

Abstract
The ester bond of peptidyl-tRNA undergoes nucleophilic attack both in solution and catalyzed by the ribosome. To characterize the uncatalyzed hydrolysis reaction - a model of peptide release - the transition state structure for hydrolysis of a peptidyl-tRNA mimic was determined. Kinetic isotope effects were measured at several atoms that potentially undergo a change in bonding at the transition state. Large kinetic isotope effects of carbonyl oxygen-18 and α-deuterium substitutions on uncatalyzed hydrolysis indicate the transition state is nearly tetrahedral. Kinetic isotope effects were also measured for aminolysis by hydroxylamine to study a reaction similar to peptide bond formation. In contrast to hydrolysis, the large leaving group oxygen-18 isotope effect indicates the C-O3′ bond has undergone significant scission in the transition state. The smaller carbonyl oxygen-18 and α-deuterium effects are consistent with a later transition state. The assay developed here can also be used to measure isotope effects for the ribosome-catalyzed reactions. These uncatalyzed reactions serve as a basis to determine what aspects of the transition states are stabilized by the ribosome to achieve a rate enhancement.
The ribosome catalyzes two chemical reactions during protein synthesis. During initiation and elongation, peptide bonds are formed by the nucleophilic attack of an aminoacyl-tRNA in the A-site on peptidyl-tRNA in the P-site. During termination, water acts as the nucleophile resulting in the release of the peptide chain from tRNA. The ribosome must be capable of selectively switching between these two catalytic activities, since hydrolysis during elongation would produce truncated proteins, and peptide bond formation in place of termination would result in read-through of stop codons.
In the last decade, a wealth of structural information has been obtained to complement biochemical study of catalysis by the ribosome. Structures of the 50S ribosome demonstrated that the active site for peptide bond formation is composed entirely of RNA(1, 2). Of the functional groups present in the active site, two have been identified as important for catalysis of peptide bond formation: the 2′-hydroxyl of A2451 in 50S RNA contributes about 10-fold(3), and the 2′-hydroxyl of the terminal adenosine of peptidyl-tRNA, which is adjacent to the leaving group, contributes at least 106-fold(4). Brönsted(5) and kinetic isotope effect studies(6) indicate that the nucleophilic nitrogen is neutral in the transition state despite at least partial peptide bond formation. More recently, structures of 70S ribosomes bound to either release factor 1 (7) or release factor 2 (8, 9) have shown the location of the conserved GGQ motif in the active site for peptide release. Mutational studies showed that both glycines are essential, while the side chain of glutamine is only moderately important(10–13).
In both peptide bond formation and hydrolysis, the electrophile is a peptidyl-tRNA bound in the P-site of the ribosome. The growing peptide chain is linked to tRNA as a carboxylic acid ester through the 3′-hydroxyl of the terminal adenosine. The reactivity of similar carboxylic acid derivatives has been studied extensively. Isotope effects(14) and pH dependence studies(15) have been used to probe proton transfer. Structure-reactivity and linear free energy relationships such as Brönsted studies have also been performed(16–18). Oxygen exchange experiments using isotopic labels have been invaluable in establishing the existence of a tetrahedral intermediate and identifying the rate-limiting step(19).
Detailed knowledge of uncatalyzed reactions serves as a basis for understanding enzymatic catalysis. The structure of the transition state is particularly informative, since enzymes increase the reaction rate by lowering the energy of this state. Because transition states cannot be viewed directly, measuring kinetic isotope effects is a valuable strategy to determine transition state structures(20, 21). The rate of a chemical reaction may change upon substitution of an atom at the reaction center with a heavier isotope. This effect is dependent on the change in bonding to that atom from the ground state to the transition state. Therefore, given a ground state structure and kinetic isotope effects for several substitutions, the structure of the transition state can be determined.
We have previously reported the synthesis of a set of molecules needed for kinetic isotope effect analysis of the reactions of a ribosomal P-site substrate(22, 23). In this study, we have determined the transition states for the uncatalyzed hydrolysis and aminolysis of this substrate. These structures indicate possible roles for the ribosome in catalyzing the two reactions of protein synthesis.
Experimental Procedures
Reactant preparation
Reactants differing only by single isotopic substitutions at the reaction center were prepared for isotope effect studies (Figure 1). The P-site mimic cytidylyl-(3′5′)-cytidylyl-(3′5′)-3′(2′)-O-(N-(6-D-(+)-biotinoylaminohexanoyl)-L-phenylalanyl)adenosine (CCApcb) was synthesized as described(22, 23) and stored at −80 °C until use. It was resuspended in 50 mM TEAA pH 6.5 and purified from hydrolyzed material (CCA) by HPLC on a C-18 column using a gradient of 0–40% acetonitrile. The HPLC peak was lyophilized, resuspended in 2 mM MES pH 5.5 and 5′-end labeled with either γ32P- or γ33P-ATP by polynucleotide kinase. Labeling reactions were done for five minutes at room temperature and pH 6.5 to reduce hydrolysis. Labeled CCApcb was purified on a pH 6.0 denaturing 15% polyacrylamide gel and gel slices eluted into 2 mM MES pH 5.5. 32P- and 33P-CCApcb were mixed at an approximately 1:4 ratio and purified on a pH 6.0 non-denaturing 12% polyacrylamide gel. Gel slices were again cut out and eluted into 2 mM MES pH 5.5. Purified mixes were used as soon as possible to minimize hydrolysis in the starting material.
Figure 1.
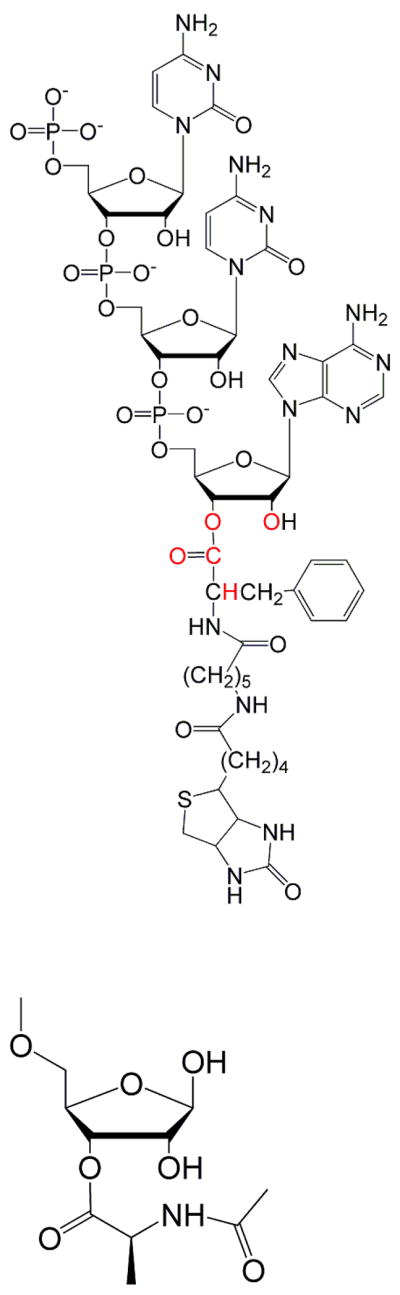
A) Structure of the ribosomal P-site substrate CCApcb with 5′-phosphate (either 32P or 33P). Positions of the individual isotopic substitutions are highlighted in red. B) Simplified model used for calculating isotope effects.
The isotopic enrichment of each sample was determined by high-resolution Fourier-transform – ion cyclotron resonance mass spectrometry. The molecule was resuspended in 1:1 water:acetonitrile and analyzed in negative-ion mode. The average value of peaks corresponding to both the -1 and -2 charge states was used. Each molecule had 95% or greater isotopic purity except for the carbonyl oxygen-18 substitution, which was 74%.
