

Abstract
The amyloid β (Aβ) and tau proteins, which misfold, aggregate, and accumulate in the Alzheimer's disease (AD) brain, are implicated as central factors in a complex neurodegenerative cascade. Studies of mutations that cause early onset AD and promote Aβ accumulation in the brain strongly support the notion that inhibiting Aβ aggregation will prevent AD. Similarly, genetic studies of frontotemporal dementia with parkinsonism linked to chromosome 17 (FTDP-17 MAPT) showing that mutations in the MAPT gene encoding tau lead to abnormal tau accumulation and neurodegeneration. Such genetic studies clearly show that tau dysfunction and aggregation can be central to neurodegeneration, however, most likely in a secondary fashion in relation to AD. Additional pathologic, biochemical and modeling studies further support the concept that Aβ and tau are prime targets for disease modifying therapies in AD. Treatment strategies aimed at preventing the aggregation and accumulation of Aβ, tau, or both proteins should therefore be theoretically possible, assuming that treatment can be initiated before either irreversible damage is present or downstream, self-sustaining, pathological cascades have been initiated. Herein, we will review recent advances and also potential setbacks with respect to the myriad of therapeutic strategies that are designed to slow down, prevent, or clear the accumulation of either “pathological” Aβ or tau. We will also discuss the need for thoughtful prioritization with respect to clinical development of the pre-clinically validated modifiers of Aβ and tau pathology. The current number of candidate therapies targeting Aβ is becoming so large that a triage process is clearly needed to insure that resources are invested in a way such that the best candidates for disease modifying therapy are rapidly moved toward clinical trials. Finally, we will discuss the challenges for an appropriate “triage” after potential disease modifying therapies targeting tau and Aβ have entered clinical trials.
Introduction
The two hallmark pathological lesions of the Alzheimer's brain, first described by Dr. Alzheimer in 1906, are senile plaques and neurofibrillary tangles (NFTs) (Terry, 1985; Alzheimer et al., 1995). Subsequent studies of the proteins that form plaques and NFTs has proven paramount to our understanding of the disease pathogenesis, and thus our hope for successful prevention or treatment of this devastating disease (Golde, 2006). The identification of Aβ and tau as the principle proteinaceous component of plaques and NFTs, respectively, coupled with genetic studies implicating these protein as triggers of neurodegeneration has validated both proteins as therapeutic targets in neurodegeneration (Selkoe, 2001a; Hardy and Selkoe, 2002). Though targeting Aβ is likely to be considered a therapeutic strategy primarily in AD, targeting tau accumulation may have therapeutic benefit in AD and other tauopathies. As neither tau nor Aβ appear to be harmful when normally folded and not aggregated, therapies targeting each are designed either to delay or prevent the formation of abnormally aggregated species or to clear such aggregates (Golde, 2006). Therefore, the guiding principles for current AD therapeutic development are i) decreasing synthesis of Aβ or tau, ii) preventing misfolding and aggregation of these proteins, iii) neutralizing or removing the toxic aggregate or misfolded forms of these proteins, or iv) some combination of these principles.
In pre-clinical mouse models of Aβ deposition (APP transgenic mice), modification of the Aβ deposition phenotype and the modest cognitive/behavioral impairment exhibited in many of these mice has proven reasonably simple (Price et al., 1998). Well over a hundred manuscripts have now been published supporting new therapeutic strategies for Aβ by demonstrating that a given factor (e.g., genetic manipulation, compound treatment, and immunotherapy) can modulate the phenotype of these APP mice, and it now seems that such manuscripts are published almost weekly. Far fewer studies identifying potential therapeutic factors in tau transgenic mice have been published; nevertheless, at least a dozen factors have been shown to modify tau pathology to date, a number that is likely to grow over the next few years with the increasing appreciation that tau is a relatively underexplored therapeutic target for AD. If one expands the list of potential tau and Aβ therapeutics to include strategies that have only been validated in culture, the list is so expansive that no single review could ever hope to cover the relative merits of each strategy. As multiple recent reviews have focused either broadly or on specific aspects of Aβ and tau therapeutics [e.g., (Golde, 2005; Schneider and Mandelkow, 2008; Lee and Trojanowski, 2006; Golde et al., 2009; Morgan, 2006)], we will limit this review to updating progress and barriers with respect to Aβ and tau therapies that have either been studied in humans or subjectively appear to represent novel approaches with a clear route towards the development of clinical leads in terms of a small molecule or biologic agent (See Figure 1). Finally, we will conclude with a discussion of the challenges faced in clinical testing of disease modifying therapies targeting tau and Aβ
Figure 1. Aβ and Tau Therapies in the clinic.
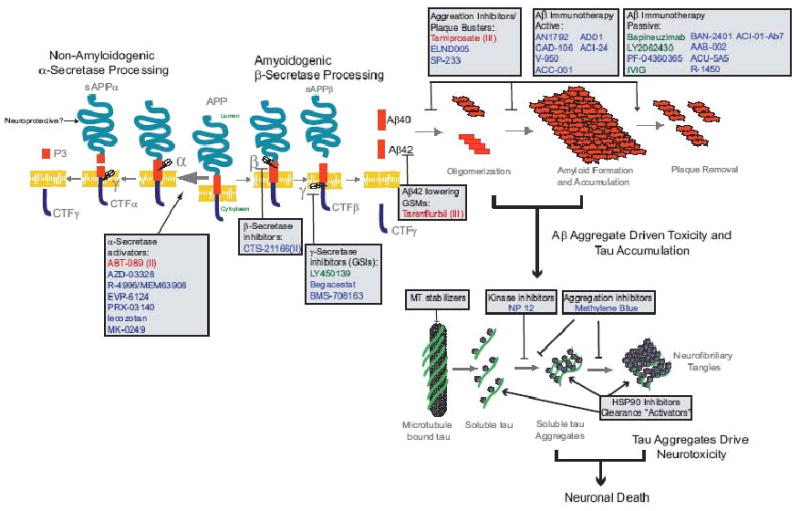
Aβ and tau therapeutic targets and therapeutics in the clinic are depicted the context of a schematic of the amyloid cascade hypothesis (Hardy and Higgins, 1992). Classes of therapeutics are shown in boxes and below the class in are therapeutics that are in, have completed, or have announced human trials (as of June 2009). Red text denotes therapies for which trials either failed to meet endpoints or were terminated for other reasons. In parenthesis is the stage of trial that the therapy completed. Green text indicates therapies in phase III trials as of June 2009. Blue text indicates therapies in early pahse trials (Phase I or II). Basic APP processing, Aβ aggregation and Tau aggregation schematics are shown.
Secretase inhibitors and modulators that target Aβ production
Rationale for the approach
There is no evidence that monomeric Aβ is the pathological entity in AD as only aggregated Aβ has pathophysiological properties. Aβ aggregates into a variety of oligomers (dimers, trimers, tetramers, dodecamers to 50-100 mers), soluble protofibrils, actively growing fibrils, and mature fibrils that have been implicated as potential pathologic entities in AD (Selkoe, 2001a; Hardy and Selkoe, 2002; Glabe, 2005; Tanzi, 2005; Klein et al., 2004; Lesne et al., 2006; Shankar et al., 2008; Klyubin et al., 2008; Lauren et al., 2009). The hypothesis that soluble assemblies of Aβ are the primary toxic entities in AD has garnered many advocates with supportive data suggesting that these soluble oligomeric assemblies mediate i) some behavioral deficits observed in APP transgenic mice and ii) the inhibition of long term potentiation (LTP) in vitro and acutely in vivo (Shankar et al., 2008; Cheng et al., 2007; Dodart et al., 2002; Walsh and Selkoe, 2004a). It remains an open question as to whether oligomeric Aβ can promote neurodegeneration. Indeed, the relationship of oligomeric assemblies to cognitive deficits in humans remains to be established (Walsh and Selkoe, 2004b; Glabe, 2006). As all Aβ aggregates form in a concentration dependent fashion, decreasing monomeric Aβ by decreasing its production will slow the rate of aggregate formation (Kelly, 2005; Jarrett et al., 1993). From this perspective, intervention that is aimed at decreasing Aβ production is attractive, as it is not dependent upon a definitive identification of the toxic Aβ aggregates.
Aβ peptides are produced from the amyloid β protein precursor (APP) through sequential proteolysis by the β- and γ-secretases (Golde et al., 2000). A 40 amino acid form of Aβ (Aβ40) is the major secreted product of these cleavages, whereas the minor 42 amino acid form of Aβ (Aβ42) that contains two additional residues at its carboxyl terminus has been implicated as the initiating molecule in the pathogenesis of AD (Younkin, 1998). Indeed, recent mouse modeling and human genetic studies suggest that, at least with respect to initial seeding of Aβ aggregation, Aβ40 may actually be protective (McGowan et al., 2003; Kim et al., 2007; Wang et al., 2006). Finally, in addition to Aβ40 and Aβ42, a number of smaller Aβ peptides (Aβ34, 36, 37, 38, 39) are normally produced (Wang et al., 1996). These peptides are hypothesized to behave like Aβ40 and inhibit Aβ42 aggregation (Kim et al., 2007), but this has not been tested in vivo.
There are currently three direct strategies targeting Aβ production that have moved to the clinical: β- secretase inhibitors and γ-secretase inhibitors and modulators (GSMs). Both β-and γ-secretase inhibitors (GSIs) block production of all species of Aβ (Vassar, 2002; Wolfe, 2008), whereas GSMs are unique in that they shift cleavage by γ-secretase resulting in an altered profile of Aβ peptides. Aβ42 lowering GSMs are of therapeutic interests and the “signature” of these compounds is that they decrease Aβ42 production and increase Aβ peptides shorter than Aβ1-40 (Weggen et al., 2001). Recently, it has been shown that certain GSMs do not bind γ-secretase but instead target substrate at a site that lies within the Aβ peptide (Kukar et al., 2008). This substrate targeting property means that select GSMs can potentially also act as inhibitors of Aβ aggregation.
Progress on β-secretase inhibitors
Though development of β-secretase inhibitors has been a major focus of both industry and academic laboratories, progress has been slow as it has proven difficult to develop potent β-secretase inhibitors that cross the blood brain barrier (Ghosh et al., 2008). One orally bioavailable β-secretase inhibitor (CTS-21166, Comentis,) has been evaluated in a Phase I study in humans (Strobel, 2008a). Although it was reported to be well tolerated and lower plasma Aβ, CNS levels of the drug and effects on CSF Aβ were not evaluated. As animal modeling studies suggest that CNS inhibition of β-secretase is needed for efficacy with respect to reducing Aβ accumulation, future data from ongoing human studies with CTS-21166 is eagerly awaited (Cai et al., 2001). Given the massive investment by pharmaceutical companies and others in β-secretase inhibitor development, it is likely that brain penetrant, orally bioavailable, selective β-secretase inhibitors will be developed in the next few years. Whether, long-term treatment with β-secretase inhibitors will be well tolerated remains an open question. Recent studies do suggest that BACE1 plays a key role in developmental myelination; however, it is not clear if inhibition will result in myelination deficits in the adult (Hu et al., 2006; Willem et al., 2006).
