unable of generating tumors [51]. These findings support the notion that combination of genetic lesions is required for the induction of endogenous GBM in mice. Additionally, combined genetic aberrations can be targeted to specific cell populations by the development of transgenic mice that express the retroviral receptor under the control of cell-type specific promoters, such as the progenitor nestin promoter or the astrocyte GFAP promoter [51, 52]. This system is very functional because it allows cell-type-specific transfer of oncogenes expressed within retroviral vectors under any type of promoter.
Lentiviral vectors have recently been employed to deliver oncogenes into the mouse brain. Considering that lentiviral vectors can transduce both dividing and non-dividing cells, these vectors constitute an attractive vehicle to deliver oncogenes to the brain of adult rodents [53]. In order to recapitulate the initiation of GBM, which is thought to arise upon genetic mutations in a few cells, oncogenic transgenes were delivered in a small population of cells in adult mouse brain by region-specific injection of lentiviral vectors encoding H-Ras or AKT [54]. To target astrocytes the Cre-LoxP-controlled lentiviruses were injected in the cortex, hippocampus and subventricular zone of GFAP-Cre mice. Again, administration of single oncogenes did not induce formation of tumors for up to 10 months. However, when Ras and AKT were delivered together in the hippocampal area ~30% of mice exhibited brain tumors that exhibit a high degree of invasiveness within 3-5 months post injection. Only one mouse developed a tumor following transduction in the sub-ventricular zone, and no animals had tumors following transduction into the cortex. Combined delivery of H-Ras and AKT into p53 KO mice greatly increased the tumorigenesis of these vectors leading to 75 and 100% of the mice injected in the subventricular zone and hippocampus, respectively. These tumors also exhibited a much shorter tumor latency with many histopathological characteristics found in human GBM [54]. These findings indicate that lentiviral vectors are useful tools to induce endogenous GBM in adult mice when several genetical abnormalities are induced in combination in the appropriate area of the brain.
Another recent approach to induce endogenous GBM in mice is the use of the Sleeping Beauty (SB) transposable element to achieve integration of oncogenes in the genome of brain cells of neo-natal immune competent mice [55, 56]. SB is a synthetic transposable element composed of a transposon DNA substrate and a transposase enzyme. SB transposase mediates excision and insertion of transposon DNA into the host genome, leading to long term expression [57]. Spontaneous brain tumors were induced by injecting SB-dependent plasmid harboring up to three genetic alterations (AKT, N-RAS, EGRFvIII, and/or shRNA specific for p53) into the lateral cerebral ventricle of neonatal mice of three different strains [55]. The histological characteristics of the tumors were dependant of the combination of genetic lesions introduced to the mice, although most resembled human astrocytoma or GBM. In some mice, multifocal tumors, another hallmark of human GBM, was observed. The combination of N-RAs, EGFRvIII, and p53 silencing was the most robust combination of genes with a 100% penetrance and a median survival of 83 days. These tumors were highly invasive and immunoreactive for nestin and GFAP indicating heterogeneity in the tumor mass. The SB is a very attractive and versatile system to induce endogenous brain tumors, allowing integration of large transposons (<10 kb) into the genome of many strains of mice [55].
In summary, endogenous rodent brain tumor models that recapitulate the genetic aberrations found in human GBM are very useful for the study of gliomagenesis; however, their variable tumor formation rate and long latency limits their use for testing preclinical treatments. Nevertheless, the use of imaging techniques to confirm tumor formation before the treatment would allow rigorous evaluation of novel therapies in these models, which resemble hystologically and genetically the human disease.
Spontaneous GBM in Dogs
Dogs bearing spontaneous GBM constitute a valuable tool in preclinical brain cancer research. GBM is the most common primary brain tumor in dogs, and brachycephalic breeds such as Boston terriers and Boxers [58-60] are predisposed to develop spontaneous GBM [61, 62]. Dog GBM exhibits the same histopathological characteristics of the human disease, including necrosis with pseudopalizading, neovascularization and endothelial proliferation [9]. The presence of pseudopalisading necroses and endothelial proliferation that closely resemble those found in human GBMs suggest the presence of a hypoxic environment in dog GBM, as described in human patients [63-65]. Importantly, canine GBM is highly invasive and exhibits the classical patterns of human GBM invasion [9], which makes it a very valuable tool to test not only the efficacy of novel therapies, but also their toxicity to the normal brain. The large size of the dog brain would be useful for preclinical assessment of doses and volumes in order to optimize treatment protocols before the translation into the clinic. Also, the detection of therapy-induced toxicity and side effects, as well as behavioral abnormalities are technically very well developed in dogs and constitutes a routine assessment in clinical veterinary practice. Moreover, the individual variability of outbreed dogs could help to better predict the clinical outcomes in human patients.
Clinical signs and prognosis of dogs with spontaneous GBM are very similar to those in human, and there is a high correlation of neuro-imaging features seen with MRI in canine and human GBM, which is also used as a diagnostic tool for canine GBM [66, 67]. The standard care of treatment in dogs with GBM is very similar to that used in human patients, consisting of surgical resection followed by radiation therapy and chemotherapy which leads to a median survival of 8.5-10 months [68]. This allows performing preclinical trials that will mimic more closely the clinical scenario, in which new therapies are applied in patients that simultaneously undergo traditional treatment. We and others have previously demonstrated the feasibility of delivering therapeutic transgenes to dog GBM cells in vitro and dog brain cells in vivo upon intracranial injection of gene therapy vectors, such as type 5 adenoviral vectors [69-71], adeno-associated viral vectors [72], plasmid DNA/polyethylenimine (PEI) complexes [71], which suggests that dogs bearing spontaneous GBM would be a suitable model to test novel gene therapy approaches. Importantly, the availability of canine GBM J3T [70, 73] and W&W [74] cell lines allows in vitro screening of novel therapeutic agents before moving to preclinical trials in dogs bearing spontaneous GBM. Also, the characterization of cancer stem cells from a GBM in a Boxer has been recently reported [75]. These cells exhibit cancer stem markers and have highly proliferative rate, and ability of self-renewal and differentiation. In vitro they form neurospheres and in vivo they growth intracranially in the brain of nude mice, forming GBMs that exhibit histopathological features of dog GBM [75].
In summary, canine GBM emerges as an attractive animal model for testing novel therapies in a spontaneous tumor in the context of a large brain. The features of dog GBM make it a unique large animal model for preclinical cancer research with therapeutic outcomes which could better predict their efficacy in human trials. In spite of these attractive features, dogs are very expensive to treat and scarce, therefore the routine testing on novel therapeutics in these animals would be unfeasible.
NON-INVASIVE SURROGATE ENDPOINTS TO ASSESS TUMOR PROGRESSION AND TREATMENT EFFICACY
Behavioral Tests Measuring Gene Therapy Efficacy on Brain Tumors
To evaluate therapeutic efficacy in pre-clinical models of brain cancer following gene therapy, it is not only necessary to monitor survival of the tumor bearing, treated animals, but to assess whether neurobiobehavioral function has returned to its baseline, pre-tumor state. This can be determined non-invasively by utilizing behavioral tests that assess general sensorimotor function and recovery from asymmetrical motor abnormalities resulting from tumor progression. While techniques to determine morphological changes have been well established, the introduction of behavioral testing to evaluate treatment efficacy on brain tumors is a fairly new concept.
Unilateral brain tumor formation produces distinct motor function asymmetries that can be detected and measured against the tumor’s progression. Of particular interest are tests that compare ipsilateral versus contralateral use of movement relative to the tumor-implanted hemisphere. Three general properties determine validity of behavioral test batteries developed for brain tumor models: ability to detect sensorimotor changes, ability to track tumor progression, and ability to assess recovery following treatment. Several established behavioral models originally developed to evaluate unilateral sensorimotor function in various rat models of central nervous system injury [76, 77] serve as appropriate measures for these properties.
The forelimb-use asymmetry test for rats, or “cylinder” test, which compares left-right weight bearing or shifting forelimb placement while rearing, has a high level of sensitivity for detecting motor function asymmetry [76, 77]. We have used this test to identify marked sensorimotor asymmetry in rats with striatal CNS-1 GBM 14-days following tumor implantation [78]. The test has also been used to evaluate treatment efficacy following gene therapy in the GBM model approximately 6 months following contralateral tumor re-implantation. Repeatability of the test also allows for its use in measuring tumor progression although evidence has not been presented to suggest it is capable of differentiating changes in magnitude based on severity of the tumor [79].
The somatosensory asymmetry test which uses bilateral tactile stimulation is also sensitive to the unilateral neurological changes associated with brain tumor induction and progression and is designed to measure magnitude changes in the impairment [76, 77, 80, 81]. This two part test is designed to elicit the natural tendency of rats to groom in the process of removing a foreign object that has been attached to their fur. The first part of the test consists of placing equally sized adhesive labels on the ventral forelimbs over repeated trials and determining whether an ipsilateral fore-limb preference with the tumor-bearing side is present. Once a unilateral sensorimotor impairment is detected the magnitude of the tumor impairment can be measured by varying the size of the bilateral stimuli until a “shift” in preference is observed. The test has been used to measure 9L-induced gliosarcoma impairment and progression [79] but its use in repeated trials to assess recovery has yet to be determined.
The vibrissae-elicited forelimb placing test, which can also be used to effectively quantify neurologically lateralized impairment, is based on the innate primary response of rats to use their vibrissae in gathering tactile information about their environment [77]. This test assesses unilateral sensorimotor damage by determining the percentage of trials in which a rat will use a particular forepaw to reach for a surface when it is partially suspended based on the notion that stimulation of the vibrissae against a surface elicits a placing response [76, 82]. The test has currently been used to assess the presence and development of impairment induced by brain tumors [79] or tumor-like brain compression [83]. While it has not been used in demonstrating treatment efficacy, the test produces a nearly perfect score in the absence of impairment making it highly sensitive to changes in tumor progress.