The equilibrium ratio of 2′- to 3′-linked ester was determined by 1H-NMR. CCApcb was suspended in 5 mM Tris pH 8.5, lyophilized, and resuspended in 99.990% D2O at approximately 500 mM. A 1D spectrum was collected immediately. The sample was then allowed to hydrolyze over several days, and another spectrum was collected to assist in peak identification. Two peaks in the adenosine H1′ region were present in the unhydrolyzed but not the hydrolyzed sample. Based on previous studies and the chemical shift rule the downfield peak was assigned to be the 2′-linked isomer(24–26) and the ratio of the two peaks was used to determine the isomeric ratio.
Identity of Nucleophile in Hydroxylamine Reaction
The reaction of CCApcb with hydroxylamine was observed by 13C NMR. CCApcb labeled at the carbonyl carbon was suspended in 10% D2O to approximately 2 mM. Hydroxylamine was diluted to a 5 M stock solution and brought to pH 8.5 with HCl. Hydroxylamine was added to a final concentration of 250 mM and the reaction was quenched with 5% formic acid at approximately 1 minute. Alternatively, 500 mM hydroxylamine or 50 mM hepes (pH 8.5) was added and these reactions were quenched with 5% formic acid after 1 day. The 13C spectrum of each sample as well as starting material (again with 5% formic acid) was collected for 12 hours.
Samples of CCApcb before and after reaction with hydroxylamine were subjected to a ferric chloride test for hydroxamic acid. To approximately 2 mM CCApcb, 1/10 volume of either 5 M hydroxylamine (pH 8.5) or water was added. HCl and FeCl3 were added to each sample to 100 mM and 0.05%, respectively and the absorbance spectra were collected with a NanoDrop 2000c spectrophotometer.
Hydrolysis Isotope Effect Reactions
Reactions were started by adding 1/10 volume of 10x ribosome reaction buffer (final concentrations 25 mM HEPES, 10 mM MES, 10 mM MOPS, 200 mM NH4Cl, 7 mM MgCl2, pH 8.5) at 25 °C. Most of the reaction was quenched at approximately 20% reacted (approximately 2 hours) by adding three volumes of low pH formamide loading buffer (FLB) and freezing at −20 °C. The remainder was allowed to react for at least 36 hours to reach completion. Both timepoints were run on a pH 6.0 denaturing 15% polyacrylamide gel to separate substrate from product.
Substrate and product bands were visualized using a Storm 840 PhosphorImager with a two-ply sheet of duck tape between the gel and screen to block 33P emission. The fraction of 32P-labeled substrate reacted could then be determined. The ratio of 32P to 33P was determined by scintillation counting. Each product band was excised from the gel and eluted into 1 mL of water overnight. This water was then added to 13 mL of Optima Gold scintillation fluid and counted for thirty minutes along with 32P and 33P standards.
Aminolysis Isotope Effect Reactions
Reactions with hydroxylamine were carried out as described for the hydrolysis reaction with the following exceptions. Reactions were initiated by adding 1/10 volume of 5 M hydroxylamine (pH 8.5). Timepoints were quenched after approximately 5 minutes by adding 2.5 volumes of FLB and immediately loading onto a pre-running gel. The remainder of the reaction was allowed to react for 1 to 2 hours.
Data Analysis
Counts per minute were divided into two channels, 0–400 keV and 400–2000 keV. Approximately 70% of the 32P standard was detected in the high energy channel, and greater than 99% of the 33P sample was detected in the low energy channel. The ratio of 32P to 33P in each sample could be determined using equation 1:
| (1) |
where A is the counts per minute in the low energy channel, B is the counts per minute in the high energy channel, and r is the fraction of total emission of a 32P standard detected in the low energy channel. The observed isotope effect was then determined from the ratio in the midpoint and endpoint samples and the fraction reacted using equation 2:
| (2) |
where f is the fraction reacted, Rp is the isotope ratio in the product at that fraction reacted, and R0 is the isotope ratio in the product at the reaction endpoint. For high fraction reacted samples (greater than 50%) the isotope effect was also determined from substrate and endpoint samples using equation 3:
| (3) |
where Rs is the isotope ratio in the remaining substrate at f fraction reacted, and R0 is as above. This value was corrected for incomplete isotopic incorporation as determined by mass spectrometry with equation 4:
| (4) |
where e is the isotopic enrichment of the heavy sample. This equation assumes a negligible amount of heavy isotope in the light sample(27).
Since kinetic isotope effects are ratios of rates, the geometric mean and standard deviation are the correct statistics to describe the data. However, the arithmetic and geometric means and standard errors were equivalent for these sets of data, and for clarity the arithmetic versions are presented. The means and standard errors were similar whether each measurement was treated as independent, or whether multiple trials with the same reactant mix at the same time were averaged and treated as a single measurement. The total number of trials is listed with each value.
Hydrolysis Kinetics
The rate effect of the 2′-deoxyadenosine substitution in CCApcb was determined in side-by-side reactions. Reactions were started by adding 1/10 volume of 10x ribosome reaction buffer. The reaction was quenched by adding four volumes of FLB.
The overall solvent isotope effect was determined by replacing H2O in the reaction with D2O. Since the radiolabeled CCApcb and 10x reaction buffer were kept in H2O, the reaction contained 85% D2O.
In all cases, plots of fraction reacted versus time fit to a single exponential (equation 5):
| (5) |
Where f is the fraction reacted at time t, f∞ is the fraction reacted extrapolated to infinite time, f0 is the fraction reacted at zero time, and kobs is the observed rate of hydrolysis. For the 2′-deoxy reactant, less than 20% reacted within approximately 3 days. Therefore the fraction reacted at infinite time was fixed to the same value as for the 2′-ribo reactant.
Computation of Transition States
Transition state structures for the hydrolysis and aminolysis reactions that reproduced the experimental KIEs were determined in vacuo using hybrid density functional methods implemented in Gaussian03(28) using the model tetrahydro-4,5-dihydroxy-2-(methoxymethyl)furan-3-yl 2-acetamidopropanoate. Structures of the transition states were optimized and the frequencies were computed on the optimized structures using the three-parameter Becke (B3) exchange functional, the LYP correlation functional and the standard 6-31G (d,p) basis set. The 5′-methoxy group and the reaction center were constrained during the optimization and many of these constraints were modified to match the experimental KIEs.
Kinetic and equilibrium isotope effects (KIEs and EIEs, respectively) were calculated from the computed frequencies using ISOEFF 98(29). KIEs and EIEs were calculated at the temperature of 298 K and the frequencies were scaled using a factor of 0.964, corresponding to B3LYP/6-31G(d, p). All vibrational modes were used to calculate isotope effects. Geometric and the electrostatic models were generated by iteratively optimizing the transition states by modifying the applied constraints until the computed isotope effects closely match the experimental KIEs. Isoeff98 uses an imaginary frequency of 50i cm−1 as a cutoff for calculating KIEs vs EIEs. KIEs were calculated when the magnitude of the imaginary frequency which contained some contribution from the reaction coordinate was greater than 50i cm−1, indicating partial bond orders. Conversely when significant or full bond orders were observed at the transition state either a small (imaginary frequency less then 50i cm−1) or no imaginary frequency was observed corresponding to the reaction coordinate. In this case the transition state was treated as an intermediate and equilibrium isotope effects were calculated. Experimental KIEs predicted full bond orders for the hydrolysis reaction with the transition state resembling the tetrahedral intermediate. The hydrolysis reaction was therefore modeled as an intermediate and EIEs were matched to the experimental KIEs. The aminolysis reaction had experimental KIEs that suggested partial bond orders; therefore it was modeled as a transition state and KIEs were calculated. The final model that is consistent with the experimental KIEs had multiple imaginary frequencies. All frequencies were used for calculating KIEs.
