

Abstract
We evaluated the association between mini‐mental status examination (MMSE) scores proximal to death and the values of 43 different clinical and pathological parameters. Studies were performed using data from 334 elderly, longitudinally evaluated research subjects who had undergone autopsy and satisfied inclusion criteria from an initial study group of 501. Interindividual variance in MMSE scores was used as a surrogate for the severity of cognitive impairment linked to aging (CILA). A statistical linear regression‐based model provided a framework for assessing the parameters with significant, direct impact on CILA severity. Strong association between CILA and Alzheimer's disease (AD) pathology, especially isocortical neurofibrillary tangles, was evident. The pattern of association between AD lesion densities with cognitive impairment severity was biologically informative, with neuritic plaques having more impact in relatively high‐functioning individuals. Abundant isocortical Lewy bodies tended to be an additive pathology correlating with final MMSE scores approximately 10 points lower. In a subset of cases we found evidence for association between TDP‐43‐related pathology and CILA severity, independent of AD or hippocampal sclerosis. There was no support for independent association between CILA severity and most evaluated indices including diffuse plaques, argyrophilic grains, heart disease, education level, apolipoprotein E alleles or diabetes.
Keywords: ApoE, cognition, human, stroke, DLB, hippocampal sclerosis
INTRODUCTION
Cognitive impairment linked to aging (CILA), a leading cause of morbidity and mortality, spans a continuum from subtle cognitive changes to end‐stage dementia. We used the term “CILA” to include cognitive impairment in an aging population regardless of diagnoses or of predicted eventual clinical course. Multiple etiologies contribute to CILA at a population level and coincident pathologies are frequently observed in aged individuals 11, 18, 87, 89, 109. As prospects for specific brain disease therapies improve 45, 50, it is increasingly important to know the relative weights of the distinct contributing factors in CILA.
Rubrics are lacking to relate the severity of CILA to the presence and/or extent of most brain diseases. Alzheimer's disease (AD) has a relatively well‐documented association between the amount of neuropathology and the severity of ante–mortem cognitive impairment 6, 10, 14, 20, 48, 52, 85, yet controversy remains about the contribution(s) of amyloid plaques and neurofibrillary tangles (NFTs) to clinical deterioration 24, 76, 87. Pathological processes also linked to CILA include cerebrovascular disease (CVD), dementia with Lewy bodies, argyrophilic grains (AGs), frontotemporal dementias (FTDs) and hippocampal sclerosis (HS) 9, 11, 14, 24, 66, 72, 74, 78, 86, 87, 91, 108, 109, 120. In addition, there are clinical comorbidities that may contribute to CILA despite the absence of definite neuropathological hallmarks to support these associations 9, 74, 90. Examples of clinical features that have been associated with worsened CILA include diabetes 40, 41, 54, coronary artery bypass graft 17, 44, 97, and “extreme old age”63, 65, 125. In contrast, there is an association with improved cognition, or decreased CILA, in persons with high levels of education and/or early linguistic ability 74, 82, 96, 108.
How can we weigh the importance of each disease mechanism in CILA? Despite great scientific progress in recent years, both normal and disease mechanisms cannot be modeled with certainty outside the aging human brain. Hence, careful clinicopathological correlations must be performed on relatively large human cohorts to tease out the importance of the factors involved in CILA. This is a formidable challenge. Precise correlation of ante–mortem cognitive measures, clinical variables and post‐mortem neuropathology requires a long‐term, prospective approach involving a multidisciplinary team. Patients must be recruited, tested longitudinally at regular intervals with thorough collection of demographic and clinical data, evaluated proximal to death, and then, upon autopsy, assessed systematically for neuropathology. A large number of patients must be studied to enable adequate statistical testing. Finally, there must be statistical tools to analyze how the ante–mortem parameters correlate to the neuropathology observed after autopsy, to produce data that are clinically and biologically relevant.
The University of Kentucky Alzheimer's Disease Center (UK ADC) studies older persons with a full gamut of cognitive impairment (annual target n = 700 subjects). Along with thorough longitudinal mental status evaluations, many clinical and demographic data are obtained on every clinical visit. All subjects agreed to brain donation following death. To date, the autopsy rate on this cohort is over 90%. To evaluate the contributions of clinical, demographic and neuropathological factors to CILA, we utilized a broad strategy to assess quantitatively which factors are most directly associated with CILA in this large autopsy convenience cohort. Methodology included statistical regression modeling of 43 different clinical and pathological variables in relation to the severity of ante–mortem cognitive impairment. This approach provides the basis for some unforeseen conclusions.
MATERIALS AND METHODS
Subjects
Patients who had come to autopsy from the UK ADC cohorts (total n = 501) were the basis for the study. Research protocols were approved by the University of Kentucky Institutional Review Board. Details of the UK ADC recruitment were described previously 78, 87. Initially non‐demented subjects (“initially normal” cohort) comprised individuals (n = 212) from the UK Biologically Resilient Adults in Neurological Studies (BRAiNS) cohort (101). The “dementia clinic” group (n = 122) derived from the patients followed longitudinally in the UK dementia clinic, who had presented with cognitive symptoms. Characteristics of these groups are shown in Table 1.
Table 1.
Cohort demographics and numbers. Data from two different research clinics show distinct demographic profiles in the groups. Although some Alzheimer's disease‐type pathology was found in many cases from both clinics, other concomitant neuropathological features including infarcts and Lewy bodies (LBs) were also quite common. Abbreviations: ApoE = apolipoprotein E; MMSE = mini‐mental status examination.
| Research clinic | n | Sex (% F) | Formal education, years | ApoE allele 2/3/4% | Age at death | Final MMSE score | Years since final MMSE | % with infarcts (all types) | % with cortical LBs |
|---|---|---|---|---|---|---|---|---|---|
| Initially normal | 212 | 58 | 16.0 | 8/77/15 | 85.9 | 26.3 | 0.82 | 55.2 | 8.0 |
| (standard deviation) | — | — | ±2.4 | — | ±7.7 | ±5.4 | ±0.6 | — | — |
| Dementia clinic | 122 | 61 | 12.9 | 3/64/33 | 80.8 | 10.4 | 1.55 | 35.2 | 26.2 |
| (standard deviation) | — | — | ±3.1 | — | ±7.0 | ±7.4 | ±1.2 | — | — |
| Combined/total | 334 | 59 | 14.8 | 6/71/23 | 84.0 | 20.4 | 1.09 | 48.0 | 14.7 |
Criteria for exclusion involved either inadequate clinical documentation (see further discussion) or the presence of pathology for which statistical power were lacking to model their impact on cognition (Table 2); that is, low patient numbers for rare or idiosyncratic diseases. Thus, the following were excluded: primary or metastatic neoplasm in brain parenchyma including pituitary adenomas; >2 cm contusions in the brain parenchyma; hydrocephalus (except ex vacuo type); infectious agents identified in the brain parenchyma; and a history of left‐sided frontal and/or temporal/parietal infarct with clinically documented aphasia that would seriously confound the results of standard mental status evaluation. Only patients who had MMSE scores within 4 years of death were used in this project [as in (92)]. Inadequate or non‐recent MMSE data (n = 120 cases) were the criteria that most frequently led to exclusion from this study. Our sample had insufficient patients with FTDs and/or tauopathies (corticobasal degeneration, Pick's disease, FTD‐motor neuron disease, progressive supranuclear palsy, other tauopathies and dementia lacking distinct histopathology; n = 17 total) to provide statistical power for a linear regression‐based study.
Table 2.
Exclusion criteria and the numbers excluded from each research clinic according to the reason for exclusion. From the total cases that came to autopsy at the University of Kentucky Alzheimer's Disease Center (n = 501), a total of 167 were excluded according to the criteria shown. By far the most frequent criterion for exclusion were undocumented mini‐mental status examination (MMSE) scores (n = 94) or cases for whom MMSE scores were documented >4 years prior to death (n = 17), which were most often seen in the high‐dementia clinic. Abbreviations: FTD = frontotemporal dementia.
| Reason for exclusion from study | n (cohort) | Total | |
|---|---|---|---|
| Initially normal | Dementia clinic | ||
| Chronic infection | 2 | 0 | 2 |
| Subdural/chronic hematoma | 3 | 2 | 5 |
| Contusions | 1 | 4 | 5 |
| Tumor (including adenomas) | 9 | 1 | 10 |
| Hydrocephalus (not ex vacuo) | 1 | 1 | 2 |
| Infarcts + aphasia | 2 | 1 | 3 |
| Undocumented MMSE scores | 1 | 93 | 94 |
| >4 years since final MMSE score | 2 | 27 | 29 |
| FTDs, all subtypes | 2 | 15 | 17 |
| Total | 23 | 144 | 167 |
Testing
All initially normal individuals were contacted at 6‐month intervals, had detailed mental status testing, and we attempted to conduct neurological and physical examinations annually. For persons with clinical symptoms of dementia, we attempted to obtain mental status testing and neurological examinations annually. Mental status testing of our subjects has been described previously 78, 87. The present study focuses on the MMSE conducted closest to the date of death as the “severity metric” for cognition. MMSE was chosen as it was the most consistently available measure obtained from both normal and demented subjects over the course of their evaluations (and see Discussion).
Neuropathology
Most aspects of pathological assessments were as described in detail previously 78, 87. Briefly, a total of at least 25 sections were taken from each brain that included the middle frontal gyrus (MFG; area 9), superior and middle temporal gyri (SMT; areas 21 and 22), inferior parietal lobule (IPL; areas 39 and 40) and the occipital lobe including primary visual area (Occ; areas 17 and 18). Amyloid plaques were separated into plaques without neurites (DPs) and plaques with degenerating neurites (NPs) in each region as described previously (87). An arithmetic mean was calculated from the count of the five most involved fields for DPs (number of DPs per 2.35 mm2), NPs (number of NPs per 2.35 mm2), and NFTs (number of NFTs per 0.586 mm2) for each region.
