

Abstract
The roles of Arg548 and Gln552 residues in the active site of the carboxyl transferase domain of Rhizobium etli pyruvate carboxylase were investigated using site-directed mutagenesis. Mutation of Arg548 to alanine or glutamine resulted in the destabilisation of the quaternary structure of the enzyme, suggesting that this residue has a structural role. The mutations R548K, Q552N and Q552A resulted in loss of the ability to catalyse pyruvate carboxylation, biotin-dependent decarboxylation of oxaloacetate and proton exchange between pyruvate and water. These mutants retained the ability to catalyse reactions that occur at the active site of the biotin carboxylase domain, i.e. bicarbonate-dependent ATP cleavage and ADP phosphorylation by carbamoyl phosphate. The effects of oxamate on the catalysis in the biotin carboxylase domain by the R548K and Q552N mutants were similar to those on the catalysis of reactions by the wild-type enzyme. However, the presence of oxamate had no effect on the reactions catalysed by the Q552A mutant. We propose that Arg548 and Gln552 facilitate the binding of pyruvate and subsequent proton transfer between pyruvate and biotin in the partial reaction catalysed in the active site of the carboxyl-transferase domain of Rhizobium etli pyruvate carboxylase.
Pyruvate carboxylase (PC) (EC 6.4.1.1), a biotin-containing enzyme, catalyses pyruvate carboxylation through a two-step reaction shown in Figure 1 (1, 2). In reaction [1] (Fig. 1), the carboxylation of biotin in the biotin carboxylase (BC) domain is thought to proceed by an initial reaction between HCO3− and MgATP to form MgADP and a reactive carboxyphosphate intermediate (1, 3, 4). The reaction most likely proceeds via the decarboxylation of this intermediate to form CO2, which can then carboxylate the 1′-N position of biotin. (1, 3).
Figure 1.

Reaction scheme showing the two partial reactions that comprise the overall pyruvate carboxylation reaction. Reaction [1] is the ATP-dependent carboxylation of biotin by bicarbonate that occurs at the BC domain. Reaction [2] is the reaction in which the carboxy group is transferred from carboxybiotin to pyruvate in the CT domain.
Recently, structures of several pyruvate carboxylases have been determined (5-7). Hybrid tetramers, composed of mutated subunits of Rhizobium etli (RePC), that either lack biotin or have a carboxyl-transferase (CT) domain mutation, which greatly reduces its ability to catalyse the full forward reaction, nevertheless retain the ability to catalyse the carboxylation of pyruvate, albeit at a reduced rate as compared to wild-type RePC (5), demonstrating that reaction [1] occurs in the BC domain of one subunit whilst reaction [2] occurs in the CT domain of its partner subunit on the same face of the tetrameric enzyme.
Previously, the catalytic mechanism and those residues involved in the reactions of both the BC and CT domains have been studied by mutating amino acid residues that are conserved in homologous enzymes (8, 9) where the structures of the subunits of these enzymes, such as the biotin carboxylase subunit of acetyl-CoA carboxylase (10) and the 5S subunit of transcarboxylase (11) have been determined. However, with the recent determination of several structures of the PC holoenzyme from various organisms (5-7), a more structure-directed mutational analysis to determine the detailed mechanism of PC has commenced (12).
Based on kinetic isotope effect studies, the catalytic reaction in the CT domain is proposed to involve the stepwise removal of a proton from pyruvate, a carboxyl group transfer and subsequent re-protonation of biotin (13, 14). The crystal structure of Staphylococcus aureus PC, which contains both pyruvate and biotin bound in the active site of the CT domain (6), suggests that both Arg548 and Gln552 interact with the substrate of the carboxyl-transferase reaction. These residues are similarly arranged in the crystal structures that have either pyruvate, oxaloacetate or α-ketoglutarate bound in the active site of the 5S subunit of transcarboxylase (5S), which is functionally homologous to the CT domain of PC, (11). Furthermore these two residues are completely conserved in sequence alignments of PC sequences from many different organisms as well as in transcarboxylase and oxaloacetate decarboxylase (2). Figure 2 shows the relative positioning of the Arg548 and Gln552 (RePC numbering) residues in the CT domain active site of Staphylococcus aureus PC, with respect to pyruvate, biotin and another catalytically important residue, Thr882. The amide group of Gln552 is positioned approximately 2.9 Å from the carboxyl oxygen of pyruvate and could potentially form a hydrogen-bonding interaction, which would promote substrate binding. In addition, both the amide group of Gln552 and the guanidinyl group of Arg548 are positioned proximal to the carbonyl oxygen of pyruvate, which could assist in its enolization. Figure 2 also shows the positioning of Thr882 between the methyl group of pyruvate and the 1’-nitrogen of biotin, ready to act in its proposed proton transfer role (7, 12) and the metal ion positioned in proximity to the carbonyl oxygen of pyruvate to assist in its enolization1.
Figure 2.

Stereoview of the active site of PC from Staphylococcus aureus (PDB accession code 3BG5; Xiang and Tong, 2008) with residues numbered according to the primary sequence in Rhizobium etli PC. Hydrogen bonds are indicated with dashed lines and the distances are indicated in Angstroms.
In this work, we have performed site-directed mutagenesis of two residues, Arg548 and Gln552, located in CT domain of RePC and have performed detailed kinetic analyses and characterization of these mutants to investigate the roles of these residues in the catalytic mechanism of the reaction that occurs in the CT domain (reaction [2]).
MATERIALS AND METHODS
Construction of wild-type PC and mutants
Mutagenesis was carried out on the 1.0 kb BamHI-XhoI PC gene fragment (15) corresponding to the CT domain. Mutagenic reactions were performed and the T882A mutant was prepared as described previously (12). The mutations were verified by DNA sequencing. The primers used to generate R548K, R548Q, R548A, Q552N and Q552A mutants are shown in Table S1 (see supplementary material). The equivalent fragment of the wild-type RePC gene in the expression clone (5, 12) was then replaced with the mutagenised fragments.
PC expression and purification
The BL21 (DE3) Escherichia coli, containing the pCY216 plasmid (16) which encoded the E. coli BirA gene, transformed with wild-type RePC or a mutant were grown in 8 L Luria Bertani broth supplemented with 6.25 g/L arabinose, 10 mg/L biotin, 200 mg/L ampicillin and 30 mg/L chloramphenicol. The cultures were grown at 37°C until an OD600 of 1.0-1.2 was reached. RePC expression was subsequently induced with the addition of 0.1 mM IPTG at 16°C for approximately 36 h. Cells were harvested by centrifugation at 4,000 rpm at 4°C for 15 min. The harvested cells were disrupted by incubation with 1 mg/mL lysozyme, followed by lysis using a Bead-Beater™ (Biospec). Nucleic acids were removed from the lysate by protamine sulfate precipitation as previously described (7). The proteins in the lysate were precipitated with 36 % (w/v) saturated ammonium sulfate and resuspended in loading buffer (50 mM NaH2PO4, 300 mM NaCl, 10 mM imidazole, pH 7.4) prior to loading onto a 20 ml column of HisPur™ cobalt resin (Thermo Scientific, Australia). PC was eluted from the column using 50 mM NaH2PO4, 300 mM NaCl, 150 mM imidazole, pH 7.4. Purified PC was stored at −80°C in storage buffer containing 30 % (v/v) glycerol, 0.1 M Tris-Cl, pH 7.8, and 1 mM DTE (17).