The natural bond orbital (NBO) calculations were performed on optimized structures by including the pop = nbo keyword in the route section of input files. The molecular electrostatic potential (MEP) surfaces were calculated by the CUBE subprogram of Gaussian03. The formatted checkpoint files used in the CUBE subprogram were generated by constrained geometry optimization at the B3LYP level of theory with the 6-31G** basis set. MEP surfaces of the substrate and the transition states were visualized using Molekel4.0 at a density of 0.008 electrons/bohr3.
Results
Measurement of Isotope Effects by Remote Label Method
Kinetic isotope effects can be used to determine the structure of the transition state of a chemical reaction (reviewed in (20, 21)). To carry out these studies, reactants must be prepared that differ only in the isotope at a particular position of interest. The rate effect for a single isotopic substitution is related to the change in bond order of that atom between the ground state and the transition state. If several substitutions are made individually, the change in bond order of each atom can be determined and a complete map of the transition state can be constructed. A synthetic strategy amenable to isotopic substitution was developed for a compound that mimics the peptidyl-tRNA that is bound to the ribosome during protein synthesis(22, 23). To better understand its reactivity to nucleophilic attack, kinetic isotope effects were determined for the uncatalyzed hydrolysis and hydroxylaminolysis of this substrate. These reactions mimic peptide release and peptide elongation reactions catalyzed by the ribosome, respectively.
Because heavy atom kinetic isotope effects are generally very small (usually a few percent or less) it was impractical to determine them by independently measuring the reaction rates of the substituted and unsubstituted compounds. Instead, a competitive reaction was employed, where the two compounds were mixed and the isotope effect was determined by the relative enrichment of one compound in either the substrate or product. The compound with the faster-reacting isotope was enriched in the product and depleted in the substrate, and the opposite was true for the slower-reacting isotope.
Since the two compounds were present in the same reaction, a remote radiolabel was used to differentiate them. The substituted and unsubstituted molecules were labeled at the 5′-end with a phosphate containing either 32P or 33P. Since the emission energies of the two radiolabels differ by approximately seven-fold, they can be distinguished by scintillation counting. An added benefit of this strategy was the flexibility of labeling either compound with either radioisotope. Therefore the isotope effect was measured by pairing the light isotope with 32P and the heavy isotope with 33P, and again by pairing the heavy isotope with 32P and the light isotope with 33P. This radiolabel pair was previously used to determine a kinetic isotope effect on a 4-thiouridine synthase ribozyme(30). The ease of labeling with polynucleotide kinase and the availability of two radioisomers of phosphorus make this radiolabel pair particularly useful for the determination of kinetic isotope effects for enzymes with oligonucleotide substrates.
To evaluate the precision and reproducibility of the assay, the primary isotope effect (13C substitution at the carbonyl carbon) was determined thirty-three times for the aminolysis reaction. A histogram of the data is consistent with a log-normal distribution as expected for repeated measurements of ratios (Figure 2). Furthermore, repeated trials on different days gave the same effect within error. Measurements at different fractions reacted, as well as using either substrate or product to determine the isotope effect, all gave the same effect. Finally, the isotope effects due to the purification and counting procedure or for the remote label were also within the error of the experiment.
Figure 2.

Histogram of 13C kinetic isotope effects on aminolysis. The dataset fits to either a log-normal (shown) or normal distribution.
Reaction Pathway of Hydrolysis of CCApcb
A simple mechanism for hydrolysis involving only the formation and breakdown of a tetrahedral intermediate was used as a framework for interpreting isotope effects (Figure 3). The observed isotope effect is dependent on the individual effects on k1, k−1, and k2, as well as the partitioning of the intermediate (k2/k−1) (equation 6)(31)):
| (6) |
where KIEn is the isotope effect on step n. If the intermediate partitions exclusively forward, then the observed isotope effect is equal to the isotope effect on k1. If the intermediate usually partitions back to reactants, the observed effect is equal to the equilibrium effect (KIE1/KIE−1) multiplied by the kinetic effect on breakdown (KIE2). While it is possible that the reaction mechanism is more complicated (especially for aminolysis, below) kinetic isotope effects only report on the highest-energy transition states. Therefore, information on multiple steps can only be obtained if they are at least partially rate-limiting.(32)
Figure 3.

Possible reaction mechanisms of CCApcb. A) Hydrolysis. Attack by hydroxide ion or base-catalyzed attack of water results in the formation of a tetrahedral intermediate. Breaking the ester C-O bond resolves the intermediate into products. In some cases the intermediate resolves back into reactants. B) Aminolysis. Amines can attack as neutral species, and the zwitterionic state may be a stable intermediate as shown. Alternatively, one or more steps may be concerted. Because NRH2 is generally a better leaving group than RO−, the intermediates will often partition back to substrates.
Kinetic Isotope Effects on Hydrolysis of CCApcb
Kinetic isotope effects on hydrolysis were determined at pH 8.5 for substitution at the carbonyl carbon and oxygen, the 2′- and 3′-oxygens, and the hydrogen of the amino acid α–carbon (Figure 4a). The measured kinetic isotope effect for the carbonyl oxygen-18 substitution is 4.0%. This effect reports on the tetrahedral character of the transition state; the carbonyl bond changes from near double bond order to near single bond order in a tetrahedral intermediate. To our knowledge, this is the largest carbonyl oxygen-18 effect reported for a reaction of a carboxylic acid ester. For the hydrolysis of methyl formate, methyl benzoate, and p-nitrophenylacetate, carbonyl oxygen-18 isotope effects near zero have been observed(31–33). However, effects of 1.8 and 2.4% have been reported for the hydrazinolysis of methyl benzoate(31) and methanolysis of phenyl benzoate(34), respectively. Isotope effects at other positions indicated those transition states were relatively late. Therefore, the isotope effects would be expected to be larger for a nearly tetrahedral transition state. Calculations for a model substrate indicate the isotope effect reaches a maximum of approximately 5% for the tetrahedral intermediate. This strongly suggests that the transition state for CCApcb hydrolysis is highly tetrahedral.
Figure 4.

Measured isotope effects. Shown are the means and standard deviations, with the number of trials in parentheses. A) Hydrolysis by water. B) Aminolysis by hydroxylamine.
The observed effect for deuterium substitution at the amino acid α-carbon is 4.1% inverse (the heavy isotope reacts faster). This effect is also largely dependent on the tetrahedral character of the transition state. The effect is derived from the loss of hyperconjugation between the π electrons of the carbonyl double bond and the σ* antibonding orbital of the C-H bond. Loss of the π electrons in a tetrahedral-like transition state increases the strength of the C-H bond and leads to an inverse isotope effect. Previous calculations for addition to an aldehyde estimated the most inverse isotope effect is approximately 4% for an extremely tetrahedral transition state (corrected to one deuterium substitution)(35). The calculations performed here yield a similar result, with the most inverse value of 4.9%. The comparatively large inverse isotope effect measured here indicates a significant tetrahedral character to the transition state in agreement with the carbonyl oxygen-18 effect.
Interpretation of the 2′- and 3′-oxygen-18 effects is complicated by isomerization of the ester between 2′- and 3′-linkages. By NMR, the ratio of 2′- to 3′-linked ester was determined to be approximately 3:7, in agreement with previous measurements for similar compounds(26, 36). Therefore, the 3′-oxygen-18 effect is primarily derived from being the leaving group, while the 2′-oxygen-18 effect is primarily from being vicinal to the leaving group. If the hydrolysis rate of the 2′- and 3′-linked isomers is the same, then the 3′-oxygen-18 isotope effect is a weighted average of the leaving group and neighboring group effects. The leaving- and neighboring-group effects can then be deconvoluted from the 2′- and 3′-effects and the equilibrium ratio determined by NMR.