TDP‐43 immunohistochemistry (IHC) was performed on hippocampal and superior and middle temporal (SMT) gyri sections from a subset of cases (n = 49). We hypothesized that aberrant TDP‐43 IHC (abTDP‐43) might be found disproportionately in patients whose final MMSE scores were lower than predicted by the statistical model because the pathology may contribute to their lower MMSE scores. We selected cases at random, except that we included 10 cases with the largest deviation from the mean such that (Predicted MMSE‐Actual MMSE) was greatest. Sections were cut on to Probe‐On slides at 5 micron thickness. Rabbit polyclonal anti‐TDP‐43 (1:500; ProteinTech, Chicago, IL, USA) was used following antigen retrieval (Trilogy pH 8.0, Cell Marque, Austin, TX, USA) in a Decloaking Chamber (Biocare Medical, Concord, CA, USA) and then formic acid pretreatment (3 minutes). Secondary antibody reaction employed the Vectastain ABC kit (Vector Labs, Burlingame, CA, USA). TDP‐43 IHC was evaluated by a neuropathologist blinded to the case identifications, and graded semiquantitively (see Analyses section and Supporting Information Table S1).
Analyses
The UK ADC database was built from in‐depth patient interviews, formal longitudinal cognitive and capacity assessments, pre‐ and post‐mortem chart reviews, extensive neuropathological investigation that includes periodic audits, and back‐maintenance on clinical and pathological data that extend from 1984. Unknown medical history items were assumed to indicate absence only after rigorous data extraction from clinical and research charts (otherwise, data were imputed as described in the following discussion). The earliest autopsy used in this series was performed in 1988. Forty‐three different clinical and pathological variables were abstracted, to encompass most of the prevalent factors that have shown associations with CILA (Table 3). Variables were operationalized to determine the relative weight of many potential clinical and pathological contributors to CILA.
Table 3.
The 43 clinical and pathological variables used in the multiple regression algorithm to find which indices are associated with variability in mini‐mental status examination scores proximal to death. Items are operationalized as described in the Supporting Information. Most of the clinical variables are dichotomous (1/0), whereas most of the pathological variables were scored on a continuum from zero to one by graded increments. “Advanced coronary artery disease” is defined by having documented myocardial infarction, coronary artery bypass graft and/or coronary artery angioplasty. Abbreviations: ApoE = apolipoprotein E; AFib = atrial fibrillation.
| Clinical indices | Pathological indices |
|---|---|
| Age at death | |
| Sex | Macroinfarct(s)—cortical |
| Education | Cerebral amyloid angiopathy |
| ApoE alleles | Arteriolosclerosis |
| AFib/other cardiac arrhythmia* | Number of microinfarcts |
| Hypertension* | Number of pale infarcts |
| Body mass index | Number of hemorrhagic infarcts |
| Transient ischemic attack(s)* | Number of lacunar infarcts |
| Head trauma* | Subcortical non‐lacunar infarcts |
| Diabetes/takes insulin* | Argyrophilic grains |
| Seizures/epilepsy* | Lewy bodies—isocortical |
| Smoking* | Lewy bodies—brainstem |
| Peripheral vascular disease* | Lewy bodies—medial temporal lobe |
| Advanced coronary artery disease* | Lewy bodies—amygdala only* |
| Cancer* | Isocortical neurofibrillary tangles (NFTs) |
| Anxiety* | Isocortical neuritic plaques (NPs) |
| Depression/antidepressant meds* | Isocortical diffuse plaques (DPs) |
| Number of documented drugs | Mesial temporal lobe NFTs |
| Opiates* | Mesial temporal lobe NPs |
| Antipsychotics* | Mesial temporal lobe DPs |
| Barbiturates* | Hippocampal sclerosis (HS) unilateral* |
| Benzodiazepenes* | HS bilateral* |
Dichotomous (1 or 0).
Each variable was rendered along a severity continuum from zero to one in a manner hypothesized to be biologically and clinically relevant. “Isocortical” pathology refers to pathology in the MFG, IPL, SMT, and Occ cortices as described earlier. Medial temporal pathology is from amygdala plus CA1, subiculum and entorhinal cortex of the hippocampal formation. The criteria and methods for each variable operationalization are outlined in Supporting Information Table S1. Briefly, some of the variables were dichotomous (0 or 1; for example, history of seizures) whereas for others the severity continua was textured. For example, operationalization of AGs was graded from 0 to 1 proportional to a scoring system wherein AGs were scored semiquantitatively (0–3) from three areas: entorhinal cortex; subiculum; and cornu ammonis fields. AGs have been posited to develop first in the ambient gyrus 99, 100; however, sampling the ambient gyrus dependably (particularly in cases of end‐stage dementia) is a challenge, and the immediately adjacent entorhinal cortex is a reliable area for detecting early‐stage AGs (21). We treated apolipoprotein E (ApoE) genotype as ApoE 2/2 (reference, 0 point), ApoE 2/3 (0.25 point), ApoE 2/4 or 3/3 (0.5 point), ApoE 3/4 (0.75 point) or ApoE 4/4 (1 point).
A subset of 54 cases (14.9% of the cohort overall) had a documentation of demographic information, and neuropathology was performed, but medical records were not available. Missing items for these cases were multiply imputed using the logistic regression method in SAS 9.1.3® PROC MI (Chicago, IL, USA). Covariates were used to predict the missing items include age at death, sex, number of infarcts and number of prescription drugs taken. Grading of arteriolosclerosis severity at autopsy was missing in 28% of cases. These missing data were multiply imputed using the logistic regression method in PROC MI. Covariates were used to predict the missing items include age at death, total number of macro infarcts and total number of micro infarcts from the other cases where records were complete. Four additional items were also multiply imputed: mesial temporal cortex Lewy bodies (0.3%); amygdala‐only Lewy bodies (2.2%); ApoE (4.96%); and body mass index (27.0%). These items were multiply imputed using the MCMC method in PROC MI. The fully imputed data were analyzed using PROC REG, and the results were combined using PROC MIANALYZE. Imputation and modeling were performed with SAS 9.1.3®. For additional information about SAS and also about the results of model‐checking procedures, including delta–betas and backward selection, please see Supporting Information Table S2.
Stratified analyses were conducted based on initial group membership (“initially normal” or “dementia clinic” cohorts). Linear models were fitted with adjusted MMSE as the dependent variable, and all the demographic, clinical and pathological variables as predictors to find the negative regressors (Table 4). Stepwise model selection was used to determine the subset of regressors that best predicts the final MMSE at the 0.10 level of significance. Because multiple imputation methods for binary data may produce biased estimates, models for the “dementia clinic” group were fit both excluding the 54 cases where all clinical data were missing and including the imputed data. Because the results did not appreciably differ, the fully imputed data were used in the final analyses that are presented.
Table 4.
Results of multiple variable linear regression analyses using all clinical and pathological variables for each research cohort separately. The research cohorts were analyzed separately first. Note that the linear regression found large differences between the cohorts in the y‐intercept, and in each case the isocortical NFTs was the largest parameter estimate. Other regressors differed, which may indicate biological differences in the parameters that affect relatively low‐CILA and high‐CILA individuals. Regressors with positive parameter estimates were not listed on this table. There are no such regressors in the “initially normal” group but ApoE and presence of diabetes were positive parameter estimates in the “dementia clinic” group. Abbreviations: ApoE = apolipoprotein E; CILA = cognitive impairment linked to aging; HS = hippocampal sclerosis; LB = Lewy body; NFT = neurofibrillary tangle; TIA = transient ischemic attack.
| Parameter | Parameter estimate | Standard error | Pr > |t| |
|---|---|---|---|
| “Initially normal” group n = 212 | |||
| Intercept | 29.49 | 0.31 | <0.0001 |
| NFTs, isocortex | −24.93 | 4.30 | <0.0001 |
| NFTs, mesial temporal | −16.10 | 2.09 | <0.0001 |
| HS—unilateral | −12.53 | 1.48 | <0.0001 |
| HS—bilateral | −5.50 | 1.65 | 0.001 |
| LBs, mesial temporal | −5.61 | 1.56 | 0.0004 |
| LBs, amygdala only | −3.73 | 1.51 | 0.0146 |
| History of TIA | −1.55 | 0.72 | 0.0334 |
| “Dementia clinic” group, n = 122 | |||
| Intercept | 10.47 | 2.05 | <0.0001 |
| NFTs, isocortex | −16.42 | 2.42 | <0.0001 |
| LBs, neocortex | −5.07 | 2.10 | 0.0177 |
| Antipsychotics | −4.61 | 2.01 | 0.0237 |
| Seizures | −4.31 | 1.91 | 0.0257 |
| Cancer | −2.84 | 1.40 | 0.0447 |
To assess the effect of the clinical predictors on final MMSE scores without the neuropathological predictors, and vice versa, stepwise models using only clinical or pathological variable predictors were fit using the fully imputed, combined data (Table 5). Lastly, in addition to the stratified analyses, a model was fit combining both groups using the pool of significant negative regressors (Table 5). Severity of isocortical NPs and lacunar infarcts were included because they were significantly associated with CILA in the pathology‐only regression. ApoE alleles were included in the regression because they were significantly associated with CILA in the clinical‐only regression.
Table 5.
Regression modeling of parameters from entire research cohort (n = 334)—results of multiple variable linear regression analyses using entire cohort. Some of the included parameters are shown only to demonstrate their lack of independent association, but were also present in prior analyses. Note, in the combined model, ApoE parameter estimates are opposite in the different cohorts (positive in “dementia clinic”, negative in “initially normal”). For the overall group, ApoE did not have a significant effect. If the different cohort‐specific effects were taken into account, then, NPs, isocortex, would lose statistical significance but all the others are roughly the same. This is discussed in the Supporting Information. Abbreviations: ApoE = apolipoprotein E; HS = hippocampal sclerosis; LB = Lewy body; NFT = neurofibrillary tangle; NP = neuritic plaques.
| Parameter | Parameter Estimate | Standard error | Pr > |t| |
|---|---|---|---|
| Clinical parameters only | |||
| Intercept | 10.86 | 2.06 | <0.0001 |
| ApoE alleles | −8.73 | 2.45 | 0.0004 |
| Pathology parameters | |||
| Intercept | 28.50 | 0.47 | <0.0001 |
| NFTs, isocortex | −21.78 | 2.30 | <0.0001 |
| NFTs, mesial temporal | −14.48 | 1.87 | <0.0001 |
| LBs, isocortex | −13.95 | 1.67 | <0.0001 |
| NPs, isocortex | −8.81 | 1.87 | <0.0001 |
| HS—bilateral | −7.81 | 2.10 | 0.0244 |
| HS—unilateral | −6.33 | 2.82 | 0.0256 |
| Macroinfarcts | −6.09 | 2.83 | 0.0323 |
| Lacunar infarcts | −4.81 | 2.51 | 0.0559 |
| All significant parameters combined | |||
| Intercept | 24.32 | 1.07 | <0.0001 |
| NFTs, isocortex | −19.66 | 2.27 | <0.0001 |
| LBs, isocortex | −12.47 | 1.67 | <0.0001 |
| HS—unilateral | −8.58 | 1.82 | <0.0001 |
| NFTs, mesial temporal | −12.44 | 1.87 | <0.0001 |
| HS—bilateral | −7.84 | 2.04 | 0.0001 |
| NPs, isocortex | −6.61 | 2.71 | 0.0152 |
| Lacunar infarcts | −5.15 | 2.46 | 0.037 |
| Age | >0.1 | ||
| LBs, amygdala only | >0.1 | ||
| Sex | >0.1 | ||
The average number of years between the final MMSE test and death for all cases was 1.09 years and the median was 0.82 years. Thirteen cases in the dataset had intervals between 3.5 and 4 years, and these were predominantly end‐stage dementia patients. As patients' MMSE scores would have declined in the interim presumably 23, 81, 82, 95, 111, we also performed the linear regression model using an MMSE correction. In this model, for patients with final MMSE scores less than 28, 2.5 points were deducted from the MMSE score for every 12‐month duration between the final MMSE evaluation and death. For the average patient in this study, this involved a “correction” of less than 2.5 MMSE points (median 1.1; range 0–12.1). In this model, the findings were broadly similar to the data reported without the MMSE correction (data not shown).
