

Abstract
A number of studies suggest an association between Alzheimer's disease (AD) and diabetes: AD patients show impaired insulin function, whereas cognitive deficits and increased risk of developing AD occur in diabetic patients. The reasons for the increased risk are not known. Recent studies of disturbances in the insulin-signaling pathway have revealed new perspectives on the links between AD and Type 1 diabetes with a particular focus on glycogen synthase-kinase-3 (GSK3). We have therefore characterized a mouse model of combined insulin-deficient diabetes and AD and find that diabetes exaggerated defects in the brain of APP transgenic mice. Mice with combined APP overexpression and diabetes showed a decreased insulin receptor activity and an increased GSK3β activity. Concomitantly, tau phosphorylation and number of Aβ plaques, the two pathologic hallmarks of AD, were increased in the brain of diabetic-APP transgenic mice. Our results indicate that the pathologic features of AD are exaggerated in the brain of APP transgenic mice that have concurrent insulin-deficient diabetes, and underscore a possible mechanism of brain dysfunction common to AD and diabetes.
Keywords: Alzheimer's disease, Diabetes, Neurodegenerative diseases, Insulin receptor, Glycogen Synthase-Kinase-3, Amyloid, Tau
Introduction
Alzheimer's disease (AD) is the leading cause of dementia in the aging population and is characterized by neurodegeneration affecting the cortex and the limbic system, along with deposition of amyloid β (Aβ) and intraneuronal neurofibrillary tangles (Terry, 2006). Despite several theories, the precise pathologic mechanisms leading to neurodegeneration in AD are not yet clear. Accumulating evidence suggests that disruption of insulin-signaling in the brain may contribute to the pathology of AD (Gasparini, et al., 2002). Several studies have reported reduced insulin levels and insulin receptor expression in AD brains (Frolich, et al., 1998, Steen, et al., 2005), while other studies have emphasized insulin resistance (Craft, 2007), but all converge to a disruption of the insulin-signaling pathway. In addition to regulation of food intake and energy homeostasis, insulin and insulin receptors in the brain play a role in cognitive function (Zhao, et al., 2004). A recent study demonstrated that severity of dementia and decline in cognitive performance were associated with decreased insulin area under the curve (AUC), not glucose AUC, after a glucose tolerance test in patients with early stage AD (Burns, et al., 2007). Insulin, acting at insulin receptors, activates signal transduction via the phosphatidylinositol 3-kinase (PI3-K)-protein kinase B (PKB or Akt) pathway. Down-stream of this pathway lies glycogen synthase kinase-3 (GSK3), the activity of which is down-regulated by phosphorylation at serine 21 and serine 9 of the 2 isoforms GSK3α and GSK3β, respectively (Sutherland, et al., 1993). GSK3 has been suggested to play a role in AD pathology by modulation of amyloid β (Aβ) formation and tau phosphorylation (Jope and Johnson, 2004), both major hallmarks of AD. In the AD brain, active GSK3β is colocalized with abnormally phosphorylated tau in pre-tangle neurons (Pei, et al., 1999), and GSK3 activity is increased in AD brains (Steen, et al., 2005). GSK3 activity is also increased in the brain of the transgenic mThy1-hAPP751 (hAPP) mice that express human amyloid precursor protein (APP) 751 containing the London and Swedish mutations and which develop amyloid plaques in the frontal cortex by 3 months of age, along with learning and memory deficits (Rockenstein, et al., 2003, Rockenstein, et al., 2001).
Diabetes affects an estimated 24 million patients in the United States and is characterized by disturbances of the insulin-signaling pathway. Theses disturbances result from hypoinsulinemia in the case of Type 1 diabetes, or from insulin resistance and impaired insulin secretion in Type 2 diabetes. A number of studies have reported that diabetes mellitus is associated with an increased risk of developing AD (Leibson, et al., 1997, Ott, et al., 1996, Xu, et al., 2007). Other studies have reported no clear association (Curb, et al., 1999, MacKnight, et al., 2002) but underscore that diabetes should be considered as a potential risk factor for cognitive impairment, dementia and AD (Akomolafe, et al., 2006).
Disturbance of the insulin-signaling pathway is emerging as a common feature of both AD and diabetes and, to date, attention has largely focused on Type 2 diabetes with hyperinsulinemia and insulin resistance being the primary insults (Ho, et al., 2004). In contrast, sparse data are available on associations between Type 1 diabetes and AD, although hypoinsulinemia evokes a similar impairment of insulin signaling. Nevertheless, cognitive deficits, such as impaired learning, memory, problem solving, and mental flexibility have been recognized as being more common in Type 1 diabetic subjects than in the general population (Biessels, et al., 2008, Ryan, et al., 1985), suggesting a detrimental effect of cerebral hyperglycemia, and/or hypoinsulinemia. Along with these deficits, degeneration of cerebral cortex (Reske-Nielsen and Lundbaek, 1963), and neuronal loss (DeJong, 1977) are observed at autopsy and are more pronounced in patients suffering from Type 1 diabetes than in age-matched non-diabetic patients. Moreover, we have recently demonstrated the presence of learning deficits associated with increased GSK3 activity, increased tau phosphorylation and increased Aβ protein levels in the brain of a mouse model of Type 1 diabetes (Jolivalt, et al., 2008). Similar changes also occur in a rat model of spontaneous Type 1 diabetes (Li, et al., 2007). Insulin therapy partially prevented behavioral and biochemical changes observed in the Type 1 diabetic mouse model (Jolivalt, et al., 2008), suggesting that insulin deficiency plays a pathogenic role in the development of AD-like features in the brain. To further investigate the roles of insulin deficiency and hyperglycemia on CNS dysfunction and pathology, we have now studied the impact of diabetes on AD progression, by inducing insulin-deficient diabetes in a mouse model of AD. Specifically, we investigated cognitive performance and changes in phosphorylation of proteins of the insulin-signaling pathway in parallel with changes in tau phosphorylation and Aβ expression, the main neuropathological hallmarks of AD.
Methods
Animals
All studies were performed using the well-characterized transgenic hAPP mice (Rockenstein, et al., 2003, Rockenstein, et al., 2001, Rockenstein, et al., 2007) and their littermates. The hAPP transgenic mice express mutated (London V717I and Swedish K670M/N671L) hAPP751 under the control of the neuronal murine (m)Thy-1 promoter (Rockenstein, et al., 2001). This transgenic mouse model was selected because of the high levels of Aβ1-42 produced and relatively early appearance of performance deficits in the water maze, synaptic damage, and plaque formation. Transgenic lines were maintained by crossing heterozygous transgenic mice with non-transgenic wild type (WT) C57BL/6 × DBA/2 F1 breeders. All mice were heterozygous with respect to the transgene. Animals were housed 4-5 per cage with free access to food and water and maintained in a vivarium approved by the American Association for the Accreditation of Laboratory Animal Care. All animal studies were carried out according to protocols approved by the Institutional Animal Care and Use Committee of the University of California San Diego. Five to 8 mice were used per group.
Induction of diabetes
Insulin-deficient diabetes was induced in 4 months old mice following an overnight fast by intraperitoneal (i.p.) injection of streptozotocin (STZ, Sigma. St, Louis, MO) at 90 mg/kg dissolved in 0.9% sterile saline, on 2 successive days. Hyperglycemia was confirmed using a strip-operated reflectance meter in a blood sample obtained by tail prick four days after STZ injection and in another sample collected at the conclusion of the study.
Accelerating rotarod task
Mice were place on a rotarod (TSE systems, Midland, MI, USA) that accelerated from 4 to 40 rpm over 5 minutes. Latency to loss of balance was recorded. The test was performed once on day 5 after the Barnes circular maze task.