The 3′-oxygen-18 effect on hydrolysis is 0.6%; this corresponds to a 0.8% leaving group effect. This effect is near the maximum isotope effect expected for the leaving group if formation of the tetrahedral intermediate is rate-limiting(31). There is some double bond character in the ester bond due to resonance with the carbonyl oxygen; the loss of that resonance in a tetrahedral transition state leads to as much as a 1% isotope effect. Effects ranging from 0.1% to 0.9% have been observed in systems where formation of the tetrahedral intermediate is the dominant rate-limiting step(31, 37). The comparatively large value observed indicates the transition state is largely tetrahedral, in agreement with the other KIEs measured. The maximum isotope effect from breakdown of the tetrahedral intermediate has been estimated to be 6–8%(31, 33). Therefore this step must not contribute substantially to the observed isotope effect, or the transition state for this step is early in breakdown, close to the tetrahedral intermediate.
The 2′-oxygen-18 effect is primarily derived from its role as the neighboring hydroxyl group. This hydroxyl group increases the rate of hydrolysis of CCApcb by approximately 30-fold (data not shown), similar to the rate enhancement in similar molecules(15, 38). This effect was not observed for lysine-tRNA, possibly because of the different aminoacyl group(39). The contribution of the 2′-hydroxyl could be due to orientation effects, proton transfer, or stabilization of the transition state through hydrogen bonds(38, 40). The 2′-oxygen-18 effect is 0.4% which corresponds to a 0.3% neighboring group effect.
The primary carbonyl 13C isotope effect is 4.5%. This effect is derived from a number of factors – a decrease in ester C-O bond order, a decrease in carbonyl C=O bond order, and the formation of the C-O bond to water. Even the formation of the C-O bond to water can lead to a normal effect due to reaction coordinate motion. The large normal effect is inconsistent with an early transition state, before there is significant bond order to the nucleophile.
To assess the movement of protons at the transition state, the effect of substituting H2O with a high concentration of D2O was determined. The measured solvent isotope effect (kH2O/kD2O) at 85% D2O was 1.6 ± 0.5. Transition state hydrogen bonds in the reactions of serine proteases, amidohydrolases, and papain range from approximately 1.7 to 4.0(41). Therefore the effect observed here, while small, could be derived from a hydrogen bond that is stronger in the transition state than the ground state. The overall effect was too small to determine the solvent isotope effect as a function of D2O to the accuracy required to determine the number of protons involved.
As a complementary approach, isotope effects were also correlated to the structure of the transition state by starting with a hypothetical structure and predicting isotope effects computationally. The structure was then varied until the calculated isotope effects match the measured ones. The transition state of the hydrolysis reaction was determined using the model shown in Figure 1b. The small 3′-oxygen-18 experimental KIE of 0.6% for the hydrolysis reaction suggests that the O3′-C bond is fully intact at the transition state and the near maximum oxygen-18 experimental KIE of 4.0% for the carbonyl oxygen indicates substantial bond order to the nucleophile. This suggests that transition state of the hydrolysis reaction is close to the tetrahedral intermediate. To create a model which matched the measured isotope effects, the transition state was constrained such that there is full bond order between the primary carbon and nucleophile. Therefore the models of the transition state for the hydrolysis reaction were generated by optimizing structures as intermediates. The transition state models corresponding to tetrahedral or near tetrahedral transition states gave no imaginary frequencies corresponding to the reaction coordinate. The single imaginary frequency that was observed corresponded to movement of atoms at the 5′ end of ribose, well removed from the reaction center. In subsequent optimizations the transition state was modeled as an intermediate and EIEs were calculated. The applied constraints were modified further to obtain EIEs that closely matched the experimental KIEs. The transition state structure obtained for the hydrolysis reaction by this method closely resembled the tetrahedral intermediate with full bond order to the nucleophile and no cleavage of the 3′O-C bond (Figure 5a). The transition state structure has a C-OH bond length of 1.61 Å (Table 1). The primary carbon is sp4.81 hybridized and the carbonyl bond mimics a single bond with a length of 1.39 Å. Concomitant delocalization of the carbonyl π electrons results in a significant increase in negative charge at the carbonyl oxygen. The bond between the primary carbon and the 3′-O is slightly elongated; the O3′-C bond length is 1.43 Å at the TS compared with 1.35 Å in the ground state.
Figure 5.

Transition state structures. Electrostatic and geometric models of each state are shown side-by-side in the same orientation. A single transition state is shown for clarity; the actual reaction pathway is likely to be more complicated. A) Hydrolysis of CCApcb goes through a transition state strongly resembling the tetrahedral intermediate, with an increase in negative charge on the carbonyl oxygen. B) The aminolysis transition state is during breakdown of the tetrahedral intermediate, with partial C-O bond breakage and an increase in negative charge at the carbonyl and 3′-oxygens.
Table 1.
Comparison of experimental and theoretical isotope effects and geometric and electronic changes in representative models of the reactant and the transition state for the hydrolysis reaction calculated using B3LYP/6-31G**
| Isotope Effects | Bond Properties | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Bond Type | Bond Length | Bond order changea Δ(σ-σ*) | RS hybridb | Carbon cont. (%)c | TS hybridb | Carbon cont. (%)c | ||||
| Position | Experimental | Calculated | RS | TS | ||||||
| Carbonyl carbon | 1.045 ± 0.003 | 1.045 | C-OH | - | 1.61 | - | - | - | sp4.81 | 29.5 |
| Carbonyl oxygen | 1.040 ± 0.005 | 1.040 | C-O | 1.21 | 1.39 | −0.896 | sp1.99 | 33.9 | sp2.49 | 36.8 |
| 3′-oxygen | 1.006 ± 0.003 | 1.006 | C-O3′ | 1.35 | 1.43 | −0.036 | sp2.56 | 30.5 | sp3.45 | 29.0 |
| α- hydrogen | 0.959 ± 0.002 | 0.959 | C-H | 1.10 | 1.10 | 0.075 | sp3.10 | 64.3 | sp3.95 | 60.1 |
Calculated by subtracting the number of electrons occupying the σ* orbital from the number occupying the σ orbital and listed as change between the reactant state (RS) and transition state (TS). For the carbonyl bond in the RS electrons in both σ and π bonds were added.
Hybridization of the carbon atom.
Contribution of the carbon atom to the bond in percent.
The validity of this computational approach has been quantitatively examined for the epoxidation of alkenes(42). While calculations of non-stationary states produce errors in frequency determinations, the average deviation between experimental and calculated bond distances was 0.05 Å. It was proposed that the low frequency modes were most likely to be in error, and these modes do not contribute significantly to the isotope effect. In the case of the hydrolysis reaction here, a single imaginary frequency of 74i cm−1 was observed corresponding to the movement of the 5′-methoxy functional group of ribose. This frequency most likely arises from constraining this part of the molecule and does not contribute significantly to the calculated isotope effects (supporting information). The lack of an imaginary frequency corresponding to the reaction coordinate will introduce some error into primary isotope effects (for instance the carbonyl carbon effect determined here). However the structure determined by calculation fits the qualitative aspects identified by comparison with previously studied reactions. Therefore the true transition state is likely to be similar to the structure calculated here.