Following the derivation of parameter estimates, we used the regression equation and the covariates to predict the MMSE score. These were arrayed on a Microsoft Excel spreadsheet. Using this database we modeled the contribution of the different clinical and pathological variables to CILA over our entire cohort (n = 334). This also allowed for comparison between the actual final MMSE scores vs. “predicted” MMSE scores based on the parameter estimates.
RESULTS
Based on the inclusion criteria described earlier, 334 patients from the UK ADC autopsy series served as the research cohort for a detailed analysis of the clinical and pathological parameters that underlie variations in ante–mortem MMSE scores. As shown in the demographics in Table 1, this cohort comprises predominantly highly educated elderly persons. All cases except one were Caucasian. Over half of the patients had some form of cerebrovascular pathology; of patients beyond 80 years of age, more than 66% had some cerebrovascular pathology. Many of the patients died with minimal CILA, or none, as detected by the MMSE scores. In all, 11% of the cohort had final MMSE scores of 29 or 30.
In the first level of analyses, the total cohort was stratified by the group in which the subjects had initially enrolled. The demographics of these two groups differed in some aspects from each other (Table 1). Stratified statistical analyses identified the determinants of MMSE variability with R 2 correlation coefficient of >0.70. In addition to running the multiple linear regression with the parameters mentioned earlier, additional analyses were ran using only the clinical and demographic variables (n = 22), or only the pathological variables (n = 21). When only clinical variables were queried, the impact of ApoE (P = 0.0004) was manifest. Another model was fit using the data combined from both groups. All the parameters were included from the stratified models, and this final model serves as the source of the results described in the following discussion.
Clinical conditions, neuropathology and CILA
Parameter estimates provide a numerically weighted indicator of how each clinical and pathological variable correlates to interindividual differences in MMSE scores in our sample. The extent of interpersonal differences in MMSE scores can be predicted referent to each parameter and for every patient. For example, all other variables being equal, the presence of HS is associated with eight fewer MMSE score points.
Evaluating parameter estimates and frequencies reveals that more than two‐thirds of the MMSE variability in our study sample was associated with the amount of Alzheimer‐type pathology, NFTs and NPs. This held true whether in the context of “pure” or mixed‐type pathology (for example, with cortical Lewy bodies or cerebrovascular pathology). The AD‐related pathologies associated with CILA were isocortical and mesial temporal NFTs and isocortical NPs [the latter only statistically significant when the whole cohort (n = 334) was analyzed]. The contribution to CILA from AD‐type pathology appeared to differ based on disease severity. In patients with higher MMSE scores, most of the CILA was associated with mesial temporal NFTs and isocortical NPs, whereas with more severe CILA, the isocortical NFTs accounted for a much larger proportion of the cognitive impairment (Figure 1).
Figure 1.
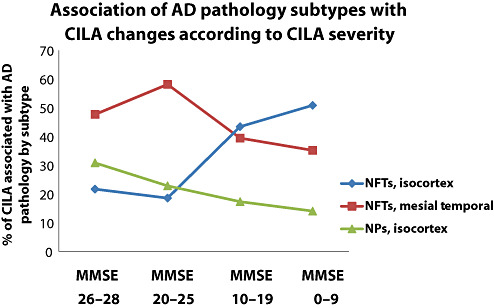
The association of particular subtypes and anatomical areas of Alzheimer's disease (AD) pathology with cognitive impairment linked to aging (CILA) severity changes according to the different stages of cognitive impairment. This chart shows data from modeling on the entire cohort using the statistically significant parameter coefficients as shown in Table 5. Cases are binned by mini‐mental status examination (MMSE) scores: MMSE 26–28 (n = 83); MMSE 20–26 (n = 38); MMSE 10–20 (n = 65); and MMSE 0–10 (n = 62). Note that as cognition declines, the apparent impact of isocortical neurofibrillary tangles (NFTs) becomes greater, and the relative importance of mesial temporal lobe NFTs and isocortical NPs declines. Nonetheless, each subtype of AD‐type pathology is associated independently with significant amount of CILA.
Isocortical LBs tended to be associated with lower MMSE scores (Table 5). However, even advanced LB pathology alone tended not to be associated with end‐stage dementia. The presence of isocortical‐pattern dementia with Lewy bodies sometimes coincided with AD pathology and these pathological processes appear to have an additive clinical effect (Table 6). On average, isocortical LBs correlated with lower MMSE scores by approximately 10 points. These results bear out the regression modeling where the parameter estimate for isocortical LBs in the most severe cases (maximally severe involvement in all four isocortical regions tested) correlate with an approximately 13‐point loss in MMSE score (Table 5).
Table 6.
Final mini‐mental status examination (MMSE) scores proximal to death in patients with extensive isocortical Lewy bodies (LBs) or no isocortical LBs, stratified by Braak stages. Moderate‐to‐severe alpha‐synuclein pathology in neocortex is an additive pathology in the University of Kentucky Alzheimer's Disease Center research cohorts. In these patients (“extensive isocortical LBs”); the frequency of concomitant Alzheimer's disease (AD) was approximately the same as in cases without isocortical LBs. Isocortical LB and AD pathologies appear additive with respect to clinical symptoms. Stage for stage, the presence of isocortical LBs appears to correlate with a decrease of approximately 10 points in MMSE scores, consistent with the statistically derived parameter estimates (4, 5). The few cases with “pure” isocortical LBs (minimal Alzheimer‐type pathology or infarcts) did not have end‐stage dementia.
| Braak stages—average MMSE (n) | |||
|---|---|---|---|
| 0–II | III, IV | V, VI | |
| Cases with extensive isocortical LBs, n = 28 | 17.1 (10) | 11.8 (4) | 7.1 (14) |
| Cases without isocortical LBs, n = 281 | 27.6 (106) | 25.3 (65) | 13.3 (110) |
The majority of the 43 clinical and pathological variables showed no statistically significant contribution to CILA in our cohort. The stepwise linear regression algorithm flags those variables most directly correlating to changes in MMSE scores. It is presumed that those variables that contribute to cognitive impairment through stimulating other pathological processes would not distinguish themselves as long as the “downstream” factor was also evaluated. A presumptive example of this phenomenon is ApoE. ApoE alleles were not associated with significant independent contribution to MMSE variability unless the pathological variables were not present also as regressors.
AGs by pathology were not associated statistically with the severity of CILA in our sample. This was not an artifact of having FTD cases excluded from analyses, because only three FTD cases had minimal AGs, and none of these cases had sufficient amounts to merit the diagnosis of argyrophilic grain disease. Seventy‐five out of 334 (22%) cases had some AGs as assessed using Gallyas silver impregnation. P‐values for the association of AGs pathology to CILA severity were always >0.5. The operationalization technique allowed a graded score of AGs reflecting the distribution and severity (see Analysis section). Of the seven cases with the highest score of AGs, four had final MMSE scores of 29 or higher, and none had MMSE scores less than 24. These additional analyses (Figure 2) again provide some validation for the statistical modeling data.
Figure 2.

Statistical methods indicated that argyrophilic grains (AGs) are not associated with CILA severity in our research cohorts. To evaluate these data, we studied separately the 75 cases with AGs in our sample. A chart (A) shows how the actual pre‐mortem mini‐mental status examination (MMSE) scores for patients correlated to the amount of AG pathology. The AG score is proportional to the severity of AGs in entorhinal cortex, subiculum and cornu ammonis hippocampal fields. Note that there is no decrease in MMSE scores as AG pathology worsens. A photomicrograph of Gallyas‐stained cornu ammonis (B) from the individual in our autopsy cohort with the most severe AG pathology, a 91‐year old woman who died without any cognitive impairment (MMSE score = 30 less than 8 months prior to death). Scale bar = 50 µm.
A subset of cases with aberrant TDP‐43 IHC
To capitalize on strengths of the statistical data, a preliminary subset of cases (n = 49) was evaluated further using TDP‐43 IHC (Table 7, 3, 4) to test the hypothesis that TDP‐43 proteinopathy may help to account for the CILA in some cases. Patients with actual MMSE more than 10 points lower than predicted MMSE (using our statistical model) were included, as were patients with actual MMSE higher than the actual MMSE. Thus, this group included cases with the highest degree of “unaccounted‐for” CILA and the rest were chosen at random for this preliminary study. TDP‐43 IHC was performed on superior and middle temporal gyri as well as hippocampal formation to encompass areas where TDP proteinopathy has been described (3). IHC‐positive lesions (abTDP‐43) were scored blindly and included either aberrant TDP‐43(+) NFT‐like structures with neuropil thread‐like profiles (Figure 4) or TDP‐43(+) inclusions in dentate granule cells (Figure 4C). As shown in Figure 4, TDP‐43 immunoreactivity could be a challenge to interpret, because the large majority of immunoreactive cells are probably not associated with pathology per se. Further, some staining irregularities may have only reflected the impact of antigen retrieval on tissue as “regularly irregular” staining. As a group, cases with abTDP‐43 showed a tendency to have a lower actual MMSE; all other things being equal (intercepts differ by linear regression at P = 0.04). These data indicate that there is an association between the presence of TDP‐43 pathology and the severity of CILA independent of other known clinical or pathological variables. These analyses represented a pilot study that we will follow up on in a separate report.