Biotin content determination
In triplicate, 5 μl aliquots of the enzyme were mixed with 85 μl of 0.2 M potassium phosphate buffer pH 7.2 followed by the addition of 10 μl of 2% (w/v) chymotrypsin (Sigma). The solutions were incubated at 37 °C for 24 h. Pronase E (Sigma) was then added to the solutions to a final concentration of 0.45% (w/v). The solutions were further incubated for 48 h at 37 °C prior to performing the biotin assay as described by Rylatt et al. (18).
Sedimentation velocity analysis of the quaternary structure
Sedimentation velocity analytical centrifugation was performed essentially as described by Jitrapakdee et al. (19) with a Beckman ProteomeLab XL-A (Beckman-Coulter, Palo Alto, CA, USA) ultracentrifuge using the absorbance optics system to visualize the protein. The wavelengths used for analysis were 278 – 280 nm. Two-sector cells were used and data were acquired every 0.003 cm. Data were collected as 300 absorbance scans with a nominal time increment of 1 minute at 30°C, at a speed of 40,000 rpm. In all cases, enzyme samples were prepared in 0.1 M Tris-HCl (pH 7.8), 20 mM NaHCO3, 10 mM pyruvate, 5 mM MgCl2, 0.1 mM acetyl-CoA, and 1mM DTE. Enzyme concentrations in the samples were 0.2 mg/ml. The computer-captured data were analysed by SEDFIT (20). The enzymes’ partial specific volumes were calculated from the amino acid composition using SEDNTERP (RASMB, http://www.bbri.org/RASMB) (20). The density of the Tris buffer, 1.005 g/ml, was assumed to be the density of the enzyme solutions.
Pyruvate carboxylation assay
Pyruvate carboxylating activity was determined in triplicate using a coupled spectrophotometric assay in which the oxaloacetate produced was converted to malate in a reaction catalysed by malate dehydrogenase (7, 19). The reactions were performed at 30°C in a 1 ml reaction mixture containing 0.1 M Tris-HCl pH 7.8, 10 mM pyruvate, 2.5 mM ATP, 0.25 mM acetyl-CoA, 0.3 mM NADH, 10 mM NaHCO3, 8.0 mM MgCl2 and 5 units of malate dehydrogenase (Sigma). Assays were started by the addition of RePC (50-200 μg). Apparent kcat values were calculated by dividing the measured reaction velocity by the biotin concentration of the PC used in the assay.
Biotin-dependent and oxamate-induced oxaloacetate decarboxylation
The determination of biotin-dependent oxaloacetate decarboxylation was carried out as previously described by Attwood and Cleland (21). For wild-type RePC and each of the mutants, three solutions were prepared as follows in 400 μl of 0.1 M Tris-HCl, pH 7.8: (1) 1 mg enzyme and 2 mg avidin; (2) 1 mg enzyme; (3) 2 mg avidin. The solutions were incubated at 30°C for 1.5 h. An aliquot of solution (1) was assayed for pyruvate carboxylating activity to verify its complete inhibition by avidin. 20 μl aliquots of each solution were then assayed for oxaloacetate decarboxylating activity at 25 μM, 100 μM and 200 μM oxaloacetate. Other reaction components were 100 mM Tris-HCl pH 7.8, 0.25 mM acetyl-CoA, 0.2 mM NADH and 4 units of lactate dehydrogenase. Aliquots of solution (2) were also assayed for oxaloacetate decarboxylating activity at 200 μM oxaloacetate in the presence of 0.5 mM oxamate. Apparent kcat values were calculated by dividing the measured reaction velocity by the biotin concentration of the RePC used in the assay.
Bicarbonate-dependent ATP cleavage assay
ATP cleavage activities were determined by measuring the amount of inorganic phosphate (Pi) released using glucose-6-phosphate dehydrogenase, phosphoglucomutase and phosphorylase a coupled enzyme system as described by Zeczycki et al. (12). Assays were performed at 30°C and initiated by the addition of 50 μg RePC. The reactions contained 0.1 M Tris-HCl, pH 7.8, 2.5 mM MgATP, 2.5 mM MgCl2, 0.33 mM NADP, 0.25 mM acetyl-CoA, 5 μM α-glucose-1-phosphate, 1 mg/ml glycogen, 2 units of phosphorylase a, 3 units of phosphoglucomutase and 3 units of glucose-6-phosphate dehydrogenase. Production of NADPH was determined by measuring the increase in absorbance at 340 nm. The effects of oxamate on the activities of ATP cleavage were determined by measuring the initial rate of Pi release at various concentrations of oxamate (0-20 mM). Apparent kcat values were calculated by dividing the measured reaction velocity by the biotin concentration of the RePC used in the assay.
Assay of MgADP Phosphorylation using Carbamoyl Phosphate
The initial rates of ATP synthesis from the RePC-catalysed ADP phosphorylation reaction using carbamoyl phosphate were determined spectrophotometrically with the coupled enzyme system of hexokinase and glucose-6-phosphate dehydrogenase (22), where the production of NADPH was determined at 30°C by measuring the increase in absorbance at 340 nm. Reactions contained 0.1 M Tris-HCl, pH 7.8, 20 mM carbamoyl phosphate, 3.0 mM MgADP, 5 mM MgCl2, 0.25 mM acetyl-CoA, 0.24 mM NADP, 0.20 mM glucose, 3 units of glucose-6-phosphate dehydrogenase and 1 unit of hexokinase. Reactions were performed in the presence or absence of 20 mM oxamate. Apparent kcat values were calculated by dividing the measured reaction velocity by the biotin concentration of the RePC used in the assay.
Determination of hydrogen-deuterium (H-D) exchange activity by 1H NMR
[3-2H3]pyruvate was prepared by dissolving oxaloacetic acid in D2O to a final concentration of 10 mM. The solution was incubated for 30 minutes prior to adjusting the pH to 6.0 with 1 M NaOH prepared in D2O. The solution was sealed in a vial and incubated at 30°C for up to 36 h until a spectrophotometric assay using malate dehydrogenase indicated that there was no residual oxaloacetate left in the solution (13). The remaining solution was then lyophilized, and the resultant powder was stored at 4 °C. When solutions containing [3-2H3]pyruvate were used, the concentration of pyruvate was determined spectrophotometrically using lactate dehydrogenase.