General locomotor activity behaviors in the presence of a brain tumor insult and treatment can also reveal potentially useful information on the progression of the disease. However, the innate tendency of laboratory rats to remain docile and inactive particularly during the light-phase complicates activity observations over longer time periods. The amphetamine-induced rotational behavior test, originally developed to look at Parkinson’s-like abnormalities in rats, uses amphetamine to stimulate activity over long periods and elicit biased ipsilateral rotational behavior in the presence of unilateral impairment [84-87]. As expected, tumor formations produce rotational asymmetry in a direction ipsilateral to the corresponding hemisphere making this a highly relevant test in the assessment of brain tumor impairment, progress, and recovery. We have recently demonstrated this by assessing the impaired behavioral profile of a GBM rat model both during tumor development and reversal of the effects 60 days later following gene therapy [78].
In addition to tests that directly measure the sensorimotor impairment and recovery associated with tumor formation and treatment, respectively, behavioral tests developed for this purpose should also include a general screen of locomotor activity and basic neurological function (i.e. auditory, visual, tactile). This can help determine if any residual damage exists due to development of the brain tumor mass and also detect potential abnormalities secondary to tumor regression upon treatment.
Assessment of Circulating Biomarkers
Efforts are ongoing to identify a quantitative circulating biomarker to monitor tumor progression in GBM patients in an earlier, faster and less invasive way. In a small study in GBM patients the methylation status of the promoters for p16, MGMT, p73, and RARβ was determined in glioma tissue and plasma [88]. Total plasma DNA content was found to be elevated in all the patients and 6/9 contained the same methylated promoters in plasma and in the tumor tissue, suggesting that plasma DNA analysis could be developed to monitor GBM molecular make up and tumor progression [88].
Insulin-like growth factor binding protein-2 (IGFBP-2) is a carrier protein for IGF that is overexpressed in GBM and has been postulated to increase malignancy [89]. Plasma levels of IGFBP-2 have been measured in ~200 glioma patients and 50 healthy volunteers [89]. Plasma IGFBP-2 levels were higher in high grade-glioma patients than in low-grade glioma or healthy donors and they correlated with recurrence and disease-free survival, indicating that circulating levels of this protein could be exploited as an assessment of therapeutic efficacy and tumor progression.
Considering that GBM progression requires recruitment of bone marrow-derived vascular precursors to sustain angiogenesis, the presence of endothelial progenitor cells and angiogenic activity was studied in blood of glioma patients [90]. Circulating CD133+ VEGFR+ cells and plasma proangiogenic activity were higher in GBM patients than in low-grade glioma patients [90] or healthy donors [91]. In GBM patients, the presence of circulating endothelial precursor cells was correlated with higher tumor blood vessel density [91] and an inverse correlation was found between percentage of these cells in blood and the survival after resection of GBM [90]. Thus, the levels of circulating endothelial precursors could be useful to assess GBM angiogenicity and tumor progression, as well as to monitor the efficacy of antiangiogenic therapy [90].
We have recently identified the circulating levels of the alarmin, high mobility group box 1 (HMGB1) protein as a potential biomarker of therapeutic efficacy for GBM [15, 20, 21]. HMGB1 is a nuclear protein that acts as a cytokine when released to the extracellular milieu by dying or inflammatory cells [92]. We found that circulating levels of HMGB1 increased in parallel with the efficacy of the treatment in several rodent GBM models, such as CNS-1, F98 and 9L rat brain tumor models and GL26, GL261 and B16 mouse brain tumor models [15, 20, 21]. The highest circulating levels of HMGB1 were reached when tumor-bearing animals are treated with a cytotoxic-immunotherapeutic gene therapy approach that led to tumor regression and long term survival [15, 20, 21], as described below (Fig. 2). In fact, the release of HMGB1 from dying tumor cells has been postulated to skew the immune response to dying tumor cells, which is critical for the outcome of anticancer therapies [21, 92-94]. HMGB1 release was also observed upon cytotoxic insult to human GBM cell lines and primary GBM cell cultures obtained from surgical biopsies [20], suggesting that this molecule could be used as a pharmacodynamic predictor of tumor regression in GBM patients, this awaits
Fig. (2). Combined cytotoxic-immunotherapy induces tumor regression and immunological memory.
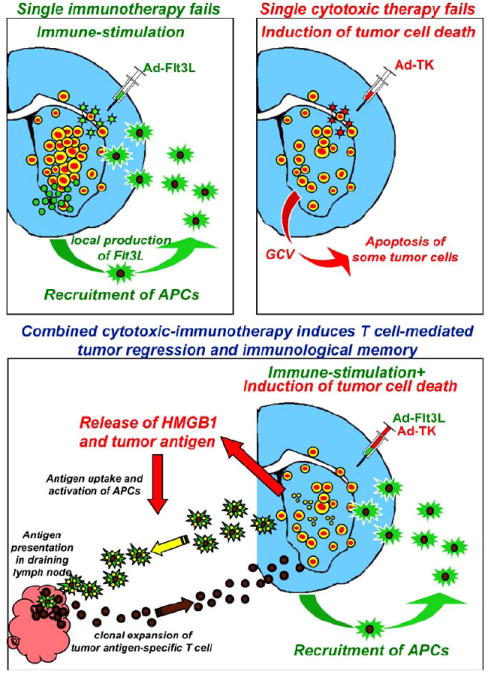
While single immunotherapy or cytotoxic therapy fails in inducing GBM regression, combination therapy leads to long term survival and immunological memory. We developed a gene therapeutic approach that combines one adenovirus that expresses Flt3L, which acts to increase the number of dendritic cells within the tumor microenvironment, with a second adenovirus, which expresses thymidine kinase, that together with the prodrug ganciclovir, kills brain tumor cells, releasing tumor antigens and inflammatory molecules from dying tumor cells. These tumor antigens are then taken up by the infiltrating dendritic cells, transported to the lymph nodes, where T cells are primed to induce a cytotoxic T cell response, which leads to tumor regression and immunological memory
Imaging
The current routine technique for tumor monitoring in glioblastoma patients is contrast-enhanced magnetic resonance imaging (MRI). However, this technique exhibits a relatively low resolution threshold of 400 tumor cells/mm2 for tumor detection [95]. Also, the contrast enhancement is heterogeneous amongst patients and can be affected by the concurrent administration of routinely used drugs, such as dexamethasone and antiangiogenic agents, making difficult the accurate determination of tumor size [96]. Current efforts are aimed at increasing the resolution of imaging techniques and enhance their ability to better discern between tumoral tissue and peritumoral edema [97]. Characteristic features of GBM, such as increased vascularization, proliferation and hypoxia, are being exploited to better delineate the tumor area using diffusion MRI, perfusion MRI and targeted tracers for positron emission tomography (PET) [96]. Diffusion-weighted imaging monitors cell density using apparent diffusion coefficient (ADC) maps, in which high ADC values correspond to areas of low cell density, where water can freely diffuse, while low ADC values correspond to increased cell density [98, 99]. Arterial spin labeling (ASL), a novel method that measures blood flow without using contrast agents, detects changes in the magnetic resonance signal emitted by the molecules of water when passing through the tumor mass [100]. Thus, ASL allows to monitor changes in the tumor vascularizacion and permeability upon treatment with antiangiogenic agents [101]. Promising PET tracers include the thymidine analog 30-deoxy-30-18F-fluorothymidine (FLT), which uptake is correlated to mitotic activity, and 18F-fluoromisonidazole (FMISO), which is trapped in hypoxic cells [96]. Treatment of GBM patients with antiangiogenic therapy led to 25% reduction in FLT uptake, which was associated with longer overall survival [102]. On the other hand, increased uptake of FMISO was found to be associated with poorer survival of GBM patients [103].
ACTIVATION OF INNATE IMMUNE RECEPTORS IS ESSENTIAL TO STIMULATE STRONG, CLINICALLY EFFECTIVE, ANTI-BRAIN TUMOR IMMUNITY
Immune privilege, tumor immune evasion and a lack of dendritic cells (DC) in the normal brain parenchyma, all contribute to immunological ignorance against glioblastoma multiforme (GBM) antigens [104, 105]. Current GBM treatments involve surgery, radiotherapy and chemotherapy, which attempt to eliminate these large tumors by directly killing tumor cells. Nevertheless, median survival is still under 18 months for patients with the most aggressive type of primary brain tumor, GBM [106].
As the normal brain parenchyma lacks dendritic cells, it is virtually impossible to prime an adaptive immune response against antigens appearing exclusively in the brain, because there are no cells available to transport tumor antigens to the draining lymph nodes where the priming of adaptive immune responses occurs. Thus, we developed a novel strategy to combat these deadly brain tumors which involves engineering the brain tumor microenvironment utilizing adenoviruses to express therapeutic transgenes. One adenovirus expresses a conditionally cytotoxic gene, i.e., herpes simplex type 1-thymidine kinase (TK), which, together with the pro-drug ganciclovir (GCV), kills brain tumor cells [18]. This releases tumor antigens into the surrounding tumor environment [21]. A second adenovirus expresses fms-like tyrosine kinase-3 ligand (Flt3L), which acts to increase the number of dendritic cells within the tumor microenvironment [16, 21, 107]. The released tumor antigens are then taken up by the infiltrating dendritic cells, transported to the draining lymph nodes, where T cells are primed to induce a cytotoxic T cell response which is able to induce brain tumor regression and immunological memory (Fig. 2) [21].
An important new step which was discovered recently by us, and published in PLoS Medicine in January 2009, indicates that activation of Toll-like receptors (TLR) is essential to induce the effective anti-tumor immune response. In the absence of TLR2 signaling, DCs fail to infiltrate the tumor; further, DCs lacking TLR2 failed to stimulate the proliferation and activation of tumor antigen specific T cells. In the absence of TLR2, CD8+ T cell dependent tumor regression was ablated [21]. Our data thus demonstrated that activation of a receptor of the innate immune system is necessary to stimulate an effective anti-brain tumor immune response directly from within the brain tumor microenvironment.
These results beg the question of how is an innate immune receptor activated in the absence of known ligands that normally stimulate the innate immune response. Further experiments then demonstrated that brain tumor cell death induced by TK/GCV causes the release of the protein high-mobility-group box 1 (HMGB1) [21]. HMGB1 is normally bound tightly to nuclear chromosomes [108-110], but is released upon tumor cell death [93, 111-113]. Released, HMGB1 then acts as an endogenous ligand for Toll-like receptor type 2 (TLR) located on bone marrow-derived dendritic cells, thus activating them [21]. However, if we neutralize HMGB1 with specific antibodies or glycyrrhizin, both of which bind HMGB1 and thus block its capacity to activate TLR2 receptors, the combined cytotoxic/immunetherapy fails [21]. Thus, tumor cell death is necessary for the release of an essential TLR2 ligand, the endogenous protein HMGB1, which, through the activation of infiltrating DCs, leads to the stimulation of an effective anti-brain tumor cytotoxic T cell response.