Table 7.
A subset of cases evaluated for TDP‐43 immunohistochemistry (IHC): demographics and mini‐mental status examination (MMSE) data (actual and predicted results). Demographics and MMSE data from the 49 cases stained for TDP‐43 IHC. Cases with abTPD‐43 had aberrant IHC in dentate granule cells (n = 18) and/or neurofibrillary tangle (NFT)‐like and neuropil thread‐like TDP‐43 immunoreactive profiles in the hippocampal formation (n = 21). Note that the actual MMSE score proximal to death was lower for the cases with abTDP‐43(+), and was even lower than the MMSE scores predicted using the regression algorithm that includes the known clinical and pathological risk factor for cognitive impairment linked to aging.
| n | Formal education, years | Braak stage | Age at death, years | Final MMSE score | Final predicted MMSE score | |
|---|---|---|---|---|---|---|
| Cases with abTDP‐43 IHC | 25 | 15.6 | 4.7 | 85.0 | 9.1 | 16.0 |
| Cases without abTDP‐43 IHC | 24 | 15.9 | 4.2 | 85.0 | 17.0 | 20.7 |
Figure 3.
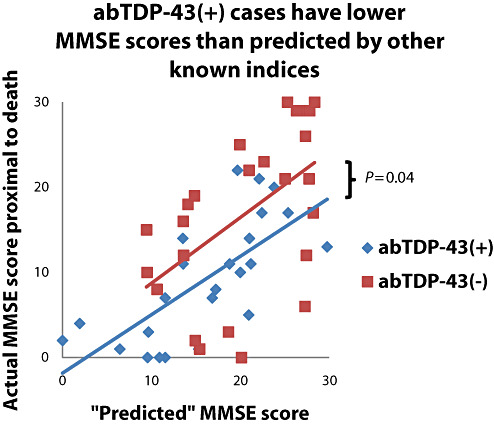
The multiple variable linear regression algorithm allows the prediction of mini‐mental status examination (MMSE) for each case based on the parameter coefficients. The algorithm was applied using the parameter coefficients from the combined cohort as shown in Table 5 for the 49 cases stained for TDP‐43 immunohistochemistry. The results indicate that the abTDP‐43(+) show a significant downward shift such that the “actual” MMSE score was lower than the abTDP‐43(−) cases (difference of intercept P = 0.04), indicating that abTDP‐43 is associated with cognitive impairment linked to aging severity independent of other known risk factors such as Alzheimer's disease or hippocampal sclerosis pathology.
Figure 4.

Photomicrographs of TDP‐43 immunohistochemistry (IHC) show abnormal (A,C) and normal (B,D) staining, which can be a challenge to discriminate. Unlike most neurodegeneration‐related immunostains, TDP‐43 IHC is strongly positive in normal tissues. In some end‐stage AD cases we interpreted to be abTDP‐43(−) there were “regularly irregular” staining in the dentate granule cells (B) and hyperchromatic cells (D, horizontal arrows) that were different from the normal‐appearing cells (vertical arrow) but distinct also from the aberrant intracellular inclusions (C). AbTDP‐43 staining in CA1 (C) showing the neurofibrillary tangle‐like (arrow) and neuritic thread‐like structures (arrows). Immunopositivity was scored blindly. Scale bars = 30 µm (A,B) or 50 µm (C,D).
DISCUSSION
Data from a large autopsy series were used to model the association of 43 clinical and pathological variables with CILA. Our results underscore the fundamental importance of AD pathology in our research cohorts, and indicate that many clinical variables (including but not limited to ApoE allele subtype) probably influence dementia if at all through altering “downstream” pathology such as NFTs. Most of the remainder of CILA in our cohort is associated with the presence of synucleinopathy, HS and cerebrovascular pathology.
Our strategy provides insights into disease mechanisms that are associated with cognitive impairment in older persons. It has been known for some time that dementia is a syndrome with many contributing pathologies. However, there have been few studies that involve weighing the direct associations (rather than relative risks) of clinical and pathological variables with cognitive impairment in patients who died encompassing a full range of cognitive abilities. Three goals motivated our analytic strategy:
-
(i)
Provide a framework to know how adequately our current understanding of aging‐related brain diseases correlates with actual cognitive decline in humans. With the caveat that “correlation is not causation,” we highlight a handful of clinical and pathological variables that are associated with a significant majority of the variability of MMSE scores in our cohort.
-
(ii)
Highlight the specific clinical and pathological variables that are most directly associated with CILA in a quantitative, statistical framework. Specifically, AD‐related pathologies are by far most important (at least in this cohort), whereas some other variables such as AGs are not associated with CILA severity.
-
(iii)
Identify groups of individuals that are apparent “discrepancies” or outliers. These cases may either have unrecognized clinical contributors to CILA, or have pathological processes that are not currently recognized. In some patients with more CILA than predicted by other clinical and pathological indices, abTDP‐43 pathology is observed disproportionately.
The clinical and pathological indices most strongly associated with CILA in the present study are compatible with prior research but also provide new insights. The data underscore the central importance of AD pathology in CILA, which has been described by a number of researchers previously 5, 6, 7, 8, 10, 14, 15, 16, 20, 47, 48, 59, 63, 87, 89, 119. Of the clinical and pathological variables, AD‐type pathology was associated with greater than two‐thirds of the variability in MMSE scores in our research group. In addition, the data had implications about the pathological contribution of the different subtypes and the neuroanatomical distribution of AD‐type pathology.
Our statistical model supports an independent role for the contribution of NFTs in the isocortex (sampled six‐layered cerebral cortex comprising Brodmann areas 9, 17, 18, 21, 22, 39 and 40), NPs in the isocortex and NFTs in the mesial temporal lobe structures (CA1, subiculum and entorhinal cortex of the hippocampal formation, and amygdala). In persons with milder CILA, the predominant contribution to MMSE loss was NFTs in the mesial temporal lobe; however, in more demented patients, the isocortical NFTs contributed most profoundly. These findings are compatible with Braak staging 19, 20.
We (87) and others [for example, 6, 10, 85] have previously interpreted clinicopathological data to argue against the importance of amyloid plaques in contributing directly to cognitive impairment, focusing instead on isocortical NFTs. This interpretation derived from the relatively poor associations between cerebral cortical NP densities found at autopsy and the severity of ante–mortem cognitive impairment 6, 87. However, the present study involves a larger cohort than our prior study and reflects a new approach that allows relatively unbiased modeling of the association of multiple variables with CILA. These data show an independent association between the density of isocortical NPs and CILA severity, although this only became statistically significant after both research cohorts were combined. The parameter coefficient for isocortical NP pathology is less than that for NFTs and these data should be interpreted with caution. However, the overall apparent contribution of isocortical NPs to CILA was statistically significant, accounting for approximately 20% of the total MMSE score variability in the sample. These data support the hypothesis that NPs themselves can contribute directly to cognitive impairment. Thus, isocortical NPs and medial temporal lobe NFTs—although their densities correlate poorly at death with ante–mortem cognitive impairment—nonetheless may contribute significantly to CILA. This is true especially at the earlier stages of the disease and in patients with mixed pathology. This theoretical point, which is harmonious with prior literature 56, 115, is illustrated in Figure 5. By contrast, our data suggest that ApoE alleles do not contribute directly to CILA. Rather, ApoE alleles probably act by differentially triggering the formation of NPs and NFTs in AD as suggested by Bennett et al (12). The interactions between ApoE and plaque formation is underscored by the fact that when ApoE is included in the combined model, the statistically significant association between isocortical NPs and MMSE scores becomes non‐significant; no other parameters are likewise affected. It should also be noted that other clinical parameters may also impact AD via NPs and NFTs, but these indirect interactions or “upstream” modifiers would not be detected in the present study.
Figure 5.
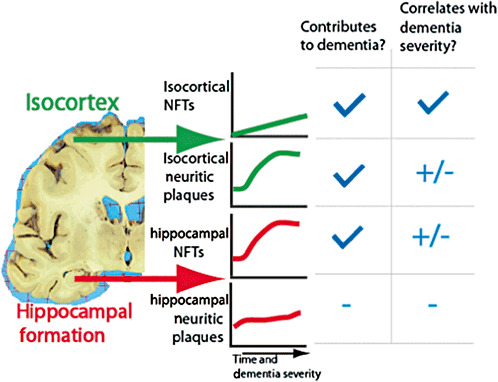
A theoretical model that harmonizes data from the present study and prior clinico‐pathological correlation research. Because isocortical neuritic plaques (NPs) and hippocampal neurofibrillary tangles (NFTs) have significant “floor” and “ceiling” effects in relationship to the course of dementia, they are not strongly related to the cognitive impairment linked to aging (CILA) severity prior to autopsy (87). However, they may still play a strong pathogenetic role and contribute directly to CILA particularly in the earliest stages of Alzheimer's disease.
Multivariable correlation also allowed for a delineation of the associative impact of HS on CILA. In our sample, the presence of bilateral or of unilateral HS were associated, on average, with patients whose final MMSE scores were ∼8 points lower. These data are important for a number of reasons. First, we confirm the biological importance of HS as reported previously 27, 30, 38, 83, 91, and we assessed quantitatively the associative impact of HS in cognitive impairment. Second, as HS is associated with TDP‐43 proteinopathy 2, 3, 28, 93, TDP‐43 pathology is apparently an important contributor to CILA in human populations through multiple means.
Previous studies have shown that TDP‐43 proteinopathy is also present in relatively rare subtypes of FTD 3, 4, 32, 77, 88, 105, 110, 127. It has also been shown previously that a subset of AD patients have abTDP‐43, and cases with abTDP‐43 may have a faster disease course and more hippocampal atrophy (67). A subset of patients in our study had a degree of cognitive impairment that is not “explained” by an association with any of the clinical or pathological variables, and abTDP‐43 is present in a significant proportion of these. This is the first formal demonstration of evidence for a role for TDP‐43 proteinopathy in CILA outside of the studies mentioned earlier.