1H NMR experiments were performed on a Bruker AV500 NMR spectrometer. The initial rates of proton exchange catalysed by wild-type and mutant RePC were determined in two different ways, using either [3-2H3]pyruvate in 95% H2O as the substrate or [3-1H3]pyruvate in 90% D2O as the substrate. When [3-2H3]pyruvate was used as the substrate, the newly emerging peaks in the 1H NMR spectra, which corresponded to the formation of the various (mono-, di-, and tri-) protonated species of pyruvate, were integrated at each time point. The reactions were monitored at 30° C in a 95% H2O and 5% D2O solution containing 20 mM ammonium bicarbonate and 10 mM [3-2H3]pyruvate in a total volume of 1 ml. Reactions were initiated by the addition of 10 μg of wild-type RePC or 250 μg of the mutant RePC enzyme. A control was also performed where the reaction mixture did not contain enzyme, there was no measurable rate of proton exchange over the time-course of the reaction. In the experiment where the reaction mix (minus enzyme) was preincubated at 30°C for 30 min with 5 units of malate dehydrogenase and 0.2 mM NADH to remove trace amounts of oxaloacetate remaining from the synthesis of the [3-2H3]pyruvate, the reaction was subsequently initiated by addition of 250 μg wild-type RePC.
The water signal was suppressed using the WATERGATE pulse sequence and 16 scans were acquired for each measurement. The integral of 10 mM non-deuterated pyruvate signals in the absence of enzyme was used as an external standard for the calculation of the concentrations of the emerging protonated species of pyruvate. The initial solution of [3-2H3]pyruvate contained 98.3 % [3-2 H3]pyruvate. The remaining 1.7% of the total pyruvate concentration consisted of a mixture of the mono-, di- and tri-protonated species. The rate of proton exchange was calculated from the rate of increase in the sum of the concentrations of the three protonated species.
When [3-1H3]pyruvate was used as a substrate, the reaction was initiated by addition of 250 μg wild-type RePC, other reaction conditions were as described above. Proton exchange reactions catalysed by wild-type RePC, determined by either method were also performed in the presence of 20 mM oxamate as a competitive inhibitor with respect to pyruvate. The reaction mixtures used to measure proton exchange reactions (with [3-1H3]pyruvate as the substrate) catalysed by R548K and Q552N in the presence of the substrates of the biotin carboxylation reaction, also contained 1 mM MgATP, 5 mM MgCl2 and 0.25mM acetyl CoA. A control was also performed where the reaction mixture did not contain enzyme, the rates of proton exchange in the presence of R548K and Q552N were then corrected the rate of proton exchange measured in the control.
In the experiments using [3-1H3]pyruvate as the substrate in 90% D2O solvent suppression was not required. Typically ten 1H NMR spectra were recorded over the time course of each experiment. The 1H spectra employed a 30° pulse angle, with16 scans being acquired for each measurement. After baseline correction, the [3-1H3]pyruvate signal in each spectrum was simulated using Bruker deconvolution software. The change in the [3-1H3]pyruvate signal during the course of each experiment was determined by comparison of the areas of the simulated signals. The computer simulation technique was employed to ensure that only the area due to the [3-1H3]pyruvate signal was measured so as to avoid inclusion of signal contributions from other species. The initial rate of proton exchange was calculated from the rate of disappearance of the [3-1H3]pyruvate signal. In one experiment with wild-type RePC, the exchange reaction was followed for 48 hours, at which time 60% of the [3-1H3]pyruvate (6 mM) had been converted to deuterated species.
The data were analysed by linear regression to determine the initial proton exchange rate, which, following correction for any non-enzymic proton exchange, was divided by the enzymic biotin concentration to obtain the apparent first order rate constant.
Data analysis
Substrate inhibition data with oxamate for the ATP cleavage activity were analysed by non-linear least squares regression fits of the data to equation (1) as described by Zeczycki et al. (12). Where A is the concentration of oxamate, Ka is the activation constant and Ki is the inhibition constant. The apparent kcat is the apparent first order rate constant at each concentration of oxamate and kcat is the maximum calculated apparent first-order rate constant.
| (1) |
RESULTS
Mutation of Arg548 or Gln552 in the CT domain active site completely abolishes pyruvate carboxylating activity
Pyruvate carboxylating activities for wild-type and mutant forms of RePC were determined at saturating levels of substrates and acetyl-CoA. The kcat for wild-type RePC was determined to be 834 ± 39 min−1, whilst that for R548K was 17 ± 3 min−1 (2% of wild-type), and no activity was detected for any of the other four mutants.
Quaternary structure of wild-type RePC and CT domain mutants
Since PC is only active as a tetramer (23-25), structural perturbations resulting from mutations which destabilise the quaternary structure of the enzyme may be responsible for the observed abolition of pyruvate carboxylating ability in the RePC mutants. To ascertain whether the lack of activity arising from these mutations can be attributed to a catalytic or mechanistic effect, rather than an effect on the quaternary structures of the enzyme, analytical ultracentrifugation sedimentation velocity analysis was performed. As shown in Figure 3, the main species present in wild-type RePC appears to have a sedimentation coefficient between 15 and 19, corresponding to an enzyme species with a molecular weight of 472 ± 52 kDa, which is equivalent to that of the RePC tetramer. The R548K, Q552N and Q552A mutants produced three peaks, which exhibited sedimentation coefficients corresponding to molecular weights of monomeric, dimeric and tetrameric RePC, with the major species being tetrameric (Table 1). The percentage of the tetrameric species in the R548K, Q552N and Q552A mutants was 74, 76 and 73, respectively, similar to the percentage observed for the wild-type RePC. The R548Q and R548A mutants appear to have much lower proportions of tetrameric RePC compared to the other mutants, with tetramers comprising only 49 % and 34 % of the total enzyme species, respectively. Since a large proportion of the loss of enzymic activity as compared to the wild-type enzyme could be ascribed to the loss of tetrameric enzyme structure, no further the kinetic analyses of the R548Q and R548A mutants were performed.
Figure 3.
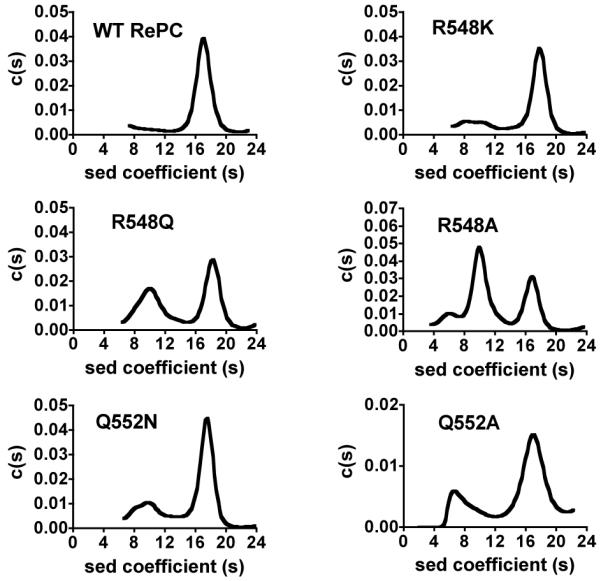
Sedimentation velocity analyses of wild-type RePC and the mutants R548K, R548Q, R548A, Q552N and Q552A. Experiments were performed as described in Materials and Methods. Plots of derived sedimentation coefficient distribution (c(s)) vs. sedimentation coefficient obtained from the application of SEDFIT and maximum entropy methods of analysis to the original apparent sedimentation coefficient distribution vs. apparent sedimentation coefficient data (g(s*) vs. s* plots).