Progress on GSIs
Several GSIs are now being tested in the clinic. The most advanced of these is LY450139 (Eli Lily), a non-selective GSI that is currently in a Phase III study (Bateman et al., 2009; Fleisher et al., 2008; Siemers et al., 2007; Siemers et al., 2006; Siemers et al., 2005). Published Phase II data show that the steady state CSF Aβ was not significantly reduced by these doses though a trend towards lowering was observed for Aβ40 (Fleisher et al., 2008). Additionally, there was no impact on cognition during the 14 week study. Although the drug was reported to be generally well-tolerated, there were some adverse reactions reported. Of note, the hair color changes reported in this trial are often observed in mice on chronic GSI treatment. In a more recent study using stable isotope labeling combined with CSF sampling to measure the synthesis rate of Aβ in healthy men, a single oral dose of either 100,140 or 280 mg of this compound lowered CSF Aβ production by 47, 52, and 84%, respectively (Bateman et al., 2009; Bateman et al., 2006). Notably, ELISA analyses of Aβ in the same study showed that significant decrease in total Aβ were appreciable in the 280 mg treatment group; however, the 100 and 140mg doses only yielded a trend toward reduced Aβ levels.
A potential concern regarding the first generation GSIs is that their limited selectivity may result in target-mediated toxicity that could appear due to inhibition of Notch signaling (De Strooper et al., 1999; Searfoss et al., 2003). In animal models, there is very narrow therapeutic window that allows one to significantly reduce Aβ levels in the brain without observing these side effects. Recently two Notch-sparing GSIs have been developed (Begacestat; Wyeth; BMS-708163, Bristol-Myers Squib) (Fagan, 2008a; Mayer et al., 2008). Begacestat is reported to have a ∼14-fold and BMS-708163 over 100-fold selectivity for inhibiting γ-secretase cleavage of APP over Notch. Both of these compounds were identified through cell based screens for selective inhibitors of APP versus Notch, and subsequent medicinal chemistry around the lead compounds. Notch-sparing pharmacologic selectivity had been conceptually proven possible by several academic groups; though the compounds they identified were of low potency (Fraering et al., 2005; Netzer et al., 2003). The original Notch-sparing GSIs are thought to bind an adenosine triphosphate site on γ-secretase (Fraering et al., 2005), the mechanism of Notch-sparing of either Begacestat or BMS-708163 in is either not known or has not been disclosed. These compounds are in or completed Phase I human studies, and it is hoped that they will have a larger therapeutic safety margin. BMS-708163 lowered plasma and CSF Aβ levels in Phase I human studies (Fagan, 2008a). Mechanistically, it has been suggested that these compounds work by binding substrate docking sites on γ-secretase that are distinct between Notch and APP; however, this hypothesis has not been formally proven. It is also unclear as to what extent these compounds inhibit cleavage of other γ-secretase substrates, which could lead to long-term toxicities. Given that Notch toxicities are initially seen in peripheral organs, it is also possible that pharmacokinetic properties of the compounds could result in Notch related toxicities even though the therapeutic window is enhanced.
Though non-selective GSIs were originally developed for AD, several are now being redirected as chemotherapeutic agents for cancers in which Notch singling is shown to play a primary (T-ALL) or secondary role (e.g., breast cancer) in the cancer's malignant phenotype (Rizzo et al., 2008). Surprisingly, a recent preclinical study in a T-ALL mouse model showed that there may be novel routes to block the gastrointestinal (GI) toxicity, frequently the initial dose-limiting side effect of GSI treatment (Searfoss et al., 2003). In this study, co-administration of a GSI with dexamethasone produced a synergist anti-tumor effect over the single agents alone, and the addition of dexamethasone treatment to GSI treatment blocked the GI toxicity observed with the GSI alone (Real et al., 2009). These observations suggest that it may be possible to use non-selective GSI in AD in combination with compounds like dexamethasone that block the GI toxicity.
Progress on GSMs
An 18 month double blind placebo controlled Phase III study of the GSM Tarenflurbil (Flurizan, R-Flurbiprofen; Myriad Genetics) in patients with mild AD showed no benefit on cognitive or functional outcomes. The program was discontinued following this disappointing result (Fagan, 2008b). Approximately 1000 subjects completed the well designed and conducted Phase III study. When the final data are published the results should provide a superb benchmark for trial design for potential disease modification with standard clinical measures (ADAScog, CDRsb, ADCS Activities of Daily Living) in populations with a high percentage of subjects already on cholinesterase inhibitors or memantine. Notably, tarenflurbil is a weak GSM with low CNS penetrance; thus, it is impossible to make inferences based on this trial with respect to clinical efficacy of more potent, CNS penetrant GSMs (Kukar and Golde, 2008; Eriksen et al., 2003). Indeed a number of companies have ongoing GSM programs. Chiesi has developed a more potent brain penetrant GSM derivative of flurbiprofen, while Eisai, Merck, Cellzome and Torrey Pines, and Satori pharmaceuticals have also reported GSMs that have increased potency relative to tarenflurbil of ∼100-1000 fold (Imbimbo et al., 2007; Pissarnitski, 2007).
Targeting Aβ: Immunotherapy
Rationale for the approach
A variety of mechanisms have been postulated regarding how Aβ immunotherapy might attenuate or in some circumstances clear Aβ from the brain; however, no mechanism has been definitively proven or disproven to date [reviewed in (Golde et al., 2009)]. Nevertheless, the therapeutic potential of altering Aβ deposition by inducing a humoral immune response to fibrillar Aβ42 (fAβ42) or passively administering anti-Aβ antibodies (Abs) has been the most extensively validated anti-Aβ approach in pre-clinical studies (Morgan, 2006).
Progress on Active Immunotherapy
The original active vaccination with fibrillar Aβ42+QS21 adjuvant, known as the AN1792 vaccine trial, was halted in Phase II (January, 2002) due to induction of meningoencephalitis in ∼6% of the vaccinated patients (Nicoll et al., 2003; Fox et al., 2005; Gilman et al., 2005; Orgogozo et al., 2003). Though the meningoencephalitis has been attributed to an autoimmune T-cell reaction, the exact nature of the side-effect has not been definitively established (Schenk et al., 2005). Individuals in the trial who showed an antibody reponse, representing up to 19% of those receiving the vaccine in the Phase II study, had a paradoxical decrease in brain volume (Fox et al., 2005; Gilman et al., 2005). Though cognitive improvement has been claimed in some studies of the antibody responders, the post-hoc nature of the analyses and the small sample sizes warrant a cautious interpretation of this claim (Gilman et al., 2005). Subsequent follow-up of a subset of the subjects enrolled in the Phase I AN1792 trial has revealed no significant alteration in long-term survival and cognition. Post-mortem analysis of 12 subjects in the AN-1792 trial, including 2 who suffered meningoencephalitis, provides data to support the claim that there is clearance of deposited Aβ as assessed solely by histochemical analysis of Aβ (Nicoll et al., 2003; Holmes et al., 2008; Masliah et al., 2005; Bombois et al., 2007). For most of these subjects, biochemical data has not been reported. In another study of two patients from the AN-1792 trials, biochemical and histochemical data suggested that some parenchymal amyloid may have been cleared, but cerebrovascular amyloid (CAA) deposits may have increased (Patton et al., 2006). At face value, such data does seem to support clearance of Aβ following active immunotherapy. However, the patchy nature of the clearance in at least some cases raises the question as to whether the clearance of deposited Aβ is antibody or cell mediated (Golde et al., 2009).
Despite setbacks in the AN-1792 trial, a number of alternative active anti-Aβ vaccination approaches are in early phase human testing or likely to begin human testing in the near future. Two general strategies are being employed in the effort to make a potentially safer vaccine by avoiding harmful T-cell activation. First, based on evidence in mice that full length Aβ contains an amino-terminal B-cell epitope (residues 1-14) and a carboxyl-terminal T-cell epitope (residues 15-42), a number of vaccines comprising the amino-terminus of Aβ coupled to T-cell epitopes or conjugated to a virus-like particle have been developed (ACC-001, Elan/Wyeth; CAD-106, Novartis; V950, Merck; ACI-24, AC Immune) (Schenk et al., 2005; Lee et al., 2005; Agadjanyan et al., 2005). Second, rather than immunize with the exact sequence of Aβ, Aβ peptide mimetics are also being developed (AD01, Affiris GmbH) (Schneeberger et al., 2009). Although these 2nd generation anti-Aβ vaccines are promising, little information is currently available regarding their immunogenicity in humans and the exact nature of the humoral response induced by these vaccines. Moreover, given that the heterogeneity of the immune response in humans, there is no guarantee that such vaccines will not induce a harmful T-cell response in some individuals. Even approaches such as those employed by Affiris GmbH to develop peptide mimetics as active vaccines are not guaranteed to be safe. It is well established that antigenic cross-talk between a pathogen encoded protein and a human protein can induce autoimmunity (Oldstone, 1998).
Progress on Passive Immunotherapy
Humanized monoclonal antibodies targeting Aβ and IVIG are being actively tested in humans. The most advanced of these is the bapineuzumab (AAB-001, Elan/Wyeth) a humanized monoclonal antibody that targets the amino-terminus of Aβ and is capable of binding monomeric, oligomeric and fibrillar Aβ (Strobel, 2008b). A 238 subject Phase II study has been completed, and although a post-hoc subgroup analysis suggested that some patients who completed the course of injections showed cognitive improvement, the primary clinical endpoint of the trial showed no treatment effect. Although a number of typical side-effects associated with recombinant antibody administration were reported, transient vasogenic edema (VE) was also noted in a subset of patients, and the incidence was higher in APOE4 carriers. For this reason, separate Phase III trials with different dosing regimens are underway, with a lower maximal doing for the APOE4 versus APOE4 no carriers. Notably, despite these separate trials, Elan/Wyeth recently announced that they were discontinuing the highest dose in the non-carriers based on the incidence of VE. In most cases, the VE appears to be clinically silent and transient, though some subjects do demonstrate transient neurologic symptoms associated with the VE. Nevertheless, as VE indicates brain edema and is indirect evidence for local inflammation or loss of blood brain barrier integrity, it is a concerning radiographic finding.
LY2062430 is a humanized monoclonal antibody based on the mouse monoclonal m266 that is directed to the mid-domain of Aβ targeting Aβ16-23. In contrast to bapineuzimab, it preferentially targets monomeric soluble Aβ and is not plaque binding. The binding specificity with respect to oligomers is not known (Strobel, 2008c). A Phase II study of 52 AD patients has been completed. In this study, the antibody was dosed at 100 mg or 400 mg every week or every 4 weeks for 12 weeks. Data from a planned one year follow-up period to assess cognition has not been released, but biomarker data provided strong evidence that the antibody was hitting target in that plasma and CSF. Aβ levels increased due to complexing with the antibody. No significant adverse effects have been reported and a Phase III trial is reported to be launched in the near future. Pfizer has several early phase trials with PF-04360365, a humanized antibody reported to selectively target Aβ1-40. No information regarding outcomes has been released to date. A number of other companies are active in this landscape, but there is limited information regarding the nature of the monoclonal antibodies being used.