Barnes circular maze task
The Barnes circular maze consists of an illuminated white circular platform with 20 holes (5 cm diameter) equally spaced and located 5 cm from the perimeter. A black escape box was placed under one of the holes. A cue was placed behind the hole with the escape box. On the first day of testing, the mouse was placed in the escape box for one minute before the trial began. The mouse was then placed in the middle of the platform and allowed to explore the maze. Timing of the session ended when the mouse found the box or after 5 minutes had elapsed. At the end of the session, the mouse was left or placed in the escape box for one additional minute. Mice were tested, starting at 11 weeks of diabetes, once a day, for 5 consecutive days (Monday through Friday) for the learning phase and after 3 days without testing at day 9 for the memory phase of the test.
Neuropathological analysis and detection of Aβ deposits
After 12 weeks of diabetes, mice were sacrificed by decapitation after brief isoflurane anesthesia in accordance with NIH guidelines for the humane treatment of animals. Caution was taken to remove the brain within a minute of decapitation to preserve phosphorylation status. Brains were removed and divided sagitally. One hemibrain was post-fixed in phosphate-buffered 4% paraformaldehyde (pH 7.4) at 4°C for 48 hr and sectioned at 40 μm with a Vibratome 200 (Leica, Germany) while the other hemibrain was prepared for western blot analysis (see next section). Forty μm thick vibratome sections from mouse brains fixed in 4% paraformaldehyde were immunolabeled with the mouse monoclonal antibody against Aβ and tau phosphorylation, as previously described (Rockenstein, et al., 2002). Briefly, vibratome sections were incubated overnight at 4°C with the mouse monoclonal antibody 6E10 (1:1000, Covance/Signet Laboratories, Berkeley, CA, USA), which specifically recognizes Aβ and the PH1 antibody that recognizes phosphorylated tau (generously provided by Dr. Peter Davies, Albert Einstein Institute), followed by avidin-biotin-conjugated anti-mouse IgG (Novo red chromogen: Vector Laboratories). Slides were analyzed by an observer who was unaware of the animal group using an Olympus BX51 microscope configured for bright field. For each animal (3 sections), 4 images of the hippocampus were obtained and analyzed for levels of phosphorylated tau immunoreactivity with the ImageQuant program. Results were averaged and expressed as mean per animal. The number of amyloid β-immunoreactive plaques were counted and normalized to surface area in mm2. Additional characterization of plaques was performed using Thioflavine-S as previously described (Lewis, et al., 1987) and imaged with a laser-scanning confocal microscope (MRC1024, BioRad, Hercules, CA, USA). Digital images were then analyzed with the NIH Image 1.43 program to determine the percent area occupied by Aβ deposits. Three immunolabeled sections were analyzed per mouse and the average of individual measurements was used to calculate group means. Another set of slides were immunolabeled with an antibody against NeuN (general neuronal marker, 1:1,000,Chemicon) and reacted with diamino-benzidine (DAB). Sections were analyzed with the Stereo-Investigator Software (MBF Biosciences) and images collected according to the optical disector method were analyzed as previously described (Ubhi, et al., 2009). Three immunolabeled sections were analyzed per mouse and the average of individual measurements was used to calculate group means.
Tissue preparation for western blot analysis
Hemibrains (without cerebellum) were homogenized in buffer (50 mM Tris-HCl pH7.4, 150 mM NaCl, 0.5% Triton X, 1 mM EDTA, protease inhibitor cocktail). Homogenates were centrifuged at 13,000g for 30 min and supernatants are stored in aliquots at -80°C. A fraction of the homogenate was boiled for 5 minutes under detergent-free conditions, and insoluble material removed from the heat-stable supernatant by centrifugation for 30 minutes. Heat-stable supernatants (about 7μg protein) were prepared with an equal volume of Laemmli SDS sample buffer for Western-blot analysis for tau. Protein concentration was assessed using the bicinchoninic acid method (BCA protein assay kit, Pierce, Rockford, IL, USA).
Western blotting
Brain tissue homogenates were centrifuged (13,000g) and aliquots of the clear extract boiled in Laemmli LDS sample buffer (Invitrogen, Carlsbad, CA, USA). Seven to 20 μg of total extract protein were separated on 4-12% SDS-PAGE Bis-Tris gels (Novex, Invitrogen, Carlsbad, CA, USA) and immunoblotted on nitrocellulose. Blots were incubated with antibodies against phospho-insulin receptor (phosphorylated Ser 972, 1/1200, Upstate, Temecula, CA, USA), insulin receptor (1/200, Chemicon International, Temecula, CA, USA), phospho-GSK3β (phospho-Ser9; 1/1000, Cell Signaling technology, USA), GSK3α/β (1/5000, Chemicon International, Temecula, CA, USA), phospho-tau and Tau-5 (phospho-Thr 231, 1/3000, Biosource, Camarillo, CA, USA), full-length (FL) APP (mouse monoclonal, clone 22C11, 1/20,000, Chemicon International, Temecula, CA, USA), Aβ (mouse monoclonal, clone 6E10, 1/1000, Covance/Signet Laboratories, Berkeley, CA, USA), APP C-terminal fragments (CTFs, rabbit polyclonal CT15, 1/2500, courtesy of Dr. E. Koo, UCSD), Insulin Degrading Enzyme (IDE, 1/5000, Calbiochem, NJ, USA), synaptophysin (1/10000, Chemicon International, Temecula, CA, USA) followed by secondary antibodies tagged with horseradish peroxidase (HRP, 1/5000 or 1/10000, Santa Cruz Biotechnology, Inc., CA, USA). Blots were developed as previously described (Jolivalt et al., 2008). Quantification of immunoreactivity was performed by densitometric scanning using Quantity One software (BioRad, San Diego, CA, USA). For each animal, band intensities were normalized by calculating the ratio of the intensity of the band corresponding to primary antigen of interest to the intensity of the band corresponding to total protein (non-phosphorylated) and/or to actin. To allow grouping of samples run on different gels (gel/blots were repeated 3 to 5 times for each protein of interest), actin-normalized densitometric measures of band intensity for each animal were then expressed as a percentage of the group mean of all samples from control mice present on the same gel.
Determination of Aβ levels by ELISA
Quantification of the levels of human Aβ1-42, human Aβ1-40 and mouse Aβ1-42 in whole brain homogenenates was performed using commercially available Elisa kits (Invitrogen, Carlsbad, CA, USA) following the manufacturer's instructions.
Statistical analysis
Behavioral data are expressed as group median ± interquartiles and the difference between groups was analyzed using Kruskal-Wallis test for non-parametric data. All other data are expressed as group mean ± SEM and differences between groups were analyzed using one-way ANOVA followed by Bonferroni's post-hoc test for more than 2 groups comparison or Student t-test when only 2-group comparison.
Results
Diabetes
Mice were injected with STZ (90mg/kg i.p.) after overnight fast on two consecutive days and exhibited hyperglycemia (blood sugar >270 mg/dl) 4 days after STZ injection. Twelve weeks later, blood glucose levels for diabetic mice were significantly higher than for non-diabetic mice (Table 1). All mice maintained a healthy weight over the 12-week period (Table 1). Mice were sacrificed at 7-8 months of age, after 3 months of diabetes. The study was initiated with 8 WT, 8 STZ, 6 APP and 7 APP-STZ mice, however during the course of the study, prior to behavior tests, 1 APP and 2 APP-STZ mice died. One additional APP-STZ mice died after the series of behavior tests prior the scheduled termination, bringing the numbers to 8 WT, 8 STZ, 5 APP and 4 APP-STZ mice for western blot analysis.
Table 1.