Kinetic Isotope Effects on Hydroxylaminolysis of CCApcb
The full set of kinetic isotope effects were also determined for the nucleophilic attack of hydroxylamine on CCApcb at pH 8.5 (Figure 4b). The amino group of hydroxylamine is a much better nucleophile than water, and a significant increase in the rate of CCApcb decomposition is indeed observed. It has been reported that the hydroxyl group of hydroxylamine can also act as a nucleophile(43, 44). To assess this possibility, 13C NMR spectra under several reaction conditions were collected (Figure 6a). At long timescales, virtually all CCApcb is converted to a product at 170 ppm, with the remainder being hydrolyzed by water. Even if the hydrolysis product were initially formed, it is expected that over this timescale it would all convert to the aminolysis product, which is thermodynamically preferred. A ferric chloride test was used to confirm that this this is the case (Figure 6b). The aminolysis product is a hydroxamate, which forms a complex with iron upon addition of ferric chloride(45, 46). The characteristic absorbance of this complex in the 450–500 nm range is indeed observed. The same 13C NMR peak is also observed at a short timepoint, indicating that both the initial (kinetic) product and the final (thermodynamic) products are from aminolysis. The product of hydrolysis by water is also observed but there is no evidence of attack by the oxygen of hydroxylamine. This is consistent with a previous report of a single product observed by thin layer electrophoresis whose mobility was consistent with the aminolysis product(13). At the concentrations of hydroxylamine used in this study aminolysis occurs more than 35-fold more rapidly than hydrolysis by water(J Shaw and R Green, personal communication). Therefore the isotope effects were derived overwhelmingly from the aminolysis reaction.
Figure 6.

A)13C-NMR of hydroxylamine reaction. 1- Starting material (CCApcb) in 5% formic acid. A doublet is expected from the 2′- and 3′-isomers. The two smaller peaks appear at low pH. 2- CCApcb allowed to hydrolyze at pH 8.5 for one day before addition of 5% formic acid. 3-CCApcb reacted with 500 mM hydroxylamine for one day before addition of 5% formic acid. 4- CCApcb reacted with 250 mM hydroxylamine for approximately one minute before quenching with 5% formic acid. All peaks in spectrum 4 are identified as reactant, the water hydrolysis product, or the hydroxylamine aminolysis product. B) Identification of the 170 ppm peak as the aminolysis product by a ferric chloride test. Spectra of unreacted CCApcb (green) and the product of CCApcb and hydroxylamine (blue) after addition of ferric chloride. The peak for ferric chloride alone (red) is ~350 nm. The aminolysis product of CCApcb is a hydroxamate, which forms an iron complex with characteristic absorbance around 450 nm.
A plausible mechanism for aminolysis is shown in Figure 3. An extra step is depicted since amines can attack as a neutral species, forming a stable zwitterionic intermediate. Because hydroxylamine is a better leaving group than the 3′-oxygen, the tetrahedral intermediate could resolve back into reactants instead of products(32). If the partitioning ratio strongly favors breakdown into reactants, the observed isotope effect is equal to the product of the equilibrium effect on formation of the tetrahedral intermediate and the kinetic isotope effect on its breakdown. This behavior has been observed for other aminolysis reactions with good nucleophiles(31, 37).
The 3′-oxygen-18 effect was determined to be 2.9% (Figure 4b). Since the 3′-oxygen is the leaving group for only one of the two isomers of CCApcb, this number represents a lower bound for the leaving group oxygen-18 effect. Regardless, this effect is significantly larger than that for hydrolysis and is too large to be due to the decrease in bond order in a tetrahedral transition state. Instead this effect must be derived from breaking the ester C-O bond. Leaving group isotope effects of 4.1% and 6.2% have been measured for hydrazinolysis of methyl benzoate and methyl formate, respectively(31, 37). In both cases breakdown of the intermediate was rate-limiting. The large normal effect indicates that either C-O bond breakage is concerted with C-N bond formation (unlike the mechanism shown in Figure 3), or that breakdown of a tetrahedral intermediate is rate-limiting.
A small normal effect of 0.7% was observed for the 2′-oxygen-18 substitution. Since this includes a large normal factor from when the 2′-oxygen is the leaving group, it is likely that the neighboring group is inverse. The vicinal hydroxyl effect determined from deconvolution is inverse 0.9%. This is a small effect relative to the error in the measurement (0.4%), and the rates of aminolysis of the two isomers may not be equal. However, this result may indicate a prominent role for the 2′-hydroxyl in the aminolysis reaction.
The carbonyl oxygen-18 and α-deuterium isotope effects are 3.7% and inverse 3.8% respectively. While somewhat less than the maximum expected for a tetrahedral transition state, these effects demonstrate there is substantial sp3 character to the aminolysis transition state. This is inconsistent with a concerted reaction. The combination of the leaving group oxygen-18, carbonyl oxygen-18, and α-deuterium isotope effects indicate that significant but not complete breakdown of the tetrahedral intermediate has occurred at the transition state for aminolysis.
The carbonyl carbon isotope effect of 2.7% is consistent with the other isotope effects measured. The large normal effect for bond-breaking outweighs the small inverse equilibrium effect on formation of the tetrahedral intermediate.
As with the hydrolysis reaction, a transition state model was varied until the calculated isotope effect matched the experimentally-determined ones. In contrast to hydrolysis, the aminolysis reaction has a large 3′-oxygen-18 KIE of 2.9%, suggesting that there is significant cleavage to the 3′O-C bond at the transition state. Matching calculated KIEs to the experimental values gave a transition state model for the aminolysis reaction that occurs late along the reaction coordinate (Figure 5b). There is full bond formation between the primary carbon and the attacking nucleophile with a bond length of 1.33 Å (Table 2). The rate-limiting step is cleavage of the 3′O-C bond, which has a Pauling bond order of 0.73 at the transition state.
Table 2.
Comparison of experimental and theoretical isotope effects and geometric and electronic changes in representative models of the reactant and the transition state for the hydroxylaminolysis reaction calculated using B3LYP/6-31G**
| Isotope Effects | Bond Properties | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Bond Type | Bond Length | Bond order changea Δ(σ-σ*) | RS hybridb | Carbon cont. (%)c | TS hybridb | Carbon cont. (%)c | ||||
| Position | Experimental | Calculated | RS | TS | ||||||
| Carbonyl carbon | 1.027 ± 0.005 | 1.030 | C-N | - | 1.330 | - | - | - | sp2.47 | 37.3 |
| Carbonyl oxygen | 1.037 ± 0.007 | 1.037 | C-O | 1.21 | 1.370 | −0.898 | sp1.95 | 33.8 | sp2.70 | 37.5 |
| 3′-oxygen | 1.029 ± 0.006 | 1.029 | C-O3′ | 1.36 | 1.643 | −0.217 | sp2.59 | 30.3 | sp5.67 | 26.5 |
| α-hydrogen | 0.962 ± 0.008 | 0.964 | C-H | 1.10 | 1.10 | 0.008 | sp3.42 | 63.2 | sp3.32 | 62.4 |
Calculated by subtracting the number of electrons occupying the σ* orbital from the number occupying the σ orbital and listed as change between the reactant state (RS) and transition state (TS). For the carbonyl bond in the RS electrons in both σ and π bonds were added.
Hybridization of the carbon atom.
Contribution of the carbon atom to the bond in percent.
Several imaginary frequencies were observed for this model which did not correspond to reaction coordinate motion. As discussed above, these frequencies are likely due to the use of external constraints. Most of these modes do not contribute significantly to the observed isotope effect; however the largest imaginary frequency has a significant effect on the predicted carbonyl carbon-13 KIE and was included in the KIE calculations (supporting information)(42).
Discussion
Transition States for Hydrolysis and Aminolysis of CCApcb
To construct a map of the transition state of the uncatalyzed hydrolysis reaction, kinetic isotope effects were determined at several positions at the reaction center. Two different methods were used to correlate observed kinetic isotope effects with transition state structure. First, the observed effects were compared with previously studied reactions. Second, the observed effects were matched to calculated effects for a given transition state structural model. Based on these methods, the transition state for hydrolysis of the ribosomal substrate CCApcb very closely resembles the tetrahedral intermediate.
