

Abstract
We previously found that myocardial ischemia/reperfusion (I/R) initiates expression of tumor necrosis factor-α (TNF) leading to coronary endothelial dysfunction. However, it is not clear whether there is a direct relationship between levels of TNF expression and endothelial dysfunction in reperfusion injury. We studied levels of TNF expression by using different transgenic animals expressing varying amounts of TNF in I/R. We crossed TNF overexpression (TNF++/++) with TNF knockout (TNF−/−) mice; thus we have a heterozygote population of mice with the expression of TNF “in between” the TNF−/− and TNF++/++ mice. Mouse hearts were subjected to 30 min of global ischemia followed by 90 min of reperfusion and their vasoactivity before and after I/R was examined in wild type (WT), TNF−/−, TNF++/++ and TNF heterozygote (TNF−/++, cross between TNF−/− and TNF++/++) mice. In heterozygote TNF−/++ mice with intermediate cardiac-specific expression of TNF, acetylcholine-induced or flow-induced endothelial-dependent vasodilation following I/R was between TNF++/++ and TNF−/− following I/R. Neutralizing antibodies to TNF administered immediately before the onset of reperfusion-preserved endothelial-dependent dilation following I/R in WT, TNF−/++ and TNF++/++ mice. In WT, TNF−/++ and TNF++/++ mice, I/R-induced endothelial dysfunction was progressively lessened by administration of free-radical scavenger TEMPOL immediately before initiating reperfusion. During I/R, production of superoxide (O2·−) was greatest in TNF++/++ mice as compared to WT, TNF−/++ and TNF−/− mice. Following I/R, arginase mRNA expression was elevated in the WT, substantially elevated in the TNF−/++ and TNF++/++ mice and not affected in the TNF−/− mice. These results suggest that the level of TNF expression determines arginase expression in endothelial cells during myocardial I/R, which is one of the mechanisms by which TNF compromises coronary endothelial function in reperfusion injury.
Keywords: Keywords Coronary artery disease, Ischemia, NO, Microcirculation, Vasodilation
Introduction
Cytokines [e.g., tumor necrosis factor-α (TNF), interlukin-1 (IL-1) and IL-2] are postulated as mediators of ischemia/reperfusion (I/R) [5, 21, 34, 42, 45]. These factors are of direct relevance to endothelial dysfunction, since cytokines exert a prominent anti-endothelium-derived relaxing factor effect in blood vessels [2, 21]. Ischemia alone induces TNF gene expression and peptide synthesis in the myocardium, that is, associated with nuclear factor-κB (NF-κB) activation [2, 21, 41, 44]. Superoxide (O2·−) may be dismutated (spontaneously or enzymatically) to hydrogen peroxide (H2O2), which is another oxidant, and, therefore, another possible mediator of oxidant injury. The reaction of nitric oxide (NO) and O2·− produces the formation of peroxynitrite (ONOO−) which is a potent oxidant [28, 41, 42, 44, 45], and a previous study has shown that TNF inhibits NO-mediated endothelium-dependent vasorelaxation in small coronary arteries via sphingomyelinase activation and consequent O2·− production in endothelial cells (ECs) [4, 30, 47]. However, it has not been elucidated whether there is a direct relationship between levels of TNF expression and endothelial dysfunction in reperfusion injury.
Many in vivo and in vitro studies show endothelial-dependent dilation to agonists, e.g., acetylcholine (ACh), is impaired in I/R injury. We have reported that TNF inhibits endothelium-dependent NO-mediated dilation of coronary arterioles via production of O2·− in porcine coronary arterioles [44]. We also found that myocardial I/R initiates expression of TNF, which induces activation of xanthine oxidase and production of O2·−, leading to coronary endothelial dysfunction [44]. ACh-induced NO-dependent dilation was found to be impaired following I/R in WT mice and this deficit was ameliorated by neutralizing antibodies against TNF (Fig. 1) [46]. We previously demonstrated that TNF upregulates expression of arginase in ECs, which leads to O2·− production, then induces endothelial dysfunction in I/R injury [9]. We also observed greater endothelial injury in TNF++/++ animals as compared to WT animals after I/R and less damage after I/R in TNF−/− animals as compared to WT mice [9]. Thus, we hypothesized that endothelial function following I/R in TNF heterozygote (TNF−/++) mice would be intermediate between that of TNF overexpression (TNF++/++) and TNF knockout (TNF−/−) mice. The purpose of our study was to evaluate whether a direct relationship between levels of TNF expression and endothelial dysfunction occurred in reperfusion injury by determining NO-mediated vasodilation to ACh in isolated and pressurized coronary arterioles and O2·− production and expression of arginase in wild type (WT), TNF−/−, TNF−/++ and TNF++/++ mice (Fig. 1).
Fig. 1.
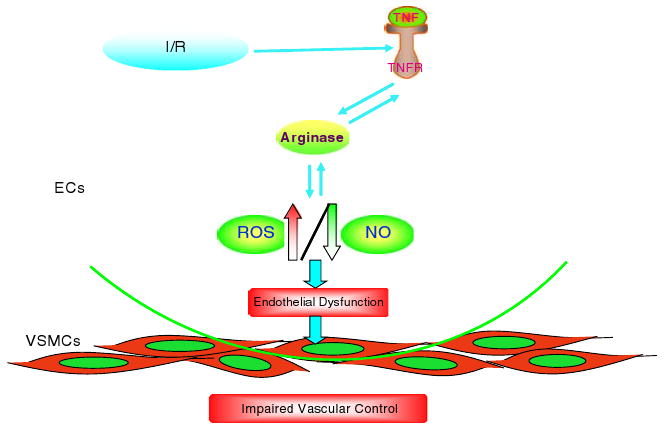
The schematic figure shows that TNF expression compromises coronary endothelial function by increasing arginase expression that results in ROS generation following myocardial I/R injury
Methods
Murine I/R model
The procedures followed were in accordance with approved guidelines set by the Laboratory Animal Care Committee at University of Missouri–Columbia. This study utilized 12- to 15-week-old, 25- to 35-g mice of either sex. We used three different strains of WT mice to match the backgrounds of the knockout (TNF−/−), transgenic (TNF++/++) and heterozygote (TNF−/++) mice. For the TNF−/− mice (Parental strain B6, 129S-Tnftm1Gkl, stock number is 003008. Jackson Laboratory, Bar Harbor, ME), the WT1 is strain B6, 129SF2/J (stock number is 101045, Jackson Laboratory, Bar Harbor, ME). Mice homozygous (TNF++/++) for the TNFtmIGkl-targeted mutation are viable, fertile and show no apparent phenotypic abnormalities. TNF++/++ mice completely lack splenic primary B-cell follicles and cannot form organized follicular dendritic cell networks and germinal centers. For the transgenic overexpression mice TNF++/++ [46], the parental strain for this transgenic line is the FVBN, and WT2 controls for this group consist of the FVBN line. The transgenic mouse heart demonstrated a mild, diffuse, lymphohistiocytic interstitial inflammatory infiltrate. Cardiomyocyte necrosis and apoptosis were present, but not abundant. Magnetic resonance imaging showed that the transgenic mouse heart was significantly dilated with reduced ejection fraction. Heterozygote TNF−/++ mice are the crosses between TNF−/− and TNF++/++ mice with intermediate cardiac-specific expression of TNF. The control mice (WT3) are the crosses between WT1 and WT2, which have the same background with TNF−/++ mice. The surgical protocol was performed similar to methods described previously [9, 10, 46] with minor modification. After 30 min of the left anterior descending (LAD) coronary artery occlusion and 90 min of reperfusion, the heart was harvested and used for functional study or other assays. A sham group was subjected to the same surgical interventions without performing occlusions. We defined WT1, 2 and 3 as WT in this study because the results from WT1, 2 and 3 were identical.