An additional approach to passive immunotherapy currently being tested in humans is IVIG (Gammagard; Bayer). Human IVIG can contain naturally occurring anti-Aβ antibodies; however, the amount of these antibodies and their reactivity will likely be relatively low and variable (Szabo et al., 2008; Relkin et al., 2008). Nevertheless, given the long-term safety record and the possibility that IVIG may have additional immunomodulatory effects that could theoretically be beneficial in AD, IVIG is an intriguing alternative approach to anti-Aβ monoclonal antibodies. Several small early phase studies have reported improvement in cognition, decreases in CSF Aβ and increases in plasma Aβ following IVIG administration to AD patients (Relkin et al., 2008). Both Phase II and III studies of IVIG in AD are underway. Although this approach is intriguing, given that the supply of IVIG for current indications is often intermittently insufficient to meet clinical demand, it is unclear whether sufficient IVIG could be collected to support long-term treatment of patients with AD (Bayry et al., 2007).
Targeting Aβ Aggregates
Rationale for the approach
Though inhibiting Aβ production can slow the rate of aggregate formation, it is an indirect way to target the aggregates of Aβ that are largely considered by the field to be the true triggers of AD. More direct approaches are 1) to block aggregate formation using aggregation inhibitors, 2) disrupt preformed aggregates (so called “plaque-busters”) or 3) simply neutralizing the aggregate by direct binding. There are a number of theoretical concerns with such approaches steming from either perceived issues with potency and specificity of small molecules that might act as aggregation inhibitors or from the uncertainties about which aggregate to target as a therapeutic agent might convert a less toxic assembly into a more toxic one. Notably, many anti-Aβ antibodies as well as immune sera generated following active immunization with Aβ can recognize Aβ aggregates to varying extents, suggesting that anti-Aβ immunotherapeutic may work, at least in part, by targeting aggregates (Golde et al., 2009). At least in APP AD mouse models, the rapid reversal of cognitive deficits following immunization would suggest that those anti-Aβ antibodies can “neutralize” certain toxic Aβ species (Dodart et al., 2002).
Progress Aggregation Inhibitors/toxicity blockers
Tramiprosate (3-aminopropane-1-sulfonic acid, Neurochem) is a glycosaminoglycan (GAG) mimetic drug that interacts with the Aβ peptide and is reported to inhibit the transition from random structures to organized β-sheets (McLaurin et al., 1999; McLaurin et al., 2000a; Aisen, 2005). A Phase III trial of tramiprosate failed to show a significant effect on cognition (Wong, 2007). Using post-hoc analysis of the Phase III study which suggested that the patients receiving tramiprosate had less atrophy as a rationale for continued “development”, Neurochem is now pursuing sale of a nutraceutical product that contains large amounts of tramisporaste. Scyllo-inostiol is another compound that has been reported to alter Aβ aggregation (McLaurin et al., 2000b; McLaurin et al., 2006). Though it was initially reported to decrease aggregation of Aβ in vitro and in vivo and improve cognitive impairment in a mouse model of AD, subsequent studies have shown that it can directly neutralize the synaptotoxicity of oligomeric Aβ aggregates in vitro and in vivo (Townsend et al., 2006). Scyllo-inositol (ELND005, Elan) is currently in Phase II testing in humans with AD. Notably, a recent report using magnetic resonance spectroscopy shows that scyllo-inositol appears to be increased in the brain of living patients with AD (Griffith et al., 2007).
PBT2 (Prana) is a second generation compound related to clioquinol (Lannfelt et al., 2008). Clinical development of Clioquinol was halted after Phase II testing due to the reported inability to formulate it in a safe manner. Like clioquinol, PBT2 is a metal chelator that is designed to alter metal dependent Aβ aggregation and redox activity associated with Aβ-metal complexes (Bush and Tanzi, 2002; Adlard et al., 2008; Bush and Tanzi, 2008). In addition, PBT2 is claimed to promote normal cooper and zinc homeostasis in the brain. A Phase II study of PBT2 has been completed in which PBT exhibited a favorable safety profile. (Lannfelt et al., 2008). A significant reduction in Aβ42 was noted in the 250 mg dosing group compared to placebo; however, it should be noted that this drop of ∼50 pg/ml is about 10% of the total CSF Aβ42. Moreover, it is not clear whether a decrease in Aβ42, which is low in patients with AD, represents a beneficial or harmful change.
Targeting Aβ removal
Rationale
Accumulation of Aβ in the brain reflects an imbalance between production and degradation. Based on estimates of the steady state level of Aβ in control brain (∼5-10 pmoles/gm), the half-life of Aβ (∼2-4 hours), and the amount of Aβ that is present in the AD brain (∼10-20 μmoles), it appears that the amount of Aβ present in the brain in end-stage AD represents 2-5 years worth of intracranial Aβ production (Golde et al., 2009; Levites et al., 2006). Though antibodies and aggregation inhibitors might enhance clearance of Aβ, an alternative approach is to activate enzymes or cells that degrade Aβ or Aβ aggregates. The protease activation approach is theoretically attractive as activating an Aβ degrading enzyme could theoretically result in robust clearance due to the catalytic nature of the enzyme and target substrate interaction (Leissring, 2006; Selkoe, 2001b); however, there are two major concerns with these approaches. First, there are concerns that activation of proteases with small-molecules is challenging form a pharmacologic perspective. Second since there are no proteases known to selectively degrade Aβ, activating a protease may also be prone to target based toxicity. The first concern can be partially addressed by targeting an inhibitor or a receptor that regulates the expression of the protease, thereby yielding an indirect increase in protease activity. Again cell-based therapies aimed at enhanced Aβ clearance remain largely theoretical though some proof of concept for this idea has emerged form animal modeling studies. As with proteases, this approach is conceptually limited by the lack of specificity and thus the potential for toxicity.
Progress on Aβ removal
Wyeth has redirected a small molecule plasminogen activator inhibitor-1 (PAI-1) inhibitor (PAZ-417) for potential use in AD. This compound appears to be in a Phase I/II trial for AD. PAI-1 inhibits the activation of plasmin, a protease that can degrade Aβ oligomers, monomers, and fibrils. PAI-1 binds and inhibits tissue plasminogen activator (tPa), which, when active, cleaves plasminogen to plasmin (Melchor et al., 2003). The net result is that, when PAI-1 is inhibited, more plasmin is activated. A recent preclinical study demonstrates that PAZ-417 can augment the activity of tPa and plasmin in the brain of mice and decrease brain and plasma Aβ levels (Jacobsen et al., 2008). Although PAI-1 is a major tPa regulator in the periphery, neuroserpin is the major tPa regulator in the brain. A recent study examining postmortem AD brains showed that the major complexes in the AD brain are between tPa and neuroserpin and not tPa and PAI-1 (Fabbro and Seeds, 2009). Though there is significant homology between PAI-1 and neuroserpin, it is not known whether PAZ-417 is a neuroserpin inhibitor.
Indirect Approaches to altering Aβ levels
Rationale for the approach
APP processing and Aβ metabolism is complex and hundreds of regulators have been described in the literature. The general concept is that one can decrease Aβ production indirectly by altering APP trafficking or levels of factors that influence its cleavage into Aβ (e.g., secretase). Various agents, such as muscarinic agonists, serotonin, glutamate, and estrogens activate α-secretase cleavage of APP [reviewed in (Vardy et al., 2005)], often by activating PKC signaling resulting in stimulation of non-amyloidogenic α-secretase processing of APP (Gandy and Greengard, 1994) (Koo, 1997). Increased α-secretase processing of APP can result in a decrease in β-secretase cleavage and decreased Aβ production in some studies (Skovronsky et al., 2000); however, in other studies it does not (Rossner et al., 2000). Notably, sAPPα has been postulated to be neuroprotective, but it is not clear if increasing its sAPPα above its constitutive levels will enhance neuroprotection.
For a number of drugs being tested in humans or in late phase pre-clinical development, the primary action against the intended target is designed to show cognitive enhancement in a symptomatic fashion, and the effect on reducing Aβ levels is thought to be a secondary target. Developing a compound that can be approved for human use in AD based solely on a symptomatic benefit which also has the potential for disease modification by lowering Aβ is attractive, as it avoids the necessity of a long, expensive Phase III trial before approval for disease treatment. For such drugs, a two step regulatory approval is likely envisaged. The initial Phase II/III studies focus solely symptomatic benefit and biomarker data to support subsequent study as a disease modifying agent. If approved, such a therapy could then be assessed for their ability to modify disease in a phase IV trial.
Progress G-protein coupled receptor (GPCR) targets
A number of agonists or partial agonists of GPCRs are being developed that have the potential to enhance cognition either through their effects on the receptor or downstream effects on neurotransmission. These compounds in theory also alter Aβ production by activating the non-amyloidogenic α-secretase pathway of APP processing. Multiple α-7 nicotinic acetylcholine receptor (α-7NAChR) agonists are in various phases of late pre-clinical studies or in phase I/II human trials. Notably, these compounds are also being evaluated for other indications. ABT-089 (Abbott) is also being evaluated for efficacy in Attention-Deficit/Hyperactivity Disorder (ADHD). AZD0328 (AstraZeneca) and R-4996 (Memory Pharmaceuticals/Roche) are evaluating α-7NAChR agonists in schizophrenia. α-7NAChR agonists may have multiple effects on cognition, improving pre-attentive sensory processing, attention and working and recognition memory (Leiser et al., 2009). In addition to possible effects on APP processing and lowering Aβ through α-secretase (Qi et al., 2007; Mousavi and Hellstrom-Lindahl, 2009), α-7NAChR agonists have also been implicated as direct neuroprotective agents (Shimohama, 2009), and as anti-inflammatory agents in the brain (de Jonge and Ulloa, 2007). Though a body of literature exists suggesting that Aβ42 may directly interact with α-7NAChR, there are a number of conflicting reports, and it is not clear whether such interaction mediates any pathophysiologic processes relevant to AD (Wang et al., 2000; Small et al., 2007).
PRX-03140 (Epix/GSK), a 5-HT4 agonist, is currently in Phase II trials for AD. Several groups have shown that 5-HT4 agonists stimulate α-secretase cleavage of APP and also increase a neurotrophic factors (Strobel, 2008d). There is no published data showing that the compounds lower Aβ production in the brain; however, 5-HT4 agonists appear to have positive effects on learning and memory and, in some studies, have been shown to increase acetylcholine release. Etazolate (EHT 0202, Exonhit Therapeutics), a GABA A receptor modulator that also weakly inhibits phosphodiesterase 4, is another compound that is in Phase II testing in humans with AD (Marcade et al., 2008). It has been reported to stimulate α-secretase, but no data on its effect on Aβ have been reported.
Decreasing APP expression
A final way to alter Aβ production would be to decrease APP expression at either the transcriptional or translational level. Without a clear understanding of the normal function of APP in the brain, reducing APP levels could conceivably yield undesired physiological effects. Surprisingly, phenserine, a compound developed as a noncompetitive ACHE inhibitor, appears to interfere with translation of APP, and in some studies lowers Aβ production (Greig et al., 2005). However, the dose needed to elicit this action is much greater than the dose needed to elicit the anticholinesterase activity limiting the clinical potential of the compound as dual action anticholinesterase anti-Aβ therapy. A Phase III trial of phenserine failed to show efficacy in AD though the trial design may have been suboptimal. Phenserine and other compounds may be targeting an iron regulated response element in the 5′ untranslated region of the APP mRNA. Screens for compounds that reduce expression of APP by targeting this element have identified several FDA approved drugs that reduce APP expression and appear to be capable of lowering APP and Aβ levels in mice (Utsuki et al., 2006).