Physiological parameters of wild type, diabetic (STZ), APP and diabetic-APP (APP-STZ) mice before and after 12 weeks of diabetes. Data are given as mean ± sem, **p<0.01 by one-way ANOVA vs Wild type followed by Bonferroni's post hoc test.
| n | Starting weight (g) |
Final weight (g) |
Final blood sugar (mg/dl) |
|
|---|---|---|---|---|
| Wild type | 8 | 32.9 ± 1.4 | 35.9±1.3 | 158 ± 7 |
| STZ | 8 | 34.0 ± 2.2 | 33.4 ± 2.6 | 506 ± 37** |
| APP | 5 | 26.4 ± 0.9 | 29.1 ± 1.6 | 141 ± 10 |
| APP-STZ | 5 | 31.6 ± 2.9 | 27.0 ± 2.3 | 498 ± 72** |
Diabetes alone and when combined with AD worsened cognitive performance
Barnes circular maze task
The testing session was repeated for 5 consecutive days at 11 weeks of diabetes to test learning ability. On the first day of testing, all 4 groups of mice found the escape box with a similar time (group median: WT=241, APP=300, STZ= 300 APP-STZ=300). By day 4, 100% of wild type mice (WT) found the escape box with a median time of 50s, indicative of learning and memory, while the time to find the escape box for STZ mice was only marginally reduced (Fig. 1), with 4 out of 8 STZ mice finding the box within the cut-off time. By day 4, APP transgenic mice found the escape box in a time similar to that of WT mice while 4 out of 5 APP-STZ mice never found the escape box over the 5 days of the trial, suggesting learning and/or memory deficits. After 11 weeks of diabetes, diabetic mice (STZ and APP-STZ) displayed a significantly reduced ability to learn the location of the escape box (Fig. 1). The mice were then left untested for 3 days before performing one further session to test memory retention. WT mice found the escape box in a time similar to that of the last test, indicative of retained memory. APP transgenic mice found the box in a time close to the cut-off time, suggesting impaired memory. This is consistent with our prior data using the Morris water maze test (Rockenstein, et al., 2003). Similar to the learning phase, both diabetic groups (STZ and APP-STZ) maintained their performance in terms of time and number of mice finding the box (Fig. 1) but only the time of APP-STZ mice was significantly (p<0.05) different from that of WT mice with only 1 out 5 mice finding the box within the cut-off time. The between-group differences were not due to marked locomotor deficits, as all 4 groups of mice performed the accelerating rotating beam task with a similar time.
Figure 1. Barnes maze task.
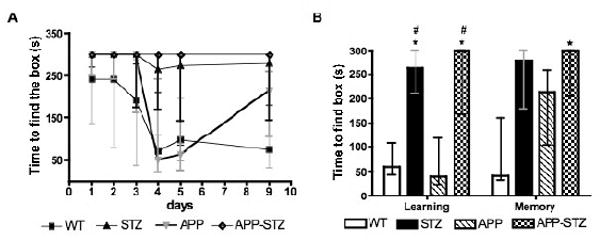
A: Time course of the learning and memory assessment using the Barnes maze test. B: Time to find the escape box at day 4 and 9: Learning was assessed on day 4 after 4 consecutive testing sessions and memory was assessed on day 9 after 3 days without testing. Data are represented as median ± interquartiles. * p< 0.05 and # p<0.05 by Kruskal-Wallis for nonparametric data followed by Dunn's post hoc test vs WT and APP group, respectively. n=8 for WT and STZ groups, n=5 for APP and APP-STZ groups.
Diabetes combined with AD reduces insulin receptor phosphorylation and exaggerates GSK3β activity
Insulin receptor phosphorylation
Phosphorylated insulin receptor levels were significantly reduced (p<0.05) in the brain of both STZ and APP-STZ mice after 3 months of diabetes when compared to WT, and significantly reduced (p<0.05) in the brain of APP-STZ mice when compared to APP mice (Fig. 2). Protein levels of insulin receptor were unchanged for all groups (Fig. 2).
Figure 2. Phosphorylation level of insulin receptor in mouse brain.
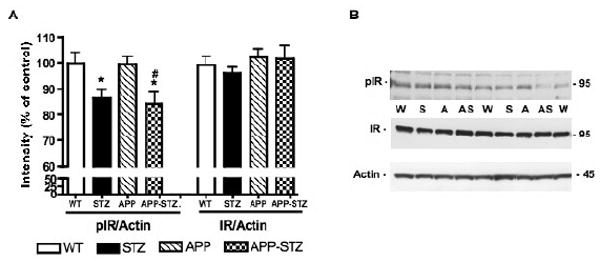
A: Intensity of bands corresponding to phosphorylated insulin receptor (pIR) and insulin receptor (IR) normalized to the intensity of bands corresponding to actin. Data are represented as mean+sem. *p<0.05 vs WT, #p<0.05 vs APP by one-way ANOVA followed by Bonferroni's post hoc test. n=8 for WT and STZ groups, n=5 for APP group and n=4 for APP-STZ group. B: Western blots of brain homogenates from Wild type (W), STZ-diabetic (S), APP (A) and APP-STZ (AS) mice for phosphorylated insulin receptor (pIR), total insulin receptor (IR) and actin.
GSK3β phosphorylation
Phosphorylation of GSK3β at the recognized inactivating site (ser 9) was significantly reduced in APP-STZ mice after 3 months of diabetes, while protein levels of total GSK3β were unchanged in all 4 groups (Fig. 3). Similarly, phosphorylation of tyrosine 216, indicative of GSK3β activity, was significantly (p<0.05) increased in the brain of APP-STZ mice when compared to WT (percent intensity for WT: 100±6, STZ: 111±6, APP: 108±5, APP-STZ: 120±8*).
Figure 3. Phosphorylation level of GSK3β in mouse brain.
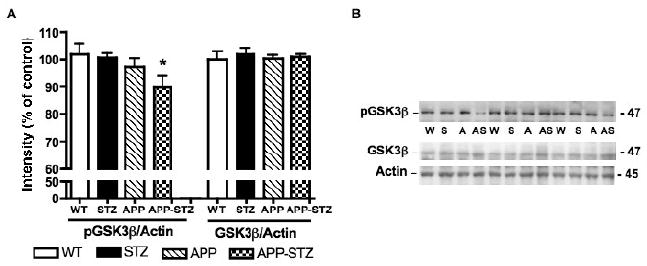
A: Intensity of bands corresponding to phosphorylated GSK3β (pGSK3β) and GSK3β normalized to the intensity of bands corresponding to actin. Data are represented as mean+sem. *p<0.05 vs WT by one-way ANOVA followed by Bonferroni's post hoc test. n=8 for WT and STZ groups, n=5 for APP group and n=4 for APP-STZ group B: Western blots of brain homogenates from Wild type (W), STZ-diabetic (S), APP (A) and APP-STZ (AS) mice for phosphorylated GSK3β (pGSK3β), total GSK3β (GSK3β) and actin.
Diabetes enhances the severity of major pathological markers of AD
Tau phosphorylation
Tau phosphorylation at the threonine 231 site that is part of the microtubule-binding domain and phosphorylated by GSK3, tended towards increase (p=0.054) in the brain of STZ mice and was significantly increased in the brain of the combined APP-STZ mice (p<0.01, Fig. 4). We also extended our analysis of tau phosphorylation using the AT8 antibody against the phosphorylated Ser199/202, which is recognized as a marker of neurofibrillary tangles and is also phosphorylated by GSK3. We found the same pattern to that seen with threonine 231, with an increase in tau phosphorylation that was significant (p<0.05) in the brain of APP-STZ mice when compared to WT mice (percent of intensity for WT: 100±5, STZ: 112±6, APP: 110±6, APP-STZ: 137±13*). Phosphorylated tau immunoreactivity was increased in hippocampal neurons of APP-STZ mice and occasionally associated with plaques (less than 10% of the plaques) in the brain of APP-STZ mice (Fig. 5). Consistent with the western blot analysis, levels of phosphorylated tau immunoreactivity were significantly increased in APP-STZ mice brain (p<0.001 when compared to all 3 groups, Fig. 5F).