Treatment with TNF and TNF neutralization
We administered TNF (5 μg/kg, i.p., R&D Systems) to WT mice for 3 days to determine whether such a lower level of TNF can inhibit coronary arterial dilations mediated by NO [27]. The neutralizing antibody to TNF (anti-TNF) used in these studies was IgG goat polyclonal antibody prepared to be essentially lipopolysaccharide free. At 12 weeks of age, all mice were administered with the neutralizing antibodies to TNF (anti-TNF IgG. i.p., 0.1 mg/mouse containing 16 mg protein/mL, immediately prior to the onset of reperfusion). This concentration was based on previous studies and sufficient to neutralize endogenous TNF [9, 10, 46].
Functional assessment of isolated coronary arterioles
The techniques for identification and isolation of coronary microvessels were described in detail previously [9, 10, 46]. After I/R, the heart was excised and immediately placed in cold (4°C) saline solution. Each coronary arteriole (50–100 μm in internal diameter) was carefully isolated and then used in the functional and molecular studies described below. To determine the response of coronary arterioles to ACh, vessels were cannulated with glass micropipettes and pressurized to 60 cm H2O intraluminal pressure. The cannulated vessel was bathed in physiological salt solution (PSS; mmol/L: NaCl 145.0, KCl 4.7, CaCl2 2.0, MgSO4 1.17, NaH2PO4 1.2, glucose 5.0, pyruvate 2.0, EDTA 0.02 and Mops 3.0) containing bovine serum albumin (1%; Amersham) at 37°C. For functional studies, vessels were cannulated with glass micropipettes and pressurized to 60 cm H2O luminal pressure without flow. After developing a stable basal tone (i.e., spontaneous constriction to 60–70% of maximal diameter), the experimental interventions were performed. The concentration–diameter relationships for ACh (0.1 nmol/L to 10 μmol/L) and sodium nitroprusside (SNP, 1 nmol/L to 1 μmol/L) were then established. We also tested the dose–response curve to flow-dependent dilation [range of ΔP (that is linearly related to flow) from 4 to 60 cm H2O], a response that is endothelial dependent, but agonist independent.
To determine whether different levels of TNF were playing a role in endothelial injury following I/R, endothelial dependent and independent dilation were assessed in coronary arterioles from anti-TNF IgG-treated mice. ACh or flow was used as an activator of endothelium-dependent NO-mediated vasodilation [9, 10, 46]. To mimic the cross of WT with the TNF null (TNF−/−) animals, we determined whether endothelial function in the presence of a lower level of TNF (5 μg/kg, i.p., 3 days, R&D Systems) in WT I/R mice treated with anti-TNF is in between of WT I/R and WT I/R mice treated with anti-TNF. To determine the role of TNF and O2·− anions in murine I/R, vasodilatory functions were examined in the presence of a membrane-permeable O2·− dismutase mimetic, TEMPOL (1 mmol/L, 60-min incubation). All drugs were administered extraluminally in these functional studies.
Plasma concentration of TNF
Tumor necrosis factor was measured using a commercial kit BIO-Plex cytokine assay (BIO-Plex Mouse 3-Plex Assay, Bio-Rad Laboratories, CA, USA). TNF concentrations were automatically calculated by BIO-Plex Manager software using a standard curve derived from a recombinant cytokine standard. Values were expressed as picogram per milliliter [46].
Measurement of O2·− by electron paramagnetic resonance spectroscopy
A 10% vessel homogenate was prepared in a 50 mmol/L phosphate buffer containing 0.01 mmol/L EDTA. The homogenate was then subjected to low-speed centrifugation (1,000g) for 10 min to remove unbroken cells and debris. The supernatants containing 2 mmol/L CP-H (1-hydrox-3 carboxypyrrolidine) were incubated for 30 min at 37°C and frozen quickly in liquid nitrogen. Electron paramagnetic resonance (EPR) spectroscopy was performed at room temperature using a Bruker EMX spectrometer and 1-mm diameter capillaries. The EPR spectrum settings were as follows: modulation amplitude 1.0 gauss, scan time 83 s, time constant 163 ms and microwave power 40 mW, field sweep 60 gauss, microwave frequency 9.78 GHz, receiver gain 5 × 103, center field 3,485 gauss. Superoxide quantitation from the EPR spectra was determined by double integration of the peaks, with reference to a standard curve generated from horseradish peroxidase generation of the anion from standard solutions of H2O2, using p-acetamidophenol as the cosubstrate, and then normalized by protein concentration.
Arginase expression by Western blotting and real-time PCR
For Western blot analysis, isolated coronary arterioles (3–4 vessels per sample) from WT, TNF−/−, TNF−/++, TNF++/++ mice before and after I/R were homogenized and sonicated in lysis buffer. Protein concentrations were assessed with the use of the BCA kit, and equal amounts of protein (40 μg) were separated by SDS–PAGE and transferred to nitrocellulose membranes (Bio-Rad). The protein expression of arginase I was tested with the use of rabbit arginase I polyclonal IgG primary antibody (Santa-Cruz Biotechnology). Horseradish peroxidase-conjugated goat anti-rabbit IgG was used as the secondary antibody (Abcam). Signals were visualized by enhanced chemiluminescence (Santa-Cruz) and quantified by Quantity One (Bio-Rad Versadoc imaging system).
Total RNA was extracted from left ventricular coronary arterioles using Trizol reagent (Life Technologies Inc.), and was processed directly to cDNA synthesis using the SuperScript™ III Reverse Transcriptase (Life Technologies Inc.) [9, 10, 46]. The 2−ΔΔCT method (ΔΔCT = CT-TNFα-CT-GAPDH) was used to analyze the change of TNF gene expression [39]. The mean threshold cycle (CT) values for both the target (arginase) and internal control (GAPDH) genes were determined. Data are presented as fold change of transcripts for the arginase gene in I/R mice normalized to GAPDH, as compared to sham mice (defined as 1.0-fold).
Assay of NOS activity and arginase activity in I/R
Nitric oxidase synthase (NOS) detection kit (Cell Technology, Inc.) was used for testing the NOS activity. Coronary arterioles (5–7 vessels/sample, 100-μm inner diameter) in WT, TNF−/−, TNF−/++ and TNF++/++ mice in sham and I/R were freshly isolated. NOS activity can be measured via fluorescence plate reader at excitation 488 nm and emission at 515 nm. Data are presented as fold change when compared with sham-control mice (defined as 1.0-fold). The urea concentration (for arginase activity assay) was determined spectrophotometrically by the absorbance at 550 nm measured with a microplate reader (Bio-Tek Instruments). The amount of urea produced, after normalization with protein, was used as an index for arginase activity.
Chemicals
All drugs were obtained from Sigma, except as specifically stated. ACh, SNP and TEMPOL were dissolved in PSS for functional studies and in phosphate-buffered saline for fluorescence detection. Vehicle control studies indicated that these final concentrations of solvent had no effect on the arteriolar function.