Targeting Aβ: Imaging agents
Rationale
The ability to detect Aβ accumulation as amyloid in the brain, while not a therapeutic agent, is likely to help guide both future trial design and be used to follow efficacy of anti-Aβ therapies (Klunk and Mathis, 2008); therefore advances with imaging agents will be briefly reviewed here. These isotopically labeled compounds bind to Aβ amyloid and can be used to assess cerebral amyloidosis using PET or SPECT scanning.
Progress
[C-11] Pittsburgh Compound-B (PIB) has been the most widely used agent for imaging Aβ amyloid. It is in use at more then 40 centers and has been tested on more than 3,000 individuals (Klunk et al., 2004). [F-18] FDDNP was the first fluorine tracer to be used in humans, but it appears to be a sub-optimal agent as it has a relatively low signal to noise ratio. (Agdeppa et al., 2003) [C-11] PIB has proven highly useful and certainly detects amyloid present in AD, MCI, and even in cognitively intact individuals (Klunk and Mathis, 2008). While cognitively normal, individuals with PIB-positive scans are expected to “convert” to AD as they are followed. Although some anecdotal data exists that this is likely to be the case, substantial cohorts of PIB-positive, cognitively intact subjects have not been followed for an extended period of time to determine the risk for conversion to AD (Fagan et al., 2005; Reiman et al., 2009; Jack et al., 2009; Mormino et al., 2008; Aizenstein et al., 2008). A myriad of new imaging agents are soon to be or currently in Phase II studies in humans including [F-18] BAY94-9172, F-18] Av-45, [F-18] AH110690, [C-11]AZD2995, and [C-11]AZD2184 [reviewed in (Klunk and Mathis, 2008)]. How these new amyloid tracers will perform relative to [C-11]PIB, will have to be empirically determined. [F-18] labeled probes do have one significant advantage in that their longer half-life (110 minutes compared to 20 minutes for [C-11]) expands the availability beyond medical centers with on-site cyclotrons (Klunk and Mathis, 2008). Given the number of probes in the clinic, it is likely that over the next few years a great deal of information will be obtained regarding both the natural history of amyloid deposition in humans and its impact upon the development and progression of AD. It is also likely that amyloid tracers will be used to evaluate effects of anti-amyloid therapy, though this will require additional validation with respect to the ability of the probe to quantitatively discriminate between varying amyloid loads in the brain.
Targeting Tau. Have we identified tractable therapeutic targets?
Accumulation of hyperphosphorylated tau as paired helical filaments within neurofibrillary tangles and in neuritic processes are intracellular hallmarks of AD pathogenesis (Selkoe, 2001a; Lee and Trojanowski, 1992). Both genetic and animal modeling studies closely link abnormal tau metabolism to neuronal dysfunction and death. The identification of mutations in the tau (MAPT) gene as the cause of frontotemporal dementia with parkinsonism linked to chromosome 17 (FTDP-17 MAPT) demonstrated that alterations in tau, by themselves, are sufficient to cause neurodegeneration (Hutton, 2000; Hutton et al., 1998; Spillantini et al., 1998; Lee et al., 2001; Boeve and Hutton, 2008). Point mutations within the tau gene as well as splice site mutations within the intron and point mutations within the coding region that also alter splicing all cause tau pathology and neurodegeneration in humans (Hutton, 2000; Goedert and Jakes, 2005). Transgenic animal models of FTDP-17 MAPT-associated mutations show that these alterations are tightly associated with NFT formation and neurodegeneration (Lewis et al., 2000; Gotz et al., 2001).
A conservative interpretation of the genetic and animal modeling data from the study of FTDP-17 MAPT would only support tau as a therapeutic target in FTDP-17 MAPT. However, given the prominence of tau pathology in the AD brain, the lack of any substantial distinguishing features between the tau pathology present in FTDP-17 MAPT and AD, and experimental evidence that Aβ can drive tau pathology in vivo, tau has become an attractive theoretical target in AD (Lee and Trojanowski, 2006; Seabrook et al., 2007). Some investigators have postulated that tau pathology arises independently of Aβ pathology, and that AD, at least in some circumstances, may represent a convergence of two distinct pathological process --an Aβ amyloidosis and a tauopathy rather then a linear cascade in which the Aβ amyloidosis drives the tauopathy (Small and Duff, 2008). As opposed to the plethora of therapeutic strategies that target Aβ, it has been much more challenging for the field to reduce tau therapeutics to practical applications that can be tested and validated in preclinical models and then advanced towards the clinic. Indeed, the relative paucity of tau therapeutics can be attributed to 1) the uncertainty regarding the relationship between specific alteration in tau and induction of neurodegeneration and 2) a paucity of clearly defined druggable targets that directly or indirectly modulate tau in a way that should be protective from neurodegeneration. Despite these uncertainties, tau therapeutics are currently being developed and several are in humans clinical trials. In the following sections, we will describe the rationale for the strategies that are likely to hold some promise in the near future.
Targeting Tau production
Rationale
Even though the exact neurotoxic species of tau has not been definitively identified, decreasing tau synthesis or a specific tau isoform is proposed to decrease the likelihood that tau will aggregate into a neurotoxic species. There is some genetic support for this concept with respect to corticobasal degeneration and progressive supranuclear palsy which are both tauopathies. In these diseases certain H1 MAPT haplotypes that may influence gene expressing, splicing or both are associated with risk for disease (Kauwe et al., 2008; Mukherjee et al., 2007; Rademakers et al., 2005). A more selective approach is based on the identification of splice site mutations in tau that cause FTDP-17 which result in an imbalance between exon 10+ (4R) and exon 10-(3R) isoforms of tau might favor tangle pathology (Hutton et al., 1998). Mutations in tau lead to increased production of exon 10 containing isoforms of tau with 4 microtubule binding domains, and these isoforms are almost exclusively deposited in tangles in many families with FTDP-17 MAPT. Thus, it may be possible to develop tau therapeutics that are designed to enhance the mRNA splicing events resulting in exclusion of exon 10.
Progress
Therapeutics targeting tau production remains in the conceptual stage, though some preclinical studies do provide proof of concept that lowering tau production can have a beneficial disease modifying effect. Suppression of mutant (P301L) tau production even after significant pathology is present in inducible tau mouse model (rTg4510) demonstrates that significant therapeutic benefit might be gained by this strategy (Santacruz et al., 2005). Cognitive deficits are greatly attenuated in the rTg4510 mice following transgene suppression even though the neurofibrillary pathology progressed. Though tau is an abundant microtubule stabilizing protein, tau knockout mice have a relatively mild phenotype indicating that tau expression could be reduced without significant side-effects (Harada et al., 1994; Takei et al., 2000; Ikegami et al., 2000). Another preclinical study in which mice lacking tau were crossed with transgenic hAPP mice demonstrated that cognitive deficits were ameliorated in the crossed mice, suggesting that reducing tau might decrease cognitive impairment induced by Aβ aggregates (Roberson et al., 2007). Though several approaches for altering exon 10 splicing and thereby increasing the ratio of 3R to 4R tau have been published; translation of these to therapeutics appears challenging. A screening method for identifying small molecule that will control exon 10 splicing by binding the 3′ tau hnRNA exon 10 stem loop has been reported, though the initial screen only yielded one hit (Donahue et al., 2006; Donahue et al., 2007).
Altering Tau Aggregation
Rationale
Several of the point mutations that cause FTLD-17 MAPT increase the propensity of the mutant tau to aggregate, both in vitro and when expressed in cells (Gamblin et al., 2003; Gamblin et al., 2000 von Bergen, 2001 #3083; Nacharaju et al., 1999). There is a good correlation between the propensity of mutant tau to aggregate and its toxicity to cells and in mice. A concern is that some in the tau field propose that misfolded non-aggregated tau might be the real toxic entity. It is therefore possible that compounds that block tau aggregation (and not misfolding) might increase toxicity.
Progress
A plethora of tau aggregation inhibitors have been identified. One of these compounds, a non-neuroleptic phenothiazine, methylene blue (Rember™, TauRx Therapeutics), has completed a Phase II trial in humans (Wischik et al., 1996; Khlistunova et al., 2006). The 24 week trial did not meet global endpoints with respect to altering cognition. Claims of efficacy following post-hoc subgroup analysis have generally been met with skepticism. Several other tau aggregation inhibitors have been identified, some of which also inhibit Aβ aggregation and α-synuclein aggregation (Masuda et al., 2006). It is quite likely that methylene blue can also inhibit Aβ aggregation. Though these compounds are effective in vitro, no proof of concept study has shown them capable of inhibiting tau aggregation in vivo. Nevertheless, the concept that a small molecule inhibitor that could block aggregation of multiple proteins involved in neurodegeneration is intriguing. Future results studying the effect of these compounds in animal models are eagerly awaited.
Altering Tau phosphorylation
Rationale
Tau contains multiple phosphorylation sites and phosphorylation appears to control the binding affinity for microtubules. Increased phosphorylation of the tau protein from an average of two to three phosphates in normal brain to an average of nine phosphates per molecules is seen in AD. (Lee et al., 2001; Gustke et al., 1992; Goedert et al., 1992; Geschwind, 2003; Friedhoff et al., 2000). By decreasing tau's affinity for microtubules, hyperphosphorylation of tau could itself play a role in the pathogenesis by promoting microtubule network breakdown. Lack of neurodegeneration in tau deficient mice indicates that such a “loss of function” may not be sufficient to drive neurodegeneration (Harada et al., 1994; Takei et al., 2000; Ikegami et al., 2000); however, interpretation of tau knockout results must consider the existence of compensatory mechanisms. Hyperphosphorylated, abnormally folded tau or tau aggregates, may exert direct toxic effects on neurons. It is still debated whether phosphorylation of tau drives aggregation, results in toxicity, or is a consequence of aggregate formation. In any case, the intriguing link between phosphorylation and tau pathology has provided the impetus to examine the role of kinase inhibitors as potential therapeutics targeting tau. The tau kinases MARK, CDK5, GSK3, PKA, and ERK1/2 have all been implicated as potential kinase targets for tau therapeutics [reviewed in (Schneider and Mandelkow, 2008)]. Because all kinases that phosphorylate tau also regulate numerous signaling pathways, kinase inhibitor approaches face challenges with respect to toxicity and specificity. Moreover, it is not clear whether targeting any single kinase or targeting multiple kinases with a semi-selective kinase inhibitor will be a better approach. As an alternative to kinase inhibition, activation of phosphatases especially protein phosphatase-2A is a theoretical strategy for reducing tau phosphorylation.