Formation of the tetrahedral intermediate is generally rate-limiting for ester hydrolysis, as observed by isotope exchange, structure-reactivity studies, and kinetic isotope effects(16, 47). If formation of the intermediate is rate-limiting in CCApcb hydrolysis as expected, the transition state determined here is extremely late in that step. This is consistent with the Hammond-Leffler postulate, since the tetrahedral intermediate is higher in energy than the reactants. However, this behavior was not observed for the alkaline hydrolysis of either methyl benzoate and methyl formate. Both transition states were early based on small deuterium and carbonyl oxygen-18 isotope effects(31, 48). This may be due to other structural differences compared with the previously studied esters, for instance the presence of a 2′-hydroxyl.
In contrast to hydrolysis, the transition state for hydroxylaminolysis of CCApcb occurs during the breakdown of the tetrahedral intermediate. This behavior has often been observed for the reaction of esters with good nucleophiles(31, 32, 37). The carbonyl oxygen-18, carbonyl carbon-13, and α-deuterium effects indicate the reaction is stepwise, with full formation of the bond to the nucleophile preceding scission of the bond to the leaving group. This is likely due to the leaving group – stepwise reactions have also been observed for methyl formate and methyl benzoate, even with hydrazine as a nucleophile(31, 37), while p-nitrophenyl acetate reactions are often concerted(32). The pKa of p-nitrophenol (approximately 7) is much lower than phenol (10), ribose (12) or methanol (15). The improved leaving group character of p-nitrophenol destabilizes the tetrahedral structure such that it is no longer a stable intermediate. This is not the case for CCApcb, which conforms to at least a two-step process even with a good nucleophile.
Implications for Release Factor-catalyzed Hydrolysis
The hydrolysis of CCApcb is chemically identical to peptide release, the final step of protein synthesis catalyzed by the ribosome. This analysis of a model hydrolysis reaction could provide insight into how this reaction is promoted on the ribosome. Recent structures of 70S ribosomes bound to release factors have shown the three-dimensional arrangement of functional groups in the product state(7–9). However, the mechanism of catalysis is still not clear. The main chain amide of glutamine in the release factor GGQ motif is within hydrogen bonding distance of the 3′-oxygen, and therefore has been hypothesized to stabilize the leaving group(7). A similar mechanism has been proposed for the Ras protein, which hydrolyzes GTP(49). Because uncatalyzed phosphodiester bond cleavage is believed to pass through a dissociative transition state(50), stabilizing the leaving group oxygen (which carries a significant negative charge in the transition state) is a potent catalytic strategy. However, the transition state for hydrolysis determined here is associative, with little charge on the leaving group and instead a buildup of negative charge on the carbonyl oxygen (Figure 5). Based on models of the 70S ribosome with a transition state inhibitor built into the active site, it was also proposed that the glutamine main-chain amide group could stabilize the carbonyl oxygen(7). Since this is the location of the buildup of negative charge in the transition state, this mechanism is more likely to be important for catalysis. This is analogous to serine proteases, where the enzyme stabilizes the transition state carbonyl oxyanion with hydrogen bonds(51–54).
Implications for Ribosomal Peptide Bond Formation
Peptide bonds are formed by the nucleophilic attack of the α-amino group of aminoacyl-tRNA on the carbonyl carbon of peptidyl-tRNA. The ribosome increases the rate of this reaction by approximately 107 over similar uncatalyzed reactions(55). Several mechanisms for this rate enhancement have been suggested, including substrate positioning, electrostatic stabilization, and a role in coordinating proton transfer(55–60). Measurement of the Brönsted coefficient and kinetic isotope effect of the nucleophile indicated the transition state for ribosomal peptide bond formation differs from uncatalyzed aminolysis reactions(5, 6). A comparison of the transition states for ribosomal peptide bond formation and uncatalyzed reactions will be necessary to determine to what extent chemical catalysis by the ribosome supplements substrate positioning to increase the reaction rate. If the ribosome is responsible for improved proton transfer or for stabilizing particular aspects of the transition state, it is expected that the structure of the transition state will be significantly altered.
The transition states for aminolysis of several esters have been determined previously(31–33, 37, 48). However, none of the esters studied contain a vicinal hydroxyl, which is crucial for ribosomal peptide bond formation. Because inclusion of this functional group increases the rate of hydrolysis, it is difficult to measure kinetic isotope effects for aminolysis by a molecule similar to the A-site tRNA. Instead a better nucleophile, hydroxylamine, was used to ensure the transition state being determined was for aminolysis and not hydrolysis. This results in a later transition state, since nucleophilic attack is not rate-limiting (as it may be for the uncatalyzed attack of A-site tRNA). While this is clearly a weakness of this model system, the measurement of a transition state for aminolysis of a true ribosomal substrate containing a vicinal hydroxyl is an important addition to the previous measurements of similar aminolysis reactions.
For uncatalyzed reactions, it is not believed that the 2′-hydroxyl participates directly in catalysis through donation or abstraction of protons. However, it is possible that in the context of the ribosome, the 2′-hydroxyl plays a more significant role that could be observed through kinetic isotope effects. In fact, the 2′-hydroxyl contributes at least 106-fold to peptide bond formation and as much as 1000-fold to peptide release in the context of the ribosome(4, 39) while uncatalyzed reaction rates are generally increased only 30-fold(15, 40).
It has been hypothesized that the ribosome promotes the use of the P-site tRNA 2′-hydroxyl as a proton shuttle(4, 56, 57). In this mechanism, the 2′-hydroxyl removes a proton from the nucleophilic nitrogen while simultaneously donating its proton to another site. If, as in hydroxylaminolysis, breakdown of the tetrahedral intermediate is rate-limiting then the proton could be directly transferred to the leaving group. Alternatively, in an earlier transition state such as that observed for hydrolysis, the proton may be transferred to the carbonyl oxygen. In either case, the atom being protonated should have a less normal isotope effect than would otherwise be expected.
In the future, kinetic isotope effects of the ribosome-catalyzed reactions can be compared with the previously studied aminolysis reactions lacking the vicinal hydroxyl and with the reaction studied here containing the vicinal hydroxyl but with a better nucleophile. Differences in the transition states of these reactions will illuminate the role of the ribosome in catalysis.
Supplementary Material
Acknowledgments
The authors thank Jared Davis for assistance with NMR, the Keck facility for mass spectrometry, and the experimental suggestions of the reviewers.
Abbreviations
- CCApcb
Cytidylyl-(3′5′)-cytidylyl-(3′5′)-3′(2′)-O-(N-(6-D-(+)-biotinoylaminohexanoyl)-L-phenylalanyl)adenosine
- KIE
kinetic isotope effect
- EIE
equilibrium isotope effect
- FLB
formamide loading buffer
Footnotes
This research was supported by NIH grant GM54839 (to SAS), NIH post-doctoral fellowship GM079980 (to DAH), and a Brown-Coxe fellowship (to VS).