These experiments uncover the basis for the proposed synergy between tumor cell killing and the stimulation of the anti-tumor immune response, and shed light on the mechanisms by which cytotoxicity contributes to specific activation of the immune system. Further, the data identifying HMGB1 as the endogenous TLR2 ligand provides experimental evidence as to why activation of an innate immune receptor normally activated by infectious agents is necessary for the effective anti-brain tumor immune response. Finally, understanding TLR2 activation and signaling in DCs could provide new tools to elicit the activation of a strong and clinically effective anti-tumor immune response. Neither cytotoxic nor immune-stimulatory therapies on their own are likely to be clinically effective, but both, in synchrony with TLR2 activation, should result in stronger anti-tumor immunity. The identification of TLR2 also paves the way to use TLR2 agonists as specific therapeutic adjuvants with concurrent chemotherapy, radiotherapy and immune-therapy approaches.
PRO-APOPTOTIC GENE THERAPY TARGETS
Induction of GBM tumor cell death is essential not only to kill tumor cells and reduce tumor burden, but also to induce the release of inflammatory molecules from dying tumor cells, which are crucial for the generation of a systemic anti-tumor immune response [20, 21]. Considering that the expression of pro-apoptotic cytokines and their receptors has been detected in human GBM, targeting of these receptors has been attempted to develop novel treatment strategies for this devastating cancer. Since pro-apoptotic cytokines released from infected cells elicit strong bystander effect, their delivery using gene therapy vectors is an attractive therapeutic modality to treat GBM. Delivery of TNF-α [20, 114], TRAIL [20, 115, 116] and FasL [20, 117, 118] into intracranial GBM in rodents has been attempted using gene therapy vectors by us and others. Although these vectors exhibit strong pro-apoptotic effects in vitro in GBM cells, in vivo they only marginally improve the survival of rodent models of GBM [20, 114, 116, 117, 119]. The presence of soluble receptors for TNF-α and FasL [120, 121], as well as the requirement of irradiation or chemotherapy to enhance expression of TRAIL receptors [116, 122-124] have been implicated in the low efficacy of these vectors. Also, pro-apoptotic cytokines that target tumor cells expressing a specific death receptor may lead to the selection of non-expressing cells that become resistant to the gene therapy.
Considering that in clinical trials gene therapy vectors are injected in the margins of tumor cavity after surgical resection of the tumor [7] it is critical to use pro-apoptotic agents whose cytotoxic effects are specific to GBM cells, in order to avoid neurological side effects. Recent results from our lab have shown that Ad-mediated delivery of TRAIL or FasL into the naïve rat brain leads to severe neuropathological side effects, including hemorrhages and significant brain tissue loss with reduction of tyrosine hydroxilase (TH) expression, demyelinization and a very strong inflammatory response [20]. These findings are in accordance with the presence of death receptors for TRAIL and FasL in neuronal cell bodies and fibers [20]. Considering that these receptors have also been detected in the normal human brain [125-127] and that there is a bystander effect exerted by the release of the pro-apoptotic cytokines from infected cells, administration of Ads encoding FasL or TRAIL into the normal brain bears a high risk of neurotoxicity.
Neurotoxicity could be reduced by delivering these powerful proapoptotic cytokines under the control of inducible promoters, which expression can be stopped by removal of the inducer [128]. The possibility of temporally and spatially controlling the expression of radio-inducible vectors using radiotherapy would make them safer and more specific vectors to deliver pro-apoptotic molecules that are potentially toxic to the surrounding brain parenchyma. Intratumoral delivery of caspase 8 under the control of a radiation-inducible early growth response gene-1 (EGR-1) promoter in combination with fractionated radiotherapy induces GBM cell death and tumor regression [129]. Expression of TRAIL under EGR-1 combined with radiotherapy leads to overexpression of TRAIL receptors and tumor cell death with synergistic reduction of GBM burden [129]. Ad-mediated delivery of TNF-α under the control of a chemo-radio-inducible promoter leads to prolonged survival of mice bearing intracranial GBM xenografts upon systemic administration of temozolomide [130]. Alternatively, the hypoxic tumor microenvironment can be exploited to increase the specificity of the pro-apoptotic gene therapy. Over-expression of the pro-apoptotic molecule Bax under the control of hypoxia responsive elements increases GBM cell death in hypoxic conditions in vitro and in vivo [131]. Nevertheless, the safety of these inducible vectors remains to be determined upon their administration into the naïve brain.
The specificity of pro-apoptotic gene therapy approaches can be enhanced by expressing the cytotoxic agents under the control of tumor-specific promoters. Overexpression of p53-upregulated modulator of apoptosis (PUMA) under the control of a tumor-specific promoter (hTERT) induces massive apoptosis in GBM cells, while sparing normal cells in surrounding non-neoplastic tissues [132]. However, it has to be taken into account that cell type-specific promoters exhibit lower transduction efficiency and expression levels than ubiquitous promoters [133], requiring higher doses to exert therapeutic efficacy, which could lead to inflammatory responses against the gene therapy vectors [18, 134].
Conditionally cytotoxic genes, such as the HSV-1 thymidine kiase (HSV1-TK), encode enzymes that are non-toxic until the administration of a chain terminator nucleotide analog [8]. The therapeutic enzyme phosphorylates the non-toxic prodrug into a highly toxic metabolite, a phosphorylated nucleotide that is included in the DNA chain during replication and can not be further elongated, leading to cell death of proliferating cells [135]. The by-stander effect of this approach relies in the passage of phosphorylated nucleotide to neighboring cells through gap junctions, amplifying the cytotoxic effect of HSV1-TK [136]. Since proliferating cells are encountered within the tumor at all stages and tumor cell replication is a requirement for tumor progression, targeting these cells with local delivery of conditionally cytotoxic molecules using gene therapy vectors is a very valuable strategy to induce apoptosis in GBM [7]. Since these agents exert their cytotoxic effect only in proliferating cells, their pro-apoptotic effects are very specific to GBM cells when delivered within the brain (Fig. 3). Also, considering the requirement of exogenous nucleotide analogs to induce cell death, the withdrawal of the pro-drug could stop the development of putative adverse side effects. Our previous results indicate that administration of Ad-TK in the naïve brain of rats [20] and dogs [69-71] does not significantly alter the structure of the normal brain and induces only a mild, transient local inflammation. Importantly, it has been demonstrated that injection of adenoviral vectors encoding HSV1-TK into the margins of the tumor cavity after GBM surgical resection followed by GCV administration was well tolerated in over 70 patients in 6 early clinical trials [2]. We recently compared the ability of Ad-TK+GCV with several Ads encoding pro-apoptotic cytokines, i.e. TNF-α, TRAIL and FasL, to employ in combination with Ad-Flt3L [20]. Delivery of HSV1-TK exerted the highest therapeutic efficacy in rats bearing small intracranial GBM (1.6 mm3), leading to over 70% long term survival. Moreover, administration of Ad-TK+GCV was the only pro-apoptotic approach that when combined with Ad-Flt3L elicited an anti-tumor immune response that led to regression of large intracranial GBM in rats (35 mm3) [20]. Importantly, efficacy of Ad-TK+GCV treatment was tested in several rat and mouse brain tumor models (Fig. 4) [15, 21]. Delivery of Ad-TK is also a very attractive gene therapy approach to use in combination with more traditional therapeutic agents, such as temozolomide, an alkylating agent routinely used in the treatment of GBM patients [5]. Phosphorylated GCV was found to inhibit DNA polymerase δ, an enzyme involved in repair of DNA cross-links, which leads to synergy between TK+GCV and temozolamide [137]. Alternatively, oncolytic adenovirus, lentivirus and HSV viral vectors have been developed that selectively replicate in tumor cells leading to their eventual death. These vector platforms are currently under evaluation in both pre-clinical and clinical trials [138-143]. This promising approach is discussed in other articles within this current issue.
Fig. (3). Proliferating cells, the target for conditionally cytotoxic approaches, are limited to the GBM mass.
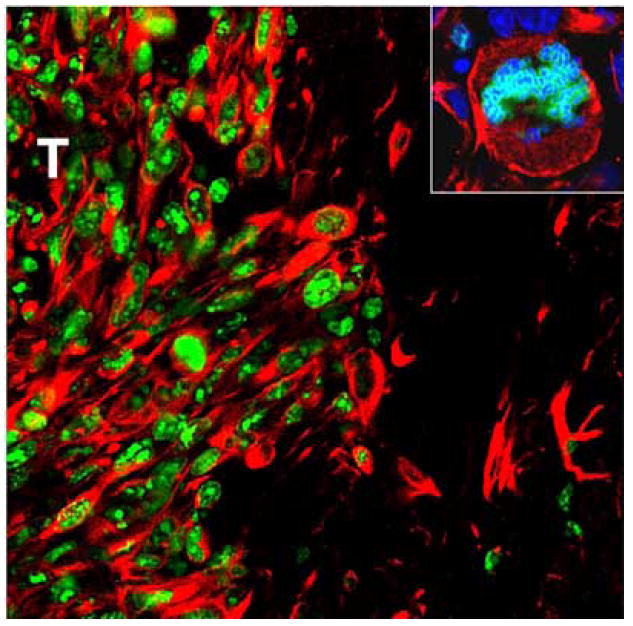
Confocal microphotographs show detection of proliferating cells stained with an anti-Ki67 antibody (green) in 9-day intracranial CNS-1 GBM in rats. Tumor cells were labeled with anti-vimentin antibodies (red). Nuclei were stained with DAPI (blue). T: tumor area. Width of illustrated field: 500 μm (Inset = 30 μm).
Fig. (4). Efficacy of combined cytotoxic-immunotherapy in several rodent models of brain cancer.