Figure 4. Phosphorylated tau in mouse brain.
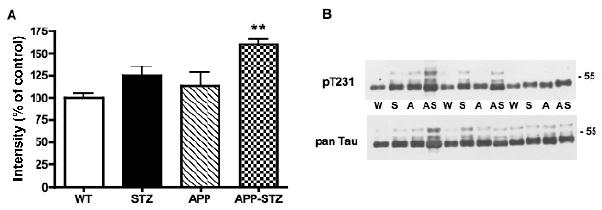
A: Intensity of bands corresponding to phosphorylated tau at threonine 231 (p T231) normalized to the intensity of bands corresponding to total tau (pan Tau). Data are represented as mean+sem. **p<0.01 vs WT by one-way ANOVA followed byBonferroni's post hoc test. n=8 for WT and STZ groups, n=5 for APP group and n=4 for APP-STZ group B: Western blots of brain homogenates from Wild type (W), STZ-diabetic (S), APP (A) and APP-STZ (AS) mice for phosphorylated tau at threonine 231 (p T231) and total tau (pan Tau).
Figure 5. Phosphorylated tau in mouse brain.
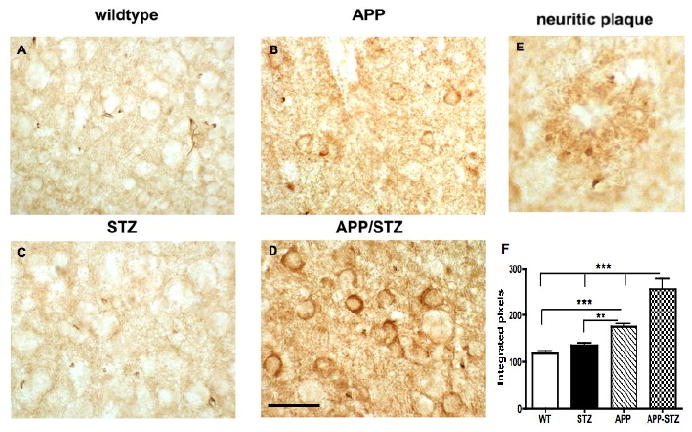
A-D: Phosphorylated tau (PHF1) immunoreactivity in mouse hippocampus illustrating somatodendritic accumulation of phosphorylated tau in APP-STZ mice brain (D), bar=40 μm. E: Phosphorylated tau immunoreactivity associated with neuritic plaque in APP-STZ mice hippocampus. F: Quantitative analysis of phosphorylated tau immunoreactivity in brain from Wild type (WT), STZ-diabetic (STZ), APP and APP-STZ mice. **p<0.01, ***p<0.01 by one-way ANOVA followed by Bonferroni's post hoc test.
Aβ
A direct comparison between non-transgenic and APP transgenic mice was not possible for Aβ using Western blots as levels of protein were drastically different due to APP overexpression in the transgenic mice. The effect of diabetes was clearly visible in the combined APP-STZ model, where soluble Aβ levels were significantly (p<0.05) increased in the brain of APP-STZ mice after 3 months of diabetes in comparison to APP transgenic mice, while levels of full-length APP (FL-APP) and C-terminal fragment (CTF-APP) were unchanged (Fig. 6A, B). Analysis of Aβ levels by ELISA showed that human Aβ1-42 was the most abundant species in mutant APP overexpressing mice (Fig. 6C, D), as previously demonstrated (Rockenstein, et al., 2001). The combination of APP overexpression and insulin-deficient diabetes led to a non-significant increase of human Aβ1-40 levels (Fig 6C) while there was a significant (p<0.05) 2-fold increase of Aβ1-42 levels in APP-STZ mice brain when compared to APP mice (Fig. 6D). Analysis of mouse Aβ1-42 by ELISA revealed detectable, however close to the limit of detection, levels in APP and APP-STZ mice brain (APP: 0.14±0.04 μg/mg protein, APP-STZ: 0.24±0.07 μg/mg protein). Absorbances for WT and STZ mice brain homogenates were below the limit of detection for the 3 ELISA assays. Immunohistochemical analysis of mouse brain demonstrated the presence of Aβ-immunoreactive plaques in APP transgenic and APP-STZ mice (Fig. 7A, B, D, E), that are similar to plaques found in the brain of a patient diagnosed with late stage AD (Fig. 7F). The number of plaques was significantly increased in the APP-STZ mice brain compared to all groups (WT: 0.1±0.1, STZ:0.1±0.1, APP:4.2±0.8%, APP-STZ: 6.8±0.9%*, p<0.001 using 2-way ANOVA followed by Bonferroni's post hoc test). Analysis of the percentage of the neuropil covered by immunoreactive plaques revealed a significant increased in the hippocampus of APP-STZ mice when compared to all groups (Fig. 7C). The increased percentage area of plaques in the combined APP-STZ model correlates with the increased levels of soluble Aβ protein detected by western blot and ELISA. Figure 7G, H illustrates neuritic plaques stained with Thioflavine-S in cortex and hippocampus of APP and APP-STZ mice.
Figure 6. Amyloid β level in mouse brain.
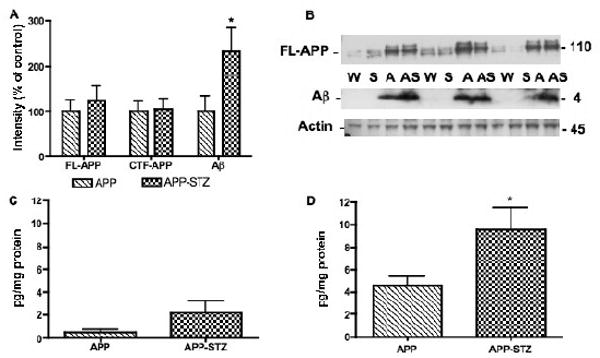
A: Intensity of bands corresponding to full length APP (FL-APP), C terminal fragment-APP (CTF-APP) and amyloid β (Aβ) normalized to the intensity of bands corresponding to actin. Data are represented as mean+sem. *p<0.05 vs APP by unpaired t test. n=5 for APP group and n=4 for APP-STZ group. B: Western blots of brain homogenates from Wild type (W), STZ-diabetic (S), APP (A) and APP-STZ (AS) mice for full length APP (FL-APP), amyloid β (Aβ) and actin. C: Aβ1-40 levels in brain homogenates from APP and APP-STZ mice using an Elisa assay for human amyloid β1–40. Data are represented as mean+sem. D: Aβ1-42 levels in brain homogenates from APP and APP-STZ mice using an Elisa assay for human amyloid β1–42. Data are represented as mean+sem. *p<0.05 vs APP by unpaired t test. n=5 for APP group and n=4 for APP-STZ group.
Figure 7. Amyloid β-immunoreactive plaques in mice hippocampus.