Supporting Information Available
Information on calculated transition state structures. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Ban N, Nissen P, Hansen J, Moore PB, Steitz TA. The Complete Atomic Structure of the Large Ribosomal Subunit at 2.4 A Resolution. Science. 2000;289:905–920. doi: 10.1126/science.289.5481.905. [DOI] [PubMed] [Google Scholar]
- 2.Nissen P, Hansen J, Ban N, Moore PB, Steitz TA. The Structural Basis of Ribosome Activity in Peptide Bond Synthesis. Science. 2000;289:920–930. doi: 10.1126/science.289.5481.920. [DOI] [PubMed] [Google Scholar]
- 3.Erlacher MD, Lang K, Shankaran N, Wotzel B, Huttenhofer A, Micura R, Mankin AS, Polacek N. Chemical engineering of the peptidyl transferase center reveals an important role of the 2′-hydroxyl group of A2451. Nucl Acids Res. 2005;33:1618–1627. doi: 10.1093/nar/gki308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Weinger JS, Parnell KM, Dorner S, Green R, Strobel SA. Substrate-assisted catalysis of peptide bond formation by the ribosome. Nat Struct Mol Biol. 2004;11:1101–1106. doi: 10.1038/nsmb841. [DOI] [PubMed] [Google Scholar]
- 5.Kingery DA, Pfund E, Voorhees RM, Okuda K, Wohlgemuth I, Kitchen DE, Rodnina MV, Strobel SA. An Uncharged Amine in the Transition State of the Ribosomal Peptidyl Transfer Reaction. Chemistry & Biology. 2008;15:493–500. doi: 10.1016/j.chembiol.2008.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Seila AC, Okuda K, Nunez S, Seila AF, Strobel SA. Kinetic Isotope Effect Analysis of the Ribosomal Peptidyl Transferase Reaction†. Biochemistry. 2005;44:4018–4027. doi: 10.1021/bi047742f. [DOI] [PubMed] [Google Scholar]
- 7.Laurberg M, Asahara H, Korostelev A, Zhu J, Trakhanov S, Noller HF. Structural basis for translation termination on the 70S ribosome. Nature. 2008;454:852–857. doi: 10.1038/nature07115. [DOI] [PubMed] [Google Scholar]
- 8.Weixlbaumer A, Jin H, Neubauer C, Voorhees RM, Petry S, Kelley AC, Ramakrishnan V. Insights into Translational Termination from the Structure of RF2 Bound to the Ribosome. Science. 2008;322:953–956. doi: 10.1126/science.1164840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Korostelev A, Asahara H, Lancaster L, Laurberg M, Hirschi A, Zhu J, Trakhanov S, Scott WG, Noller HF. Crystal structure of a translation termination complex formed with release factor RF2. Proceedings of the National Academy of Sciences. 2008;105:19684–19689. doi: 10.1073/pnas.0810953105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Frolova LY, Tsivkovskii RY, Sivolobova GF, Oparina NY, Serpinsky OI, Blinov VM, Tatkov SI, Kisselev LL. Mutations in the highly conserved GGQ motif of class 1 polypeptide release factors abolish ability of human eRF1 to trigger peptidyl-tRNA hydrolysis. RNA. 1999;5:1014–1020. doi: 10.1017/s135583829999043x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Seit Nebi A, Frolova L, Ivanova N, Poltaraus A, Kisselev L. Substitutions of the Glutamine Residue in the ubiquitous GGQ tripeptide in human eRF1 do not entirely abolish the release factor activity. Molecular Biology. 2000;34:764–765. [Google Scholar]
- 12.Zavialov AV, Mora L, Buckingham RH, Ehrenberg M. Release of Peptide Promoted by the GGQ Motif of Class 1 Release Factors Regulates the GTPase Activity of RF3. Molecular Cell. 2002;10:789–798. doi: 10.1016/s1097-2765(02)00691-3. [DOI] [PubMed] [Google Scholar]
- 13.Shaw JJ, Green R. Two Distinct Components of Release Factor Function Uncovered by Nucleophile Partitioning Analysis. Molecular Cell. 2007;28:458–467. doi: 10.1016/j.molcel.2007.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kovach IM, Belz M, Larson M, Rousy S, Schowen RL. Transition-state structures for ester aminolysis with and without rate-limiting proton transfer. Journal of the American Chemical Society. 1985;107:7360–7365. [Google Scholar]
- 15.Wolfenden R. The Mechanism of Hydrolysis of Amino Acyl RNA*. Biochemistry. 1963;2:1090–1092. doi: 10.1021/bi00905a031. [DOI] [PubMed] [Google Scholar]
- 16.Jencks WP, Gilchrist M. Nonlinear structure-reactivity correlations. The reactivity of nucleophilic reagents toward esters. Journal of the American Chemical Society. 1968;90:2622–2637. [Google Scholar]
- 17.Blackburn GM, Jencks WP. The mechanism of the aminolysis of methyl formate. Journal of the American Chemical Society. 1968;90:2638–2645. [Google Scholar]
- 18.Satterthwait AC, Jencks WP. Mechanism of the aminolysis of acetate esters. Journal of the American Chemical Society. 1974;96:7018–7031. doi: 10.1021/ja00829a034. [DOI] [PubMed] [Google Scholar]
- 19.Bender ML. Oxygen Exchange as Evidence for the Existence of an Intermediate in Ester Hydrolysis. Journal of the American Chemical Society. 1951;73:1626–1629. [Google Scholar]
- 20.Cleland WW. Enzyme Kinetics and Mechanism Part D: Developments in Enzyme Dynamics. Academic Press; 1995. [13] Isotope effects: Determination of enzyme transition state structure; pp. 341–373. [DOI] [PubMed] [Google Scholar]
- 21.Schramm VL. Enzymatic Transition State Poise and Transition State Analogues. Accounts of Chemical Research. 2003;36:588–596. doi: 10.1021/ar0200495. [DOI] [PubMed] [Google Scholar]
- 22.Zhong M, Strobel SA. Synthesis of the Ribosomal P-Site Substrate CCA-pcb. Organic Letters. 2006;8:55–58. doi: 10.1021/ol052484f. [DOI] [PubMed] [Google Scholar]
- 23.Zhong M, Strobel SA. Synthesis of Isotopically Labeled P-Site Substrates for the Ribosomal Peptidyl Transferase Reaction. The Journal of Organic Chemistry. 2008;73:603–611. doi: 10.1021/jo702070m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Griffin BE, Jarman M, Reese CB, Sulston JE, Trentham DR. Some Observations Relating to Acyl Mobility in Aminoacyl Soluble Ribonucleic Acids*. Biochemistry. 1966;5:3638–3649. [Google Scholar]
- 25.Fromageot HPM, Griffin BE, Reese CB, Sulston JE, Trentham DR. Orientation of ribonucleoside derivatives by proton magentic resonance spectroscopy. Tetrahedron. 1966;22:705–710. doi: 10.1016/0040-4020(66)80041-8. [DOI] [PubMed] [Google Scholar]
- 26.Wolfenden R, Rammler DH, Lipmann F. On the Site of Esterification of Amino Acids to Soluble RNA*. Biochemistry. 1964;3:329–338. doi: 10.1021/bi00891a006. [DOI] [PubMed] [Google Scholar]
- 27.Caldwell SR, Raushel FM, Weiss PM, Cleland WW. Transition-state structures for enzymic and alkaline phosphotriester hydrolysis. Biochemistry. 1991;30:7444–7450. doi: 10.1021/bi00244a011. [DOI] [PubMed] [Google Scholar]
- 28.Frisch M, Trucks G, Schlegel H, Scuseria G, Robb M, Cheeseman J, Montgomery J, Vreven T, Kudin K, Burant J, Millam J, Iyengar S, Tomasi J, Barone V, Mennucci B, Cossi M, Scalmani G, Rega N, Petersson G, Nakatsuji H, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Klene M, Li X, Knox J, Hratchian H, Cross J, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann R, Yazyev O, Austin A, Cammi R, Pomelli C, Ochterski J, Ayala P, Morokuma K, Voth G, Salvador P, Dannenberg J, Zakrzewski V, Dapprich S, Daniels A, Strain M, Farkas O, Malick D, Rabuck A, Raghavachari K, Foresman J, Ortiz J, Cui Q, Baboul A, Clifford S, Cioslowski J, Stefanov B, Liu G, Liashenko A, Piskorz P, Komaromi I, Martin R, Fox D, Keith T, Laham A, Peng C, Nanayakkara A, Challacombe M, Gill P, Johnson B, Chen W, Wong M, Gonzalez C, Pople J. Gaussian 03, Revision C.02 2003 [Google Scholar]