In contrast to AD‐type pathology and HS, some of the factors that have been associated with CILA in the past were not associated with lower MMSE scores in our cohort. For example, the presence of brain AGs was not associated with CILA. Prior studies have suggested that AGs may not associate strongly with CILA 68, 70. In our cohort there was not even a weak association between the presence of AGs and CILA. This finding was not because of our exclusion of FTDs from the analyses (see Results section). It is possible that the presence of AGs represent a phase of pathological development such that in later stages of dementia the AGs regress. It is also possible that had we used anti‐Tau 4R antibodies we may have had greater sensitivity for AGs 116, 117, 126, although in some cases with late‐stage AD pathology some AGs could be seen. Nonetheless, persons in our cohort did not show end‐stage dementia in the form of pure AG disease. We also confirm prior studies about the lack of impact on CILA by amygdala Lewy bodies (121). Other factors that have been previously shown to be correlated to CILA were also found not to be in our cohort. These included the number of years of formal education; whether or not patients had undergone coronary artery bypass grafts; and dozens of other clinical and pathological variables (see Table 3).
Because of the strong environmental risk factors, cerebrovascular‐related morbidity and mortality is a source for potential clinical and pathological differences between populations 87, 123, 124. Although more than two‐thirds of individuals aged 80 years and above had CVD, our sample is probably biased against high‐infarct cases [cases of infarct‐associated aphasia (n = 3) were excluded from the statistical study]. Interestingly, in our sample microinfarcts did not constitute a parameter that associated with MMSE score variability, unlike other studies 109, 123. This may be partly because other clinical and pathological variables linked to microinfarcts “washed out” the effect in our regression model. By contrast, and in agreement with prior studies 49, 104, we did find a positive impact from lacunar infarcts in the model. These data highlight the need for a clinically relevant, precise, collectively agreed‐upon classification scheme for smaller infarcts.
The presence on autopsy of large infarcts were associated with lower MMSE scores in the context of the pathology parameters alone, and history of transient ischemic attacks was linked to lower MMSE scores in the “initially normal” cohort. Our current model will be applied in the future to separate cohorts with even higher CVD incidence to enhance our understanding of clinicopathological correlation in CVD.
Aside from the inherent difficulty with studying CVD, there were other limitations and potential confounds relevant to our study. The studied group represents a convenience sample rather than an epidemiological or population‐based cohort. These UK ADC research cohorts are biased toward aged, Caucasian and highly educated individuals (Table 1). There also is a relative lack of movement disorders such as “pure” Parkinson's disease in our cohort. Although there are drawbacks to the demographic and pathological homogeneity, there are also benefits from the dampening of variability in our cohort. Athough perhaps less able to generalize conclusions to other populations, we are able to better establish relationships between the types of disease mechanisms and the severity of clinical deterioration seen in these individuals.
In the present study, MMSE scores were used as the sole indicator of cognitive impairment severity. This is an imperfect metric for a number of reasons. Perhaps most importantly, a linear regression algorithm was applied although MMSE and other tests of cognition (and indeed, “cognition” itself) do not decline in a perfectly linear fashion during brain disease 29, 82, 84, 94. This discrepancy would theoretically be reflected by artificially decreasing the degree to which accurate parameter coefficients could be established. Non‐linear regression models are available; however, they tend to have the disadvantage of providing less information about multivariable correlations. MMSE, like other cognitive assessments, also has “ceiling” and “floor” effects 39, 46, 61. Other measures such as Alzheimer's Disease Assessment Scale—cognitive subscale assessments are more sensitive tools for detecting cognitive impairment (102). Hence our analyses are not able to fully take into account subtle cognitive changes representative of “preclinical” cognitive impairment. The many patients in our cohort that achieved perfect MMSE scores of 30 prior to death may be misleading, given that our “predicted” MMSE (using our derived parameter estimates) was somewhat lower.
Despite the imperfections of using MMSE scores, there are also compelling reasons to use this tool in the particular context of multivariable clinicopathological correlation. For a variety of reasons, MMSE scores 1, 8, 22, 28, 31, 33, 35, 37, 51, 60, 62, 64, 68, 71, 73, 84, 87, 98, 112, 114, 115, 122, or the even less finely graded Clinical Dementia Rating scale 14, 15, 48, 57, 103, 107, have been used as metrics of dementia severity in many informative clinicopathological correlation studies. MMSE scores offer good sensitivity in predicting disease 53, 69, 75, with well‐known normative data. The test evaluates a number of cognitive domains, including orientation, language, attention/calculation, short‐term memory/recall and praxis, although not executive or psychiatric domains. MMSE data can be used for cross‐study comparisons and for biologically informative statistical rendering 25, 36, 43, 55, 106, 113, 115, 118. Although MMSE scores are an imperfect metric of cognition 26, 81, relatively small MMSE score changes (>2–4 points) have been found to be statistically reliable indicators of cognitive impairment 42, 58 even in many mild cognitive impairment cases 13, 34. In the present study, the statistically significant parameter estimates tended to be greater than four MMSE score points. Some short‐term, memory‐intensive cognitive tests are less accurate in the detection of cognitive changes in late‐stage cases (102). Moreover, other cognitive assessment tools have undergone repeated “fine‐tuning” over time, and—as such—cannot be used for longitudinal or retrospective comparisons over many years. In sum, MMSE scores were the best option in this context as a robust metric for multiple cognitive domains that we have documented in a consistent fashion between 1984 and 2008.
Some aspects of the statistical modeling also deserve comment. We are studying variability in MMSE scores as a surrogate for CILA severity; however, we are using a cross‐sectional approach instead of comparing the trajectory of cognitive changes in individuals. It would be desirable to have a larger sample for the statistical modeling to assess with higher resolution the contribution of each parameter in association with MMSE score variability. The present approach is tailored to finding the relatively large, more direct contributing parameters that are associated with differences seen in final MMSE scores proximal to death.
Inevitably there are both clinical and pathological variables left out of this study. For example, we do not measure synaptic density although others have found that synaptic density correlates well with CILA in AD brains 79, 80. Instead, we concentrated on the clinical and pathological variables that were available in our dataset and were then considered mostly likely to relate to CILA. Ultimately, our statistical modeling approach is helpful only to the extent that it allows one to address clinically and biologically interesting questions. It is necessary to show that the clinicopathological correlations are not merely relevant to one population group. The present study is only the first step in developing parameter estimates for clinical and pathological variables that can be applied with greater confidence to more heterogeneous populations.
Supporting information
Table S1. Operationalization of clinical and pathological parameters for Nelson PT et al “Modeling the association . . .”
Table S2. Model checking and model selection
Please note: Wiley‐Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
Supporting info item
Supporting info item
ACKNOWLEDGMENTS
We are deeply grateful to all of the participants in our longitudinal aging study and to the patients with AD in our Alzheimer's Disease Center's research clinic. We thank Ann Tudor, Paula Thomason, Dr. Huaichen Liu and Sonya Anderson for the technical support, and Gregory Cooper, MD, PhD, Nancy Stiles, MD and Allison Caban‐Holt, PhD for the clinical evaluations.
Funding/support: This study was supported by grant 5‐P30‐AG028383 and K08 NS050110 from the National Institutes of Health, Bethesda, MD, and a grant from the Healy Family Foundation.
REFERENCES
- 1. Akram A, Christoffel D, Rocher AB, Bouras C, Kovari E, Perl DP et al (2008) Stereologic estimates of total spinophilin‐immunoreactive spine number in area 9 and the CA1 field: relationship with the progression of Alzheimer's disease. Neurobiol Aging 29:1296–1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Amador‐Ortiz C, Ahmed Z, Zehr C, Dickson DW (2007) Hippocampal sclerosis dementia differs from hippocampal sclerosis in frontal lobe degeneration. Acta Neuropathol (Berl) 113:245–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Amador‐Ortiz C, Lin WL, Ahmed Z, Personett D, Davies P, Duara R et al (2007) TDP‐43 immunoreactivity in hippocampal sclerosis and Alzheimer's disease. Ann Neurol 61:435–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Arai T, Hasegawa M, Akiyama H, Ikeda K, Nonaka T, Mori H et al (2006) TDP‐43 is a component of ubiquitin‐positive tau‐negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem Biophys Res Commun 351:602–611. [DOI] [PubMed] [Google Scholar]