Data analysis
At the end of each experiment, the vessel was relaxed with 100 μmol/L SNP to obtain its maximal diameter at 60 cm H2O intraluminal pressure [9, 10, 30]. All diameter changes in response to agonists were normalized to the vasodilation in response to 100 μmol/L SNP and expressed as a percentage of maximal dilation. All data are presented as mean ± SEM. Statistical comparisons of vasomotor responses under various treatments were performed with two-way ANOVA and intergroup differences were tested with Bonferonni inequality. Significance was accepted at P < 0.05.
Results
Plasma concentration of TNF
Table 1 shows the plasma concentration of TNF in circulating levels in sham and I/R injury in WT, TNF−/−, TNF−/++ and TNF++/++ mice.
Table 1.
Levels of TNF (n = 9) at baseline (sham) and after I/R injury in WT, TNF−/++ and TNF++/++ mice
| Groups | Plasma concentration of TNF (pM/mI) | |
|---|---|---|
| Sham (baseline) | I/R | |
| WT | 1.2 ± 0.4 (9) | 5.0 ± 0.6* (9) |
| TNF−/− | Below level of detection | Below level of detection |
| TNF−/++ | 2.3 ± 0.3* (9) | 6.9 ± 0.8*,# (9) |
| TNF++/++ | 7.4 ± 1.2*,# (9) | 13.6 ± 1.8*,# (9) |
P < 0.05 versus WT-sham control and # P < 0.05 versus WT-I/R mice (n = 9)
In heterozygote TNF−/++ mice, the levels of TNF in sham and after I/R injury were in between of TNF++/++ and WT mice. In TNF−/− mice, the levels of TNF were below the level of detection; n number of mice; * P < 0.05 versus sham
Roles of dose dependency of TNF in I/R-induced vascular dysfunction
The dilation to ACh, or flow was the best in the TNF−/−, worst in the TNF++/++, and intermediate in the WT and TNF−/++ mice following I/R versus sham controls (Fig. 2). Administration of neutralizing antibodies to TNF prior to the onset of reperfusion lessened endothelial dysfunction in reperfusion injury in WT, TNF−/++ and TNF++/++ mice, but not affect responses in TNF−/− mice. Importantly, neutralizing antibodies to TNF restored flow-induced dilation (FID) to near the sham-control levels in I/R (Fig. 3).
Fig. 2.

ACh-induced vasodilation was not impaired in sham group when compared with the control, but ACh-induced vasodilation was impaired after I/R in WT mice (Fig. 2a, n = 5). Anti-TNF (n = 7) restored vasodilation to ACh in I/R versus sham. ACh-induced vasodilation was not impaired in sham group and in I/R TNF−/− mice when compared with WT mice (Fig. 2b, n = 5). Anti-TNF (n = 7) did not affect the vasodilation to ACh in I/R and sham in TNF−/− mice. In heterozygote TNF−/++ mice (Fig. 2c), ACh-induced endothelial-dependent vasodilation following I/R was between TNF++/++ (Fig. 2d) and TNF−/− following I/R. Administering neutralizing antibodies to TNF immediately prior to the onset of reperfusion-preserved endothelial-dependent dilation following I/R in WT, TNF−/++ and TNF++/++ mice; n = number of vessels; * P < 0.05 versus sham
Fig. 3.
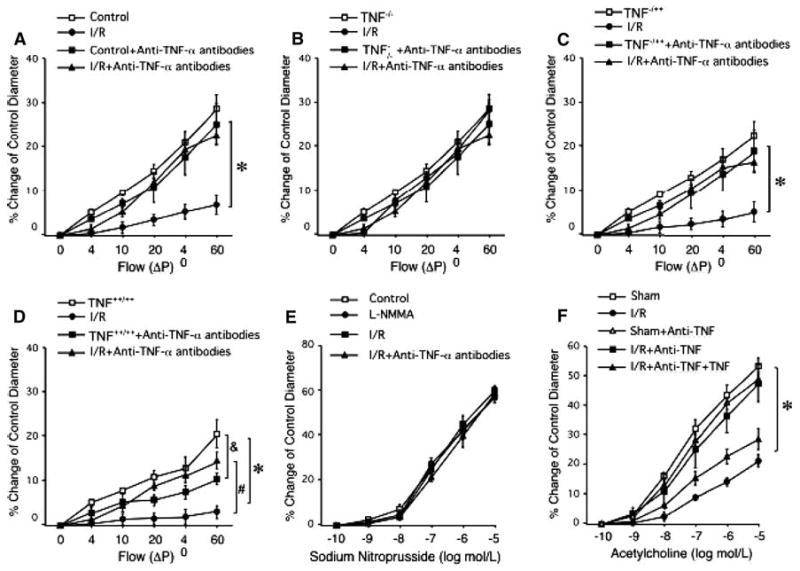
Isolated mice coronary arterioles from control mice (without I/R) dilated in a concentration-dependent manner to flow (n = 6, Fig. 3). Flow-induced vasodilation was not impaired in sham group when compared with the control, but flow-induced vasodilation was impaired after I/R in WT mice (Fig. 3a, n = 5). In heterozygote TNF−/++ mice (Fig. 3c), flow-induced endothelial-dependent vasodilation following I/R was between TNF++/++ (Fig. 3d) and TNF−/− (Fig. 3b) following I/R. Neutralizing antibodies to TNF immediately prior to the onset of reperfusion-preserved endothelial-dependent dilation following I/R in WT (Fig. 3a), TNF−/++ and TNF++/++ mice. Figure 3e shows a dose–response curve for SNP in WT control mice before and following I/R. There are no significant differences in endothelium-independent vasodilator SNP-induced vasodilation (% of control diameter) before and after I/R. Figure 3f shows that endothelial function is restored in I/R at WT mice treated with anti-TNF (mimicked the cross of WT with the null animals), endothelial function is impaired with a lower level of TNF in WT mice and endothelial function is in between of these two groups with a lower level of TNF in WT mice treated with anti-TNF; n = number of vessels; * P < 0.05 versus sham
Figure 3e shows a dose–response curve for SNP in WT control mice before and after I/R. There are no significant differences in endothelium-independent vasodilator SNP-induced vasodilation before and after I/R. These results demonstrate an unchanged response to SNP among the different groups analyzed and assure that effects are related to the endothelium rather than to vascular smooth muscle cell alterations. Figure 3f shows that endothelial function is restored in I/R at WT mice treated with anti-TNF (mimicked the cross of WT with the TNF null animals), endothelial function is impaired with a lower level of TNF in WT mice, and endothelial function is in between of these two groups with a lower level of TNF in WT mice treated with anti-TNF. These results indicated that I/R contributes endothelial dysfunction via increasing TNF level in coronary arterioles.
Roles of O2·− in I/R-induced vascular dysfunction
We incubated TEMPOL with isolated microvessels for 60 min (1 mmol/L) then examined vasodilation induced by ACh. Administration of an O2·− scavenger TEMPOL (Fig. 4) maintained vasodilation to ACh in I/R, but did not affect ACh-induced vasodilation in sham groups. TEMPOL partially restored ACh-induced vasodilation after I/R injury in WT, TNF−/++ and TNF++/++ mice, but did not affect that in TNF−/− mice.
Fig. 4.