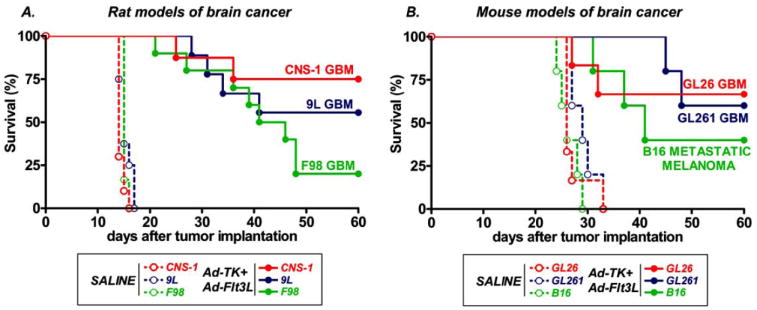
A, Kaplan Meier survival curves show the survival of rats bearing intracranial CNS-1, F98 or 9L GBM tumors that were treated at 7-9 days with saline (n=6-10) or Ad-Flt3L+Ad-TK (n=8-10) followed by GCV administration. B, Kaplan Meier survival curves show the survival of C57/B6 mice that were implanted with GL26 or GL261 GBM cells or B16 melona cells in the brain. Mice were treated 17 days later with saline (n=5-6) or Ad-Flt3L+Ad-TK (n=5-6), followed by GCV administration.
In summary, although tumor cell death is essential in the therapy of GBM, delivery of highly cytotoxic molecules can damage the brain parenchyma surrounding the tumor mass, hampering its application in patients. Thus, preclinical evaluation of pro-apoptotic gene therapy vectors needs to comprise not only efficacy assessment in relevant intracranial GBM animal models, but also meticulous evaluation of their neuropathology in the naïve brain.
THE CLINICAL SCENARIO
Considering the invasive nature of GBM, complete tumor resection is virtually impossible and the tumor recurs in spite of aggressive surgical resection. For decades GBM treatment has been approached by attempting to kill infiltrating GBM cells by administration of chemotherapy and radiotherapy following surgery. However, only modest improvement in the median survival has been achieved [1, 2]. Failure of these therapies has been attributed to the lack of specificity for neoplastic tissue, which results in dose-limiting toxicity, as well as to the intrinsic resistance of GBM cells to these therapies. Thus, immunotherapy has been proposed as a very specific anti-tumor approach that stimulates the immune system to target and kill GBM cells. However, growing evidence suggests that immunotherapies require combination with tumor cell killing strategies in order to induce anti-tumor responses. Immunogenic apoptotic tumor cell death seems to be crucial to trigger efficient anti-tumor immunity. Release of HMGB1 from dying tumor cells [20, 21, 92], as well as exposure of calreticulin in the membrane of apoptotic cells [144] have been shown to act as powerful pro-inflammatory signals that initiate anti-tumor immune responses upon apoptotic cell death. Indeed, GBM patients that received intracerebral administration of HSV1-TK during tumor resection followed by ganciclovir and radiotherapy showed an increase in the number of tumor-specific IFN-γ-producing T cells that was accompanied by a peak in serum IL-12 and FasL levels, suggesting that HSV1-TK gene therapy induces the development of a Th-1 immune response [145].
Challenging the old dogma that states that chemotherapy-induced immunosuppression abrogates the development of anti-tumor immune responses, several clinical trials actually indicate that chemotherapy have also a synergistic effect when combined with immunotherapy. Combination of high dose chemotherapy with adoptive immunotherapy was tested in a small Phase I clinical trial in 3 pediatric patients with recurrent brain tumors [146]. Patients received high-dose chemotherapy followed by adoptive transfer of peripheral blood T-cells expanded ex vivo after immunization with autologous cancer antigens obtained at tumor resection. Two of the patients exhibited markedly prolonged survival when compared to historical controls. Indeed, it has been proposed that chemotherapy-induced immunogenicity actually overcomes its immunosuppressive effect since even during chemotherapy-induced leucopenia, patients display a functional T cell system [147]. The reduction in the tumor size following chemotherapy and other tumor cell killing strategies has been postulated to also facilitate the efficacy of immunotherapy due to a higher T cell:cancer cell ratio [148].
Not only induction of tumor cell death has an immune-stimulant effect through the release of intracellular inflammatory molecules, which enhances the efficacy of immunotherapy, but also the induction of anti-tumor immune responses seems to increase the sensibility of tumor cells to tumor cell killing strategies [149]. In a Phase I clinical trial in which GBM patients were treated with dendritic cells pulsed with autologous tumor peptides followed by adjuvant therapy with chemotherapy the 2-year overall survival rate was 50%, with 2/12 patients living over 4 years. In another trial, patients bearing primary GBM that received chemotherapy after vaccination with autologous tumor antigen-pulsed DCs exhibited slower tumor progression and longer survival than those that received either chemotherapy or vaccination alone [150]. It has been proposed that targeting of the GBM antigen tyrosinase-related peptide (trp) by endogenous cytotoxic T-cells leads to an increase in the chemo-sensitivity of the tumor by reducing the number of trp-expressing cells [151], which seem to be resistant to chemotherapy and radiotherapy [152]. It has also been suggested that temozolamide could synergize with immunotherapy by selectively depleting CD4+/CD25+ regulatory T cells [148, 153].
In conclusion, growing preclinical and clinical data indicates that combination of tumor cell killing strategies with immunotherapy leads to synergism between the two approaches, which would lead to improve efficacy and reduced toxicity. This body of evidence contradicts the old dogma that stated that tumor cell killing strategies would impair the ability of the immune system to recognize and destroy a brain tumor and supports the application of combined cytotoxic-immunotherapeutic strategies in the treatment of GBM patients.
Acknowledgments
FUNDING This work is supported by National Institutes of Health/National Institute of Neurological Disorders & Stroke (NIH/NINDS) Grant 1R01 NS44556.01, Minority Supplement NS445561.01; 1R21-NSO54143.01; 1UO1 NS0524-65.01, 1 RO3 TW006273-01; 1RO1-NS 057711 to M.G.C.; NIH/NINDS Grants 1 RO1 NS 054193.01; RO1 NS 42893.01, U54 NS045309-01 and 1R21 NS047298-01 to P.R.L. The Bram and Elaine Goldsmith and the Medallions Group Endowed Chairs in Gene Therapeutics to PRL and MGC, respectively, The Linda Tallen & David Paul Kane Foundation Annual Fellowship and the Board of Governors at CSMC. M.C is supported by NIH/NINDS 1F32 NS058156.01.
References
- 1.Chinot OL, Barrie M, Fuentes S, et al. Correlation between O6-methylguanine-DNA methyltransferase and survival in inoperable newly diagnosed glioblastoma patients treated with neoadjuvant temozolomide. J Clin Oncol. 2007;25:1470–5. doi: 10.1200/JCO.2006.07.4807. [DOI] [PubMed] [Google Scholar]
- 2.Sathornsumetee S, Rich JN. Designer therapies for glioblastoma multiforme. Ann N Y Acad Sci. 2008;1142:108–32. doi: 10.1196/annals.1444.009. [DOI] [PubMed] [Google Scholar]
- 3.Lefranc F, Brotchi J, Kiss R. Possible future issues in the treatment of glioblastomas: special emphasis on cell migration and the resistance of migrating glioblastoma cells to apoptosis. J Clin Oncol. 2005;23:2411–22. doi: 10.1200/JCO.2005.03.089. [DOI] [PubMed] [Google Scholar]
- 4.Hegi ME, Diserens AC, Godard S, et al. Clinical trial substantiates the predictive value of O-6-methylguanine-DNA methyltransferase promoter methylation in glioblastoma patients treated with temozolomide. Clin Cancer Res. 2004;10:1871–4. doi: 10.1158/1078-0432.ccr-03-0384. [DOI] [PubMed] [Google Scholar]
- 5.Hegi ME, Diserens AC, Gorlia T, et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N Engl J Med. 2005;352:997–1003. doi: 10.1056/NEJMoa043331. [DOI] [PubMed] [Google Scholar]
- 6.Amberger-Murphy V. Hypoxia helps glioma to fight therapy. Curr Cancer Drug Targets. 2009;9:381–90. doi: 10.2174/156800909788166637. [DOI] [PubMed] [Google Scholar]
- 7.Pulkkanen KJ, Yla-Herttuala S. Gene therapy for malignant glioma: current clinical status. Mol Ther. 2005;12:585–98. doi: 10.1016/j.ymthe.2005.07.357. [DOI] [PubMed] [Google Scholar]
- 8.King GD, Curtin JF, Candolfi M, et al. Gene therapy and targeted toxins for glioma. Curr Gene Ther. 2005;5:535–57. doi: 10.2174/156652305774964631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Candolfi M, Curtin JF, Nichols WS, et al. Intracranial glioblastoma models in preclinical neuro-oncology: neuropathological characterization and tumor progression. J Neurooncol. 2007;85:133–48. doi: 10.1007/s11060-007-9400-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kim CH, Hong MJ, Park SD, et al. Enhancement of anti-tumor immunity specific to murine glioma by vaccination with tumor cell lysate-pulsed dendritic cells engineered to produce interleukin-12. Cancer Immunol Immunother. 2006;55(11):1309–19. doi: 10.1007/s00262-006-0134-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sampson JH, Ashley DM, Archer GE, et al. Characterization of a spontaneous murine astrocytoma and abrogation of its tumorigenicity by cytokine secretion. Neurosurgery. 1997;41:1365–72. doi: 10.1097/00006123-199712000-00024. discussion 72-3. [DOI] [PubMed] [Google Scholar]