- 5. Armstrong RA (2003) Quantifying the pathology of neurodegenerative disorders: quantitative measurements, sampling strategies and data analysis. Histopathology 42:521–529. [DOI] [PubMed] [Google Scholar]
- 6. Arriagada PV, Growdon JH, Hedley‐Whyte ET, Hyman BT (1992) Neurofibrillary tangles but not senile plaques parallel duration and severity of Alzheimer's disease. Neurology 42:631–639. [DOI] [PubMed] [Google Scholar]
- 7. Bancher C, Braak H, Fischer P, Jellinger KA (1993) Neuropathological staging of Alzheimer lesions and intellectual status in Alzheimer's and Parkinson's disease patients. Neurosci Lett 162:179–182. [DOI] [PubMed] [Google Scholar]
- 8. Bancher C, Jellinger K, Lassmann H, Fischer P, Leblhuber F (1996) Correlations between mental state and quantitative neuropathology in the Vienna Longitudinal study on Dementia. Eur Arch Psychiatry Clin Neurosci 246:137–146. [DOI] [PubMed] [Google Scholar]
- 9. Bennett DA (2006) Postmortem indices linking risk factors to cognition: results from the religious order study and the memory and aging project. Alzheimer Dis Assoc Disord 20 (3 Suppl. 2):S63–8. [DOI] [PubMed] [Google Scholar]
- 10. Bennett DA, Schneider JA, Wilson RS, Bienias JL, Arnold SE (2004) Neurofibrillary tangles mediate the association of amyloid load with clinical Alzheimer disease and level of cognitive function. Arch Neurol 61:378–384. [DOI] [PubMed] [Google Scholar]
- 11. Bennett DA, Schneider JA, Bienias JL, Evans DA, Wilson RS (2005) Mild cognitive impairment is related to Alzheimer disease pathology and cerebral infarctions. Neurology 64:834–841. [DOI] [PubMed] [Google Scholar]
- 12. Bennett DA, Schneider JA, Wilson RS, Bienias JL, Berry‐Kravis E, Arnold SE (2005) Amyloid mediates the association of apolipoprotein E e4 allele to cognitive function in older people. J Neurol Neurosurg Psychiatry 76:1194–1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Benson AD, Slavin MJ, Tran TT, Petrella JR, Doraiswamy PM (2005) Screening for early Alzheimer's disease: is there still a role for the mini‐mental state examination? Prim Care Companion J Clin Psychiatry 7:62–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Berg L, McKeel DW Jr, Miller, JP , Storandt M, Rubin EH Morris JC et al (1998) Clinicopathologic studies in cognitively healthy aging and Alzheimer's disease: relation of histologic markers to dementia severity, age, sex, and apolipoprotein E genotype. Arch Neurol 55:326–335. [DOI] [PubMed] [Google Scholar]
- 15. Bierer LM, Hof PR, Purohit DP, Carlin L, Schmeidler J, Davis KL, Perl DP (1995) Neocortical neurofibrillary tangles correlate with dementia severity in Alzheimer's disease. Arch Neurol 52:81–88. [DOI] [PubMed] [Google Scholar]
- 16. Blessed G, Tomlinson BE, Roth M (1968) The association between quantitative measures of dementia and of senile change in the cerebral grey matter of elderly subjects. Br J Psychiatry 114:797–811. [DOI] [PubMed] [Google Scholar]
- 17. Blumenthal JA, Madden DJ, Burker EJ, Croughwell N, Schniebolk S, Smith R et al (1991) A preliminary study of the effects of cardiac procedures on cognitive performance. Int J Psychosom 38:13–16. [PubMed] [Google Scholar]
- 18. Bowler JV, Munoz DG, Merskey H, Hachinski V (1998) Fallacies in the pathological confirmation of the diagnosis of Alzheimer's disease. J Neurol Neurosurg Psychiatry 64:18–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Braak H, Braak E (1995) Staging of Alzheimer's disease‐related neurofibrillary changes. Neurobiol Aging 16:271–278; discussion 8–84. [DOI] [PubMed] [Google Scholar]
- 20. Braak H, Braak E, Bohl J (1993) Staging of Alzheimer‐related cortical destruction. Eur Neurol 33:403–408. [DOI] [PubMed] [Google Scholar]
- 21. Braak H, Del Tredici K, Bohl J, Bratzke H, Braak E (2000) Pathological changes in the parahippocampal region in select non‐Alzheimer's dementias. Ann NY Acad Sci 911: 221–239. [DOI] [PubMed] [Google Scholar]
- 22. Braak H, Rub U, Jansen Steur EN, Del Tredici K, De Vos RA (2005) Cognitive status correlates with neuropathologic stage in Parkinson disease. Neurology 64:1404–1410. [DOI] [PubMed] [Google Scholar]
- 23. Carcaillon L, Peres K, Pere JJ, Helmer C, Orgogozo JM, Dartigues JF (2007) Fast cognitive decline at the time of dementia diagnosis: a major prognostic factor for survival in the community. Dement Geriatr Cogn Disord 23:439–445. [DOI] [PubMed] [Google Scholar]
- 24. Castellani RJ, Lee HG, Zhu X, Nunomura A, Perry G, Smith MA (2006) Neuropathology of Alzheimer disease: pathognomonic but not pathogenic. Acta Neuropathol (Berl) 111:503–509. [DOI] [PubMed] [Google Scholar]
- 25. Cherbuin N, Anstey KJ, Lipnicki DM (2008) Screening for dementia: a review of self‐ and informant‐assessment instruments. Int Psychogeriatr 20:431–458. [DOI] [PubMed] [Google Scholar]
- 26. Clark CM, Sheppard L, Fillenbaum GG, Galasko D, Morris JC, Koss E et al (1999) Variability in annual mini‐mental state examination score in patients with probable Alzheimer disease: a clinical perspective of data from the consortium to establish a registry for Alzheimer's disease. Arch Neurol 56:857–862. [DOI] [PubMed] [Google Scholar]
- 27. Corey‐Bloom J, Sabbagh MN, Bondi MW, Hansen L, Alford MF, Masliah E, Thal LJ (1997) Hippocampal sclerosis contributes to dementia in the elderly. Neurology 48:154–160. [DOI] [PubMed] [Google Scholar]
- 28. Counts SE, He B, Che S, Ikonomovic MD, DeKosky ST, Ginsberg SD, Mufson EJ (2007) Alpha7 nicotinic receptor up‐regulation in cholinergic basal forebrain neurons in Alzheimer disease. Arch Neurol 64:1771–1776. [DOI] [PubMed] [Google Scholar]
- 29. Crane PK, Narasimhalu K, Gibbons LE, Mungas DM, Haneuse S, Larson EB et al (2008) Item response theory facilitated cocalibrating cognitive tests and reduced bias in estimated rates of decline. J Clin Epidemiol 61:1018–1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Crystal HA, Dickson DW, Sliwinski MJ, Lipton RB, Grober E, Marks‐Nelson H, Antis P (1993) Pathological markers associated with normal aging and dementia in the elderly. Ann Neurol 34:566–573. [DOI] [PubMed] [Google Scholar]
- 31. Cummings BJ, Pike CJ, Shankle R, Cotman CW (1996) Beta‐amyloid deposition and other measures of neuropathology predict cognitive status in Alzheimer's disease. Neurobiol Aging 17:921–933. [DOI] [PubMed] [Google Scholar]
- 32. Davidson Y, Kelley T, Mackenzie IR, Pickering‐Brown S, Du Plessis D, Neary D et al (2007) Ubiquitinated pathological lesions in frontotemporal lobar degeneration contain the TAR DNA‐binding protein, TDP‐43. Acta Neuropathol 113:521–533. [DOI] [PubMed] [Google Scholar]
- 33. Davis PC, Gearing M, Gray L, Mirra SS, Morris JC, Edland SD et al (1995) The CERAD experience, Part VIII: neuroimaging‐neuropathology correlates of temporal lobe changes in Alzheimer's disease. Neurology 45:178–179. [DOI] [PubMed] [Google Scholar]
- 34. De Jager CA, Budge MM (2005) Stability and predictability of the classification of mild cognitive impairment as assessed by episodic memory test performance over time. Neurocase 11:72–79. [DOI] [PubMed] [Google Scholar]
- 35. DeKosky ST, Scheff SW (1990) Synapse loss in frontal cortex biopsies in Alzheimer's disease: correlation with cognitive severity. Ann Neurol 27:457–464. [DOI] [PubMed] [Google Scholar]
- 36. Dellasega C, Morris D (1993) The MMSE to assess the cognitive state of elders. Mini‐Mental State Examination. J Neurosci Nurs 25:147–152. [DOI] [PubMed] [Google Scholar]
- 37. Devine ME, Fonseca JA, Walker RW, Sikdar T, Stevens T, Walker Z (2007) Cerebral white matter changes and rate of progression of dementia during cholinesterase inhibitor treatment: a retrospective cohort study. Int J Geriatr Psychiatry 22:1120–1126. [DOI] [PubMed] [Google Scholar]
- 38. Dickson DW, Davies P, Bevona C, Van Hoeven KH, Factor SM, Grober E et al (1994) Hippocampal sclerosis: a common pathological feature of dementia in very old (≥80 years of age) humans. Acta Neuropathol 88:212–221. [DOI] [PubMed] [Google Scholar]
- 39. Doody RS, Strehlow SL, Massman PJ, Feher EP, Clark C, Roy JR (1999) Baylor profound mental status examination: a brief staging measure for profoundly demented Alzheimer disease patients. Alzheimer Dis Assoc Disord 13:53–59. [DOI] [PubMed] [Google Scholar]
- 40. Elias MF, Elias PK, Sullivan LM, Wolf PA, D'Agostino RB (2005) Obesity, diabetes and cognitive deficit: the Framingham heart study. Neurobiol Aging 26 (Suppl. 1):11–16. [DOI] [PubMed] [Google Scholar]
- 41. Elias PK, Elias MF, D'Agostino RB, Cupples LA, Wilson PW, Silbershatz H, Wolf PA (1997) NIDDM and blood pressure as risk factors for poor cognitive performance. The Framingham study. Diabetes Care 20:1388–1395. [DOI] [PubMed] [Google Scholar]
- 42. Eslinger PJ, Swan GE, Carmelli D (2003) Changes in mini‐mental state exam in community‐dwelling older persons over 6 years: relationship to health and neuropsychological measures. Neuroepidemiology 22:23–30. [DOI] [PubMed] [Google Scholar]
- 43. Farber JF, Schmitt FA, Logue PE (1988) Predicting intellectual level from the mini‐mental state examination. J Am Geriatr Soc 36:509–510. [DOI] [PubMed] [Google Scholar]
- 44. Folks DG, Freeman AM, 3rd , Sokol RS, Govier AV, Reves JG, Baker DM (1988) Cognitive dysfunction after coronary artery bypass surgery: a case‐controlled study. South Med J 81:202–206. [DOI] [PubMed] [Google Scholar]