Administration of a O2·− scavenger, TEMPOL (1 mmol/L, n = 7) restored vasodilation after I/R. In WT, TNF−/++ and TNF++/++ mice, I/R-induced endothelial dysfunction was lessened progressively by administration of free-radical scavenger TEMPOL immediately before initiating reperfusion; n = number of vessels; * P < 0.05 versus sham
I/R-induced O2·− production in murine coronary arterioles
Figure 5 shows the results from EPR spectroscopy to quantify the production of O2·−. O2·− production increased after I/R as compared to the sham group (P < 0.05), and importantly, administration of anti-TNF (Fig. 5a), or free-radical scavenger TEMPOL (Fig. 5b) reduced the expression of O2·− to the level observed in the sham group. O2·− production was enhanced in the TNF++/++ group before and after I/R when compared with WT groups. Production of O2·− is less in the TNF−/− mice following I/R versus I/R in WT mice. The production of O2·− was intermediate in TNF−/++ I/R mice occurring between that of TNF++/++ and TNF−/− mice following I/R.
Fig. 5.

EPR spectroscopy shows O2·− production increased in isolated coronary arteries in I/R mice as compared to the sham (P < 0.05), and importantly, administration of anti-TNFα (Fig. 5a) or TEMPOL (Fig. 5b) reduced the production of O2·− to the level observed in the sham in WT mice. During I/R, production of superoxide (O−2) was greatest in TNF++/++ mice as compared to WT, TNF−/++ and TNF−/− mice. * P < 0.05 versus sham; # P < 0.05 versus I/R
Effect of TNF on arginase I expression in coronary arterioles during I/R
The protein expression of arginase I was lower in TNF−/− mice, higher in TNF++/++ mice, and in between of TNF−/− and TNF++/++ mice in TNF−/++ mice than that in WT mice (Fig. 6a, b). The mRNA expression of arginase I was lower in TNF−/− mice and higher in TNF++/++ mice than that in WT mice (Fig. 6c), which suggested that arginase titer is inversely linked to TNF titer. Arginase expression was significantly increased in WT I/R mice, but was not changed in TNF−/− mice following I/R. I/R increased arginase expression in TNF−/++ mice, but did not increase arginase expression in TNF++/++ mice, perhaps because arginase expression mediated by TNF was already maximal or saturated in the TNF++/++ mice. We previously reported that anti-TNF decreased arginase I expression in WT-I/R versus WT-sham mice [9, 10, 46]. These results suggested that a “dose dependency” of TNF plays an important role in upregulating arginase progressively in I/R, which may then cause the impairment of NO-mediated vasodilation in I/R.
Fig. 6.
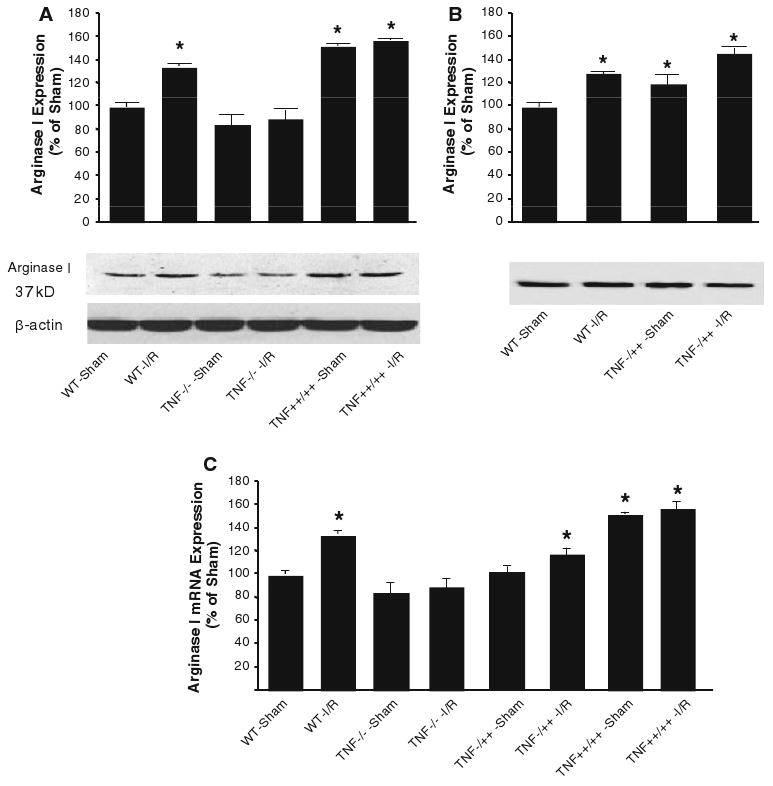
The protein expression of arginase I was lower in TNF−/− mice, higher in TNF++/++ mice, and in between of TNF−/− and TNF++/++ mice in TNF−/++ mice than that in WT mice (Fig. 6a, b). Following I/R, arginase mRNA expression (Fig. 6c) was elevated in the WT, substantially elevated in the TNF−/++ and TNF++/++ mice, and not affected in the TNF−/− mice. Antibody neutralization of TNF decreased arginase expression in the WT, TNF−/++ and TNF++/++ mice, but did not affect arginase expression in the TNF−/− mice. * P < 0.05 versus sham; # P < 0.05 versus I/R
NOS and arginase activity in I/R
Activity of NOS and arginase was assayed in the coronary arteriolar lysate to further elucidate the role of TNF-α in the upregulation of arginase in I/R (Fig. 7). We found that reduced eNOS activity (Fig. 7a) in WT, TNF−/−, TNF−/++, TNF++/++ versus WT-sham mice and arginase activity (Fig. 7b) is increased in I/R in WT-I/R, TNF−/++, TNF++/++ versus WT-sham and TNF−/− mice.
Fig. 7.
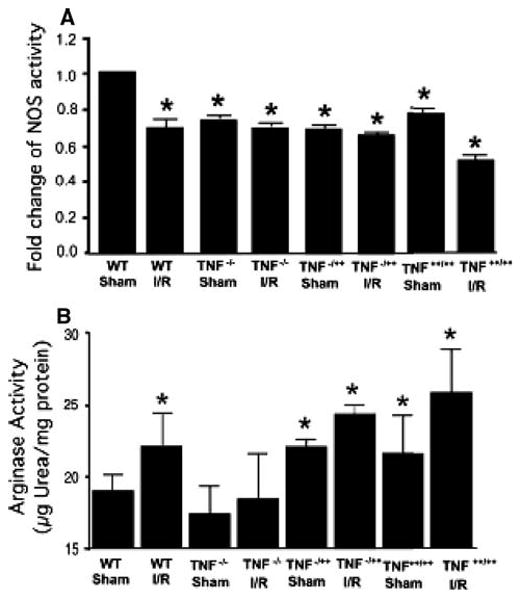
NOS activity (n = 7) and arginase activity (n = 6) were assayed in the coronary arteriolar lysate to further elucidate the role of TNF in the upregulation of arginase in I/R (Fig. 7). We found that reduced eNOS activity (Fig. 7a) in WT, TNF−/−, TNF−/++, TNF++/++ versus WT-sham mice and arginase activity (Fig. 7b) is increased in I/R in WT-I/R, TNF−/++, TNF++/++ versus WT-sham and TNF−/− mice
Discussion
Our major finding is that increasing levels of TNF expression result in dose-dependent increases in arginase expression in ECs during myocardial I/R; this is one of the mechanisms by which TNF compromises coronary endothelial function (Fig. 1). Our results suggest a direct relationship between levels of TNF expression and endothelial dysfunction in reperfusion injury. We believe that our findings further understanding of the mechanism(s) underlying endothelial dysfunction following myocardial I/R injury.