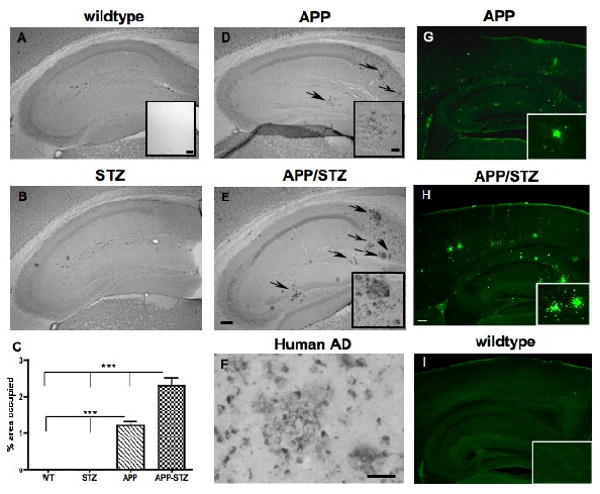
Amyloid β-immunoreactive plaques in hippocampus from Wild type (A), STZ-diabetic (STZ, B), APP (D) and diabetic-APP (APP/STZ, E) mice after 12 weeks of diabetes. Arrows point at amyloid β-immunoreactive plaques, bar=200μm. Inset in A: Specificity of immunostaining by omission of the primary antibody, bar=200 μm. Insets in D and E: magnification of amyloid β-immunoreactive plaques in APP and APP-STZ mice hippocampus, respectively, bar=40μm. C: Quantification of amyloid β-immunoreactive plaques as percent area occupied by plaques in mouse hippocampus. Data are represented as mean+sem. ***p<0.001 by one-way ANOVA followed by Bonferroni's post hoc test. n=8 for WT and STZ groups, n=5 for APP group and n=4 for APP-STZ group. F: Amyloid β-immunoreactive plaques in the brain from a patient diagnosed with late stage AD, bar=40μm. G-H: Mature plaques were intensively labeled with Thioflavine-S in hippocampus and cortex from APP and APP/STZ mice, respectively but not in Wildtype (I), bar=200 μm. Insets in G, H and I: Magnification of hippocampus containing plaques for APP (G) and APP/STZ (H) mice and no plaques for WT mouse (I).
Diabetes combined with AD reduces Insulin-Degrading Enzyme and synaptophysin protein levels
Insulin-Degrading Enzyme (IDE) protein expression was significantly reduced (p<0.05) in the combined APP-STZ group after 3 months of diabetes (Fig. 8A). Synaptophysin, a marker of synapse integrity, was quantified by Western blot. After 3 months of diabetes, synaptophysin protein levels were significantly reduced (p<0.05) in the brain of mice with combined AD and diabetes (Fig. 8B). Analysis of neuronal loss, using NeuN immunoreactivity, showed a significant (p<0.05) decrease in the hippocampus of APP-STZ mice when compared to WT mice (Number of NeuN immunolabeled cells × 102: WT: 267.5±7.5, STZ: 252.5±12.3, APP: 227.5±8.5, APP-STZ: 205±31.9*).
Figure 8. IDE and synaptophysin protein level in mouse brain.
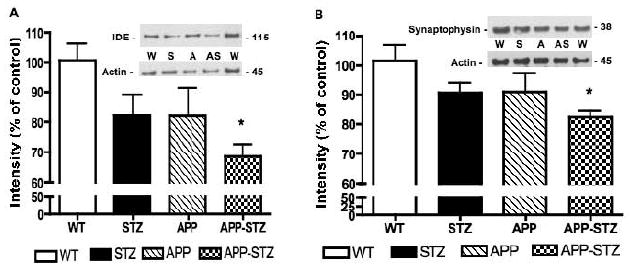
A: Intensity of bands corresponding to IDE normalized to the intensity of bands corresponding to actin. Data are represented as mean+sem. *p<0.05 vs WT by one-way ANOVA followed by Bonferroni's post hoc test. Western blots of brain homogenates from Wild type (W), STZ-diabetic (S), APP (A) and APP-STZ (AS) mice for IDE and actin. B: Intensity of bands corresponding to synaptophysin normalized to the intensity of bands corresponding to actin. Data are represented as mean+sem. *p<0.05 vs WT by one-way ANOVA followed by Bonferroni's post hoc test. Western blots of brain homogenates from Wild type (W), STZ-diabetic (S), APP (A) and APP-STZ (AS) mice for synaptophysin and actin. n=8 for WT and STZ groups, n=5 for APP group and n=4 for APP-STZ group.
Discussion
There is growing evidence of links between AD and diabetes (Leibson, et al., 1997, Ott, et al., 1996, Sima and Li, 2006, Xu, et al., 2007), with AD patients showing impaired insulin function (Frolich, et al., 1998, Steen, et al., 2005), while cognitive deficits and increased risk of developing AD occur in both Type 1 and Type 2 diabetic patients (Biessels, et al., 2008, Ryan, et al., 1985). The reasons for the increased risk are not known. Both diseases have a neurodegenerative pathology and may share common mechanisms that culminate in neurodegeneration. Recent studies of disturbances of the insulin-signaling pathway by ourselves and others (Jolivalt, et al., 2008, Li, et al., 2007) have revealed new perspectives on the links between AD and Type 1 diabetes. The systemic insulin deficiency in STZ-diabetic mice promotes reduced insulin-signaling pathway activity and increased GSK3 activity in the brain, associated with behavioral and biochemical features of AD (Jolivalt, et al., 2008). In the present study, we demonstrated similar, but exaggerated, effects in the brain of APP-STZ mice, suggesting that peripheral insulin deficiency may enhance aspects of AD pathology in the brain.
Pathological changes in AD brain include tau hyperphosphorylation and Aβ accumulation. Several pieces of evidence implicate GSK3 as playing a role in APP processing leading to the formation of Aβ, (Su, et al., 2004) (Lee, et al., 2003). The role of GSK3 in Aβ production is supported by studies using lithium, an inhibitor of GSK3 activity that showed reduced Aβ production and amyloid plaque load in hAPP transgenic mice (Phiel, et al., 2003, Su, et al., 2004). In our current study, consistent with our prior data (Rockenstein, et al., 2001), hAPP overexpressing transgenic mice displayed a significant increased of Aβ levels and Aβ-immunoreactive plaques in the hippocampus compared to WT. These increased levels of soluble Aβ, and more specifically Aβ1-42, and increased number of immunoreactive plaques were significantly exaggerated by the superimposition of insulin-deficient diabetes to mutant APP overexpression, suggesting that diabetes and the disturbances associated with it in the brain contribute to the enhanced progression of AD pathology. The contribution of local insulin deficiency was also demonstrated by studies in which STZ was injected directly into the brain, resulting in downregulation of mRNA for both insulin and the insulin receptor and a concurrent increase of GSK3 activity and Aβ accumulation in the brain (Lester-Coll, et al., 2006, Salkovic-Petrisic, et al., 2006). In addition to increased processing of APP by GSK3, the accumulation of soluble and insoluble Aβ in the diabetic-APP transgenic mice brain may result from reduced degradation. The Insulin-Degrading Enzyme (IDE) degrades insulin but also a number of other small proteins, including Aβ (Chesneau, et al., 2000). IDE protein levels and activity are reduced in AD brains (Perez, et al., 2000). Although not usually described as part of the insulin-PI3K-GSK3 pathway, IDE upregulation requires insulin-mediated AKT activation (Zhao, et al., 2004). Therefore, insulin deficiency and a decreased signaling via the PI3K pathway may contribute to decreased IDE expression and thus contribute to increased Aβ protein levels as a result of a reduced degradation.
Tau pathology is often disregarded as an initiator of AD pathology and rather considered a consequence. However, the severity and the progression of AD correlates better with tau hyperphosphorylation and tangle formation than with amyloid plaques (Braak and Braak, 1991). Moreover, a recent study demonstrated the role of tau in Aβ excitoxicity (Roberson, et al., 2007). Tau can be phosphorylated by GSK3, facilitating its accumulation into neurofibrillary tangles (Jope and Johnson, 2004). In our study, there is co-incidence of activated GSK3β and increased tau phosphorylation in the brain of mice with combined mutant APP overexpression and diabetes. Supporting the role of active GSK3 in tau phosphorylation, studies showed increased GSK3β activity associated with increased tau phosphorylation in the brain of neuron-specific insulin receptor knock-out mice (Schubert, et al., 2004), of transgenic mice overexpressing active GSK3 (Brownlees, et al., 1997), of transgenic mice conditionally overexpressing GSK3β (Lucas, et al., 2001) and of rats exposed to intracerebral STZ that exhibit increased brain GSK3β activity (Grunblatt, et al., 2007, Lester-Coll, et al., 2006). In contrast, systemic STZ injection acutely induced an increase of tau phosphorylation without increased GSK3 activity in the mice brain 3 days after STZ injection (Clodfelder-Miller, et al., 2006). Activation of several pathways may results in tau phosphorylation and to definitively link the observed increased GSK3 activity with the observed increased tau phosphorylation in our study, pharmacological studies will be necessary. However, in vivo and human studies bring additional support to our results. For example, lithium treatment to reduce GSK3 activity also reduces tau hyperphosphorylation and formation of tangle-like lesions in tau transgenic mice (Noble, et al., 2005, Perez, et al., 2003, Phiel, et al., 2003), and in the human AD brain, active GSK3 colocalizes with phosphorylated tau in pretangle neurons (Pei, et al., 1999).