Table 1.
Molecular weights and structural conformations determined by analytical ultracentrifugation of wild-type, R548K, R548Q, R548A, Q552N and Q552A RePCa
| Enzyme form | Monomerb Sedimentation Coefficient |
Molecular mass (kDa) |
% |
Dimerb Sedimentation coefficient |
Molecular mass (kDa) |
% |
Tetramerb Sedimentation cofficient |
Molecular mass (kDa) |
% |
|---|---|---|---|---|---|---|---|---|---|
| wild-type | 14% of monomers and dimersc | 17.1 ± 1.2 | 472 ± 52 | 86 | |||||
| R548K | 8.2 ± 0.8 | 183 ± 27 | 13 | 10.8 ± 1.0 | 289 ± 37 | 13 | 17.7 ± 1.2 | 591 ± 69 | 74 |
| R548Q | 7.7 ± 0.6 | 143 ± 17 | 9 | 10.6 ± 1.7 | 239 ± 55 | 42 | 18.5 ± 1.2 | 518 ± 52 | 49 |
| R548A | 5.8 ± 0.9 | 99 ± 24 | 13 | 10.1 ± 1.1 | 218 ± 43 | 53 | 16.8 ± 1.1 | 451 ± 44 | 34 |
| Q552N | 7.8 ± 0.6 | 172 ± 19 | 11 | 10.4 ± 0.9 | 267 ± 37 | 13 | 17.4 ± 1.0 | 575 ± 57 | 76 |
| Q552A | 6.8 ± 0.7 | 123 ± 21 | 15 | 9.7 ± 1.1 | 203 ± 34 | 12 | 16.9 ± 1.9 | 471 ± 75 | 73 |
Analytical ultracentrifugations were performed at 30°C in 0.1 Tris-HCl pH 7.8, 20 mM NaHCO3, 5 mM MgCl2, 10 mM pyruvate, 0.1 mM acetyl-CoA, 1 mM DTE using 0.2 mg/ml enzyme.
Listed under these headings are enzyme species that have sedimentation coefficients that are most appropriate to the particular quaternary conformation in the context of the enzyme sample analysed. The subunit molecular mass is theoretically 126,009 Da.
Unable to obtain separate estimates of dimers and monomers.
Oxaloacetate decarboxylation
Oxaloacetate decarboxylation assays for wild-type, R548K, Q552N and Q552A RePC were performed to determine the effects of the mutations on the reverse of reaction [2], which occurs in the CT domain. Biotin-dependent oxaloacetate decarboxylation involves the formation of carboxybiotin resulting from the transfer of the carboxyl group from oxaloacetate to biotin, whilst non biotin-dependent oxaloacetate decarboxylation does not involve carboxybiotin formation and is independent of the reaction mechanism (21). Biotin-dependent oxaloacetate decarboxylation was determined at various concentrations of oxaloacetate, as described by Attwood and Cleland (21). The results are shown in Table 2 and were generally comparable to those previously observed for chicken liver pyruvate carboxylase (21). At 25 μM oxaloacetate, the decarboxylating activity dependent on biotin for wild-type RePC was 66.6 % of the total decarboxylating activity, but at higher concentrations of oxaloacetate the percent of biotin-dependent decarboxylation activity was reduced. No decarboxylating activity was observed in the presence of avidin alone. None of the mutants exhibited any biotin-dependent decarboxylating activity (kcat1 was shown to be not significantly different from kcat2 at all concentrations of oxaloacetate; see Supporting Information).
Table 2.
Biotin-dependent oxaloacetate decarboxylation activities of wild-type PC in the absence of oxamate at different concentrations of oxaloacetatea
| additionb |
[oxaloacetate] (μM) |
||||
|---|---|---|---|---|---|
| wild-type PC | avidin | 25 | 100 | 200 | |
| kcat1 c | + | + | 0.24 ± 0.01 | 2.0 ± 0.2 | 2.7 ± 0.3 |
| kcat2 d | + | - | 0.7 ± 0.2 | 2.8 ± 0.1 | 3.6 ± 0.4 |
| kcat3 | − | + | 0 | 0 | 0 |
| % of activity dependent on biotinf |
66 | 28 | 24 | ||
The apparent kcat values are have units of min−1. Reported values are means ± standard deviations, of three separate determinations.
Pyruvate carboxylase and avidin used for each reaction were 50 μg and 150 μg, respectively.
kcat1 is the apparent rate constant for biotin-independent oxaloacetate decarboxylation.
kcat2 is the apparent rate constant for biotin-dependent plus biotin-independent oxaloacetate decarboxylation and
kcat3 is the apparent rate constant for oxaloacetate decarboxylation in the presence of avidin alone .
Percent of activity dependent on biotin was calculated as 100(kcat2 − kcat1 + kcat3) / kcat2 after statistical analysis had that showed kcat1 and kcat2 to be significantly different using a T-test (p < 0.05).
Oxaloacetate decarboxylation was also assayed in the presence of oxamate, a structural analogue of pyruvate. Since oxamate stimulates the rate-limiting decarboxylation of carboxybiotin in the CT active site during oxaloacetate decarboxylation, measuring oxaloacetate decarboxylation activity in the presence of oxamate enhances the rate of oxaloacetate decarboxylation and allows the activity to be independent of reaction [1] (13, 21). Our results show that the oxaloacetate decarboxylating activity of wild-type RePC increased four-fold in the presence of 0.5 mM oxamate (Table 3). However, the presence of oxamate did not enhance the oxaloacetate-decarboxylating activities of the R548K, Q552N and Q552A mutants. These results were consistent with the lack of biotin-dependency of oxaloacetate decarboxylation in the mutants, as described above.
Table 3.
Oxaloacetate decarboxylation activities in the presence or absence of oxamatea
| No oxamate (min−1) |
+ 0.5 mM oxamate (min−1) |
|
|---|---|---|
| wild-type | 3.6 ± 0.4 | 14.3 ± 0.5 |
| R548K | 3.7 ± 0.6 | 3.3 ± 0.2 |
| Q552N | 3.5 ± 0.5 | 3.5 ± 0.5 |
| Q552A | 2.2 ± 0.3 | 2.1 ± 0.3 |
Assay conditions: 100 mM Tris-HCl pH 7.8, 30°C, 0.25 mM acetyl-CoA, 0.2 mM oxaloacetate, 0.2 mM NADH. The reported errors are standard deviations of the means of three separate determinations of apparent kcat values.