- 12.Kruse CA, Molleston MC, Parks EP, et al. A rat glioma model, CNS-1, with invasive characteristics similar to those of human gliomas: a comparison to 9L gliosarcoma. J Neurooncol. 1994;22:191–200. doi: 10.1007/BF01052919. [DOI] [PubMed] [Google Scholar]
- 13.Tzeng JJ, Barth RF, Orosz CG, James SM. Phenotype and functional activity of tumor-infiltrating lymphocytes isolated from immunogenic and nonimmunogenic rat brain tumors. Cancer Res. 1991;51:2373–8. [PubMed] [Google Scholar]
- 14.Barth RF. Rat brain tumor models in experimental neuro-oncology: the 9L, C6, T9, F98, RG2 (D74), RT-2 and CNS-1 gliomas. J Neurooncol. 1998;36:91–102. doi: 10.1023/a:1005805203044. [DOI] [PubMed] [Google Scholar]
- 15.Muhammad AKMG, Candolfi M, King GD, et al. Anti-glioma immunological memory in response to conditional cytotoxic/immune-stimulatory gene therapy: humoral and cellular immunity lead to tumor regression. Clin Cancer Res. 2009 doi: 10.1158/1078-0432.CCR-09-1087. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ali S, Curtin JF, Zirger JM, et al. Inflammatory and anti-glioma effects of an adenovirus expressing human soluble Fms-like tyrosine kinase 3 ligand (hsFlt3L): treatment with hsFlt3L inhibits intracranial glioma progression. Mol Ther. 2004;10:1071–84. doi: 10.1016/j.ymthe.2004.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ali S, King GD, Curtin JF, et al. Combined immunostimulation and conditional cytotoxic gene therapy provide long-term survival in a large glioma model. Cancer Res. 2005;65:7194–204. doi: 10.1158/0008-5472.CAN-04-3434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dewey RA, Morrissey G, Cowsill CM, et al. Chronic brain inflammation and persistent herpes simplex virus 1 thymidine kinase expression in survivors of syngeneic glioma treated by adenovirus-mediated gene therapy: implications for clinical trials. Nat Med. 1999;5:1256–63. doi: 10.1038/15207. [DOI] [PubMed] [Google Scholar]
- 19.Owens GC, Orr EA, DeMasters BK, et al. Overexpression of a transmembrane isoform of neural cell adhesion molecule alters the invasiveness of rat CNS-1 glioma. Cancer Res. 1998;58:2020–8. [PubMed] [Google Scholar]
- 20.Candolfi M, Yagiz K, Foulad D, et al. Release of HMGB1 in response to proapoptotic glioma killing strategies: efficacy and neurotoxicity. Clin Cancer Res. 2009;15:4401–14. doi: 10.1158/1078-0432.CCR-09-0155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Curtin J, Liu N, Candolfi M, et al. HMGB1 mediates endogenous TLR2 activation and brain tumor regression. PLoS Med. 2009;6:e10. doi: 10.1371/journal.pmed.1000010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.King GD, Muhammad AG, Xiong W, et al. High-Capacity adenoviral vector-mediated anti-glioma gene therapy in the presence of systemic anti-adenovirus immunity. J Virol. 2008;82:4680–4. doi: 10.1128/JVI.00232-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.King GD, Muhammad AKM, Curtin JF, et al. Flt3L and TK gene therapy eradicate multifocal glioma in a syngeneic glioblastoma model. Neuro Oncol. 2008;10:19–31. doi: 10.1215/15228517-2007-045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Giannini C, Sarkaria JN, Saito A, et al. Patient tumor EGFR and PDGFRA gene amplifications retained in an invasive intracranial xenograft model of glioblastoma multiforme. Neuro Oncol. 2005;7:164–76. doi: 10.1215/S1152851704000821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sarkaria JN, Carlson BL, Schroeder MA, et al. Use of an orthotopic xenograft model for assessing the effect of epidermal growth factor receptor amplification on glioblastoma radiation response. Clin Cancer Res. 2006;12:2264–71. doi: 10.1158/1078-0432.CCR-05-2510. [DOI] [PubMed] [Google Scholar]
- 26.Cho SY, Ravasi L, Szajek LP, et al. Evaluation of (76)Br-FBAU as a PET reporter probe for HSV1-tk gene expression imaging using mouse models of human glioma. J Nucl Med. 2005;46:1923–30. [PubMed] [Google Scholar]
- 27.Kirsch M, Strasser J, Allende R, et al. Angiostatin suppresses malignant glioma growth in vivo. Cancer Res. 1998;58:4654–9. [PubMed] [Google Scholar]
- 28.Lund EL, Bastholm L, Kristjansen PE. Therapeutic synergy of TNP-470 and ionizing radiation: effects on tumor growth, vessel morphology, and angiogenesis in human glioblastoma multiforme xenografts. Clin Cancer Res. 2000;6:971–8. [PubMed] [Google Scholar]
- 29.Schmidt NO, Ziu M, Carrabba G, et al. Antiangiogenic therapy by local intracerebral microinfusion improves treatment efficiency and survival in an orthotopic human glioblastoma model. Clin Cancer Res. 2004;10:1255–62. doi: 10.1158/1078-0432.ccr-03-0052. [DOI] [PubMed] [Google Scholar]
- 30.Tentori L, Leonetti C, Scarsella M, et al. Systemic administration of GPI 15427, a novel poly(ADP-ribose) polymerase-1 inhibitor, increases the antitumor activity of temozolomide against intracranial melanoma, glioma, lymphoma. Clin Cancer Res. 2003;9:5370–9. [PubMed] [Google Scholar]
- 31.Shen S, Khazaeli MB, Gillespie GY, Alvarez VL. Radiation dosimetry of 131I-chlorotoxin for targeted radiotherapy in glioma-bearing mice. J Neurooncol. 2005;71:113–9. doi: 10.1007/s11060-004-0890-4. [DOI] [PubMed] [Google Scholar]
- 32.Kawakami K, Kawakami M, Kioi M, Husain SR, Puri RK. Distribution kinetics of targeted cytotoxin in glioma by bolus or convection-enhanced delivery in a murine model. J Neurosurg. 2004;101:1004–11. doi: 10.3171/jns.2004.101.6.1004. [DOI] [PubMed] [Google Scholar]
- 33.Cirielli C, Inyaku K, Capogrossi MC, Yuan X, Williams JA. Adenovirus-mediated wild-type p53 expression induces apoptosis and suppresses tumorigenesis of experimental intracranial human malignant glioma. J Neurooncol. 1999;43:99–108. doi: 10.1023/a:1006289505801. [DOI] [PubMed] [Google Scholar]
- 34.Jiang H, Gomez-Manzano C, Alemany R, et al. Comparative effect of oncolytic adenoviruses with E1A-55 kDa or E1B-55 kDa deletions in malignant gliomas. Neoplasia. 2005;7:48–56. doi: 10.1593/neo.04391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Aghi M, Rabkin S, Martuza RL. Effect of chemotherapy-induced DNA repair on oncolytic herpes simplex viral replication. J Natl Cancer Inst. 2006;98:38–50. doi: 10.1093/jnci/djj003. [DOI] [PubMed] [Google Scholar]
- 36.Conrad C, Miller CR, Ji Y, et al. Delta24-hyCD adenovirus suppresses glioma growth in vivo by combining oncolysis and chemosensitization. Cancer Gene Ther. 2005;12:284–94. doi: 10.1038/sj.cgt.7700750. [DOI] [PubMed] [Google Scholar]
- 37.Lamfers ML, Gianni D, Tung CH, et al. Tissue inhibitor of metalloproteinase-3 expression from an oncolytic adenovirus inhibits matrix metalloproteinase activity in vivo without affecting antitumor efficacy in malignant glioma. Cancer Res. 2005;65:9398–405. doi: 10.1158/0008-5472.CAN-04-4264. [DOI] [PubMed] [Google Scholar]
- 38.Samoto K, Ehtesham M, Perng GC, et al. A herpes simplex virus type 1 mutant with gamma 34.5 and LAT deletions effectively oncolyses human U87 glioblastomas in nude mice. Neurosurgery. 2002;50:599–605. doi: 10.1097/00006123-200203000-00031. discussion -6. [DOI] [PubMed] [Google Scholar]
- 39.Zhu Y, Parada LF. The molecular and genetic basis of neurological tumours. Nat Rev Cancer. 2002;2:616–26. doi: 10.1038/nrc866. [DOI] [PubMed] [Google Scholar]
- 40.Sanson M, Thillet J, Hoang-Xuan K. Molecular changes in gliomas. Curr Opin Oncol. 2004;16:607–13. doi: 10.1097/01.cco.0000142485.81849.cc. [DOI] [PubMed] [Google Scholar]
- 41.Nagane M, Lin H, Cavenee WK, Huang HJ. Aberrant receptor signaling in human malignant gliomas: mechanisms and therapeutic implications. Cancer Lett. 2001;162(Suppl):S17–S21. doi: 10.1016/s0304-3835(00)00648-0. [DOI] [PubMed] [Google Scholar]
- 42.Schwartzbaum JA, Fisher JL, Aldape KD, Wrensch M. Epidemiology and molecular pathology of glioma. Nat Clin Pract Neurol. 2006;2:494–503. doi: 10.1038/ncpneuro0289. quiz 1p following 516. [DOI] [PubMed] [Google Scholar]
- 43.Reilly KM. Brain tumor susceptibility: the role of genetic factors and uses of mouse models to unravel risk. Brain Pathol. 2009;19:121–31. doi: 10.1111/j.1750-3639.2008.00236.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Olson MV, Johnson DG, Jiang H, et al. Transgenic E2F1 expression in the mouse brain induces a human-like bimodal pattern of tumors. Cancer Res. 2007;67:4005–9. doi: 10.1158/0008-5472.CAN-06-2973. [DOI] [PubMed] [Google Scholar]
- 45.Begemann M, Fuller GN, Holland EC. Genetic modeling of glioma formation in mice. Brain Pathol. 2002;12:117–32. doi: 10.1111/j.1750-3639.2002.tb00428.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ding H, Roncari L, Shannon P, et al. Astrocyte-specific expression of activated p21-ras results in malignant astrocytoma formation in a transgenic mouse model of human gliomas. Cancer Res. 2001;61:3826–36. [PubMed] [Google Scholar]