Role of levels of TNF expression in I/R-induced endothelium-dependent dilation in murine coronary arterioles
We have established the critical role of TNF in I/R impaired endothelial function in coronary arterioles, and suggest that TNF plays an important role in endothelial dysfunction during I/R injury. The mechanisms of I/R injury are multifactorial and incompletely understood. Many agents have been directed at preserving cardiac myocytes, but few have targeted the endothelium. Perhaps, this is the very reason that the therapies failed because without adequate flow during reperfusion, functional recovery is likely to be poor. Our study was designed to mimic a clinically relevant paradigm in which an antibody designed to neutralize TNF could be delivered at the time of a recanalization procedure or restoration of flow following a coronary bypass. Reperfusion is associated with the rapid expression of genes for antioxidant enzymes, which may enhance reactive oxygen intermediate scavenging [6]. Herskowitz et al. [15] reported that the induction of cytokine mRNA was observed at 15 min of LAD occlusion, with levels returning to baseline by 3 h in a model of myocardial I/R. Several studies have reported reversible myocardial dysfunction in normal animals in response to systemic administration of TNF [6, 24, 35]. TNF inhibits NO-mediated endothelium-dependent vasorelaxation in small coronary arteries via sphingomyelinase activation and consequent O2·− production in ECs [47]. Gilmont et al. [12] have shown a direct effect of TNF on EC, whereby the cells are rendered more susceptible to oxidant injury accompanying reperfusion in rat pulmonary artery ECs. We previously found that I/R-induced coronary endothelial dysfunction is mediated by TNF. TNF expression in a model of partial coronary embolization can induce frank ischemia and induce dysfunction [7, 29, 32]. Thus, this cytokine may have a key role in reperfusion injury. Gurevitch et al. [13] have found in isolated, buffer-perfused rabbit hearts, anti-TNF improves functional recovery following global ischemia. Our studies indicate therapies can and should be tailored to remediate the in vivo level of TNF encountered in the pathology of endothelial injury in myocardial I/R injury.
Previous studies [14, 37, 40] have shown that I/R produces impaired vascular endothelial function as defined by abrogated endothelium-dependent dilation. This impairment has been attributed to arginase [14], depletion of tetrahydrobiopterin [33], O2·−, cytokines, proteases and lipid mediators [19]. Endothelium-derived NO is an important endogenous vasodilator that regulates microvascular tone. The endothelial release of NO underlies the mechanism of coronary arteriolar dilation to ACh [1, 3]. The endothelial NO component was evident in the present study because L-NMMA attenuated the ACh-induced vasodilation. Consistent with our previous studies, we show here that ACh evokes dilation of coronary arterioles (probably by activation of eNOS). In the present study, endothelium-dependent, ACh-induced vasodilation in mouse coronary artery was impaired after I/R, whereas endothelium-independent, SNP-induced vasodilation was normal, suggesting that I/R produces endothelial dysfunction in a murine model of reperfusion injury. Endothelial function following I/R was related to TNF expression. We studied the role of TNF by using different transgenic animals expressing varying amounts of TNF. As follows, the dilation to ACh, or flow was the best in the TNF−/−, worst in the TNF++/++ and intermediate in the WT and TNF−/++ mice. TNF++/++ mice survived well after I/R protocol, although TNF++/++ mice over-express large amounts of TNF and exhibit more severe manifestations of I/R injury, e.g., contractile dysfunction. Our results showed that the administration of neutralizing antibodies to TNF before the onset of reperfusion lessened endothelial dysfunction in reperfusion injury in WT, TNF−/++ and TNF++/++ mice, but did not affect responses in TNF−/− mice. In heterozygote TNF−/++ mice, the plasma concentration of TNF in sham and after I/R injury was between TNF++/++ and WT mice. In TNF−/− mice, the levels of TNF were below level of detection. The concentration of TNF in the heart of TNF++/++ is very high [16]. We found that endothelial function is impaired with a lower level of TNF in WT mice, and endothelial function is in between of these two groups with a lower level of TNF in WT mice treated with anti-TNF. Our results suggest that there is a dose dependency of TNF in endothelial dysfunction in I/R injury and I/R contributes endothelial dysfunction via increasing TNF level in coronary arterioles. Importantly, neutralizing antibodies to TNF restored FID to near the sham-control levels in I/R, as it did for ACh. This is consistent with the likelihood that ROS limit the bioavailability of NO during I/R injury, thus attenuating responses to the different endothelial-dependent stimuli. The rationale for this dose of neutralizing antibody to TNF (1.6 mg/kg) was sufficient to neutralize endogenous TNF based on previous studies [9, 10, 46]. We have also found this concentration effectively neutralizes the actions of TNF.
Roles of O2·− in coronary vascular function in I/R injury
Endothelial dysfunction appears to occur early after reperfusion, signaled by the endothelial generation of a large burst of superoxide radicals [8, 11, 20, 28, 29, 31, 36]. The decrease in endothelium-dependent dilation has been shown to occur soon after the generation of O2·− by the reperfused coronary endothelium [20, 31] suggesting that endothelial generation of O2·− radicals acts as a trigger mechanism for endothelial dysfunction. O2·− also reduces the bioavailability of NO, which would compromise dilation to endothelium-dependent stimuli. Our results extend this knowledge by demonstrating that the administration of an O2·− scavenger TEMPOL maintained vasodilation to ACh in I/R, but did not affect ACh-induced vasodilation in sham groups in WT mice, and that TEMPOL partially restored ACh-induced vasodilation following I/R injury in TNF−/++ and TNF++/++ mice, but did not affect that in TNF−/− mice.
A recent study has shown that TNF inhibits NO-mediated endothelium-dependent vasorelaxation in small coronary arteries via sphingomyelinase activation and consequent O2·− production in ECs [47]. The source of O2·− and the signal transduction mechanism by which TNF stimulation is linked to O2·− production was addressed using pharmacological inhibitors of enzyme systems capable of producing O2·− during I/R previously [8, 11, 28, 29, 31, 42, 45, 46]. O2·− generation impairs NO-mediated coronary arteriolar function and we have suggested that this oxidative free radical might be involved in TNF-induced vascular dysfunction [44, 46]. Although previous studies have shown that TNF increases the generation of O2·− during I/R, it is unclear whether different levels of TNF induce different levels of O2·− production in the vascular wall in microvessels after I/R. We studied the role of O2·− in the WT, TNF−/− and transgenic TNF++/++ animals in I/R endothelial injury and evaluate its production in animals under experimental conditions.