Bicarbonate-dependent ATP cleavage and ADP phosphorylation activities
The effects of mutations on reaction [1] were examined by measuring the bicarbonate-dependent ATPase activities of the wild-type and mutant forms of RePC in the presence and absence of oxamate. In the absence of oxamate, the ATPase activities of R548K, Q552N and Q552A mutants were 7.3, 7.0 and 7.9 min−1 respectively, approximately 70% of the wild-type activity (11.0 min−1) (see Fig. 3), suggesting that these CT domain mutations have only a small effect on the rates of the ATPase reaction. At low concentrations, oxamate stimulated the ATPase activity in the wild-type, R548K and Q552N RePC-catalysed reactions, but at high concentrations, the enzyme activities were inhibited by oxamate (Figure 4). These data were fitted to equation 1 and the values of the kinetic parameters derived from these fits are shown in Table 4. While the values of kcat for the ATPase reaction in the presence of oxamate are similar to those of the wild-type RePC, the activation constant for oxamate (Ka) is 1.9-2.5 fold higher for R548K and Q552N, respectively. However, the inhibition constants (Ki) of oxamate for the R548K and Q552N mutants were 53% and 42%, respectively, of that for wild-type RePC (see Table 4). Oxamate had little effect on the ATPase activity of the Q552A mutant, suggesting that oxamate does not bind properly to this mutant form of RePC.
Figure 4.
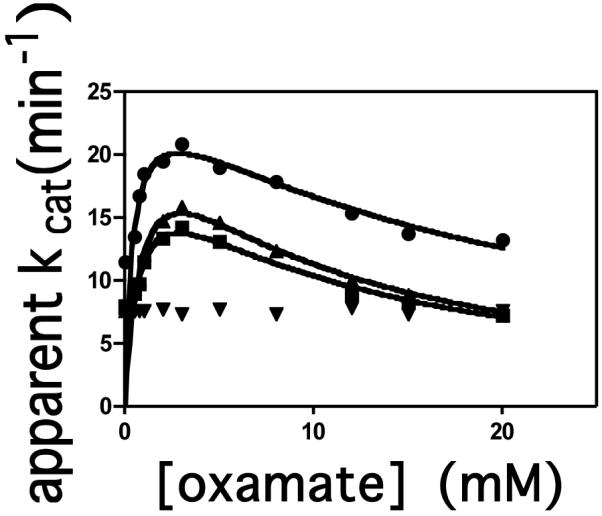
Effect of increasing oxamate concentration on the bicarbonate-dependent ATPase reaction catalysed by RePC: wild-type (●); R548K (■); Q552N (▲); Q552A (▼). Velocity (v) is expressed as μmol Pi released per μmol enzymic biotin per min. The lines represent non-linear least squares regression fits of the data to equation (1).
Table 4.
Kinetic parameters for oxamate stimulation and inhibition of ATP cleavagea
| kcat | Ka | Ki | |
|---|---|---|---|
| (min−1) | (mM) | (mM) | |
| wild-type | 26 ± 1 | 0.43 ± 0.07 | 19 ± 2 |
| R548K | 22 ± 2 | 0.8 ± 0.1 | 10 ± 2 |
| Q552N | 27 ± 2 | 1.1 ± 0.2. | 8 ± 1 |
| Q552A | - | - | - |
Assay conditions: 100 mM Tris-HCl pH 7.8, 30°C, 20 mM NaHCO3, 2.5 mM MgATP, 2.5 mM MgCl2, 0.25 mM acetyl CoA. The reported errors are standard errors of the values of the parameters calculated from the non-linear regression analysis of the data using equation (1).
The phosphorylation of ADP by carbamoyl phosphate, an analogue of the putative carboxyphosphate intermediate, does not directly involve the tethered biotin, thus allowing for the examination of the effects of the CT domain mutations on the partial reverse reaction of the BC domain. The activities of the wild-type and mutant enzymes were determined both in the presence and absence of oxamate. As shown in Table 5, in the absence of oxamate, the ADP phosphorylating activities of the R548K, Q552N and Q552A mutants were 114 %, 78% and 77%, respectively, of the wild-type ADP phosphorylating activity. The presence of 20 mM oxamate appeared to inhibit the activities of wild-type (68%), R548K (37%) and Q552N (30%) RePC, but had no significant effect on the reaction catalysed by the Q552A mutant (t-test, p>0.05).
Table 5.
ADP phosphorylation by carbamoyl phosphate in the absence and presence of oxamatea
| − Oxamate (min−1) |
% of wild-type activity |
+Oxamate (20 mM) (min−1) |
|
|---|---|---|---|
| wild-type | 1.6 ± 0.2 | (100) | 0.50 ± 0.02 |
| R548K | 1.8 ± 0.2 | 114 | 1.13 ± 0.09 |
| Q552N | 1.23 ± 0.08 | 78 | 0.86 ± 0.09 |
| Q552A | 1.2 ± 0.2 | 77 | 1.1 ± 0.1 |
Assay conditions: 100 mM Tris-HCl, pH 7.8, 0.25 mM acetyl-CoA, 20 mM carbamoyl phosphate, 3.0 mM MgADP, 0.24 mM NADP, 0.20 mM glucose. The reported errors are standard deviations of the means of three separate determinations of apparent kcat values.
Proton/deuterium exchange studies using with pyruvate
To examine the effect of the mutations on the ability of the enzyme to facilitate proton transfer and pyruvate enolization during the catalytic reaction, isotope-exchange experiments, which measured proton exchange between the methyl group of pyruvate and water, were performed using either [3-2H3]pyruvate as a substrate in H2O or non-deuterated pyruvate in 90% D2O. In the experiments with [3-2H3]pyruvate, the sum of the integrals of the 1H NMR signal intensities of the three protonated forms of pyruvate (mono-, di-, and tri-protonated) were used to calculate the total concentration of protonated pyruvate at each time point and hence, determine the extent of the enzymatic deuterium/proton exchange between D2O and pyruvate. As shown in Figure 5, the rate of the exchange was linear over time, and the rate constant determined for the wild-type RePC-catalysed exchange was 74 ± 3 min−1. This exchange reaction was completely inhibited by the addition of 20 mM oxamate (data not shown). Since oxamate has been shown to bind at the substrate binding site of the CT domain, the inhibition of the observed proton exchange reaction in the presence of oxamate indicates that it is occurring within the CT domain active site. The exchange reaction was not catalysed by the R548K, Q552N, Q552A or T882A mutants.
Figure 5.

(a) 1H-NMR spectra of protonated pyruvate species at different times after addition of wild-type RePC in a 1 ml of the reaction mixture containing 20 mM NH4HCO3, 10 mM [3-2H3]pyruvate preparation, 5% D2O, 0.01 mg ml−1 enzyme. NMR spectra were acquired at 30 °C.
*C: Spectrum of protonated pyruvate species in the absence of enzyme that corresponding to the initial amount of protonated pyruvate species (1.7% of total pyruvate).