Progress
Despite the lack of clear insight into the “best” target kinase, efforts are underway to develop CDK5 inhibitors, GSK3β inhibitors, and other tau kinase inhibitors. NP 12 (Noscira) a GSK-3β inhibitor is being tested in humans. A mixed kinase inhibitor, SRN-003-556, targeting ERK2/CDC2, GSk3β, PKA and PKC, showed efficacy in a tau mouse model (JNPL3) by reducing soluble aggregated hyperphosphorylated tau and delaying the motor phenotype (Le Corre et al., 2006). Other preclinical studies also suggest that tau phosphorylation is potentially a disease modifying target. Lithium, a dual action GSK3 inhibitor, and a second GSK3 inhibitor, AR-A014418, reduced tau phosphorylation, tau accumulation, and axonal degeneration in tau mice (Nakashima et al., 2005; Noble et al., 2005). Lithium also has been shown to modulate Aβ production, though it is not clear whether it has a beneficial or adverse effect on Aβ production. In one report, lithium was shown to reduce Aβ production through inhibition of GSK3-α (Phiel et al., 2003). A second report suggested that lithium might actually increase Aβ production independently of effects on GSK3 (Feyt et al., 2005). An open label study of lithium to evaluate safety and tolerability in patients with mild to moderate AD has been completed. 22 patients were enrolled and dosing lasted for up to 1 year. A high discontinuation rate was noted as only 8 patients completed the trial, though the side effects were mild and reversible. Efficacy was not assessed in this study (Macdonald et al., 2008).
Targeting tau Chaperones
Rationale
Chaperone proteins that that regulate the aggregation and folding of tau or potentially mediate clearance of misfolded or aggregated tau are being explored as therapeutic targets (Gendron and Petrucelli, 2009). The main challenge to this approach is that the chaperone systems need to be upregulated, and in many cases there is no known pathway that will lead to upregulation of the specific chaperone. For example, there is no known way to pharmacologically activate or increase expression of Pin1, which is an attractive target that has been reported to isomerize phosphorylated tau and restores the ability of phosphorylated tau to bind microtubules, and eventually promote dephosphorylation of tau by PP2A phosphatase (Lu et al., 1999; Zhou et al., 2000; Pastorino et al., 2006). Likewise the ubiquitin ligase CHIP can polyubiquitinate tau and appears to play crucial role in preventing accumulation of phospho-tau (Dickey et al., 2006a; Shimura et al., 2004). However, there is no proof of concept that CHIP can be aselectivly targeted.
Progress
HSP90 inhibitors that induce a heat shock response increase the degradation of phospho-tau at specific sites in vitro and in vivo (Dickey et al., 2006b; Dickey et al., 2007; Luo et al., 2007). HSP70 and HSP90 also promote tau association with microtubules and reduce phospho-tau levels (Petrucelli et al., 2004; Dou et al., 2003). The Hsp90 chaperone complex in affected areas of AD brain is more than two orders of magnitude more sensitive to HSP90 inhibition by ansamycin ATP-competitive inhibitors than the HSP90 in control tissues or unaffected areas of AD brain (Dickey et al., 2007). This strongly suggests that HSP90 inhibitors possess an exploitable therapeutic index for the treatment of tauopathies. If such inhibitors prove effective in tau mouse models, it may be possible to rapidly test such inhibitors in humans with AD or other tauopathies (Dickey et al., 2007).
Tau: Microtubule stabilizers
Rationale
Based on the notion that sequestration of tau in inclusions or in a phosphorylated state results in loss of the normal microtubule stabilizing function of tau, MT-stabilizing agents have been tested in tau mouse models.
Progress
Paclitaxel has been used clinically as a potent anticancer drug because of its antimitotic activity through MT stabilization. When administered to tau mice in a micellar formulation, it restored fast axonal transport in spinal axons, increased microtubule number and ameliorated motor impairments (Zhang et al., 2005). Epothilone, (Bristol Myers Squib) another microtubule stabilizer, has also shown promising results in initial studies in tau rTg4510 mice showing improved cognitive performance and markedly reduced tau pathology (Wartmann and Altmann, 2002; Landhuis, 2008).
Tau Immunotherapy
Rationale
Antibodies targeting the pathological conformers of tau might clear or suppress tau pathology. Though theoretically attractive, there are concerns that antibodies are not optimal agents for targeting intraneuronal proteins. Additionally, the lack of clarity regarding which conformer of tau should be targeted remains a concern.
Progress
One preclinical study in P301L (JNPL3) mice has provided some proof of concept with respect to the possibility that tau immunotherapy could have efficacy (Sigurdsson, 2008). A vaccine based on a phosopho-tau peptide was able to suppress tau accumulation and improve motor deficits present in these mice. Also direct injection of the purified mouse polyclonal antibody provided evidence that the antibody could actually get in the brain and bind to intercellular tau structures.
Magic Shotgun Therapies that could target both tau and Aβ
Rationale
It is possible that through a pleiotropic mechanism, a single agent could alter both tau and Aβ pathology. Such an approach clearly has merits as it would avoid polypharmacy in an elderly population. The main issue is that identifying compounds with this type of mechanism of action is likely to be serendipitous, at least until the signaling pathways that link Aβ aggregate accumulation to tau pathology are definitively identified.
Progress
Lithium is one such agent that has been discussed previously; however, data on its effect on Aβ are conflicting. Statins have been shown in independent studies in APP and tau mouse models to partially suppress Aβ and tau pathology (Boimel et al., 2009; Fassbender et al., 2001; Refolo et al., 2001). Fairly strong epidemiologic data that statin use is associated with decreased risk for developing AD; however, Phase II studies have failed to show efficacy in AD (Sparks et al., 2005). In 3X-TgAD mice a number of preclinical studies have identified agents that can reduce both Aβ pathology and tau pathology including ibuprofen (Caccamo et al., 2006; McKee et al., 2008; Oddo et al., 2004).
Separating wheat from chaff: the challenge of matching trial design and triage decisions regarding which therapies to move toward investigational new drugs (INDs) based on preclinical studies
The AD field can do well to consider failed efforts to develop neuroprotective drugs to treat stroke. Such failures have greatly reduced the commercial interest in developing such types of treatments despite the fact that stoke remains among the leading causes of mortality and morbidity. In 2006, O'Collins and colleagues published a report of 1,026 candidate stroke drugs, 912 of which were only tested in animal models, 97 in humans and animal models and 17 only in humans(O'Collins et al., 2006). From these therapeutic candidates, only tPA has been approved for human use. Notably, based on the animal modeling studies, O'Collins and colleagues conclude that there was little efficacy data that would have elevated tPA above the other therapies. Moreover, they make several general remarks regarding the factors that may have contributed to the failure of most stroke drugs in the clinic. With respect to relevance for tau and Aβ therapy for Alzheimer's disease, the most important of these factors that they identified for stroke drugs may be 1) the conditions of the clinical trial fail to replicate the conditions under which the drug worked in the animal model and 2) in many cases, extensive animal testing was often only performed after a failure in the clinic (often in an attempt to explain that failure) (O'Collins et al., 2006). Given that critical events in stroke occur over a few hours versus the prolonged development of Alzheimer's Disease, one could argue that the failure of drug therapies for stroke may not parallel the likely results from drug trials for AD. Similar issues; however, have been raised with respect to preclinical studies in Parkinson's disease and amyotrophic lateral sclerosis, diseases that have protracted course more akin to that of Alzheimer's Disease (Waldmeier et al., 2006; Aggarwal and Cudkowicz, 2008).
To date only two targeted anti-Aβ therapies, tarenflurbil and tamiprosate, have “failed” to show efficacy following Phase III therapeutic studies for AD. Given the time, cost, and complexity of disease modifying trials in AD, it is likely that the enthusiasm for developing disease modifying therapies targeting tau and Aβ will rapidly erode if there are continued failures in therapeutic phase III studies. Indeed, given that a single phase III therapeutic study in AD often costs upwards of a $100 million, a handful of more failures in phase III for any category (e.g. anti-Aβ, anti-tau) of disease modifying AD therapy will probably be sufficient to significantly diminish commercial interest in further pre-clinical and clinical development. As reviewed above there are several dozen anti-Aβ therapies and a few anti-tau therapies in the clinic. Yet, in the preclinical pipeline, there are probably several hundreds agents being developed. Clearly, it is worth some discussion on how we might collectively make better triage decisions regarding what therapies to move forward, what resources would ensure the best study design and, if we move them forward, how to match trial design to the conditions under which the drug worked in the animal model.
Before making some recommendations regarding triage decisions and matching trial design, we need to have real clarity on the relevance of our animal models to AD. APP transgenic mice that express mutant APPs linked to the development of human AD are good models of amyloid deposition, plaque associated inflammation, and neuritic dysfunction surrounding the plaque (Price and Sisodia, 1998; Games et al., 2006; Hsiao, 1998; Spires and Hyman, 2005). Although some of these models also show synaptic loss, they do not show the neuronal loss typical of even the earliest stages of AD and they do not show significant tau pathology. Cognitive changes present in these models are subtle and in some cases rapidly reversible. Moreover, the cognitive changes vary between each model with respect to the timing, progression and relationship to pathological features. Thus, APP mice appear to be good models of amyloid deposition and can be considered to model prodromal AD. However, it remains an open question as to whether they are models of cognitive dysfunction in AD, and they certainly are incomplete models of neurodegenerative component of AD. Thus, any study of anti-Aβ therapy is likely to predict one outcome in humans --effects on amyloid pathology. Cognitive effects and effects on neurodegeneration could follow, but that can't be inferred with certainty using such mouse models. Tau mouse models based on FTDP-17 MAPT mutations develop NFT pathology and some do show marked neurodegeneration as well as cognitive or motor changes depending on the location in which the pathology primarily arises (Hutton et al., 2001; Gotz et al., 2007). However, it should be noted that such models are truly models of tauopathy in FTDP-17 MAPT, and it is still a leap of faith (although a reasonable one, given the relative lack of alternative tauopathy models) that the tauopathy in FTDP-17 MAPT is a model of tauopathy in AD. Depending on the timing of the study, it can be argued that in mice such as the inducible tau P301L rTg4510, which exhibit tremendous neuronal cortical and hippocampal loss, one can conduct preclinical studies that are more reflective of a therapeutic trial in humans rather then a prevention trial (Santacruz et al., 2005).
If a preclinical study validating a specific anti-Aβ therapy in an APP transgenic mouse model is predictive of the drugs behavior in a primary prevention trial, then by performing therapeutic human trials of these agents, we are greatly increasing the chance that the therapy will fail to show efficacy. Though hundreds of interventions that target Aβ can be shown to suppress Aβ deposition when administered in a primary prevention paradigm (i.e., before the onset of Aβ deposition), most of these treatments actually have only modest effects on pathology with reduction reported typically less than 50%. Moreover, in the rare cases where the treatment is systematically evaluated in mice with varying degrees of amyloid deposited at the initiation of treatment, the efficacy in terms of suppression of Aβ deposition decreases with increased levels of deposits at initiation. An exception to this generalization is Aβ immunotherapy, which in certain preclinical studies can have up to 90% reduction amyloid loads in a prevention setting and may result in clearance of preexisting deposits. Nevertheless, even Aβ immunotherapy appears to be much more effective in mice if started prior to the onset of Aβ deposition.
A resource that would greatly enhance drug trials in animal models and their translation into the clinic would be the availability of a robust, reliable, non-invasive biomarker for AD. While imaging Aβ amyloid is now reliably used in many centers (Klunk and Mathis, 2008), animal imaging during pre-clinical trials would only be possible for a small number of groups. Ideally, pre-clinical efficacy for AD therapeutics could be repeatedly assessed within the same trial by a non-invasive, low cost, reliable biomarker. Such a biomarker could then be used as a standard measure across different trials and models, allowing a more informative comparison of potential agents in model systems. Importantly, the ideal biomarker would be equally usable in clinical trials, allowing direct comparisons between the pre-clinical and clinical outcomes within a shorter time period. Although notable efforts are underway to utilize plasma Abeta as a reliable biomarker, this assay is not yet in routine clinical use (Graff-Radford et al., 2007).