- 47.Weissenberger J, Steinbach JP, Malin G, et al. Development and malignant progression of astrocytomas in GFAP-v-src transgenic mice. Oncogene. 1997;14:2005–13. doi: 10.1038/sj.onc.1201168. [DOI] [PubMed] [Google Scholar]
- 48.Assanah M, Lochhead R, Ogden A, et al. Glial progenitors in adult white matter are driven to form malignant gliomas by platelet-derived growth factor-expressing retroviruses. J Neurosci. 2006;26:6781–90. doi: 10.1523/JNEUROSCI.0514-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Uhrbom L, Hesselager G, Nister M, Westermark B. Induction of brain tumors in mice using a recombinant platelet-derived growth factor B-chain retrovirus. Cancer Res. 1998;58:5275–9. [PubMed] [Google Scholar]
- 50.Shih AH, Dai C, Hu X, et al. Dose-dependent effects of platelet-derived growth factor-B on glial tumorigenesis. Cancer Res. 2004;64:4783–9. doi: 10.1158/0008-5472.CAN-03-3831. [DOI] [PubMed] [Google Scholar]
- 51.Holland EC, Hively WP, DePinho RA, Varmus HE. A constitutively active epidermal growth factor receptor cooperates with disruption of G1 cell-cycle arrest pathways to induce glioma-like lesions in mice. Genes Dev. 1998;12:3675–85. doi: 10.1101/gad.12.23.3675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dai C, Celestino JC, Okada Y, et al. PDGF autocrine stimulation dedifferentiates cultured astrocytes and induces oligodendrogliomas and oligoastrocytomas from neural progenitors and astrocytes in vivo. Genes Dev. 2001;15:1913–25. doi: 10.1101/gad.903001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Singer O, Verma IM. Applications of lentiviral vectors for shRNA delivery and transgenesis. Curr Gene Ther. 2008;8:483–8. doi: 10.2174/156652308786848067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Marumoto T, Tashiro A, Friedmann-Morvinski D, et al. Development of a novel mouse glioma model using lentiviral vectors. Nat Med. 2009;15:110–6. doi: 10.1038/nm.1863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wiesner SM, Decker SA, Larson JD, et al. De novo induction of genetically engineered brain tumors in mice using plasmid DNA. Cancer Res. 2009;69:431–9. doi: 10.1158/0008-5472.CAN-08-1800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ohlfest JR, Freese AB, Largaespada DA. Nonviral vectors for cancer gene therapy: prospects for integrating vectors and combination therapies. Curr Gene Ther. 2005;5:629–41. doi: 10.2174/156652305774964749. [DOI] [PubMed] [Google Scholar]
- 57.Ohlfest JR, Lobitz PD, Perkinson SG, Largaespada DA. Integration and long-term expression in xenografted human glioblastoma cells using a plasmid-based transposon system. Mol Ther. 2004;10:260–8. doi: 10.1016/j.ymthe.2004.05.005. [DOI] [PubMed] [Google Scholar]
- 58.Heidner GL, Kornegay JN, Page RL, Dodge RK, Thrall DE. Analysis of survival in a retrospective study of 86 dogs with brain tumors. J Vet Intern Med. 1991;5:219–26. doi: 10.1111/j.1939-1676.1991.tb00952.x. [DOI] [PubMed] [Google Scholar]
- 59.Foster ES, Carrillo JM, Patnaik AK. Clinical signs of tumors affecting the rostral cerebrum in 43 dogs. J Vet Intern Med. 1988;2:71–4. doi: 10.1111/j.1939-1676.1988.tb02796.x. [DOI] [PubMed] [Google Scholar]
- 60.LeCouteur RA. Current concepts in the diagnosis and treatment of brain tumours in dogs and cats. J Small Anim Pract. 1999;40:411–6. doi: 10.1111/j.1748-5827.1999.tb03113.x. [DOI] [PubMed] [Google Scholar]
- 61.Stonehewer J, Mackin AJ, Tasker S, Simpson JW, Mayhew IG. Idiopathic phenobarbital-responsive hypersialosis in the dog: an unusual form of limbic epilepsy? J Small Anim Pract. 2000;41:416–21. doi: 10.1111/j.1748-5827.2000.tb03236.x. [DOI] [PubMed] [Google Scholar]
- 62.Stoica G, Kim HT, Hall DG, Coates JR. Morphology, immunohistochemistry, and genetic alterations in dog astrocytomas. Vet Pathol. 2004;41:10–9. doi: 10.1354/vp.41-1-10. [DOI] [PubMed] [Google Scholar]
- 63.Brat DJ, Castellano-Sanchez AA, Hunter SB, et al. Pseudopalisades in glioblastoma are hypoxic, express extracellular matrix proteases, and are formed by an actively migrating cell population. Cancer Res. 2004;64:920–7. doi: 10.1158/0008-5472.can-03-2073. [DOI] [PubMed] [Google Scholar]
- 64.Rong Y, Durden DL, Van Meir EG, Brat DJ. ‘Pseudopalisading’ necrosis in glioblastoma: a familiar morphologic feature that links vascular pathology, hypoxia, and angiogenesis. J Neuropathol Exp Neurol. 2006;65:529–39. doi: 10.1097/00005072-200606000-00001. [DOI] [PubMed] [Google Scholar]
- 65.Dong S, Nutt CL, Betensky RA, et al. Histology-based expression profiling yields novel prognostic markers in human glioblastoma. J Neuropathol Exp Neurol. 2005;64:948–55. doi: 10.1097/01.jnen.0000186940.14779.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lipsitz D, Higgins RJ, Kortz GD, et al. Glioblastoma multiforme: clinical findings, magnetic resonance imaging, and pathology in five dogs. Vet Pathol. 2003;40:659–69. doi: 10.1354/vp.40-6-659. [DOI] [PubMed] [Google Scholar]
- 67.Orrison WW, Jr, Rose DF, Hart BL, et al. Noninvasive preoperative cortical localization by magnetic source imaging. AJNR Am J Neuroradiol. 1992;13:1124–8. [PMC free article] [PubMed] [Google Scholar]
- 68.Braund K. Neoplasia of the Nervous System. In: Braund K, editor. Clinical neurology in small animals - localization, diagnosis and treatment Ithaca. New York: International Veterinary Information Service; 2003. p. B02270203. www.ivis.org. [Google Scholar]
- 69.Candolfi M, Kroeger KM, Pluhar GE, et al. Adenoviral-mediated gene transfer into the canine brain in vivo. Neurosurgery. 2007;60:167–77. doi: 10.1227/01.NEU.0000249210.89096.6C. discussion 78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Candolfi M, Pluhar GE, Kroeger K, et al. Optimization of adenoviral vector-mediated transgene expression in the canine brain in vivo, and in canine glioma cells in vitro. Neuro Oncol. 2007;9:245–58. doi: 10.1215/15228517-2007-012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Oh S, Pluhar GE, McNeil EA, et al. Efficacy of nonviral gene transfer in the canine brain. J Neurosurg. 2007;107:136–44. doi: 10.3171/JNS-07/07/0136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ciron C, Desmaris N, Colle MA, et al. Gene therapy of the brain in the dog model of Hurler’s syndrome. Ann Neurol. 2006;60:204–13. doi: 10.1002/ana.20870. [DOI] [PubMed] [Google Scholar]
- 73.Rainov NG, Koch S, Sena-Esteves M, Berens ME. Characterization of a canine glioma cell line as related to established experimental brain tumor models. J Neuropathol Exp Neurol. 2000;59:607–13. doi: 10.1093/jnen/59.7.607. [DOI] [PubMed] [Google Scholar]
- 74.Garcia-Escudero V, Gargini R, Izquierdo M. Glioma regression in vitro and in vivo by a suicide combined treatment. Mol Cancer Res. 2008;6:407–17. doi: 10.1158/1541-7786.MCR-07-0243. [DOI] [PubMed] [Google Scholar]
- 75.Stoica G, Lungu G, Martini-Stoica H, et al. Identification of cancer stem cells in dog glioblastoma. Vet Pathol. 2009;46:391–406. doi: 10.1354/vp.08-VP-0218-S-FL. [DOI] [PubMed] [Google Scholar]
- 76.Schallert T, Fleming SM, Leasure JL, Tillerson JL, Bland ST. CNS plasticity and assessment of forelimb sensorimotor outcome in unilateral rat models of stroke, cortical ablation, parkinsonism and spinal cord injury. Neuropharmacology. 2000;39:777–87. doi: 10.1016/s0028-3908(00)00005-8. [DOI] [PubMed] [Google Scholar]
- 77.Schallert T, Woodlee M. Motor systems: orienting and placing. In: Whishaw I, Kolb B, editors. The Behavior of the laboratory rat: a handbook with tests. New York: Oxford University Press; 2005. pp. 129–40. [Google Scholar]
- 78.King GD, Kroeger KM, Bresee CJ, et al. Flt3L in combination with HSV1-TK mediated gene therapy reverses brain tumor induced behavioral deficits. Mol Ther. 2008;16:682–90. doi: 10.1038/mt.2008.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Yang H, Chopp M, Weiland B, et al. Sensorimotor deficits associated with brain tumor progression and tumor-induced brain plasticity mechanisms. Exp Neurol. 2007;207:357–67. doi: 10.1016/j.expneurol.2007.07.008. [DOI] [PubMed] [Google Scholar]
- 80.Schallert T. Behavioral tests for preclinical intervention assessment. NeuroRx. 2006;3:497–504. doi: 10.1016/j.nurx.2006.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Schallert T, Upchurch M, Lobaugh N, et al. Tactile extinction: distinguishing between sensorimotor and motor asymmetries in rats with unilateral nigrostriatal damage. Pharmacol Biochem Behav. 1982;16:455–62. doi: 10.1016/0091-3057(82)90452-x. [DOI] [PubMed] [Google Scholar]
- 82.Woodlee MT, Asseo-Garcia AM, Zhao X, et al. Testing forelimb placing “across the midline” reveals distinct, lesion-dependent patterns of recovery in rats. Exp Neurol. 2005;191:310–7. doi: 10.1016/j.expneurol.2004.09.005. [DOI] [PubMed] [Google Scholar]