**: The time at which the first spectrum was acquired was set as zero time. The dead time of experiments that is spent for the adjusting of NMR conditions and acquiring the first NMR scans was approximately 14 min.
(b) Time course for the H/D exchange reaction between [3-2H3]pyruvate and water as measured by 1H-NMR. [Protonated pyruvate] is determined from the sum of the concentrations of all the protonated species of pyruvate (mono-, di- and tri-protonated) derived from the integrals of the spectra similar to those shown in (a) and a standard of protonated pyruvate of known concentration. (■) Reaction performed with 10 μg wild-type RePC; (▼) reaction performed with 250 μg wild-type RePC, following pre-incubation of the reaction mixture (− RePC) for 30 min at 30°C with 5 units of malate dehydrogenase and 0.2 mM NADH. Solid lines were obtained by linear least squared regression fits of the data, from the slopes of which rate constants were calculated.
(C) Time course for the H/D exchange reaction between [3-1H3]pyruvate and D2O catalysed by (□) R548K and (●) Q552N, as measured by 1H-NMR in the presence of 1 mM MgATP, 5 mM MgATP and 0.25 mM acetyl CoA. The integrated signal for [3-1H3]pyruvate was used to calculate the concentration of this species remaining at each time point as described in materials and methods. Reactions were performed with 250 μg of each mutant enzyme at 30°C. Solid lines were obtained by linear least squared regression fits of the data, from the slopes of which rate constants were calculated.
However, the pre-incubation of the wild-type RePC reaction mixture with malate dehydrogenase and NADH to remove any possible traces of residual oxaloacetate resulted in the drastic reduction of the rate of the proton/deuterium exchange reaction to 1.4 ± 0.1 min−1 (see Fig. 5b). This suggested that the small amount of contaminating oxaloacetate present in the original reaction mixture was sufficient to carboxylate biotin in the CT domain and promote the proton exchange between [3-2H3]pyruvate and water. To confirm that 1.4 min−1 was the true rate of proton exchange between water and pyruvate in the presence of un-carboxylated biotin, the exchange reaction was performed with wild-type RePC using non-deuterated pyruvate (purchased from Sigma, not prepared from oxaloacetate) in 90% D2O. The rate of disappearance of the 1H NMR signal of [3-1H3]pyruvate, due to the proton exchange with D2O was found to be 1.2 ± 0.2 min−1 and, similar to previous experiments, the proton exchange was abolished in the presence of 20 mM oxamate.
Since R548K and Q552N did not catalyse biotin-dependent oxaloacetate decarboxylation, but still responded to the oxamate-stimulation and -inhibition of ATP cleavage, proton exchange reactions with these mutants were performed using non-deuterated pyruvate as a substrate and in the presence of MgATP and Mg2+ which promotes the carboxylation of biotin in the BC domain (see Fig 5c). Under these conditions, proton exchange between pyruvate and D2O was catalysed by these mutant enzymes, with rate constants of 3.6 ± 0.3 min−1 and 1.3 ± 0.2 min−1 for R548K and Q552N, respectively.
DISCUSSION
The proposed roles for Gln552 and Arg548 in the catalytic mechanism of reactions occurring in the CT domain (see Introduction) are supported by the effects of several mutations of these residues on the overall reaction and those of the individual domains. While individual mutations of these residues resulted in the complete loss of pyruvate carboxylating ability for majority of the mutant RePC enzymes, and a 98% loss in activity for the R548K mutant, the CT domain mutants still retained the ability to catalyse the ATP cleavage and ADP phosphorylation reactions of the BC domain. In addition, the direct effects of the mutation of these residues on reactions occurring in the CT domain were evident in the loss of the biotin-dependent oxaloacetate decarboxylation activities and a reduced ability to catalyse proton exchange between pyruvate and water.
The catalysis of the proton exchange reaction by wild-type RePC using either [3-2H3]pyruvate, from which traces of oxaloacetate had been removed, or [3-1H3]pyruvate, suggests that deprotonation and formation of the enol- pyruvate can occur in the absence of carboxyl transfer, but at a very low rate compared to the overall reaction or when biotin is carboxylated. The inhibition of the reaction with the addition of 20 mM oxamate indicates that the proton exchange between pyruvate and D2O is occurring in the CT domain active site and further signifies the high degree of coupling between the proton and carboxyl transfer. The finding that proton exchange is not catalysed by any of the mutants when biotin is not carboxylated, and at only very low rates when biotin is carboxylated at the BC domain by R548K and Q552N suggests that these mutations have compromised the ability of the enzyme to enolize pyruvate. The higher exchange rate seen with R548K compared to Q552N probably reflects the fact that R548K does retain some capacity to catalyse pyruvate carboxylation.
The fact that the presence of oxamate had no effect on the rates of ATP cleavage or on the ADP phosphorylation reactions catalysed by the Q552A mutant, coupled with the inability of the mutant to catalyse the pyruvate/H2O proton exchange, strongly suggests that this mutation resulted in an enzyme that is unable to properly bind either substrate or inhibitor in the CT domain active site. On the other hand, in the R548K- and Q552N-catalysed reactions, the effects of oxamate on both the rates of the ATP cleavage and the rates of the ADP phosphorylation reactions are generally similar to those effects observed in the reactions catalysed by wild-type RePC, indicating that these mutants still retain some ability to correctly bind pyruvate and facilitate the enolization.
Pyruvate or oxamate binding in the active site of the CT domain is thought to induce the binding of biotin/carboxybiotin at that domain (26), where carboxybiotin is induced to decarboxylate even when subsequent substrate carboxylation does not occur (26-28). This can explain the hydrolytic leak previously observed at low pyruvate concentrations, where the stoichiometry of ATP cleavage to oxaloacetate formation is greater than one (29, 30). Furthermore, it can also explain the stimulatory effect of low concentrations of oxamate on the ATPase reaction presented in the current work and observed by others (12). In the absence of a CT domain substrate, a major rate-limiting step in the steady-state is the decarboxylation of carboxybiotin in the CT domain (31, 32). Thus, the low concentrations of oxamate will stimulate the movement of carboxybiotin from the BC domain to the CT domain where decarboxylation is induced thereby enhancing the overall rate of ATP cleavage.
In non-biotinylated mutant forms of PC, the presence of free biotin has been shown to stimulate both the ATPase reaction and the ADP phosphorylation reaction with carbamoyl phosphate (12, 17), even though biotin does not play a direct role in either reaction. The inhibitory effects of high oxamate concentrations on both ATP cleavage and ADP phosphorylation observed in the current work and by others have been explained in terms of the induced movement of biotin/carboxybiotin to the active site of the CT domain. (12).