Aβ and hyperphosphorylated tau contribute to the neurodegeneration and neuronal loss that characterizes AD. In the present study both are significantly increased in the combined APP overexpression/insulin-deficient diabetes model, suggesting an enhanced neurodegeneration that was illustrated by a significant neuronal loss and significant reduced level of synaptophysin protein, a marker of synapse loss. The limited reduction of synaptophysin protein level in these relatively young hAPP transgenic mice is in agreement with prior work (Rockenstein, et al., 2003, Rockenstein, et al., 2001), which showed that a significant reduction of synaptophysin immunoreaction occurred only after 9-11 months of age. APP-STZ mice exhibited a significant reduction in brain synaptophysin protein levels by 7-8 months of age when compared to WT mice. This suggests that diabetes hastens or enhances the synaptic failure and the neuronal loss that occurs in AD brains and is consistent with the concomitant increase of Aβ accumulation and increased tau phosphorylation.
Accumulation of tau and/or Aβ has been shown to lead to cognitive impairments. Recently, reducing tau levels in the brain was shown to ameliorate Aβ-induced deficits in AD mouse model (Roberson, et al., 2007), demonstrating the interaction of both Aβ and tau in onset of behavioral deficits. In addition, Aβ oligomers injected into mouse hippocampus significantly slowed the spatial learning of mice only when concomitant with hyperglycemia induced by STZ injection (Huang, et al., 2007). Similarly, diabetes induced by STZ injection promotes impaired memory formation and retention in presymptomatic human APP/presenilin transgenic mice (Burdo, et al., 2008). In our study, both increased tau phosphorylation and Aβ accumulation were present and significantly increased in the combined disease mouse model and may contribute to cognitive impairments, that were assessed using the Barnes maze (Barnes, 1979, Jolivalt, et al., 2008). This task takes advantage of the natural preference of rodents for dark environment and relies minimally on motor skills (Pompl, et al., 1999). In the Barnes maze, WT and APP transgenic mice performed similarly in the learning phase of the test while APP transgenic mice performed significantly worse during the memory phase, consistent with their water maze performance (Rockenstein, et al., 2003). Similar results have been obtained in the Barnes maze test for other AD mice models (Pompl, et al., 1999, Reiserer, et al., 2007). Superimposition of diabetes on mutant APP overexpression induced learning deficits in the transgenic mice similar to those observed in diabetic mice when compared to WT and APP mice. Pharmacologic interventions, such as lithium, specific GSK3 inhibitors or insulin treatment, will be necessary to confirm the link between insulin-deficient diabetes, GSK3β activity, AD-like neuropathology and cognitive performance deficits in the combined APP overexpression/insulin-deficent diabetes model. Nevertheless, this association is strongly supported by our previous studies, in which APP/dominant negative GSK3 transgenic mice and APP transgenic mice treated with lithium, that inhibited GSK3β activity, displayed improved cognitive performance, reduced Aβ immunoreactive plaques, and decreased tau phosphorylation (Rockenstein, et al., 2007). Moreover, as insulin treatment prevented learning deficits, GSK3 activation and increased tau phosphorylation in STZ-diabetic mice (Jolivalt, et al., 2008), these disorders can be attributed to insulin deficiency and its consequences rather than any direct STZ neurotoxicity.
In patients with early stage AD, higher plasma insulin levels were associated with less whole brain and hippocampal atrophy and with better global cognitive performance, whereas plasma glucose levels did not correlate well (Burns, et al., 2007). Our current study shows that the severity of the pathologic features of AD is increased in APP transgenic mice that have concurrent insulin-deficient diabetes. In addition to our previous study (Jolivalt, et al., 2008), our data suggest that dysregulation of the insulin-signaling pathway during type 1 diabetes may provide a convergent mechanism of CNS damage and dysfunction in AD and diabetes that underlies reports that untreated diabetes increases the risk of AD (Xu, et al., 2009).
Acknowledgments
This work was supported by a grant from the Institute for the Study of Aging (ISOA) and a Career Development Award from the Juvenile Diabetes Research Foundation (JDRF) to C.G.J. and by NIH grants AG5131, AG18440, AG10435, AG022074 and NS057096 to E.M. The authors would like to thank Anthony Adame for technical assistance and Dr. Nigel Calcutt for fruitful discussions on diabetes models.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Akomolafe A, Beiser A, Meigs JB, Au R, Green RC, Farrer LA, Wolf PA, Seshadri S. Diabetes mellitus and risk of developing Alzheimer disease: results from the Framingham Study. Arch Neurol. 2006;63:1551–1555. doi: 10.1001/archneur.63.11.1551. [DOI] [PubMed] [Google Scholar]
- 2.Barnes CA. Memory deficits associated with senescence: a neurophysiological and behavioral study in the rat. J Comp Physiol Psychol. 1979;93:74–104. doi: 10.1037/h0077579. [DOI] [PubMed] [Google Scholar]
- 3.Biessels GJ, Deary IJ, Ryan CM. Cognition and diabetes: a lifespan perspective. Lancet Neurol. 2008;7:184–190. doi: 10.1016/S1474-4422(08)70021-8. [DOI] [PubMed] [Google Scholar]
- 4.Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991;82:239–259. doi: 10.1007/BF00308809. [DOI] [PubMed] [Google Scholar]
- 5.Brownlees J, Irving NG, Brion JP, Gibb BJ, Wagner U, Woodgett J, Miller CC. Tau phosphorylation in transgenic mice expressing glycogen synthase kinase-3beta transgenes. Neuroreport. 1997;8:3251–3255. doi: 10.1097/00001756-199710200-00013. [DOI] [PubMed] [Google Scholar]
- 6.Burdo JR, Chen Q, Calcutt NA, Schubert D. The pathological interaction between diabetes and presymptomatic Alzheimer's disease. Neurobiol Aging. 2008 doi: 10.1016/j.neurobiolaging.2008.02.010. [DOI] [PubMed] [Google Scholar]
- 7.Burns JM, Donnelly JE, Anderson HS, Mayo MS, Spencer-Gardner L, Thomas G, Cronk BB, Haddad Z, Klima D, Hansen D, Brooks WM. Peripheral insulin and brain structure in early Alzheimer disease. Neurology. 2007;69:1094–1104. doi: 10.1212/01.wnl.0000276952.91704.af. [DOI] [PubMed] [Google Scholar]