- 45. Forte A, Cipollaro M, Cascino A, Galderisi U (2005) Small interfering RNAs and antisense oligonucleotides for treatment of neurological diseases. Curr Drug Targets 6:21–29. [DOI] [PubMed] [Google Scholar]
- 46. Galasko DR, Gould RL, Abramson IS, Salmon DP (2000) Measuring cognitive change in a cohort of patients with Alzheimer's disease. Stat Med 19:1421–1432. [DOI] [PubMed] [Google Scholar]
- 47. Giannakopoulos P, Gold G, Kovari E, Von Gunten A, Imhof A, Bouras C, Hof PR (2007) Assessing the cognitive impact of Alzheimer disease pathology and vascular burden in the aging brain: the Geneva experience. Acta Neuropathol (Berl) 113:1–12. [DOI] [PubMed] [Google Scholar]
- 48. Gold G, Bouras C, Kovari E, Canuto A, Glaria BG, Malky A et al (2000) Clinical validity of braak neuropathological staging in the oldest‐old. Acta Neuropathol (Berl) 99:579–582, discussion 83–4. [DOI] [PubMed] [Google Scholar]
- 49. Gold G, Kovari E, Herrmann FR, Canuto A, Hof PR, Michel JP et al (2005) Cognitive consequences of thalamic, basal ganglia, and deep white matter lacunes in brain aging and dementia. Stroke 36:1184–1188. [DOI] [PubMed] [Google Scholar]
- 50. Gonzalez‐Alegre P (2007) Therapeutic RNA interference for neurodegenerative diseases: from promise to progress. Pharmacol Ther 114:34–55. [DOI] [PubMed] [Google Scholar]
- 51. Green MS, Kaye JA, Ball MJ (2000) The Oregon brain aging study. Neuropathology accompanying healthy aging in the oldest old. Neurology 54:105–113. [DOI] [PubMed] [Google Scholar]
- 52. Grober E, Dickson D, Sliwinski MJ, Buschke H, Katz M, Crystal H, Lipton RB (1999) Memory and mental status correlates of modified Braak staging. Neurobiol Aging 20:573–579. [DOI] [PubMed] [Google Scholar]
- 53. Grut M, Fratiglioni L, Viitanen M, Winblad B (1993) Accuracy of the mini‐mental status examination as a screening test for dementia in a Swedish elderly population. Acta Neurol Scand 87:312–317. [DOI] [PubMed] [Google Scholar]
- 54. Guccione AA, Felson DT, Anderson JJ, Anthony JM, Zhang Y, Wilson PW et al (1994) The effects of specific medical conditions on the functional limitations of elders in the Framingham Study. Am J Public Health 84:351–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Han L, Cole M, Bellavance F, McCusker J, Primeau F (2000) Tracking cognitive decline in Alzheimer's disease using the mini‐mental state examination: a meta‐analysis. Int Psychogeriatr 12:231–247. [DOI] [PubMed] [Google Scholar]
- 56. Haroutunian V, Perl DP, Purohit DP, Marin D, Khan K, Lantz M et al (1998) Regional distribution of neuritic plaques in the nondemented elderly and subjects with very mild Alzheimer disease. Arch Neurol 55:1185–1191. [DOI] [PubMed] [Google Scholar]
- 57. Haroutunian V, Purohit DP, Perl DP, Marin D, Khan K, Lantz M et al (1999) Neurofibrillary tangles in nondemented elderly subjects and mild Alzheimer disease. Arch Neurol 56:713–718. [DOI] [PubMed] [Google Scholar]
- 58. Hensel A, Angermeyer MC, Riedel‐Heller SG (2007) Measuring cognitive change in older adults: reliable change indices for the mini‐mental state examination. J Neurol Neurosurg Psychiatry 78:1298–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Hogervorst E, Bandelow S, Combrinck M, Irani SR, Smith AD (2003) The validity and reliability of 6 sets of clinical criteria to classify Alzheimer's disease and vascular dementia in cases confirmed post‐mortem: added value of a decision tree approach. Dement Geriatr Cogn Disord 16:170–180. [DOI] [PubMed] [Google Scholar]
- 60. Holtzer R, Irizarry MC, Sanders J, Hyman BT, Wegesin DJ, Riba A et al (2006) Relation of quantitative indexes of concurrent alpha‐synuclein abnormalities to clinical outcome in autopsy‐proven Alzheimer disease. Arch Neurol 63:226–230. [DOI] [PubMed] [Google Scholar]
- 61. Ihl R, Frolich L, Dierks T, Martin EM, Maurer K (1992) Differential validity of psychometric tests in dementia of the Alzheimer type. Psychiatry Res 44:93–106. [DOI] [PubMed] [Google Scholar]
- 62. Ikonomovic MD, Mufson EJ, Wuu J, Bennett DA, DeKosky ST (2005) Reduction of choline acetyltransferase activity in primary visual cortex in mild to moderate Alzheimer's disease. Arch Neurol 62:425–430. [DOI] [PubMed] [Google Scholar]
- 63. Imhof A, Kovari E, Von Gunten A, Gold G, Rivara CB, Herrmann FR et al (2007) Morphological substrates of cognitive decline in nonagenarians and centenarians: a new paradigm? J Neurol Sci 257:72–79. [DOI] [PubMed] [Google Scholar]
- 64. Iraizoz I, Guijarro JL, Gonzalo LM, De Lacalle S (1999) Neuropathological changes in the nucleus basalis correlate with clinical measures of dementia. Acta Neuropathol 98:186–196. [DOI] [PubMed] [Google Scholar]
- 65. Jellinger KA (2000) Clinical validity of Braak staging in the oldest‐old. Acta Neuropathol (Berl) 99:583–584. [DOI] [PubMed] [Google Scholar]
- 66. Jicha GA, Parisi JE, Dickson DW, Johnson K, Cha R, Ivnik RJ et al (2006) Neuropathologic outcome of mild cognitive impairment following progression to clinical dementia. Arch Neurol 63:674–681. [DOI] [PubMed] [Google Scholar]
- 67. Josephs KA, Whitwell JL, Knopman DS, Hu WT, Stroh DA, Baker M et al (2008) Abnormal TDP‐43 immunoreactivity in AD modifies clinicopathologic and radiologic phenotype. Neurology 70:1850–1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Josephs KA, Whitwell JL, Parisi JE, Knopman DS, Boeve BF, Geda YE et al (2008) Argyrophilic grains: a distinct disease or an additive pathology? Neurobiol Aging 29:566–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Kahle‐Wrobleski K, Corrada MM, Li B, Kawas CH (2007) Sensitivity and specificity of the mini‐mental state examination for identifying dementia in the oldest‐old: the 90+ study. J Am Geriatrics Soc 55:284–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Knopman DS, Parisi JE, Salviati A, Floriach‐Robert M, Boeve BF, Ivnik RJ et al (2003) Neuropathology of cognitively normal elderly. J Neuropathol Exp Neurol 62:1087–1095. [DOI] [PubMed] [Google Scholar]
- 71. Koepsell TD, Kurland BF, Harel O, Johnson EA, Zhou XH, Kukull WA (2008) Education, cognitive function, and severity of neuropathology in Alzheimer disease. Neurology 70:1732–1739. [DOI] [PubMed] [Google Scholar]
- 72. Kosaka K, Iseki E (1998) Recent advances in dementia research in Japan: non‐Alzheimer‐type degenerative dementias. Psychiatry Clin Neurosci 52:367–373. [DOI] [PubMed] [Google Scholar]
- 73. Kraybill ML, Larson EB, Tsuang DW, Teri L, McCormick WC, Bowen JD et al (2005) Cognitive differences in dementia patients with autopsy‐verified AD, Lewy body pathology, or both. Neurology 64:2069–2073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Kryscio RJ, Schmitt FA, Salazar JC, Mendiondo MS, Markesbery WR (2006) Risk factors for transitions from normal to mild cognitive impairment and dementia. Neurology 66:828–832. [DOI] [PubMed] [Google Scholar]
- 75. Kukull WA, Larson EB, Teri L, Bowen J, McCormick W, Pfanschmidt ML (1994) The mini‐mental state examination score and the clinical diagnosis of dementia. J Clin Epidemiol 47:1061–1067. [DOI] [PubMed] [Google Scholar]
- 76. Lee HG, Castellani RJ, Zhu X, Perry G, Smith MA (2005) Amyloid‐beta in Alzheimer's disease: the horse or the cart? Pathogenic or protective? Int J Exp Path 86:133–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Mackenzie IR, Rademakers R (2007) The molecular genetics and neuropathology of frontotemporal lobar degeneration: recent developments. Neurogenetics 8:237–248. [DOI] [PubMed] [Google Scholar]
- 78. Markesbery WR, Schmitt FA, Kryscio RJ, Davis DG, Smith CD, Wekstein DR (2006) Neuropathologic substrate of mild cognitive impairment. Arch Neurol 63:38–46. [DOI] [PubMed] [Google Scholar]
- 79. Masliah E (1995) Mechanisms of synaptic dysfunction in Alzheimer's disease. Histol Histopathol 10:509–519. [PubMed] [Google Scholar]
- 80. Masliah E, Terry RD, DeTeresa RM, Hansen LA (1989) Immunohistochemical quantification of the synapse‐related protein synaptophysin in Alzheimer disease. Neurosci Lett 103:234–239. [DOI] [PubMed] [Google Scholar]
- 81. McCarten JR, Rottunda SJ, Kuskowski MA (2004) Change in the mini‐mental state exam in Alzheimer's disease over 2 years: the experience of a dementia clinic. J Alzheimers Dis 6:11–15. [DOI] [PubMed] [Google Scholar]
- 82. Mendiondo MS, Ashford JW, Kryscio RJ, Schmitt FA (2000) Modelling mini mental state examination changes in Alzheimer's disease. Stat Med 19:1607–1616. [DOI] [PubMed] [Google Scholar]
- 83. Miller LA, Munoz DG, Finmore M (1993) Hippocampal sclerosis and human memory. Arch Neurol 50:391–394. [DOI] [PubMed] [Google Scholar]
- 84. Mungas D, Reed BR, Ellis WG, Jagust WJ (2001) The effects of age on rate of progression of Alzheimer disease and dementia with associated cerebrovascular disease. Arch Neurol 58:1243– 1247. [DOI] [PubMed] [Google Scholar]
- 85. Nagy Z, Esiri MM, Jobst KA, Morris JH, King EM, McDonald B et al (1995) Relative roles of plaques and tangles in the dementia of Alzheimer's disease: correlations using three sets of neuropathological criteria. Dementia (Basel, Switzerland) 6: 21–31. [DOI] [PubMed] [Google Scholar]
- 86. Nagy Z, Esiri MM, Jobst KA, Morris JH, King EM, McDonald B et al (1997) The effects of additional pathology on the cognitive deficit in Alzheimer disease. J Neuropathol Exp Neurol 56: 165–170. [DOI] [PubMed] [Google Scholar]
- 87. Nelson PT, Jicha GA, Schmitt FA, Liu H, Davis DG, Mendiondo MS et al (2007) Clinicopathologic correlations in a large Alzheimer disease center autopsy cohort: neuritic plaques and neurofibrillary tangles “do count” when staging disease severity. J Neuropathol Exp Neurol 66:1136–1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT et al (2006) Ubiquitinated TDP‐43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science (New York, N.Y.) 314:130–133. [DOI] [PubMed] [Google Scholar]