These studies established the link between TNF and ROS in the induction of endothelial injury following myocardial I/R. Administration of an antibody to TNF at the time of reperfusion prevented ROS generation [10], and mimics a clinically relevant paradigm in which an antibody designed to neutralize TNF could be delivered at the time of a recanalization procedure or restoration of flow following a coronary bypass. We also established the link between TNF and the activation of O2·− generating enzymes using EPR spectroscopy to quantitatively measure O2·− production from coronary arterioles during exposure to varying doses of TNF. Our results indicated that O2·− production increased following I/R as compared to the sham group, and importantly, administration of anti-TNF or TEMPOL reduced the expression of O2·− to the level observed in the sham group. The finding of Pereda et al. [26] shows that simultaneous inhibition of TNF production and xanthine oxidase activity greatly reduced local systemic inflammatory response in acute pancreatitis. Our previous study [46] shows that I/R injury increased the xanthine oxidase activity, and antibody to TNF and xanthine oxidase inhibitor, allopurinol, separately attenuated the xanthine oxidase activity in I/R injury. Our results demonstrate the production of TNF is essential in eliciting this oxidative stress.
In the present study, we show that O2·− production is enhanced in the TNF++/++ group before and after I/R compared with WT groups; the production of O2·− is decreased in the TNF−/− mice following I/R. The production of O2·− in TNF−/++ I/R mice is between TNF++/++ and TNF−/− mice following I/R. This suggests there is a dose dependency of TNF in I/R-induced O2·− production. This identifies a mechanism for and mechanisms by which TNF produces endothelial dysfunction during I/R injury.
Roles of arginase in coronary vascular function in I/R injury
The overexpression of arginase by limiting the arginine pool would convert activated eNOS (or any NOS) into an enzyme that produces O2·− instead of NO. Vasquez-Vivar et al. [38] reported that eNOS can switch from an NO generating NAD(P)H oxidase to an O2·− generating enzyme. We have found in previous studies [9, 10, 46] that TNF appears to be linked to endothelial dysfunction and ROS generation in I/R injury. We also demonstrated that I/R inhibits NO-mediated dilation of coronary arterioles by increasing arginase activity in ECs, which limits the availability of l-arginine to NOS for NO production [14]. Arginase shares a common substrate, l-arginine, with NOS. Molecular evidence in the porcine coronary arterioles from our previous study indicated that arginase I, but not arginase II, mRNA and arginase I protein were expressed in the coronary arterioles [43]. Arginase II is the main isoform detected in mitochondria [9, 14, 43], but we do not observe it in our preparation. Perhaps, it can be found in mitochondria of other tissues because it is encoded by the nuclear genome and, therefore, may show some tissue-specific expression. The arginase expressed in the endothelium plays a counteracting role in the stimulated NO production, and thus in NO-mediated vasodilatory function. It is possible that increased cytokines and/or reactive oxygen species mediate the arginase activation. Both these factors are generated during myocardial I/R [17, 18] and have been shown to inhibit endothelium-dependent dilation in the coronary circulation [22]. Thus, we postulated a link between TNF and arginase expression during I/R injury. We showed that TNF expression is linked to arginase expression following cardiac I/R by demonstrating that O2·− production was enhanced in the TNF++/++ group before and after I/R when compared with WT groups. The production of O2·− is less in the TNF−/− mice following I/R versus the WT mice following I/R. The production of O2·− was intermediate in TNF−/++ I/R mice when compared with the TNF++/++ and TNF−/− mice following I/R. We previously showed that antibody neutralization of TNF reduced arginase expression following I/R [9]. Our current findings indicate that TNF increases arginase expression (mRNA and protein) in ECs and represents one of the mechanisms by which TNF compromises endothelial function. The results from NOS and arginase activity (Fig. 7) indicates that TNF increases the levels of O2·− due to both increased arginase and decreased NOS activity scavenged NO and reduced its bioavailability during I/R injury. Although we do not have a precise mechanism for the alterations in NOS activity in the various lines, we speculate that the oxidative stress induced by TNF overexpression can limit NOS activity by reducing levels of BH4, which is sensitive to levels of ROS.
In conclusion, our results demonstrate that endothelial dysfunction occurring subsequent to I/R injury is directly related to the level of TNF expression, which results in the production of O2·− via increasing arginase expression. This suggests that there is a direct relationship between levels of TNF expression and endothelial dysfunction in reperfusion injury. New animal models, diagnostic techniques and therapeutic agents are expected to be developed from this work. Knowledge gained from these studies provides a better understanding of the mechanism(s) underlying endothelial dysfunction, defined as blunted endothelial dilation of coronary arterioles, following myocardial I/R injury. The results of this study may contribute to the development of novel adjunctive therapies using TNF antibodies or soluble receptors to prevent microvascular endothelial dysfunction following I/R injury and/or during thrombolytic therapies. The early appearance of TNF in the disease process and its association with subsequent inflammation indicates that the role of TNF warrants further study. Understanding endothelial dysfunction is critical because this condition precedes the development of coronary disease; thus, if endothelial dysfunction can be rectified, the progression of vascular disease may be halted [42]. Selective modulation of ROS signaling may provide a novel therapeutic target to prevent or alleviate coronary diseases associated with TNF activation.
Acknowledgments
This study was supported by Grants from American Heart Association Grant-in-Aid (0455435B), Atorvastatin Research Award (2004-37), American Heart Association SDG (110350047A) and NIH Grants (RO1-HL077566 and RO1-HL085119) to Dr. C Zhang.
Contributor Information
Cuihua Zhang, Email: ZhangCu@missouri.edu, Department of Internal Medicine, Medical Pharmacology and Physiology and Nutritional Sciences, Dalton Cardiovascular Research Center, University of Missouri, Columbia, MO 65211, USA.
Junxi Wu, Department of Internal Medicine, Medical Pharmacology and Physiology and Nutritional Sciences, Dalton Cardiovascular Research Center, University of Missouri, Columbia, MO 65211, USA.
Xiangbin Xu, Department of Internal Medicine, Medical Pharmacology and Physiology and Nutritional Sciences, Dalton Cardiovascular Research Center, University of Missouri, Columbia, MO 65211, USA.
Barry J. Potter, Department of Physiology, LSU Health Sciences Center, 1901 Perdido Street, New Orleans, LA 70112, USA
Xue Gao, Department of Internal Medicine, Medical Pharmacology and Physiology and Nutritional Sciences, Dalton Cardiovascular Research Center, University of Missouri, Columbia, MO 65211, USA.