So how might we make more informed decision about which therapeutic approaches targeting Aβ and tau we should advance towards the clinic targeting Aβ and tau? First, we might want to consider that the animal model studies should reflect the nature of the human trial. Such an alignment could work in one of two ways. It could mean that for therapeutic trials, we are testing the drug in mouse models with significant preexisting pathology, and won't move them forward in a therapeutic trial for AD unless they show efficacy against the target (tau or Aβ) in that setting. Alternatively, it could mean that, if the therapy is working in the pre-clinical study in a prevention or early intervention paradigm, then the therapy needs to be tested in a prevention study in humans, or at a minimum tested in a mild-cognitive impairment of the AD type population. Second, we need ways to better compare the efficacy of various therapies in the preclinical models. Often for Aβ therapies, the preclinical validation consists of a single disease modification study in a single mouse model with relatively low group sizes. This paradigm is likely to be the case for preclinical testing of tau therapies as well. There are also variations in the time of initiation, duration of treatment, and the methodologies for analysis. For a number of reasons, it is highly unlikely that a truly systemic approach to preclinical studies can be instituted. Some useful guidelines would be to insure that the study is sufficiently powered, reproducible in at least two independent experiments (and preferably in two different models), and tested in different paradigms with respect to extent of pathology at initiation of treatment. The magnitude of the effect should also be considered, though given the lack of systematic and uniform methodology for preclinical studies, inferring relative efficacy will be confounded. Nevertheless, the larger the effect and the longer the treatment can be extended and still show efficacy in the mouse model would seem to bode well for human studies. Third, we should use the animal models to prove definitively that therapy effectively hits the target. If this requires brain penetration, there should be sufficient and compelling data on the pharmacokinetics to insure that effective concentrations of the therapeutic are present in the appropriate site.
Despite these concerns, there are several reasons that we probably should remain more optimistic about disease modifying strategies for AD than stroke therapy. The first is that we can design trials which control many more variables then those in acute stroke. Even with fairly restrictive inclusion criteria, it is possible to recruit sufficient subjects to fully power an AD study. Though the tarenflurbil (Flurizan) trial failed from the standpoint that it did not show efficacy of tarenflurbil versus placebo, the trial was clearly sufficiently powered and conducted in away that if a relatively small alteration in rate of decline was induced by tarenflurbil it would have been detectable. Furthermore, even though there are huge obstacles to primary prevention studies in AD, there is growing movement in the field that recognizes that those obstacles must be overcome (Khachaturian et al., 2009; Golde, 2009). Even if drugs fail in the therapeutic setting, if they are proven safe, we may be able to test them for efficacy in a primary prevention setting. The second reason is that we have the ability to monitor multiple biomarkers that provide a number of important readouts (Blennow, 2004). As noted above, we now have the ability to image amyloid in the brain. We also have the ability to monitor brain volume loss over time using serial registered MRI (Klunk and Mathis, 2008; Fox et al., 2001; Jack et al., 2002). Though not validated in the setting of response to a disease modifying agent, a number of biomarkers are emerging that may be useful with respect to providing surrogate measures of disease modification. At least for therapies targeting Aβ production, it will also be possible to evaluate a given therapies effect on Aβ in plasma and CSF. The Alzheimer's Disease Neuroimaging Initiative (ADNI), a major international effort, is underway to evaluate both imaging and biomarkers for AD (Jack et al., 2009; Mueller et al., 2005; Leow et al., 2009; Shaw et al., 2009).
If we have learned any lessons in early years of testing potential disease modifying therapies for AD, it would be that we must be cautious about claims of efficacy in potentially disease modifying Phase II therapeutic studies in AD (Khachaturian et al., 2009; Schneider and Lahiri, 2009). We must be especially cautious if the global pre-specified efficacy endpoint is not met. Though subgroup analysis may be useful in terms of guiding future trial design, it appears that such analysis can also be very misleading. Hopefully, as we become more sophisticated with respect to the interpretation of imaging and biomarker studies, we can infer with a greater degree of certainty whether a given therapy is in fact hitting target and potentially having a disease modifying effect.
Conclusions
The holy grail of AD therapy is “disease modification”. Current therapies, cholinesterase inhibitors and memantine, provide temporary symptomatic benefit to some individuals, but their overall efficacy is very modest, and there is almost no evidence that they modify the disease course. A plethora of anti-Aβ and a few anti-tau therapies have entered clinic trials for AD with several anti-Aβ therapies have failing to show disease modifying effects. However, both of these agents, tarenflurbil and tamiprosate, were certainly not optimal drugs with respect to ability to actually hit their intended target in humans. Tarenfluribil is a very weak GSM relative to other GSMs that have been recently reported, and tamiprosate is not a potent Aβ aggregation inhibitor. Furthermore, there was no solid evidence that either hit target in the human studies. Though a small but vocal group of investigators in the field argue that such studies do invalidate Aβ as a target, these failed studies simply do not provide a rigorous test of the hypothesis that targeting Aβ will have a therapeutic effect in AD. Even if additional failures of anti-Aβ and even anti-tau therapy occur in the therapeutic setting, we must, as a field acknowledge that these therapies that target disease triggers are much more likely to be effective when given to asymptomatic at risk individuals (Golde, 2009).
Reflecting on the history of failed therapeutic development for stroke, we must also be willing to critically examine our own preclinical studies in partial animal models of human AD, and use them to optimally design clinical trials that are more likely to show success (O'Collins et al., 2006). It may be that therapies targeting tau and Aβ will only work in a prophylactic fashion. As we have little certainty about what pathways downstream of Aβ and tau to target and therefore a very poor roadmap for developing alternative disease modifying therapies, we must work collectively to develop creative paradigms that are ethically acceptable that will enable prophylactic testing of such therapies.
Acknowledgments
This work was supported by Mayo Clinic (to TG, LP, and JL), National Institutes of Health/National Institute on Aging Grants RO1AG18454 (T.G.), RO1AG29886, P01AG25531, R01AG026251 (L.P.) and P01-AG17216-08 (L.P.), National Institutes of Health/National Institute of Neurological Disorders and Stroke Grant RO1NS39072 (T.G.) R21NS055698 (L.P.). and R21NS059363 (L.P.). National Institute of Neurological Diseases and Stroke R01-NS046355(J.L.), Johnnie B. Byrd Sr. Alzheimer's Research Institute and Center 2005A108 (J.L.), the Alzheimer's Association IIRG-06-27277-1 (to J.L.). T.G. is supported by Rotarians in South Carolina, Georgia and North Carolina through the CART fund.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Terry RD. Alzheimer's disease. In: Davis RL, Robertson DM, editors. Textbook of Neuropathology. Williams & Wilkins; Baltimore: 1985. pp. 824–841. [Google Scholar]
- Alzheimer A, Stelzmann RA, Schnitzlein HN, Murtagh FR. An English translation of Alzheimer's 1907 paper, “Uber eine eigenartige Erkankung der Hirnrinde”. Clin Anat. 1995;8:429–431. doi: 10.1002/ca.980080612. [DOI] [PubMed] [Google Scholar]
- Golde TE. Disease modifying therapy for AD? J Neurochem. 2006;99:689–707. doi: 10.1111/j.1471-4159.2006.04211.x. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ. Alzheimer's disease: genes, proteins, and therapy. Physiol Rev. 2001a;81:741–766. doi: 10.1152/physrev.2001.81.2.741. [DOI] [PubMed] [Google Scholar]
- Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science. 2002;297:353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- Price DL, Sisodia SS, Borchelt DR. Genetic neurodegenerative diseases: the human illness and transgenic models. Science. 1998;282:1079–1083. doi: 10.1126/science.282.5391.1079. [DOI] [PubMed] [Google Scholar]
- Golde TE. The Abeta hypothesis: leading us to rationally-designed therapeutic strategies for the treatment or prevention of Alzheimer disease. Brain Pathol. 2005;15:84–87. doi: 10.1111/j.1750-3639.2005.tb00104.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider A, Mandelkow E. Tau-based treatment strategies in neurodegenerative diseases. Neurotherapeutics. 2008;5:443–457. doi: 10.1016/j.nurt.2008.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee VM, Trojanowski JQ. Progress from Alzheimer's tangles to pathological tau points towards more effective therapies now. J Alzheimers Dis. 2006;9:257–262. doi: 10.3233/jad-2006-9s328. [DOI] [PubMed] [Google Scholar]
- Golde TE, Das P, Levites Y. Quantitative and mechanistic studies of abeta immunotherapy. CNS Neurol Disord Drug Targets. 2009;8:31–49. doi: 10.2174/187152709787601830. [DOI] [PubMed] [Google Scholar]
- Morgan D. Immunotherapy for Alzheimer's disease. J Alzheimers Dis. 2006;9:425–432. doi: 10.3233/jad-2006-9s348. [DOI] [PubMed] [Google Scholar]