- 89. Neuropathology Group of the Medical Research Council Cognitive Function and Ageing Study (MRC CFAS) (2001) Pathological correlates of late‐onset dementia in a multicentre, community‐based population in England and Wales. Neuropathology Group of the Medical Res Council Cognitive Function and Ageing Study (MRC CFAS). Lancet 357:169–175. [DOI] [PubMed] [Google Scholar]
- 90. Patterson C, Feightner JW, Garcia A, Hsiung GY, MacKnight C, Sadovnick AD (2008) Diagnosis and treatment of dementia: 1. Risk assessment and primary prevention of Alzheimer disease. CMAJ 178:548–556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Petersen RC, Parisi JE, Dickson DW, Johnson KA, Knopman DS, Boeve BF et al (2006) Neuropathologic features of amnestic mild cognitive impairment. Arch Neurol 63:665–672. [DOI] [PubMed] [Google Scholar]
- 92. Petrovitch H, Ross GW, He Q, Uyehara‐Lock J, Markesbery W, Davis D et al (2008) Characterization of Japanese‐American men with a single neocortical AD lesion type. Neurobiol Aging 29:1448–1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Probst A, Taylor KI, Tolnay M (2007) Hippocampal sclerosis dementia: a reappraisal. Acta Neuropathol 114:335–345. [DOI] [PubMed] [Google Scholar]
- 94. Proust‐Lima C, Letenneur L, Jacqmin‐Gadda H (2007) A nonlinear latent class model for joint analysis of multivariate longitudinal data and a binary outcome. Stat Med 26:2229–2245. [DOI] [PubMed] [Google Scholar]
- 95. Rascovsky K, Salmon DP, Lipton AM, Leverenz JB, DeCarli C, Jagust WJ et al (2005) Rate of progression differs in frontotemporal dementia and Alzheimer disease. Neurology 65:397–403. [DOI] [PubMed] [Google Scholar]
- 96. Riley KP, Snowdon DA, Desrosiers MF, Markesbery WR (2005) Early life linguistic ability, late life cognitive function, and neuropathology: findings from the Nun study. Neurobiol Aging 26:341–347. [DOI] [PubMed] [Google Scholar]
- 97. Royter V, Bornstein NM, Russell D (2005) Coronary artery bypass grafting (CABG) and cognitive decline: a review. J Neurol Sci 229–230:65–67. [DOI] [PubMed] [Google Scholar]
- 98. Sabbagh MN, Corey‐Bloom J, Tiraboschi P, Thomas R, Masliah E, Thal LJ (1999) Neurochemical markers do not correlate with cognitive decline in the Lewy body variant of Alzheimer disease. Arch Neurol 56:1458–1461. [DOI] [PubMed] [Google Scholar]
- 99. Saito Y, Yamazaki M, Kanazawa I, Murayama S (2002) Severe involvement of the ambient gyrus in a case of dementia with argyrophilic grain disease. J Neurol Sci 196:71–75. [DOI] [PubMed] [Google Scholar]
- 100. Saito Y, Ruberu NN, Sawabe M, Arai T, Tanaka N, Kakuta Y et al (2004) Staging of argyrophilic grains: an age‐associated tauopathy. J Neuropathol Exp Neurol 63:911–918. [DOI] [PubMed] [Google Scholar]
- 101. Schmitt FA, Wetherby MM, Wekstein DR, Dearth CM, Markesbery WR (2001) Brain donation in normal aging: procedures, motivations, and donor characteristics from the Biologically Resilient Adults in Neurological Studies (BRAiNS) project. Gerontologist 41:716–722. [DOI] [PubMed] [Google Scholar]
- 102. Schmitt FA, Cragar D, Ashford JW, Reisberg B, Ferris S, Mobius HJ, Stoffler A (2002) Measuring cognition in advanced Alzheimer's disease for clinical trials. J Neural Transm (Suppl. 62):135–148. [DOI] [PubMed] [Google Scholar]
- 103. Schnaider Beeri M, Silverman JM, Schmeidler J, Wysocki M, Grossman HZ, Purohit DP et al (2008) Clinical dementia rating performed several years prior to death predicts regional Alzheimer's neuropathology. Dement Geriatr Cogn Disord 25:392–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Schneider JA, Boyle PA, Arvanitakis Z, Bienias JL, Bennett DA (2007) Subcortical infarcts, Alzheimer's disease pathology, and memory function in older persons. Ann Neurol 62:59–66. [DOI] [PubMed] [Google Scholar]
- 105. Seelaar H, Schelhaas HJ, Azmani A, Kusters B, Rosso S, Majoor‐Krakauer D et al (2007) TDP‐43 pathology in familial frontotemporal dementia and motor neuron disease without Progranulin mutations. Brain 130:1375–1385. [DOI] [PubMed] [Google Scholar]
- 106. Sheng B, Cheng LF, Law CB, Li HL, Yeung KM, Lau KK (2007) Coexisting cerebral infarction in Alzheimer's disease is associated with fast dementia progression: applying the national Institute for Neurological Disorders and Stroke/Association Internationale pour la Recherche et l'Enseignement en Neurosciences Neuroimaging criteria in Alzheimer's disease with concomitant cerebral infarction. J Am Geriatrics Soc 55:918–922. [DOI] [PubMed] [Google Scholar]
- 107. Silver MH, Newell K, Brady C, Hedley‐White ET, Perls TT (2002) Distinguishing between neurodegenerative disease and disease‐free aging: correlating neuropsychological evaluations and neuropathological studies in centenarians. Psychosom Med 64:493–501. [DOI] [PubMed] [Google Scholar]
- 108. Snowdon DA, Kemper SJ, Mortimer JA, Greiner LH, Wekstein DR, Markesbery WR (1996) Linguistic ability in early life and cognitive function and Alzheimer's disease in late life. Findings from the Nun study. JAMA 275:528–532. [PubMed] [Google Scholar]
- 109. Sonnen JA, Larson EB, Crane PK, Haneuse S, Li G, Schellenberg GD et al (2007) Pathological correlates of dementia in a longitudinal, population‐based sample of aging. Ann Neurol 62:406–413. [DOI] [PubMed] [Google Scholar]
- 110. Spina S, Murrell JR, Huey ED, Wassermann EM, Pietrini P, Grafman J, Ghetti B (2007) Corticobasal syndrome associated with the A9D progranulin mutation. J Neuropathol Exp Neurol 66:892–900. [DOI] [PubMed] [Google Scholar]
- 111. Suh GH, Ju YS, Yeon BK, Shah A (2004) A longitudinal study of Alzheimer's disease: rates of cognitive and functional decline. Int J Geriatr Psychiatry 19:817–824. [DOI] [PubMed] [Google Scholar]
- 112. Sze CI, Troncoso JC, Kawas C, Mouton P, Price DL, Martin LJ (1997) Loss of the presynaptic vesicle protein synaptophysin in hippocampus correlates with cognitive decline in Alzheimer disease. J Neuropathol Exp Neurol 56:933–944. [DOI] [PubMed] [Google Scholar]
- 113. Teipel SJ, Mitchell AJ, Moller HJ, Hampel H (2007) Improving linear modeling of cognitive decline in patients with mild cognitive impairment: comparison of two methods. J Neural Transm (Suppl. 72):241–247. [DOI] [PubMed] [Google Scholar]
- 114. Tiraboschi P, Hansen LA, Alford M, Merdes A, Masliah E, Thal LJ, Corey‐Bloom J (2002) Early and widespread cholinergic losses differentiate dementia with Lewy bodies from Alzheimer disease. Arch Gen Psychiatry 59:946–951. [DOI] [PubMed] [Google Scholar]
- 115. Tiraboschi P, Hansen LA, Thal LJ, Corey‐Bloom J (2004) The importance of neuritic plaques and tangles to the development and evolution of AD. Neurology 62:1984– 1989. [DOI] [PubMed] [Google Scholar]
- 116. Togo T, Sahara N, Yen SH, Cookson N, Ishizawa T, Hutton M et al (2002) Argyrophilic grain disease is a sporadic 4‐repeat tauopathy. J Neuropathol Exp Neurol 61:547–556. [DOI] [PubMed] [Google Scholar]
- 117. Tolnay M, Clavaguera F (2004) Argyrophilic grain disease: a late‐onset dementia with distinctive features among tauopathies. Neuropathology 24:269–283. [DOI] [PubMed] [Google Scholar]
- 118. Tombaugh TN, McIntyre NJ (1992) The mini‐mental state examination: a comprehensive review. J Am Geriatrics Soc 40:922–935. [DOI] [PubMed] [Google Scholar]
- 119. Tomlinson BE, Blessed G, Roth M (1970) Observations on the brains of demented old people. J Neurol Sci 11:205–242. [DOI] [PubMed] [Google Scholar]
- 120. Tsuang DW, Riekse RG, Purganan KM, David AC, Montine TJ, Schellenberg GD et al (2006) Lewy body pathology in late‐onset familial Alzheimer's disease: a clinicopathological case series. J Alzheimers Dis 9:235–242. [DOI] [PubMed] [Google Scholar]
- 121. Uchikado H, Lin WL, DeLucia MW, Dickson DW (2006) Alzheimer disease with amygdala Lewy bodies: a distinct form of alpha‐synucleinopathy. J Neuropathol Exp Neurol 65: 685–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Wang DS, Bennett DA, Mufson E, Cochran E, Dickson DW (2004) Decreases in soluble alpha‐synuclein in frontal cortex correlate with cognitive decline in the elderly. Neurosci Lett 359:104–108. [DOI] [PubMed] [Google Scholar]
- 123. White L, Petrovitch H, Hardman J, Nelson J, Davis DG, Ross GW et al (2002) Cerebrovascular pathology and dementia in autopsied Honolulu‐Asia Aging Study participants. Ann NY Acad Sci 977:9–23. [DOI] [PubMed] [Google Scholar]
- 124. White L, Small BJ, Petrovitch H, Ross GW, Masaki K, Abbott RD et al (2005) Recent clinical‐pathologic research on the causes of dementia in late life: update from the Honolulu‐Asia aging study. J Geriatr Psychiatry Neurol 18:224–227. [DOI] [PubMed] [Google Scholar]
- 125. Xuereb JH, Brayne C, Dufouil C, Gertz H, Wischik C, Harrington C et al (2000) Neuropathological findings in the very old. Results from the first 101 brains of a population‐based longitudinal study of dementing disorders. Ann NY Acad Sci 903:490–496. [DOI] [PubMed] [Google Scholar]
- 126. Yoshida M (2006) Cellular tau pathology and immunohistochemical study of tau isoforms in sporadic tauopathies. Neuropathology 26:457–470. [DOI] [PubMed] [Google Scholar]
- 127. Zhang H, Tan CF, Mori F, Tanji K, Kakita A, Takahashi H, Wakabayashi K (2008) TDP‐43‐immunoreactive neuronal and glial inclusions in the neostriatum in amyotrophic lateral sclerosis with and without dementia. Acta Neuropathol 115:115–122. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Operationalization of clinical and pathological parameters for Nelson PT et al “Modeling the association . . .”
Table S2. Model checking and model selection
Please note: Wiley‐Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
Supporting info item
Supporting info item