Our results on the effects of oxamate on ATP cleavage are consistent with the findings of Zeczycki et al. (12) for wild-type RePC and indicate that substrate or inhibitor binding in the CT domain is not greatly affected in the R548K and Q552N mutant forms of the RePC enzyme. The lower Ki values for oxamate, as compared to wild-type RePC, determined for these mutants suggest that whilst the mutants may show somewhat improved abilities to signal the movement of carboxybiotin formed at the BC domain active site to the CT domain active site upon oxamate binding, the ability of these mutants to induce its decarboxylation is somewhat diminished, as reflected in the higher Ka values. The mutation of Thr882, which is essential for proton transfer between the substrate and biotin (12), results in a mutant where oxamate, even at low concentrations, had no stimulatory effect on the ATP cleavage, and, in fact, partially inhibited the reaction. Zeczycki et al. (12) proposed that the essential role Thr882 in facilitating the proton transfer between substrate/inhibitor and carboxybiotin, which is highly coupled to the decarboxylation of carboxybiotin, in the CT domain, explains the inhibition of the ATPase reaction of the BC domain in the T882A-catalysed reaction. Namely, the T882A mutant is able to bind oxamate and trigger the movement of carboxybiotin to the CT domain, but is unable to stimulate the decarboxylation and release of biotin, thereby inhibiting its ability to return to the BC domain where it would further stimulate the ATP cleavage reaction.
Our results indicate that while the R548K and Q552N mutants can effectively bind substrates or inhibitors in the CT domain so as to induce the movement of biotin away from the BC domain, but the inability of these mutants to catalyse the biotin-dependent oxaloacetate decarboxylation, pyruvate carboxylation and the proton exchange reaction between pyruvate and water indicates that these residues may play a catalytic role in facilitating the enolization of pyruvate. This is most likely accomplished through the stabilisation of the developing negative charge at the carbonyl oxygen of the substrate through the formation of hydrogen-bonding interactions. In addition, the positive charge of Arg548 could also electrostatically interact with the carbonyl oxygen, further promoting the formation of the enol-pyruvate intermediate. Furthermore, it is likely that these residues also participate in correctly positioning the substrate relative to Thr882. Thr882 has been proposed to facilitate the transfer a proton between the substrate and biotin (7, 12), which is further supported by our finding that the T882A mutant does not catalyse proton exchange between pyruvate and water.
Our ultracentrifugation results indicate that the quaternary structures of R548Q and R548A were greatly perturbed, suggesting that Arg458 may also play a role in maintaining and stabilising the overall active tetrameric structure of RePC. Apart from hydrogen-bonding interactions with both the substrate and Gln552, the guanidinyl group of Arg548 is positioned near the carboxyl group of Glu582 (2.70 Å). The mutation of the corresponding glutamate residue in Bacillus thermodenitrificans PC (Glu576) by Yong-Biao et al. (9), also resulted in the destabilisation of the quaternary structure of the enzyme, alluding to the possible importance of the interaction between Arg548 and Glu582 in stabilising the quaternary structure of the enzyme, although it is currently not clear how this stabilisation would occur.
The results presented in the current work in tandem with the previous evidence of the role of Thr882 in the reaction [2] (7, 12), provides a more comprehensive view of the catalytic mechanism of the carboxyl-transferase domain reaction. Arg548 and Gln552 are proposed to position the substrate in a catalytically competent orientation in the active site of the CT domain, and their interactions with the carbonyl oxygen of the substrate are vital, in concert with the proton removal by Thr882, to the induction of enolization of pyruvate.
Supplementary Material
Acknowledgements
SD was a M.Sc. student supported by the DPST scholarship, Thailand and the Endeavour Asia-Australia Postgraduate and Postdoctoral Research Fellowship from the Department of Education, Science and Training (DEST), Australia.
Abbreviations
- PC
pyruvate carboxylase
- RePC
Rhizobium etli pyruvate carboxylase
- BC
biotin carboxylase
- CT
carboxyl transferase
Footnotes
This work was supported by the National Institute of Health grant GM070455 to WWC, JCW, PVA and MSt.M and, an Australian Research Council Discovery Grant DP0988153 to JCW and PVA and a Career Establishment Grant, the Faculty of Science, Mahidol University to SJ.
The precise orientation of pyruvate in the active site of PC remains poorly defined. The resolution limit of current crystal structures precludes an absolute identification of the position of the pyruvate atoms. While the absolute binding configuration of pyruvate has not been clearly determined, its relative position is unambiguous and can be confidently placed in close proximity to Arg548 and Gln552
References
- 1.Attwood PV, Wallace JC. Chemical and catalytic mechanisms of carboxyl transfer reactions in biotin-dependent enzymes. Acc.Chem. Res. 2002;35:113–120. doi: 10.1021/ar000049+. [DOI] [PubMed] [Google Scholar]
- 2.Jitrapakdee S, St. Maurice M, Rayment I, Cleland WW, Wallace JC, Attwood PV. Structure, mechanism and regulation of pyruvate carboxylase. Biochem. J. 2008;413:369–387. doi: 10.1042/BJ20080709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Knowles JR. The mechanism of biotin-dependent enzymes. Annu. Rev. Biochem. 1989;58:195–221. doi: 10.1146/annurev.bi.58.070189.001211. [DOI] [PubMed] [Google Scholar]
- 4.Jitrapakdee S, Wallace JC. The biotin enzyme family: conserved structural motifs and domain rearrangements. Curr. Protein Pept. Sci. 2003;4:217–229. doi: 10.2174/1389203033487199. [DOI] [PubMed] [Google Scholar]
- 5.St. Maurice M, Reinhardt L, Surinya KH, Attwood PV, Wallace JC, Cleland WW, Rayment I. Domain architecture of pyruvate carboxylase, a biotin-dependent multifunctional enzyme. Science. 2007;317:1076–1079. doi: 10.1126/science.1144504. [DOI] [PubMed] [Google Scholar]
- 6.Xiang S, Tong L. Crystal structures of human and Staphylococcus aureus pyruvate carboxylase and molecular insights into the carboxyltransfer reaction. Nat. Struct. Mol. Biol. 2008;25:295–302. doi: 10.1038/nsmb.1393. [DOI] [PubMed] [Google Scholar]