- Glabe CC. Amyloid accumulation and pathogensis of Alzheimer's disease: significance of monomeric, oligomeric and fibrillar Abeta. Subcell Biochem. 2005;38:167–177. doi: 10.1007/0-387-23226-5_8. [DOI] [PubMed] [Google Scholar]
- Tanzi RE. The synaptic Abeta hypothesis of Alzheimer disease. Nat Neurosci. 2005;8:977–979. doi: 10.1038/nn0805-977. [DOI] [PubMed] [Google Scholar]
- Klein WL, Stine WB, Jr, Teplow DB. Small assemblies of unmodified amyloid beta-protein are the proximate neurotoxin in Alzheimer's disease. Neurobiol Aging. 2004;25:569–580. doi: 10.1016/j.neurobiolaging.2004.02.010. [DOI] [PubMed] [Google Scholar]
- Lesne S, Koh MT, Kotilinek L, Kayed R, Glabe CG, Yang A, Gallagher M, Ashe KH. A specific amyloid-beta protein assembly in the brain impairs memory. Nature. 2006;440:352–357. doi: 10.1038/nature04533. [DOI] [PubMed] [Google Scholar]
- Shankar GM, Li S, Mehta TH, Garcia-Munoz A, Shepardson NE, Smith I, Brett FM, Farrell MA, Rowan MJ, Lemere CA, Regan CM, Walsh DM, Sabatini BL, Selkoe DJ. Amyloid-beta protein dimers isolated directly from Alzheimer's brains impair synaptic plasticity and memory. Nat Med. 2008;14:837–842. doi: 10.1038/nm1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klyubin I, Betts V, Welzel AT, Blennow K, Zetterberg H, Wallin A, Lemere CA, Cullen WK, Peng Y, Wisniewski T, Selkoe DJ, Anwyl R, Walsh DM, Rowan MJ. Amyloid beta protein dimer-containing human CSF disrupts synaptic plasticity: prevention by systemic passive immunization. J Neurosci. 2008;28:4231–4237. doi: 10.1523/JNEUROSCI.5161-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauren J, Gimbel DA, Nygaard HB, Gilbert JW, Strittmatter SM. Cellular prion protein mediates impairment of synaptic plasticity by amyloid-beta oligomers. Nature. 2009;457:1128–1132. doi: 10.1038/nature07761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng IH, Scearce-Levie K, Legleiter J, Palop JJ, Gerstein H, Bien-Ly N, Puolivali J, Lesne S, Ashe KH, Muchowski PJ, Mucke L. Accelerating amyloid-beta fibrillization reduces oligomer levels and functional deficits in Alzheimer disease mouse models. J Biol Chem. 2007;282:23818–23828. doi: 10.1074/jbc.M701078200. [DOI] [PubMed] [Google Scholar]
- Dodart JC, Bales KR, Gannon KS, Greene SJ, DeMattos RB, Mathis C, DeLong CA, Wu S, Wu X, Holtzman DM, Paul SM. Immunization reverses memory deficits without reducing brain Abeta burden in Alzheimer's disease model. Nat Neurosci. 2002;5:452–457. doi: 10.1038/nn842. [DOI] [PubMed] [Google Scholar]
- Walsh DM, Selkoe DJ. Deciphering the molecular basis of memory failure in Alzheimer's disease. Neuron. 2004a;44:181–193. doi: 10.1016/j.neuron.2004.09.010. [DOI] [PubMed] [Google Scholar]
- Walsh DM, Selkoe DJ. Oligomers on the brain: the emerging role of soluble protein aggregates in neurodegeneration. Protein Pept Lett. 2004b;11:213–228. doi: 10.2174/0929866043407174. [DOI] [PubMed] [Google Scholar]
- Glabe CG. Common mechanisms of amyloid oligomer pathogenesis in degenerative disease. Neurobiol Aging. 2006;27:570–575. doi: 10.1016/j.neurobiolaging.2005.04.017. [DOI] [PubMed] [Google Scholar]
- Kelly JW. Attacking amyloid. N Engl J Med. 2005;352:722–723. doi: 10.1056/NEJMcibr044231. [DOI] [PubMed] [Google Scholar]
- Jarrett JT, Berger EP, Jr, L PT. The carboxy terminus of β amyloid protein is critical for the seeding of amyloid formation: Implications for pathogenesis of Alzheimer's disease. Biochem. 1993;32:4693–4697. doi: 10.1021/bi00069a001. [DOI] [PubMed] [Google Scholar]
- Golde TE, Eckman CB, Younkin SG. Biochemical detection of Abeta isoforms: implications for pathogenesis, diagnosis, and treatment of Alzheimer's disease. Biochim Biophys Acta. 2000;1502:172–187. doi: 10.1016/s0925-4439(00)00043-0. [DOI] [PubMed] [Google Scholar]
- Younkin SG. The role of A beta 42 in Alzheimer's disease. Journal of Physiology, Paris. 1998;92:289–292. doi: 10.1016/s0928-4257(98)80035-1. [DOI] [PubMed] [Google Scholar]
- McGowan E, Onstead L, Pickford F, Kim J, Beard J, Murphy MP, Yu C, Das P, Golde TE. Selective Overexpression of Abeta42, but not Abeta40, in the secretory pathway is sufficient for plaque deposition in mice. Society for Neuroscience Abstracts. 2003 Program No. 877.14. [Google Scholar]
- Kim J, Onstead L, Randle S, Price R, Smithson L, Zwizinski C, Dickson DW, Golde T, McGowan E. Abeta40 inhibits amyloid deposition in vivo. J Neurosci. 2007;27:627–633. doi: 10.1523/JNEUROSCI.4849-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang R, Wang B, He W, Zheng H. Wild-type presenilin 1 protects against Alzheimer disease mutation-induced amyloid pathology. J Biol Chem. 2006;281:15330–15336. doi: 10.1074/jbc.M512574200. [DOI] [PubMed] [Google Scholar]
- Wang R, Sweeney D, Gandy SE, Sisodia SS. The profile of soluble amyloid beta protein in cultured cell media. Detection and quantification of amyloid beta protein and variants by immunoprecipitation-mass spectrometry. Journal of Biological Chemistry. 1996;271:31894–31902. doi: 10.1074/jbc.271.50.31894. [DOI] [PubMed] [Google Scholar]
- Vassar R. Beta-secretase (BACE) as a drug target for Alzheimer's disease. Adv Drug Deliv Rev. 2002;54:1589–1602. doi: 10.1016/s0169-409x(02)00157-6. [DOI] [PubMed] [Google Scholar]
- Wolfe MS. Inhibition and modulation of gamma-secretase for Alzheimer's disease. Neurotherapeutics. 2008;5:391–398. doi: 10.1016/j.nurt.2008.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weggen S, Eriksen JL, Das P, Sagi SA, Wang R, Pietrzik CU, Findlay KA, Smith TE, Murphy MP, Bulter T, Kang DE, Marquez-Sterling N, Golde TE, Koo EH. A subset of NSAIDs lower amyloidogenic Abeta42 independently of cyclooxygenase activity. Nature. 2001;414:212–216. doi: 10.1038/35102591. [DOI] [PubMed] [Google Scholar]
- Kukar TL, Ladd TB, Bann MA, Fraering PC, Narlawar R, Maharvi GM, Healy B, Chapman R, Welzel AT, Price RW, Moore B, Rangachari V, Cusack B, Eriksen J, Jansen-West K, Verbeeck C, Yager D, Eckman C, Ye W, Sagi S, Cottrell BA, Torpey J, Rosenberry TL, Fauq A, Wolfe MS, Schmidt B, Walsh DM, Koo EH, Golde TE. Substrate-targeting gamma-secretase modulators. Nature. 2008;453:925–929. doi: 10.1038/nature07055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh AK, Gemma S, Tang J. beta-Secretase as a therapeutic target for Alzheimer's disease. Neurotherapeutics. 2008;5:399–408. doi: 10.1016/j.nurt.2008.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strobel G. Keystone Drug News: CoMentis BACE Inhibitor Debuts. 2008a available at http://www.alzforum.org/new/detail.asp?id=1790.
- Cai H, Wang Y, McCarthy D, Wen H, Borchelt DR, Price DL, Wong PC. BACE1 is the major beta-secretase for generation of Abeta peptides by neurons. Nat Neurosci. 2001;4:233–234. doi: 10.1038/85064. [DOI] [PubMed] [Google Scholar]
- Hu X, Hicks CW, He W, Wong P, Macklin WB, Trapp BD, Yan R. Bace1 modulates myelination in the central and peripheral nervous system. Nat Neurosci. 2006;9:1520–1525. doi: 10.1038/nn1797. [DOI] [PubMed] [Google Scholar]
- Willem M, Garratt AN, Novak B, Citron M, Kaufmann S, Rittger A, DeStrooper B, Saftig P, Birchmeier C, Haass C. Control of peripheral nerve myelination by the beta-secretase BACE1. Science. 2006;314:664–666. doi: 10.1126/science.1132341. [DOI] [PubMed] [Google Scholar]
- Bateman RJ, Siemers ER, Mawuenyega KG, Wen G, Browning KR, Sigurdson WC, Yarasheski KE, Friedrich SW, Demattos RB, May PC, Paul SM, Holtzman DM. A gamma-secretase inhibitor decreases amyloid-beta production in the central nervous system. Ann Neurol. 2009 doi: 10.1002/ana.21623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleisher AS, Raman R, Siemers ER, Becerra L, Clark CM, Dean RA, Farlow MR, Galvin JE, Peskind ER, Quinn JF, Sherzai A, Sowell BB, Aisen PS, Thal LJ. Phase 2 safety trial targeting amyloid beta production with a gamma-secretase inhibitor in Alzheimer disease. Arch Neurol. 2008;65:1031–1038. doi: 10.1001/archneur.65.8.1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siemers ER, Dean RA, Friedrich S, Ferguson-Sells L, Gonzales C, Farlow MR, May PC. Safety, tolerability, and effects on plasma and cerebrospinal fluid amyloid-beta after inhibition of gamma-secretase. Clin Neuropharmacol. 2007;30:317–325. doi: 10.1097/WNF.0b013e31805b7660. [DOI] [PubMed] [Google Scholar]
- Siemers ER, Quinn JF, Kaye J, Farlow MR, Porsteinsson A, Tariot P, Zoulnouni P, Galvin JE, Holtzman DM, Knopman DS, Satterwhite J, Gonzales C, Dean RA, May PC. Effects of a gamma-secretase inhibitor in a randomized study of patients with Alzheimer disease. Neurology. 2006;66:602–604. doi: 10.1212/01.WNL.0000198762.41312.E1. [DOI] [PubMed] [Google Scholar]
- Siemers E, Skinner M, Dean RA, Gonzales C, Satterwhite J, Farlow M, Ness D, May PC. Safety, tolerability, and changes in amyloid beta concentrations after administration of a gamma-secretase inhibitor in volunteers. Clin Neuropharmacol. 2005;28:126–132. doi: 10.1097/01.wnf.0000167360.27670.29. [DOI] [PubMed] [Google Scholar]
- Bateman RJ, Munsell LY, Morris JC, Swarm R, Yarasheski KE, Holtzman DM. Human amyloid-beta synthesis and clearance rates as measured in cerebrospinal fluid in vivo. Nat Med. 2006 doi: 10.1038/nm1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Strooper B, Annaert W, Cupers P, Saftig P, Craessaerts K, Mumm JS, Schroeter EH, Schrijvers V, Wolfe MS, Ray WJ, Goate A, Kopan R. A presenilin-1-dependent gamma-secretase-like protease mediates release of Notch intracellular domain. Nature. 1999;398:518–522. doi: 10.1038/19083. [DOI] [PubMed] [Google Scholar]
- Searfoss GH, Jordan WH, Calligaro DO, Galbreath EJ, Schirtzinger LM, Berridge BR, Gao H, Higgins MA, May PC, Ryan TP. Adipsin, a biomarker of gastrointestinal toxicity mediated by a functional gamma-secretase inhibitor. J Biol Chem. 2003;278:46107–46116. doi: 10.1074/jbc.M307757200. [DOI] [PubMed] [Google Scholar]
- Fagan T. DC: New γ Secretase Inhibitors Hit APP, Spare Notch. 2008a avaiable at http://www.alzforum.org/new/detail.asp?id=1992.