References
- 1.Ammar RF, Jr, Gutterman DD, Brooks LA, Dellsperger KC. Free radicals mediate endothelial dysfunction of coronary arterioles in diabetes. Cardiovasc Res. 2000;47:595–601. doi: 10.1016/s0008-6363(00)00094-8. [DOI] [PubMed] [Google Scholar]
- 2.Aoki N, Siegfried M, Lefer AM. Anti-EDRF effect of tumor necrosis factor in isolated, perfused cat carotid arteries. Am J Physiol. 1989;256:H1509–H1512. doi: 10.1152/ajpheart.1989.256.5.H1509. [DOI] [PubMed] [Google Scholar]
- 3.Bagi Z, Koller A, Kaley G. PPAR-g activation, by reducing oxidative stress, increases NO bioavailability in coronary arterioles of mice with type 2 diabetes. Am J Physiol Heart Circ Physiol. 2004;286:H742–H748. doi: 10.1152/ajpheart.00718.2003. [DOI] [PubMed] [Google Scholar]
- 4.Bose D, von Birgelen C, Zhou XY, Schmermund A, Philipp S, Sack S, Konorza T, Mohlenkamp S, Leineweber K, Kleinbongard P, Wijns W, Heusch G, Erbel R. Impact of atherosclerotic plaque composition on coronary microembolization during percutaneous coronary interventions. Basic Res Cardiol. 2008;103:587–597. doi: 10.1007/s00395-008-0745-9. [DOI] [PubMed] [Google Scholar]
- 5.Bramos D, Ikonomidis I, Tsirikos N, Kottis G, Kostopoulou V, Pamboucas C, Papadopoulou E, Venetsanou K, Giatrakos N, Yang GZ, Nihoyannopoulos P, Toumanidis S. The association of coronary flow changes and inflammatory indices to ischaemia–reperfusion microvascular damage and left ventricular remodelling. Basic Res Cardiol. 2008;103:345–355. doi: 10.1007/s00395-008-0720-5. [DOI] [PubMed] [Google Scholar]
- 6.Chandrasekar B, Colston JT, Freeman GL. Induction of proinflammatory cytokine and antioxidant enzyme gene expression following brief myocardial ischemia. Clin Exp Immunol. 1997;108:346–351. doi: 10.1046/j.1365-2249.1997.d01-1017.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dorge H, Schulz R, Belosjorow S, Post H, van de Sand A, Konietzka I, Frede S, Hartung T, Vinten-Johansen J, Youker KA, Entman ML, Erbel R, Heusch G. Coronary microembolization: the role of TNF-alpha in contractile dysfunction. J Mol Cell Cardiol. 2002;34:51–62. doi: 10.1006/jmcc.2001.1489. [DOI] [PubMed] [Google Scholar]
- 8.Fischer P, Hilfiker-Kleiner D. Survival pathways in hypertrophy and heart failure: the gp130-STAT axis. Basic Res Cardiol. 2007;102:393–411. doi: 10.1007/s00395-007-0674-z. [DOI] [PubMed] [Google Scholar]
- 9.Gao X, Xu X, Belmadani S, Park Y, Tang Z, Feldman AM, Chilian WM, Zhang C. TNF-alpha contributes to endothelial dysfunction by upregulating arginase in ischemia/reperfusion injury. Arterioscler Thromb Vasc Biol. 2007;27:1269–1275. doi: 10.1161/ATVBAHA.107.142521. [DOI] [PubMed] [Google Scholar]
- 10.Gao X, Zhang H, Belmadani S, Wu J, Xu X, Elford H, Potter BJ, Zhang C. Role of TNF-alpha-induced reactive oxygen species in endothelial dysfunction during reperfusion injury. Am J Physiol Heart Circ Physiol. 2008;295:H2242–H2249. doi: 10.1152/ajpheart.00587.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Garcia SC, Pomblum V, Gams E, Langenbach MR, Schipke JD. Independency of myocardial stunning of endothelial stunning? Basic Res Cardiol. 2007;102:359–367. doi: 10.1007/s00395-007-0657-0. [DOI] [PubMed] [Google Scholar]
- 12.Gilmont RR, Dardano A, Engle JS, Adamson BS, Welsh MJ, Li T, Remick DG, Smith DJJ, Rees RS. TNF-alpha potentiates oxidant and reperfusion-induced endothelial cell injury. J Surg Res. 1996;61:175–182. doi: 10.1006/jsre.1996.0101. [DOI] [PubMed] [Google Scholar]
- 13.Gurevitch J, Frolkis I, Yuhas Y, Lifschitz-Mercer B, Berger E, Paz Y, Matsa M, Kramer A, Mohr R. Anti-tumor necrosis factor-alpha improves myocardial recovery after ischemia and reperfusion. J Am Coll Cardiol. 1997;30:1554–1561. doi: 10.1016/s0735-1097(97)00328-8. [DOI] [PubMed] [Google Scholar]
- 14.Hein TW, Zhang C, Wang W, Chang CI, Thengchaisri N, Kuo L. Ischemia–reperfusion selectively impairs nitric oxide-mediated dilation in coronary arterioles: counteracting role of arginase. FASEB J. 2003;17:2328–2330. doi: 10.1096/fj.03-0115fje. [DOI] [PubMed] [Google Scholar]
- 15.Herskowitz A, Choi S, Ansari AA, Wesselingh S. Cytokine mRNA expression in postischemic/reperfused myocardium. Am J Pathol. 1995;146:419–428. [PMC free article] [PubMed] [Google Scholar]
- 16.Huber SA, Feldman AM, Sartini D. Coxsackie virus B3 induces T regulatory cells, which inhibit cardiomyopathy in tumor necrosis factor-alpha transgenic mice. Circ Res. 2006;99:1109–1116. doi: 10.1161/01.RES.0000249405.13536.49. [DOI] [PubMed] [Google Scholar]
- 17.Kupatt C, Habazettl H, Goedecke A, Wolf D, Zahler S, Boekstegers P, Kelly R, Becker B. Tumor necrosis factor-alpha contributes to ischemia- and reperfusion-induced endothelial activation in isolated hearts. Circ Res. 1999;84:392–400. doi: 10.1161/01.res.84.4.392. [DOI] [PubMed] [Google Scholar]
- 18.Lawson DL, Mehta JL, Nichols WW. Coronary reperfusion in dogs inhibits endothelium-dependent relaxation: role of superoxide radicals. Free Radic Biol Med. 1990;8:373–380. doi: 10.1016/0891-5849(90)90103-p. [DOI] [PubMed] [Google Scholar]
- 19.Lefer AM, Lefer DJ. Pharmacology of the endothelium in ischemia–reperfusion and circulatory shock. Annu Rev Pharmacol Toxicol. 1993;33:71–90. doi: 10.1146/annurev.pa.33.040193.000443. [DOI] [PubMed] [Google Scholar]
- 20.Lefer AM, Ma X. Cytokines and growth factors in endothelial dysfunction. Crit Care Med. 1993;21(Suppl):S9–S14. doi: 10.1097/00003246-199302001-00003. [DOI] [PubMed] [Google Scholar]
- 21.Lefer AM, Ma XL. Cytokines and growth factors in endothelial dysfunction. Crit Care Med. 1993;21:S9–S14. doi: 10.1097/00003246-199302001-00003. [DOI] [PubMed] [Google Scholar]
- 22.Lefer AM, Tsao PS, Ma XL. Shock- and ischemia-induced mechanisms of impairment of endothelium-mediated vasodilation. Chest. 1991;100:160S–163S. [PubMed] [Google Scholar]
- 23.Levine B, Kalman J, Mayer L, Fillit HM, Packer M. Elevated circulating levels of tumor necrosis factor in severe chronic heart failure. N Engl J Med. 1990;323:236–241. doi: 10.1056/NEJM199007263230405. [DOI] [PubMed] [Google Scholar]
- 24.Murray DR, Freeman GL. Tumor necrosis factor-α induces a biphasic effect on myocardial contractility in conscious dogs. Circ Res. 1996;78:154–160. doi: 10.1161/01.res.78.1.154. [DOI] [PubMed] [Google Scholar]