- 7.Yu LP, Xiang S, Lasso G, Gil D, Valle M, Tong L. A symmetrical tetramer for S. aureus pyruvate carboxylase in complex with coenzyme A. Structure. 2009;17:823–832. doi: 10.1016/j.str.2009.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Branson JP, Nezic M, Jitrapadkee S, Wallace JC, Attwood PV. Kinetic characterization of yeast pyruvate carboxylase isoenzyme Pyc1 and the Pyc1 mutants, C249A. Biochemistry. 2004;43:1075–1081. doi: 10.1021/bi035575y. [DOI] [PubMed] [Google Scholar]
- 9.Yong-Biao J, Islam MN, Sueda S, Kondo H. Identification of the catalytic residues involved in the carboxyl transfer of pyruvate carboxylase. Biochemistry. 2004;43:5912–5920. doi: 10.1021/bi035783q. [DOI] [PubMed] [Google Scholar]
- 10.Waldrop GL, Rayment I, Holden HM. Three-dimensional structure of the biotin carboxylase subunit of acetyl-CoA carboxylase. Biochemistry. 1994;33:10249–10256. doi: 10.1021/bi00200a004. [DOI] [PubMed] [Google Scholar]
- 11.Hall PR, Zheng R, Antony L, Pusztai-Carey M, Carey PR, Yee VC. Transcarboxylase 5S structures: Assembly and catalytic mechanism of a multienzyme complex subunit. EMBO J. 2004;23:3621–3631. doi: 10.1038/sj.emboj.7600373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zeczycki TN, St Maurice M, Jitrapakdee S, Wallace JC, Attwood PV, Cleland WW. Insight into the carboxyl transferase domain mechanism of pyruvate carboxylase from Rhizobium etli. Biochemistry. 2009;48:4305–4313. doi: 10.1021/bi9003759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Attwood PV, Tipton PA, Cleland WW. Carbon-13 and deuterium isotope effects on oxaloacetate decarboxylation by pyruvate carboxylase. Biochemistry. 1986;25:8197–8205. doi: 10.1021/bi00373a012. [DOI] [PubMed] [Google Scholar]
- 14.O’Keefe SJ, Knowles JR. Enzymatic biotin-mediated carboxylation is not a concerted process. J. Am. Chem. Soc. 1986;108:328–329. [Google Scholar]
- 15.Dunn MF, Encarnacion S, Araiza G, Vargas MC, Davalos A, Peralta H, Mora Y, Mora J. Pyruvate carboxylase from Rhizobium etli: Mutant characterization, nucleotide sequence, and physiological role. J. Bacteriol. 1996;178:5960–5970. doi: 10.1128/jb.178.20.5960-5970.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chapman-Smith A, Turner DL, Cronan JE, Jr., Morris TW, Wallace JC. Expression, biotinylation and purification of a biotin-domain peptide from the biotin carboxy carrier protein of Escherichia coli acetyl CoA carboxylase. Biochem. J. 1994;302:881–887. doi: 10.1042/bj3020881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Adina-Zada A, Jitrapakdee S, Surinya KH, Mcldowie MJ, Piggott MJ, Cleland WW, Wallace JC, Attwood PV. Insight into the mechanism and regulation of pyruvate carboxylase by characterization of a biotin deficient mutant of the Bacillus thermodenitrificans enzyme. Int. J. Biochem. Cell Biol. 2008;40:1743–1752. doi: 10.1016/j.biocel.2008.01.001. [DOI] [PubMed] [Google Scholar]
- 18.Rylatt DB, Keech DB, Wallace JC. Pyruvate carboxylase: Isolation of the biotin-containing tryptic peptide and the determination of its primary sequence. Arch. Biochem. Biophys. 1977;183:113–122. doi: 10.1016/0003-9861(77)90425-8. [DOI] [PubMed] [Google Scholar]
- 19.Jitrapakdee S, Surinya KH, Adina-Zada A, Polyak SW, Stojkoski C, Smyth R, Booker GW, Cleland WW, Attwood PV, Wallace JC. Conserved Glu40 and Glu433 of the biotin carboxylase domain of yeast pyruvate carboxylase I isoenzyme are essential for the association of tetramers. Int. J. Biochem. Cell Biol. 2007;39:2130–2134. doi: 10.1016/j.biocel.2007.06.006. [DOI] [PubMed] [Google Scholar]
- 20.Schuck P. Size distribution analysis of macromolecules by sedimentation velocity ultracentrifugation and Lamm equation modeling. Biophys. J. 2000;78:1606–1619. doi: 10.1016/S0006-3495(00)76713-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Attwood PV, Cleland WW. Decarboxylation of oxaloacetate by pyruvate carboxylase. Biochemistry. 1986;25:8191–8196. doi: 10.1021/bi00373a011. [DOI] [PubMed] [Google Scholar]
- 22.Attwood PV, Graneri BDLA. Pyruvate carboxylase catalysis of phosphate transfer between carbamoyl phosphate and ADP. Biochem. J. 1991;273:443–448. doi: 10.1042/bj2730443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Khew-Goodall YS, Johannssen W, Attwood PV, Wallace JC, Keech DB. Studies on dilution inactivation of sheep liver pyruvate carboxylase. Arch. Biochem. Biophys. 1991;284:98–105. doi: 10.1016/0003-9861(91)90269-o. [DOI] [PubMed] [Google Scholar]
- 24.Attwood PV, Johannssen W, Chapman-Smith A, Wallace JC. The existence of multiple tetrameric conformers of chicken liver pyruvate carboxylase and their roles in dilution inactivation. Biochem J. 1993;290:583–590. doi: 10.1042/bj2900583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Attwood PV, Geeves MA. Changes in catalytic activity and association state of pyruvate carboxylase which are dependent on enzyme concentration. Arch. Biochem. Biophys. 2002;401:63–72. doi: 10.1016/S0003-9861(02)00039-5. [DOI] [PubMed] [Google Scholar]
- 26.Goodall GJ, Baldwin GS, Wallace JC, Keech DB. Factors that influence the translocation of the N-carboxybiotin moiety between the two sub sites of pyruvate carboxylase. Biochem. J. 1981;199:603–609. doi: 10.1042/bj1990603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Attwood PV, Wallace JC, Keech DB. The carboxybiotin complex of pyruvate carboxylase: a kinetic analysis of the effects of Mg2+ ions on its stability and on its reaction with pyruvate. Biochem. J. 1984;219:243–251. doi: 10.1042/bj2190243. Keech. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Attwood PV, Wallace JC. The carboxybiotin complex of chicken liver pyruvate carboxylase: A kinetic analysis of the effects of acetyl-CoA, Mg2+ ions and temperature on its reaction with 2-oxobutyrate. Biochem. J. 1986;235:359–364. doi: 10.1042/bj2350359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Easterbrook-Smith SB, Hudson PJ, Goss NH, Keech DB, Wallace JC. Pyruvate carboxylase: mechanism of the second partial reaction. Arch. Biochem. Biophys. 1976;176:709–720. doi: 10.1016/0003-9861(76)90215-0. [DOI] [PubMed] [Google Scholar]
- 30.Wallace JC, Phillips NB, Snoswell MA, Goodall GJ, Attwood PV, Keech DB. Pyruvate carboxylase - mechanisms of partial reactions. Ann. N.Y. Acad. Sci. 1985;447:169–188. doi: 10.1111/j.1749-6632.1985.tb18436.x. [DOI] [PubMed] [Google Scholar]
- 31.Branson JP, Attwood PV. Effects of Mg2+ on the pre-steady-state kinetics of the biotin-carboxylation reaction of pyruvate carboxylase. Biochemistry. 2000;39:7480–7491. doi: 10.1021/bi992825v. [DOI] [PubMed] [Google Scholar]
- 32.Branson JP, Nezic M, Wallace JC, Attwood PV. Kinetic characterisation of yeast pyruvate carboxylase iosozyme Pyc1. Biochemistry. 2002;41:4459–4466. doi: 10.1021/bi011888m. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.