- 29.Anisimov V, Paneth P. ISOEFF98. A program for studies of isotope effects using Hessian modifications. Journal of Mathematical Chemistry. 1999;26:75–86. [Google Scholar]
- 30.Unrau PJ, Bartel DP. An oxocarbenium-ion intermediate of a ribozyme reaction indicated by kinetic isotope effects. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:15393–15397. doi: 10.1073/pnas.2433147100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.O’Leary MH, Marlier JF. Heavy-atom isotope effects on the alkaline hydrolysis and hydrazinolysis of methyl benzoate. Journal of the American Chemical Society. 1979;101:3300–3306. [Google Scholar]
- 32.Hengge AC, Hess RA. Concerted or Stepwise Mechanisms for Acyl Transfer Reactions of p-Nitrophenyl Acetate? Transition State Structures from Isotope Effects. Journal of the American Chemical Society. 1994;116:11256–11263. [Google Scholar]
- 33.Marlier JF. Heavy-atom isotope effects on the alkaline hydrolysis of methyl formate: the role of hydroxide ion in ester hydrolysis. Journal of the American Chemical Society. 1993;115:5953–5956. [Google Scholar]
- 34.Mitton CG, Schowen RL. Oxygen isotope effects by a noncompetitive technique: The transition-state carbonyl stretching frequency in ester cleavage. Tetrahedron Letters. 1968;9:5803–5806. [Google Scholar]
- 35.Hogg JL, Rodgers J, Kovach I, Schowen RL. Kinetic isotope-effect probes of transition-state structure. Vibrational analysis of model transition states for carbonyl addition. Journal of the American Chemical Society. 1980;102:79–85. [Google Scholar]
- 36.Khym JX, Cohn WE. Identification of the Purine Nucleotides a and b as the 2′- and 3′-Phosphoribosides, Respectively1. Journal of the American Chemical Society. 1954;76:1818–1823. [Google Scholar]
- 37.Sawyer CB, Kirsch JF. Kinetic isotope effects for reactions of methyl formate-methoxyl-18O. Journal of the American Chemical Society. 1973;95:7375–7381. doi: 10.1021/ja00840a065. [DOI] [PubMed] [Google Scholar]
- 38.Bruice TC, Fife TH. Hydroxyl Group Catalysis. III. 1 The Nature of Neighboring Hydroxyl Group Assistance in the Alkaline Hydrolysis of the Ester Bond. Journal of the American Chemical Society. 1962;84:1973–1979. [Google Scholar]
- 39.Brunelle JL, Shaw JJ, Youngman EM, Green R. Peptide release on the ribosome depends critically on the 2′ OH of the peptidyl–tRNA substrate. RNA. 2008;14:1526–1531. doi: 10.1261/rna.1057908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kupchan SM, Slade P, Young RJ, Milne GWA. Intramolecular catalysis--IV,: Facilitation of alkaline hydrolysis of alycyclic 1,2-diol monoesters. Tetrahedron. 1962;18:499–506. [Google Scholar]
- 41.Cook PF. Enzyme mechanism from isotope effects. CRC; 1991. [Google Scholar]
- 42.Hirschi JS, Takeya T, Hang C, Singleton DA. Transition-State Geometry Measurements from 13C Isotope Effects. The Experimental Transition State for the Epoxidation of Alkenes with Oxaziridines. Journal of the American Chemical Society. 2009;131:2397–2403. doi: 10.1021/ja8088636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jencks WP. The Reaction of Hydroxylamine with Activated Acyl Groups. I. Formation of O-Acylhydroxylamine1. Journal of the American Chemical Society. 1958;80:4581–4584. [Google Scholar]
- 44.Mazera DJ, Gesser JC, Pliego JR. On the mechanism of the reaction between aryl acetates and hydroxylamine. ARKIVOC. 2007;15:199–214. [Google Scholar]
- 45.Lipmann F, Tuttle LC. A specific micromethod for the determination of acyl phosphates. Journal of Biological Chemistry. 1945;159:21. [Google Scholar]
- 46.Hulcher FH. Isolation and characterization of a new hydroxamic acid from Pseudomonas mildenbergii. Biochemistry. 1982;21:4491–4495. doi: 10.1021/bi00261a046. [DOI] [PubMed] [Google Scholar]
- 47.Bender ML. Mechanisms of Catalysis of Nucleophilic Reactions of Carboxylic Acid Derivatives. Chemical Reviews. 1960;60:53–113. [Google Scholar]
- 48.Bilkadi Z, De Lorimier R, Kirsch JF. Secondary. alpha. -deuterium kinetic isotope effects and transition-state structures for the hydrolysis and hydrazinolysis reactions of formate esters. Journal of the American Chemical Society. 1975;97:4317–4322. [Google Scholar]
- 49.Maegley KA, Admiraal SJ, Herschlag D. Ras-catalyzed hydrolysis of GTP: a new perspective from model studies. Proceedings of the National Academy of Sciences of the United States of America. 1996;93:8160–8166. doi: 10.1073/pnas.93.16.8160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Du X, Black GE, Lecchi P, Abramson FP, Sprang SR. Kinetic isotope effects in Ras-catalyzed GTP hydrolysis: Evidence for a loose transition state. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:8858–8863. doi: 10.1073/pnas.0401675101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Henderson R. Structure of crystalline [alpha]-chymotrypsin: IV. The structure of indoleacryloyl-[alpha]-chymotrypsin and its relevance to the hydrolytic mechanism of the enzyme. Journal of Molecular Biology. 1970;54:341–354. doi: 10.1016/0022-2836(70)90434-1. [DOI] [PubMed] [Google Scholar]
- 52.Robertus JD, Kraut J, Alden RA, Birktoft JJ. Subtilisin. Stereochemical mechanism involving transition-state stabilization. Biochemistry. 1972;11:4293–4303. doi: 10.1021/bi00773a016. [DOI] [PubMed] [Google Scholar]
- 53.Bryan P, Pantoliano MW, Quill SG, Hsiao HY, Poulos T. Site-directed mutagenesis and the role of the oxyanion hole in subtilisin. Proc Natl Acad Sci U S A. 1986;83:3743–3745. doi: 10.1073/pnas.83.11.3743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Joseph S, Whirl ML, Kondo D, Noller HF, Altman RB. Calculation of the relative geometry of tRNAs in the ribosome from directed hydroxyl-radical probing data. RNA. 2000;6:220–232. doi: 10.1017/s1355838200992112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sievers A, Beringer M, Rodnina MV, Wolfenden R. The ribosome as an entropy trap. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:7897–7901. doi: 10.1073/pnas.0402488101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Das GK, Bhattacharyya D, Burma DP. A Possible Mechanism of Peptide Bond Formation on Ribosome without Mediation of Peptidyl Transferase. Journal of Theoretical Biology. 1999;200:193–205. doi: 10.1006/jtbi.1999.0987. [DOI] [PubMed] [Google Scholar]
- 57.Trobro S, Åqvist J. Mechanism of peptide bond synthesis on the ribosome. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:12395–12400. doi: 10.1073/pnas.0504043102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Trobro S, Aqvist J. Analysis of Predictions for the Catalytic Mechanism of Ribosomal Peptidyl Transfer†. Biochemistry. 2006;45:7049–7056. doi: 10.1021/bi0605383. [DOI] [PubMed] [Google Scholar]
- 59.Erlacher MD, Lang K, Wotzel B, Rieder R, Micura R, Polacek N. Efficient Ribosomal Peptidyl Transfer Critically Relies on the Presence of the Ribose 2′-OH at A2451 of 23S rRNA. Journal of the American Chemical Society. 2006;128:4453–4459. doi: 10.1021/ja0588454. [DOI] [PubMed] [Google Scholar]
- 60.Schmeing TM, Huang KS, Kitchen DE, Strobel SA, Steitz TA. Structural Insights into the Roles of Water and the 2′ Hydroxyl of the P Site tRNA in the Peptidyl Transferase Reaction. Molecular Cell. 2005;20:437–448. doi: 10.1016/j.molcel.2005.09.006. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.