- 25.Oral H, Dorn GW, 2nd, Mann DL. Sphingosine mediates the immediate negative inotropic effects of tumor necrosis factor-alpha in the adult mammalian cardiac myocyte. J Biol Chem. 1997;272:4836–4842. doi: 10.1074/jbc.272.8.4836. [DOI] [PubMed] [Google Scholar]
- 26.Pereda J, Sabater L, Cassinello N, Gomez-Cambronero L, Closa D, Folch-Puy E, Aparisi L, Calvete J, Cerda M, Lledo S, Vina J, Sastre J. Effect of simultaneous inhibition of TNF-alpha production and xanthine oxidase in experimental acute pancreatitis: the role of mitogen activated protein kinases. Ann Surg. 2004;240:108–116. doi: 10.1097/01.sla.0000129343.47774.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Picchi A, Gao X, Belmadani S, Potter BJ, Focardi M, Chilian WM, Zhang C. Tumor necrosis factor-alpha induces endothelial dysfunction in the prediabetic metabolic syndrome. Circ Res. 2006;99:69–77. doi: 10.1161/01.RES.0000229685.37402.80. [DOI] [PubMed] [Google Scholar]
- 28.Schmidt A, Geigenmuller S, Volker W, Buddecke E. The antiatherogenic and antiinflammatory effect of HDL-associated lysosphingolipids operates via Akt >NF-kappaB signalling pathways in human vascular endothelial cells. Basic Res Cardiol. 2006;101:109–116. doi: 10.1007/s00395-005-0582-z. [DOI] [PubMed] [Google Scholar]
- 29.Schulz R, Aker S, Belosjorow S, Heusch G. TNFalpha in ischemia/reperfusion injury and heart failure. Basic Res Cardiol. 2004;99:8–11. doi: 10.1007/s00395-003-0431-x. [DOI] [PubMed] [Google Scholar]
- 30.Schulz R, Heusch G. Tumor necrosis factor-alpha and its receptors 1 and 2: Yin and Yang in myocardial infarction? Circulation. 2009;119:1355–1357. doi: 10.1161/CIRCULATIONAHA.108.846105. [DOI] [PubMed] [Google Scholar]
- 31.Tanner FC, van der Loo B, Shaw S, Greutert H, Bachschmid MM, Berrozpe M, Rozenberg I, Blau N, Siebenmann R, Schmidli J, Meyer P, Luscher TF. Inactivity of nitric oxide synthase gene in the atherosclerotic human carotid artery. Basic Res Cardiol. 2007;102:308–317. doi: 10.1007/s00395-007-0650-7. [DOI] [PubMed] [Google Scholar]
- 32.Thielmann M, Dorge H, Martin C, Belosjorow S, Schwanke U, van De Sand A, Konietzka I, Buchert A, Kruger A, Schulz R, Heusch G. Myocardial dysfunction with coronary microembolization: signal transduction through a sequence of nitric oxide, tumor necrosis factor-alpha, and sphingosine. Circ Res. 2002;90:807–813. doi: 10.1161/01.res.0000014451.75415.36. [DOI] [PubMed] [Google Scholar]
- 33.Tiefenbacher CP, Chilian WM, Mitchell M, DeFily DV. Restoration of endothelium-dependent vasodilation after reperfusion injury by tetrahydrobiopterin. Circulation. 1999;94:1423–1429. doi: 10.1161/01.cir.94.6.1423. [DOI] [PubMed] [Google Scholar]
- 34.Tracey KJ, Beutler B, Lowry SF, Merryweather J, Wolpe S, Milsark IW, Hariri RJ, Fahey TJ, 3rd, Zentella A, Albert JD, et al. Shock and tissue injury induced by recombinant human cachectin. Science. 1986;234:470–474. doi: 10.1126/science.3764421. [DOI] [PubMed] [Google Scholar]
- 35.Tracey KJ, Vlassara H, Cerami A. Cachectin/tumor necrosis factor. N Engl J Med. 1987;316:379–385. doi: 10.1056/NEJM198702123160705. [DOI] [PubMed] [Google Scholar]
- 36.Tsao PS, Aoki N, Lefer DJ, Johnson G, III, Lefer AM. Time course of endothelial dysfunction and myocardial injury during myocardial ischemia and reperfusion in the cat. Circulation. 1990;82:1402–1412. doi: 10.1161/01.cir.82.4.1402. [DOI] [PubMed] [Google Scholar]
- 37.Tsao PS, Ma X, Lefer AM. Activated neutrophils aggravate endothelial dysfunction after reperfusion of the ischemic feline myocardium. Am Heart J. 1992;123:1464–1471. doi: 10.1016/0002-8703(92)90796-x. [DOI] [PubMed] [Google Scholar]
- 38.Vasquez-Vivar J, Kalyanaraman B, Martasek P, Hogg N, Masters BS, Karoui H, Tordo P, Pritchard KA., Jr Superoxide generation by endothelial nitric oxide synthase: the influence of cofactors. Proc Natl Acad Sci USA. 1998;95:9220–9225. doi: 10.1073/pnas.95.16.9220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Weyrich AS, Ma XL, Lefer AM. The role of l-arginine in ameliorating reperfusion injury after myocardial ischemia in the cat. Circulation. 1992;86:279–288. doi: 10.1161/01.cir.86.1.279. [DOI] [PubMed] [Google Scholar]
- 40.Yamamoto S, Matsui K, Itoh N, Ohaashi N. The effect of an Na+/H+ exchange inhibitor, SM-20550, on ischemia/reperfusion-induced endothelial dysfunction in isolated perfused rat hearts. Int J Tissue React. 2001;23:1–7. [PubMed] [Google Scholar]
- 41.Yang J, Park Y, Zhang H, Xu X, Laine GA, Dellsperger KC, Zhang C. Feed-forward signaling of TNF-alpha and NF-kappaB via IKK-beta pathway contributes to insulin resistance and coronary arteriolar dysfunction in type 2 diabetic mice. Am J Physiol Heart Circ Physiol. 2009;296:H1850–H1858. doi: 10.1152/ajpheart.01199.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhang C. The role of inflammatory cytokines in endothelial dysfunction. Basic Res Cardiol. 2008;103:398–406. doi: 10.1007/s00395-008-0733-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhang C, Hein TW, Wang W, Chang CI, Kuo L. Constitutive expression of arginase in microvascular endothelial cells counteracts nitric oxide-mediated vasodilatory function. FASEB J. 2001;15:1264–1266. doi: 10.1096/fj.00-0681fje. [DOI] [PubMed] [Google Scholar]
- 44.Zhang C, Hein TW, Wang W, Ren Y, Shipley RD, Kuo L. Activation of JNK and xanthine oxidase by TNF-alpha impairs nitric oxide-mediated dilation of coronary arterioles. J Mol Cell Cardiol. 2006;40:247–257. doi: 10.1016/j.yjmcc.2005.11.010. [DOI] [PubMed] [Google Scholar]
- 45.Zhang C, Park Y, Picchi A, Potter BJ. Maturation-induces endothelial dysfunction via vascular inflammation in diabetic mice. Basic Res Cardiol. 2008;103:407–416. doi: 10.1007/s00395-008-0725-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhang C, Xu X, Potter BJ, Wang W, Kuo L, Michael L, Bagby GJ, Chilian WM. TNF-alpha contributes to endothelial dysfunction in ischemia/reperfusion injury. Arterioscler Thromb Vasc Biol. 2006;26:475–480. doi: 10.1161/01.ATV.0000201932.32678.7e. [DOI] [PubMed] [Google Scholar]
- 47.Zhang DX, Yi FX, Zou AP, Li PL. Role of ceramide in TNF-alpha-induced impairment of endothelium-dependent vasorelaxation in coronary arteries. Am J Physiol Heart Circ Physiol. 2002;283:H1785–H1794. doi: 10.1152/ajpheart.00318.2002. [DOI] [PubMed] [Google Scholar]