- 8.Chesneau V, Vekrellis K, Rosner MR, Selkoe DJ. Purified recombinant insulin-degrading enzyme degrades amyloid beta-protein but does not promote its oligomerization. Biochem J. 2000;351(Pt 2):509–516. [PMC free article] [PubMed] [Google Scholar]
- 9.Clodfelder-Miller BJ, Zmijewska AA, Johnson GV, Jope RS. Tau is hyperphosphorylated at multiple sites in mouse brain in vivo after streptozotocin-induced insulin deficiency. Diabetes. 2006;55:3320–3325. doi: 10.2337/db06-0485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Craft S. Insulin resistance and Alzheimer's disease pathogenesis: potential mechanisms and implications for treatment. Curr Alzheimer Res. 2007;4:147–152. doi: 10.2174/156720507780362137. [DOI] [PubMed] [Google Scholar]
- 11.Curb JD, Rodriguez BL, Abbott RD, Petrovitch H, Ross GW, Masaki KH, Foley D, Blanchette PL, Harris T, Chen R, White LR. Longitudinal association of vascular and Alzheimer's dementias, diabetes, and glucose tolerance. Neurology. 1999;52:971–975. doi: 10.1212/wnl.52.5.971. [DOI] [PubMed] [Google Scholar]
- 12.DeJong RN. CNS manifestations of diabetes mellitus. Postgrad Med. 1977;61:101–107. doi: 10.1080/00325481.1977.11714510. [DOI] [PubMed] [Google Scholar]
- 13.Frolich L, Blum-Degen D, Bernstein HG, Engelsberger S, Humrich J, Laufer S, Muschner D, Thalheimer A, Turk A, Hoyer S, Zochling R, Boissl KW, Jellinger K, Riederer P. Brain insulin and insulin receptors in aging and sporadic Alzheimer's disease. J Neural Transm. 1998;105:423–438. doi: 10.1007/s007020050068. [DOI] [PubMed] [Google Scholar]
- 14.Gasparini L, Netzer WJ, Greengard P, Xu H. Does insulin dysfunction play a role in Alzheimer's disease? Trends Pharmacol Sci. 2002;23:288–293. doi: 10.1016/s0165-6147(02)02037-0. [DOI] [PubMed] [Google Scholar]
- 15.Grunblatt E, Salkovic-Petrisic M, Osmanovic J, Riederer P, Hoyer S. Brain insulin system dysfunction in streptozotocin intracerebroventricularly treated rats generates hyperphosphorylated tau protein. J Neurochem. 2007;101:757–770. doi: 10.1111/j.1471-4159.2006.04368.x. [DOI] [PubMed] [Google Scholar]
- 16.Ho L, Qin W, Pompl PN, Xiang Z, Wang J, Zhao Z, Peng Y, Cambareri G, Rocher A, Mobbs CV, Hof PR, Pasinetti GM. Diet-induced insulin resistance promotes amyloidosis in a transgenic mouse model of Alzheimer's disease. FASEB J. 2004;18:902–904. doi: 10.1096/fj.03-0978fje. [DOI] [PubMed] [Google Scholar]
- 17.Huang HJ, Liang KC, Chen CP, Chen CM, Hsieh-Li HM. Intrahippocampal administration of A beta(1-40) impairs spatial learning and memory in hyperglycemic mice. Neurobiol Learn Mem. 2007;87:483–494. doi: 10.1016/j.nlm.2006.11.006. [DOI] [PubMed] [Google Scholar]
- 18.Jolivalt CG, Lee CA, Beiswenger KK, Smith JL, Orlov M, Torrance MA, Masliah E. Defective insulin signaling pathway and increased glycogen synthase kinase-3 activity in the brain of diabetic mice: Parallels with Alzheimer's disease and correction by insulin. J Neurosci Res. 2008 doi: 10.1002/jnr.21787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jope RS, Johnson GV. The glamour and gloom of glycogen synthase kinase-3. Trends Biochem Sci. 2004;29:95–102. doi: 10.1016/j.tibs.2003.12.004. [DOI] [PubMed] [Google Scholar]
- 20.Lee MS, Kao SC, Lemere CA, Xia W, Tseng HC, Zhou Y, Neve R, Ahlijanian MK, Tsai LH. APP processing is regulated by cytoplasmic phosphorylation. J Cell Biol. 2003;163:83–95. doi: 10.1083/jcb.200301115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Leibson CL, Rocca WA, Hanson VA, Cha R, Kokmen E, O'Brien PC, Palumbo PJ. Risk of dementia among persons with diabetes mellitus: a population-based cohort study. Am J Epidemiol. 1997;145:301–308. doi: 10.1093/oxfordjournals.aje.a009106. [DOI] [PubMed] [Google Scholar]
- 22.Lester-Coll N, Rivera EJ, Soscia SJ, Doiron K, Wands JR, de la Monte SM. Intracerebral streptozotocin model of type 3 diabetes: relevance to sporadic Alzheimer's disease. J Alzheimers Dis. 2006;9:13–33. doi: 10.3233/jad-2006-9102. [DOI] [PubMed] [Google Scholar]
- 23.Lewis DA, Campbell MJ, Terry RD, Morrison JH. Laminar and regional distributions of neurofibrillary tangles and neuritic plaques in Alzheimer's disease: a quantitative study of visual and auditory cortices. J Neurosci. 1987;7:1799–1808. doi: 10.1523/JNEUROSCI.07-06-01799.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li ZG, Zhang W, Sima AA. Alzheimer-like changes in rat models of spontaneous diabetes. Diabetes. 2007;56:1817–1824. doi: 10.2337/db07-0171. [DOI] [PubMed] [Google Scholar]
- 25.Lucas JJ, Hernandez F, Gomez-Ramos P, Moran MA, Hen R, Avila J. Decreased nuclear beta-catenin, tau hyperphosphorylation and neurodegeneration in GSK-3beta conditional transgenic mice. EMBO J. 2001;20:27–39. doi: 10.1093/emboj/20.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.MacKnight C, Rockwood K, Awalt E, McDowell I. Diabetes mellitus and the risk of dementia, Alzheimer's disease and vascular cognitive impairment in the Canadian Study of Health and Aging. Dement Geriatr Cogn Disord. 2002;14:77–83. doi: 10.1159/000064928. [DOI] [PubMed] [Google Scholar]
- 27.Noble W, Planel E, Zehr C, Olm V, Meyerson J, Suleman F, Gaynor K, Wang L, LaFrancois J, Feinstein B, Burns M, Krishnamurthy P, Wen Y, Bhat R, Lewis J, Dickson D, Duff K. Inhibition of glycogen synthase kinase-3 by lithium correlates with reduced tauopathy and degeneration in vivo. Proc Natl Acad Sci U S A. 2005;102:6990–6995. doi: 10.1073/pnas.0500466102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ott A, Stolk RP, Hofman A, van Harskamp F, Grobbee DE, Breteler MM. Association of diabetes mellitus and dementia: the Rotterdam Study. Diabetologia. 1996;39:1392–1397. doi: 10.1007/s001250050588. [DOI] [PubMed] [Google Scholar]
- 29.Pei JJ, Braak E, Braak H, Grundke-Iqbal I, Iqbal K, Winblad B, Cowburn RF. Distribution of active glycogen synthase kinase 3beta (GSK-3beta) in brains staged for Alzheimer disease neurofibrillary changes. J Neuropathol Exp Neurol. 1999;58:1010–1019. doi: 10.1097/00005072-199909000-00011. [DOI] [PubMed] [Google Scholar]
- 30.Perez A, Morelli L, Cresto JC, Castano EM. Degradation of soluble amyloid beta-peptides 1-40, 1-42, and the Dutch variant 1-40Q by insulin degrading enzyme from Alzheimer disease and control brains. Neurochem Res. 2000;25:247–255. doi: 10.1023/a:1007527721160. [DOI] [PubMed] [Google Scholar]
- 31.Perez M, Hernandez F, Lim F, Diaz-Nido J, Avila J. Chronic lithium treatment decreases mutant tau protein aggregation in a transgenic mouse model. J Alzheimers Dis. 2003;5:301–308. doi: 10.3233/jad-2003-5405. [DOI] [PubMed] [Google Scholar]