- 83.Yang H, Zhang X, Chopp M, Jiang F, Schallert T. Local fluorouracil chemotherapy interferes with neural and behavioral recovery after brain tumor-like mass compression. Behav Brain Res. 2006;172:80–9. doi: 10.1016/j.bbr.2006.04.023. [DOI] [PubMed] [Google Scholar]
- 84.Ebert AD, Beres AJ, Barber AE, Svendsen CN. Human neural progenitor cells over-expressing IGF-1 protect dopamine neurons and restore function in a rat model of Parkinson’s disease. Exp Neurol. 2008;209:213–23. doi: 10.1016/j.expneurol.2007.09.022. [DOI] [PubMed] [Google Scholar]
- 85.Torres EM, Dunnett SB. Amphetamine induced rotation in the assessment of lesions and grafts in the unilateral rat model of Parkinson’s disease. Eur Neuropsychopharmacol. 2007;17:206–14. doi: 10.1016/j.euroneuro.2006.04.009. [DOI] [PubMed] [Google Scholar]
- 86.Glick SD, Shapiro RM, Drew KL, Hinds PA, Carlson JN. Differences in spontaneous and amphetamine-induced rotational behavior, and in sensitization to amphetamine, among Sprague-Dawley derived rats from different sources. Physiol Behav. 1986;38:67–70. doi: 10.1016/0031-9384(86)90133-2. [DOI] [PubMed] [Google Scholar]
- 87.Robinson TE, Becker JB, Presty SK. Long-term facilitation of amphetamine-induced rotational behavior and striatal dopamine release produced by a single exposure to amphetamine: sex differences. Brain Res. 1982;253:231–41. doi: 10.1016/0006-8993(82)90690-4. [DOI] [PubMed] [Google Scholar]
- 88.Weaver KD, Grossman SA, Herman JG. Methylated tumor-specific DNA as a plasma biomarker in patients with glioma. Cancer Invest. 2006;24:35–40. doi: 10.1080/07357900500449546. [DOI] [PubMed] [Google Scholar]
- 89.Lin Y, Jiang T, Zhou K, et al. Plasma IGFBP-2 levels predict clinical outcomes of patients with high-grade gliomas. Neuro Oncol. 2009 doi: 10.1215/15228517-2008-114. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Greenfield JP, Jin DK, Young LM, et al. Surrogate markers predict angiogenic potential and survival in patients with glioblastoma multiforme. Neurosurgery. 2009;64:819–26. doi: 10.1227/01.NEU.0000343742.06625.DB. discussion 26-7. [DOI] [PubMed] [Google Scholar]
- 91.Rafat N, Beck GC, Schulte J, Tuettenberg J, Vajkoczy P. Circulating endothelial progenitor cells in malignant gliomas. J Neurosurg. 2009 doi: 10.3171/2009.5.JNS081074. In press. [DOI] [PubMed] [Google Scholar]
- 92.Palumbo R, Sampaolesi M, De Marchis F, et al. Extracellular HMGB1, a signal of tissue damage, induces mesoangioblast migration and proliferation. J Cell Biol. 2004;164:441–9. doi: 10.1083/jcb.200304135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Apetoh L, Ghiringhelli F, Tesniere A, et al. Toll-like receptor 4-dependent contribution of the immune system to anticancer chemotherapy and radiotherapy. Nat Med. 2007;13:1050–9. doi: 10.1038/nm1622. [DOI] [PubMed] [Google Scholar]
- 94.Kepp O, Tesniere A, Schlemmer F, et al. Immunogenic cell death modalities and their impact on cancer treatment. Apoptosis. 2009;14:364–75. doi: 10.1007/s10495-008-0303-9. [DOI] [PubMed] [Google Scholar]
- 95.Swanson KR, Bridge C, Murray JD, Alvord EC., Jr Virtual and real brain tumors: using mathematical modeling to quantify glioma growth and invasion. J Neurol Sci. 2003;216:1–10. doi: 10.1016/j.jns.2003.06.001. [DOI] [PubMed] [Google Scholar]
- 96.Gerstner ER, Sorensen AG, Jain RK, Batchelor TT. Advances in neuroimaging techniques for the evaluation of tumor growth, vascular permeability, and angiogenesis in gliomas. Curr Opin Neurol. 2008;21:728–35. doi: 10.1097/WCO.0b013e328318402a. [DOI] [PubMed] [Google Scholar]
- 97.Jensen TR, Schmainda KM. Computer-aided detection of brain tumor invasion using multiparametric MRI. J Magn Reson Imaging. 2009;30:481–9. doi: 10.1002/jmri.21878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Higano S, Yun X, Kumabe T, et al. Malignant astrocytic tumors: clinical importance of apparent diffusion coefficient in prediction of grade and prognosis. Radiology. 2006;241:839–46. doi: 10.1148/radiol.2413051276. [DOI] [PubMed] [Google Scholar]
- 99.Nelson SJ, Cha S. Imaging glioblastoma multiforme. Cancer J. 2003;9:134–45. doi: 10.1097/00130404-200303000-00009. [DOI] [PubMed] [Google Scholar]
- 100.Warmuth C, Gunther M, Zimmer C. Quantification of blood flow in brain tumors: comparison of arterial spin labeling and dynamic susceptibility-weighted contrast-enhanced MR imaging. Radiology. 2003;228:523–32. doi: 10.1148/radiol.2282020409. [DOI] [PubMed] [Google Scholar]
- 101.Wong ET, Brem S. Antiangiogenesis treatment for glioblastoma multiforme: challenges and opportunities. J Natl Compr Canc Netw. 2008;6:515–22. doi: 10.6004/jnccn.2008.0039. [DOI] [PubMed] [Google Scholar]
- 102.Chen W, Delaloye S, Silverman DH, et al. Predicting treatment response of malignant gliomas to bevacizumab and irinotecan by imaging proliferation with [18F] fluorothymidine positron emission tomography: a pilot study. J Clin Oncol. 2007;25:4714–21. doi: 10.1200/JCO.2006.10.5825. [DOI] [PubMed] [Google Scholar]
- 103.Spence AM, Muzi M, Swanson KR, et al. Regional hypoxia in glioblastoma multiforme quantified with [18F]fluoromisonidazole positron emission tomography before radiotherapy: correlation with time to progression and survival. Clin Cancer Res. 2008;14:2623–30. doi: 10.1158/1078-0432.CCR-07-4995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Vega EA, Graner MW, Sampson JH. Combating immunosuppression in glioma. Future Oncol. 2008;4:433–42. doi: 10.2217/14796694.4.3.433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Lowenstein PR. Dendritic cells and immune responses in the central nervous system. Trends Immunol. 2002;23:70. doi: 10.1016/s1471-4906(01)02151-2. [DOI] [PubMed] [Google Scholar]
- 106.Wen PY, Kesari S. Malignant gliomas in adults. N Engl J Med. 2008;359:492–507. doi: 10.1056/NEJMra0708126. [DOI] [PubMed] [Google Scholar]
- 107.Curtin JF, King GD, Barcia C, et al. Fms-like tyrosine kinase 3 ligand recruits plasmacytoid dendritic cells to the brain. J Immunol. 2006;176:3566–77. doi: 10.4049/jimmunol.176.6.3566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Park JS, Arcaroli J, Yum HK, et al. Activation of gene expression in human neutrophils by high mobility group box 1 protein. Am J Physiol Cell Physiol. 2003;284:C870–9. doi: 10.1152/ajpcell.00322.2002. [DOI] [PubMed] [Google Scholar]
- 109.Stros M, Muselikova-Polanska E, Pospisilova S, Strauss F. High-affinity binding of tumor-suppressor protein p53 and HMGB1 to hemicatenated DNA loops. Biochemistry. 2004;43:7215–25. doi: 10.1021/bi049928k. [DOI] [PubMed] [Google Scholar]
- 110.Stros M, Ozaki T, Bacikova A, Kageyama H, Nakagawara A. HMGB1 and HMGB2 cell-specifically down-regulate the p53- and p73-dependent sequence-specific transactivation from the human Bax gene promoter. J Biol Chem. 2002;277:7157–64. doi: 10.1074/jbc.M110233200. [DOI] [PubMed] [Google Scholar]
- 111.Bianchi ME, Manfredi AA. High-mobility group box 1 (HMGB1) protein at the crossroads between innate and adaptive immunity. Immunol Rev. 2007;220:35–46. doi: 10.1111/j.1600-065X.2007.00574.x. [DOI] [PubMed] [Google Scholar]
- 112.Lotze MT, Tracey KJ. High-mobility group box 1 protein (HMGB1): nuclear weapon in the immune arsenal. Nat Rev Immunol. 2005;5:331–42. doi: 10.1038/nri1594. [DOI] [PubMed] [Google Scholar]
- 113.Lotze MT, Zeh HJ, Rubartelli A, et al. The grateful dead: damage-associated molecular pattern molecules and reduction/oxidation regulate immunity. Immunol Rev. 2007;220:60–81. doi: 10.1111/j.1600-065X.2007.00579.x. [DOI] [PubMed] [Google Scholar]
- 114.Enderlin M, Kleinmann EV, Struyf S, et al. TNF-alpha and the IFN-gamma-inducible protein 10 (IP-10/CXCL-10) delivered by parvoviral vectors act in synergy to induce antitumor effects in mouse glioblastoma. Cancer Gene Ther. 2008:149–60. doi: 10.1038/cgt.2008.62. [DOI] [PubMed] [Google Scholar]
- 115.Kim CY, Jeong M, Mushiake H, et al. Cancer gene therapy using a novel secretable trimeric TRAIL. Gene Ther. 2006;13:330–8. doi: 10.1038/sj.gt.3302658. [DOI] [PubMed] [Google Scholar]
- 116.Nagane M, Pan G, Weddle JJ, et al. Increased death receptor 5 expression by chemotherapeutic agents in human gliomas causes synergistic cytotoxicity with tumor necrosis factor-related apoptosis-inducing ligand in vitro and in vivo. Cancer Res. 2000;60:847–53. [PubMed] [Google Scholar]
- 117.Ambar BB, Frei K, Malipiero U, et al. Treatment of experimental glioma by administration of adenoviral vectors expressing Fas ligand. Hum Gene Ther. 1999;10:1641–8. doi: 10.1089/10430349950017644. [DOI] [PubMed] [Google Scholar]
- 118.Nafe C, Cao YJ, Quinones A, et al. Expression of mutant non-cleavable Fas ligand on retrovirus packaging cells causes apoptosis of immunocompetent cells and improves prodrug activation gene therapy in a malignant glioma model. Life Sci. 2003;73:1847–60. doi: 10.1016/s0024-3205(03)00542-3. [DOI] [PubMed] [Google Scholar]