- 32.Phiel CJ, Wilson CA, Lee VM, Klein PS. GSK-3alpha regulates production of Alzheimer's disease amyloid-beta peptides. Nature. 2003;423:435–439. doi: 10.1038/nature01640. [DOI] [PubMed] [Google Scholar]
- 33.Pompl PN, Mullan MJ, Bjugstad K, Arendash GW. Adaptation of the circular platform spatial memory task for mice: use in detecting cognitive impairment in the APP(SW) transgenic mouse model for Alzheimer's disease. J Neurosci Methods. 1999;87:87–95. doi: 10.1016/s0165-0270(98)00169-1. [DOI] [PubMed] [Google Scholar]
- 34.Reiserer RS, Harrison FE, Syverud DC, McDonald MP. Impaired spatial learning in the APPSwe + PSEN1DeltaE9 bigenic mouse model of Alzheimer's disease. Genes Brain Behav. 2007;6:54–65. doi: 10.1111/j.1601-183X.2006.00221.x. [DOI] [PubMed] [Google Scholar]
- 35.Reske-Nielsen E, Lundbaek K. Diabetic Encephalopathy. Diffuse and Focal Lesions of the Brain in Long-Term Diabetes. Acta Neurol Scand Suppl. 1963;39 4:273–290. [PubMed] [Google Scholar]
- 36.Roberson ED, Scearce-Levie K, Palop JJ, Yan F, Cheng IH, Wu T, Gerstein H, Yu GQ, Mucke L. Reducing endogenous tau ameliorates amyloid beta-induced deficits in an Alzheimer's disease mouse model. Science. 2007;316:750–754. doi: 10.1126/science.1141736. [DOI] [PubMed] [Google Scholar]
- 37.Rockenstein E, Adame A, Mante M, Moessler H, Windisch M, Masliah E. The neuroprotective effects of Cerebrolysin in a transgenic model of Alzheimer's disease are associated with improved behavioral performance. J Neural Transm. 2003;110:1313–1327. doi: 10.1007/s00702-003-0025-7. [DOI] [PubMed] [Google Scholar]
- 38.Rockenstein E, Mallory M, Mante M, Alford M, Windisch M, Moessler H, Masliah E. Effects of Cerebrolysin on amyloid-beta deposition in a transgenic model of Alzheimer's disease. J Neural Transm Suppl. 2002:327–336. [PubMed] [Google Scholar]
- 39.Rockenstein E, Mallory M, Mante M, Sisk A, Masliah E. Early formation of mature amyloid-b proteins deposits in a mutant APP transgenic model depends on levels of Ab1-42. J neurosci Res. 2001;66:573–582. doi: 10.1002/jnr.1247. [DOI] [PubMed] [Google Scholar]
- 40.Rockenstein E, Mallory M, Mante M, Sisk A, Masliaha E. Early formation of mature amyloid-beta protein deposits in a mutant APP transgenic model depends on levels of Abeta(1-42) J Neurosci Res. 2001;66:573–582. doi: 10.1002/jnr.1247. [DOI] [PubMed] [Google Scholar]
- 41.Rockenstein E, Mante M, Adame A, Crews L, Moessler H, Masliah E. Effects of Cerebrolysin on neurogenesis in an APP transgenic model of Alzheimer's disease. Acta Neuropathol. 2007;113:265–275. doi: 10.1007/s00401-006-0166-5. [DOI] [PubMed] [Google Scholar]
- 42.Rockenstein E, Torrance M, Adame A, Mante M, Bar-on P, Rose JB, Crews L, Masliah E. Neuroprotective effects of regulators of the glycogen synthase kinase-3beta signaling pathway in a transgenic model of Alzheimer's disease are associated with reduced amyloid precursor protein phosphorylation. J Neurosci. 2007;27:1981–1991. doi: 10.1523/JNEUROSCI.4321-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ryan C, Vega A, Drash A. Cognitive deficits in adolescents who developed diabetes early in life. Pediatrics. 1985;75:921–927. [PubMed] [Google Scholar]
- 44.Salkovic-Petrisic M, Tribl F, Schmidt M, Hoyer S, Riederer P. Alzheimer-like changes in protein kinase B and glycogen synthase kinase-3 in rat frontal cortex and hippocampus after damage to the insulin signalling pathway. J Neurochem. 2006;96:1005–1015. doi: 10.1111/j.1471-4159.2005.03637.x. [DOI] [PubMed] [Google Scholar]
- 45.Schubert M, Gautam D, Surjo D, Ueki K, Baudler S, Schubert D, Kondo T, Alber J, Galldiks N, Kustermann E, Arndt S, Jacobs AH, Krone W, Kahn CR, Bruning JC. Role for neuronal insulin resistance in neurodegenerative diseases. Proc Natl Acad Sci U S A. 2004;101:3100–3105. doi: 10.1073/pnas.0308724101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sima AA, Li ZG. Diabetes and Alzheimer's disease - is there a connection? Rev Diabet Stud. 2006;3:161–168. doi: 10.1900/RDS.2006.3.161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Steen E, Terry BM, Rivera EJ, Cannon JL, Neely TR, Tavares R, Xu XJ, Wands JR, de la Monte SM. Impaired insulin and insulin-like growth factor expression and signaling mechanisms in Alzheimer's disease--is this type 3 diabetes? J Alzheimers Dis. 2005;7:63–80. doi: 10.3233/jad-2005-7107. [DOI] [PubMed] [Google Scholar]
- 48.Su Y, Ryder J, Li B, Wu X, Fox N, Solenberg P, Brune K, Paul S, Zhou Y, Liu F, Ni B. Lithium, a common drug for bipolar disorder treatment, regulates amyloid-beta precursor protein processing. Biochemistry. 2004;43:6899–6908. doi: 10.1021/bi035627j. [DOI] [PubMed] [Google Scholar]
- 49.Sutherland C, Leighton IA, Cohen P. Inactivation of glycogen synthase kinase-3 beta by phosphorylation: new kinase connections in insulin and growth-factor signalling. Biochem J. 1993;296(Pt 1):15–19. doi: 10.1042/bj2960015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Terry RD. Alzheimer's disease and the aging brain. J Geriatr Psychiatry Neurol. 2006;19:125–128. doi: 10.1177/0891988706291079. [DOI] [PubMed] [Google Scholar]
- 51.Ubhi K, Rockenstein E, Doppler E, Mante M, Adame A, Patrick C, Trejo M, Crews L, Paulino A, Moessler H, Masliah E. Neurofibrillary and neurodegenerative pathology in APP-transgenic mice injected with AAV2-mutant TAU: neuroprotective effects of Cerebrolysin. Acta Neuropathol. 2009;117:699–712. doi: 10.1007/s00401-009-0505-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Xu W, Qiu C, Winblad B, Fratiglioni L. The effect of borderline diabetes on the risk of dementia and Alzheimer's disease. Diabetes. 2007;56:211–216. doi: 10.2337/db06-0879. [DOI] [PubMed] [Google Scholar]
- 53.Xu WL, von Strauss E, Qiu CX, Winblad B, Fratiglioni L. Uncontrolled diabetes increases the risk of Alzheimer's disease: a population-based cohort study. Diabetologia. 2009 doi: 10.1007/s00125-009-1323-x. [DOI] [PubMed] [Google Scholar]
- 54.Zhao L, Teter B, Morihara T, Lim GP, Ambegaokar SS, Ubeda OJ, Frautschy SA, Cole GM. Insulin-degrading enzyme as a downstream target of insulin receptor signaling cascade: implications for Alzheimer's disease intervention. J Neurosci. 2004;24:11120–11126. doi: 10.1523/JNEUROSCI.2860-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhao WQ, Chen H, Quon MJ, Alkon DL. Insulin and the insulin receptor in experimental models of learning and memory. Eur J Pharmacol. 2004;490:71–81. doi: 10.1016/j.ejphar.2004.02.045. [DOI] [PubMed] [Google Scholar]