- 119.Maleniak TC, Darling JL, Lowenstein PR, Castro MG. Adenovirus-mediated expression of HSV1-TK or Fas ligand induces cell death in primary human glioma-derived cell cultures that are resistant to the chemotherapeutic agent CCNU. Cancer Gene Ther. 2001;8:589–98. doi: 10.1038/sj.cgt.7700348. [DOI] [PubMed] [Google Scholar]
- 120.Roth W, Isenmann S, Nakamura M, et al. Soluble decoy receptor 3 is expressed by malignant gliomas and suppresses CD95 ligand-induced apoptosis and chemotaxis. Cancer Res. 2001;61:2759–65. [PubMed] [Google Scholar]
- 121.Chen TC, Hinton DR, Sippy BD, Hofman FM. Soluble TNF-alpha receptors are constitutively shed and downregulate adhesion molecule expression in malignant gliomas. J Neuropathol Exp Neurol. 1997;56:541–50. doi: 10.1097/00005072-199705000-00010. [DOI] [PubMed] [Google Scholar]
- 122.Rieger J, Frank B, Weller M, Wick W. Mechanisms of resistance of human glioma cells to Apo2 ligand/TNF-related apoptosis-inducing ligand. Cell Physiol Biochem. 2007;20:23–34. doi: 10.1159/000104150. [DOI] [PubMed] [Google Scholar]
- 123.Sheikh MS, Burns TF, Huang Y, et al. p53-dependent and - independent regulation of the death receptor KILLER/DR5 gene expression in response to genotoxic stress and tumor necrosis factor alpha. Cancer Res. 1998;58:1593–8. [PubMed] [Google Scholar]
- 124.Hingtgen S, Ren X, Terwilliger E, et al. Targeting multiple pathways in gliomas with stem cell and viral delivered S-TRAIL and Temozolomide. Mol Cancer Ther. 2008;7:3575–85. doi: 10.1158/1535-7163.MCT-08-0640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.de la Monte SM, Sohn YK, Wands JR. Correlates of p53- and Fas (CD95)-mediated apoptosis in Alzheimer’s disease. J Neurol Sci. 1997;152:73–83. doi: 10.1016/s0022-510x(97)00131-7. [DOI] [PubMed] [Google Scholar]
- 126.Spierings DC, de Vries EG, Vellenga E, et al. Tissue distribution of the death ligand TRAIL and its receptors. J Histochem Cytochem. 2004;52:821–31. doi: 10.1369/jhc.3A6112.2004. [DOI] [PubMed] [Google Scholar]
- 127.Dorr J, Bechmann I, Waiczies S, et al. Lack of tumor necrosis factor-related apoptosis-inducing ligand but presence of its receptors in the human brain. J Neurosci. 2002;22:RC209 1–5. doi: 10.1523/JNEUROSCI.22-04-j0001.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Curtin JF, Candolfi M, Xiong W, Lowenstein PR, Castro MG. Turning the gene tap off; implications of regulating gene expression for cancer therapeutics. Mol Cancer Ther. 2008;7:439–48. doi: 10.1158/1535-7163.MCT-07-2328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Tsurushima H, Yuan X, Dillehay LE, Leong KW. Radioresponsive tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) gene therapy for malignant brain tumors. Cancer Gene Ther. 2007;14:706–16. doi: 10.1038/sj.cgt.7701065. [DOI] [PubMed] [Google Scholar]
- 130.Yamini B, Yu X, Gillespie GY, Kufe DW, Weichselbaum RR. Transcriptional targeting of adenovirally delivered tumor necrosis factor alpha by temozolomide in experimental glioblastoma. Cancer Res. 2004;64:6381–4. doi: 10.1158/0008-5472.CAN-04-2117. [DOI] [PubMed] [Google Scholar]
- 131.Ozawa T, Hu JL, Hu LJ, et al. Functionality of hypoxia-induced BAX expression in a human glioblastoma xenograft model. Cancer Gene Ther. 2005;12:449–55. doi: 10.1038/sj.cgt.7700814. [DOI] [PubMed] [Google Scholar]
- 132.Ito H, Kanzawa T, Miyoshi T, et al. Therapeutic efficacy of PUMA for malignant glioma cells regardless of p53 status. Hum Gene Ther. 2005;16:685–98. doi: 10.1089/hum.2005.16.685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Morelli AE, Larregina AT, Smith-Arica J, et al. Neuronal and glial cell type-specific promoters within adenovirus recombinants restrict the expression of the apoptosis-inducing molecule Fas ligand to predetermined brain cell types, and abolish peripheral liver toxicity. J Gen Virol. 1999;80(Pt 3):571–83. doi: 10.1099/0022-1317-80-3-571. [DOI] [PubMed] [Google Scholar]
- 134.Gerdes CA, Castro MG, Lowenstein PR. Strong promoters are the key to highly efficient, noninflammatory and noncytotoxic adenoviral-mediated transgene delivery into the brain in vivo. Mol Ther. 2000;2:330–8. doi: 10.1006/mthe.2000.0140. [DOI] [PubMed] [Google Scholar]
- 135.Beltinger C, Fulda S, Kammertoens T, et al. Herpes simplex virus thymidine kinase/ganciclovir-induced apoptosis involves ligand-independent death receptor aggregation and activation of caspases. Proc Natl Acad Sci USA. 1999;96:8699–704. doi: 10.1073/pnas.96.15.8699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Mesnil M, Yamasaki H. Bystander effect in herpes simplex virus-thymidine kinase/ganciclovir cancer gene therapy: role of gap-junctional intercellular communication. Cancer Res. 2000;60:3989–99. [PubMed] [Google Scholar]
- 137.Rainov NG, Fels C, Droege JW, et al. Temozolomide enhances herpes simplex virus thymidine kinase/ganciclovir therapy of malignant glioma. Cancer Gene Ther. 2001;8:662–8. doi: 10.1038/sj.cgt.7700355. [DOI] [PubMed] [Google Scholar]
- 138.Ulasov IV, Tyler MA, Rivera AA, et al. Evaluation of E1A double mutant oncolytic adenovectors in anti-glioma gene therapy. J Med Virol. 2008;80:1595–603. doi: 10.1002/jmv.00000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Aghi MK, Chiocca EA. Phase ib trial of oncolytic herpes virus G207 shows safety of multiple injections and documents viral replication. Mol Ther. 2009;17:8–9. doi: 10.1038/mt.2008.275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Piao Y, Jiang H, Alemany R, et al. Oncolytic adenovirus retargeted to Delta-EGFR induces selective antiglioma activity. Cancer Gene Ther. 2009;16:256–65. doi: 10.1038/cgt.2008.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Thorne SH, Bartlett DL, Kirn DH. The use of oncolytic vaccinia viruses in the treatment of cancer: a new role for an old ally? Curr Gene Ther. 2005;5:429–43. doi: 10.2174/1566523054546215. [DOI] [PubMed] [Google Scholar]
- 142.Dalba C, Klatzmann D, Logg CR, Kasahara N. Beyond oncolytic virotherapy: replication-competent retrovirus vectors for selective and stable transduction of tumors. Curr Gene Ther. 2005;5:655–67. doi: 10.2174/156652305774964659. [DOI] [PubMed] [Google Scholar]
- 143.Collins SA, Guinn BA, Harrison PT, et al. Viral vectors in cancer immunotherapy: which vector for which strategy? Curr Gene Ther. 2008;8:66–78. doi: 10.2174/156652308784049345. [DOI] [PubMed] [Google Scholar]
- 144.Obeid M, Tesniere A, Ghiringhelli F, et al. Calreticulin exposure dictates the immunogenicity of cancer cell death. Nat Med. 2007;13:54–61. doi: 10.1038/nm1523. [DOI] [PubMed] [Google Scholar]
- 145.Rainov NG, Kramm CM, Banning U, et al. Immune response induced by retrovirus-mediated HSV-tk/GCV pharmacogene therapy in patients with glioblastoma multiforme. Gene Ther. 2000;7:1853–8. doi: 10.1038/sj.gt.3301311. [DOI] [PubMed] [Google Scholar]
- 146.Peres E, Wood GW, Poulik J, et al. High-dose chemotherapy and adoptive immunotherapy in the treatment of recurrent pediatric brain tumors. Neuropediatrics. 2008;39:151–6. doi: 10.1055/s-0028-1093333. [DOI] [PubMed] [Google Scholar]
- 147.Bruserud O, Ersvaer E, Olsnes A, Gjertsen BT. Anticancer immunotherapy in combination with proapoptotic therapy. Curr Cancer Drug Targets. 2008;8:666–75. doi: 10.2174/156800908786733496. [DOI] [PubMed] [Google Scholar]
- 148.Bruserud O, Wendelboe O. Biological treatment in acute myelogenous leukaemia: how should T-cell targeting immunotherapy be combined with intensive chemotherapy? Expert Opin Biol Ther. 2001;1:1005–16. doi: 10.1517/14712598.1.6.1005. [DOI] [PubMed] [Google Scholar]
- 149.Schor NF. Pharmacotherapy for adults with tumors of the central nervous system. Pharmacol Ther. 2009;121:253–64. doi: 10.1016/j.pharmthera.2008.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Wheeler CJ, Das A, Liu G, Yu JS, Black KL. Clinical responsiveness of glioblastoma multiforme to chemotherapy after vaccination. Clin Cancer Res. 2004;10:5316–26. doi: 10.1158/1078-0432.CCR-04-0497. [DOI] [PubMed] [Google Scholar]
- 151.Liu G, Akasaki Y, Khong HT, et al. Cytotoxic T cell targeting of TRP-2 sensitizes human malignant glioma to chemotherapy. Oncogene. 2005;24:5226–34. doi: 10.1038/sj.onc.1208519. [DOI] [PubMed] [Google Scholar]
- 152.Pak BJ, Chu W, Lu SJ, Kerbel RS, Ben-David Y. Lineage-specific mechanism of drug and radiation resistance in melanoma mediated by tyrosinase-related protein 2. Cancer Metastasis Rev. 2001;20:27–32. doi: 10.1023/a:1013175516793. [DOI] [PubMed] [Google Scholar]
- 153.Zhang L, Dermawan KT, Jin ML, Xiong SD, Chu YW. Does chemotherapy augment anti-tumor immunotherapy by preferential impairment of regulatory T cells? Med Hypotheses. 2008;71:802–4. doi: 10.1016/j.mehy.2008.06.022. [DOI] [PubMed] [Google Scholar]