- Mayer SC, Kreft AF, Harrison B, Abou-Gharbia M, Antane M, Aschmies S, Atchison K, Chlenov M, Cole DC, Comery T, Diamantidis G, Ellingboe J, Fan K, Galante R, Gonzales C, Ho DM, Hoke ME, Hu Y, Huryn D, Jain U, Jin M, Kremer K, Kubrak D, Lin M, Lu P, Magolda R, Martone R, Moore W, Oganesian A, Pangalos MN, Porte A, Reinhart P, Resnick L, Riddell DR, Sonnenberg-Reines J, Stock JR, Sun SC, Wagner E, Wang T, Woller K, Xu Z, Zaleska MM, Zeldis J, Zhang M, Zhou H, Jacobsen JS. Discovery of Begacestat, a Notch-1-Sparing gamma-Secretase Inhibitor for the Treatment of Alzheimer's Disease. J Med Chem. 2008 doi: 10.1021/jm801252w. [DOI] [PubMed] [Google Scholar]
- Fraering PC, Ye W, LaVoie MJ, Ostaszewski BL, Selkoe DJ, Wolfe MS. gamma-Secretase substrate selectivity can be modulated directly via interaction with a nucleotide-binding site. J Biol Chem. 2005;280:41987–41996. doi: 10.1074/jbc.M501368200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Netzer WJ, Dou F, Cai D, Veach D, Jean S, Li Y, Bornmann WG, Clarkson B, Xu H, Greengard P. Gleevec inhibits beta-amyloid production but not Notch cleavage. Proc Natl Acad Sci U S A. 2003;100:12444–12449. doi: 10.1073/pnas.1534745100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizzo P, Osipo C, Foreman K, Golde T, Osborne B, Miele L. Rational targeting of Notch signaling in cancer. Oncogene. 2008;27:5124–5131. doi: 10.1038/onc.2008.226. [DOI] [PubMed] [Google Scholar]
- Real PJ, Tosello V, Palomero T, Castillo M, Hernando E, de Stanchina E, Sulis ML, Barnes K, Sawai C, Homminga I, Meijerink J, Aifantis I, Basso G, Cordon-Cardo C, Ai W, Ferrando A. Gamma-secretase inhibitors reverse glucocorticoid resistance in T cell acute lymphoblastic leukemia. Nat Med. 2009;15:50–58. doi: 10.1038/nm.1900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fagan T. Research Brief—Flurizan Fails Final Hurdle, Company Discontinues Drug. 2008b available at http://www.alzforum.org/new/detail.asp?id=1871.
- Kukar T, Golde TE. Possible mechanisms of action of NSAIDs and related compounds that modulate gamma-secretase cleavage. Curr Top Med Chem. 2008;8:47–53. doi: 10.2174/156802608783334042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eriksen JL, Sagi SA, Smith TE, Weggen S, Das P, McLendon DC, Ozols VV, Jessing KW, Zavitz KH, Koo EH, Golde TE. NSAIDs and enantiomers of flurbiprofen target gamma-secretase and lower Abeta 42 in vivo. J Clin Invest. 2003;112:440–449. doi: 10.1172/JCI18162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imbimbo BP, Del Giudice E, Colavito D, D'Arrigo A, Dalle Carbonare M, Villetti G, Facchinetti F, Volta R, Pietrini V, Baroc MF, Serneels L, De Strooper B, Leon A. 1-(3′,4′-Dichloro-2-fluoro[1,1′-biphenyl]-4-yl)-cyclopropanecarboxylic acid (CHF5074), a novel gamma-secretase modulator, reduces brain beta-amyloid pathology in a transgenic mouse model of Alzheimer's disease without causing peripheral toxicity. J Pharmacol Exp Ther. 2007;323:822–830. doi: 10.1124/jpet.107.129007. [DOI] [PubMed] [Google Scholar]
- Pissarnitski D. Advances in gamma-secretase modulation. Curr Opin Drug Discov Devel. 2007;10:392–402. [PubMed] [Google Scholar]
- Nicoll JA, Wilkinson D, Holmes C, Steart P, Markham H, Weller RO. Neuropathology of human Alzheimer disease after immunization with amyloid-beta peptide: a case report. Nat Med. 2003;9:448–452. doi: 10.1038/nm840. [DOI] [PubMed] [Google Scholar]
- Fox NC, Black RS, Gilman S, Rossor MN, Griffith SG, Jenkins L, Koller M. Effects of A{beta} immunization (AN1792) on MRI measures of cerebral volume in Alzheimer disease. Neurology. 2005 doi: 10.1212/01.WNL.0000159743.08996.99. [DOI] [PubMed] [Google Scholar]
- Gilman S, Koller M, Black RS, Jenkins L, Griffith SG, Fox NC, Eisner L, Kirby L, Boada Rovira M, Forette F, Orgogozo JM. Clinical effects of A{beta} immunization (AN1792) in patients with AD in an interrupted trial. Neurology. 2005 doi: 10.1212/01.WNL.0000159740.16984.3C. [DOI] [PubMed] [Google Scholar]
- Orgogozo JM, Gilman S, Dartigues JF, Laurent B, Puel M, Kirby LC, Jouanny P, Dubois B, Eisner L, Flitman S, Michel BF, Boada M, Frank A, Hock C. Subacute meningoencephalitis in a subset of patients with AD after Abeta42 immunization. Neurology. 2003;61:46–54. doi: 10.1212/01.wnl.0000073623.84147.a8. [DOI] [PubMed] [Google Scholar]
- Schenk DB, Seubert P, Grundman M, Black R. A beta immunotherapy: Lessons learned for potential treatment of Alzheimer's disease. Neurodegener Dis. 2005;2:255–260. doi: 10.1159/000090365. [DOI] [PubMed] [Google Scholar]
- Holmes C, Boche D, Wilkinson D, Yadegarfar G, Hopkins V, Bayer A, Jones RW, Bullock R, Love S, Neal JW, Zotova E, Nicoll JA. Long-term effects of Abeta42 immunisation in Alzheimer's disease: follow-up of a randomised, placebo-controlled phase I trial. Lancet. 2008;372:216–223. doi: 10.1016/S0140-6736(08)61075-2. [DOI] [PubMed] [Google Scholar]
- Masliah E, Hansen L, Adame A, Crews L, Bard F, Lee C, Seubert P, Games D, Kirby L, Schenk D. Abeta vaccination effects on plaque pathology in the absence of encephalitis in Alzheimer disease. Neurology. 2005;64:129–131. doi: 10.1212/01.WNL.0000148590.39911.DF. [DOI] [PubMed] [Google Scholar]
- Bombois S, Maurage CA, Gompel M, Deramecourt V, Mackowiak-Cordoliani MA, Black RS, Lavielle R, Delacourte A, Pasquier F. Absence of beta-amyloid deposits after immunization in Alzheimer disease with Lewy body dementia. Arch Neurol. 2007;64:583–587. doi: 10.1001/archneur.64.4.583. [DOI] [PubMed] [Google Scholar]
- Patton RL, Kalback WM, Esh CL, Kokjohn TA, Van Vickle GD, Luehrs DC, Kuo YM, Lopez J, Brune D, Ferrer I, Masliah E, Newel AJ, Beach TG, Castano EM, Roher AE. Amyloid-beta peptide remnants in AN-1792-immunized Alzheimer's disease patients: a biochemical analysis. Am J Pathol. 2006;169:1048–1063. doi: 10.2353/ajpath.2006.060269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee M, Bard F, Johnson-Wood K, Lee C, Hu K, Griffith SG, Black RS, Schenk D, Seubert P. Abeta42 immunization in Alzheimer's disease generates Abeta N-terminal antibodies. Ann Neurol. 2005;58:430–435. doi: 10.1002/ana.20592. [DOI] [PubMed] [Google Scholar]
- Agadjanyan MG, Ghochikyan A, Petrushina I, Vasilevko V, Movsesyan N, Mkrtichyan M, Saing T, Cribbs DH. Prototype Alzheimer's disease vaccine using the immunodominant B cell epitope from beta-amyloid and promiscuous T cell epitope pan HLA DR-binding peptide. J Immunol. 2005;174:1580–1586. doi: 10.4049/jimmunol.174.3.1580. [DOI] [PubMed] [Google Scholar]
- Schneeberger A, Mandler M, Otawa O, Zauner W, Mattner F, Schmidt W. Development of Affitope Vaccines for Alzheimer's Disease (AD) - From Concept to Clinical Testing. J Nutr Health Aging. 2009;13:264–267. doi: 10.1007/s12603-009-0070-5. [DOI] [PubMed] [Google Scholar]
- Oldstone MB. Molecular mimicry and immune-mediated diseases. Faseb J. 1998;12:1255–1265. doi: 10.1096/fasebj.12.13.1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strobel G. Chicago: Bapineuzumab's Phase 2—Was the Data Better Than the Spin? 2008b avaiable online at http://www.alzforum.org/new/detail.asp?id=1894.
- Strobel G. Chicago: Lilly's Antibody Appears to Do No Harm, But Will It Help? 2008c avalible online at http://www.alzforum.org/new/detail.asp?id=1896.
- Szabo P, Relkin N, Weksler ME. Natural human antibodies to amyloid beta peptide. Autoimmun Rev. 2008;7:415–420. doi: 10.1016/j.autrev.2008.03.007. [DOI] [PubMed] [Google Scholar]
- Relkin NR, Szabo P, Adamiak B, Burgut T, Monthe C, Lent RW, Younkin S, Younkin L, Schiff R, Weksler ME. 18-Month study of intravenous immunoglobulin for treatment of mild Alzheimer disease. Neurobiol Aging. 2008 doi: 10.1016/j.neurobiolaging.2007.12.021. [DOI] [PubMed] [Google Scholar]
- Bayry J, Kazatchkine MD, Kaveri SV. Shortage of human intravenous immunoglobulin--reasons and possible solutions. Nat Clin Pract Neurol. 2007;3:120–121. doi: 10.1038/ncpneuro0429. [DOI] [PubMed] [Google Scholar]
- McLaurin J, Franklin T, Zhang X, Deng J, Fraser PE. Interactions of Alzheimer amyloid-beta peptides with glycosaminoglycans effects on fibril nucleation and growth. Eur J Biochem. 1999;266:1101–1110. doi: 10.1046/j.1432-1327.1999.00957.x. [DOI] [PubMed] [Google Scholar]
- McLaurin J, Yang D, Yip CM, Fraser PE. Review: modulating factors in amyloid-beta fibril formation. J Struct Biol. 2000a;130:259–270. doi: 10.1006/jsbi.2000.4289. [DOI] [PubMed] [Google Scholar]
- Aisen PS. The development of anti-amyloid therapy for Alzheimer's disease : from secretase modulators to polymerisation inhibitors. CNS Drugs. 2005;19:989–996. doi: 10.2165/00023210-200519120-00002. [DOI] [PubMed] [Google Scholar]
- Wong GT. FDA Deems U.S. Alzhemed Trial Results Inconclusive. 2007 avaible at http://www.alzforum.org/new/detail.asp?id=1647.
- McLaurin J, Golomb R, Jurewicz A, Antel JP, Fraser PE. Inositol stereoisomers stabilize an oligomeric aggregate of Alzheimer amyloid beta peptide and inhibit abeta -induced toxicity. J Biol Chem. 2000b;275:18495–18502. doi: 10.1074/jbc.M906994199. [DOI] [PubMed] [Google Scholar]
- McLaurin J, Kierstead ME, Brown ME, Hawkes CA, Lambermon MH, Phinney AL, Darabie AA, Cousins JE, French JE, Lan MF, Chen F, Wong SS, Mount HT, Fraser PE, Westaway D, George-Hyslop PS. Cyclohexanehexol inhibitors of Abeta aggregation prevent and reverse Alzheimer phenotype in a mouse model. Nat Med. 2006 doi: 10.1038/nm1423. [DOI] [PubMed] [Google Scholar]
- Townsend M, Cleary JP, Mehta T, Hofmeister J, Lesne S, O'Hare E, Walsh DM, Selkoe DJ. Orally available compound prevents deficits in memory caused by the Alzheimer amyloid-beta oligomers. Ann Neurol. 2006;60:668–676. doi: 10.1002/ana.21051. [DOI] [PubMed] [Google Scholar]
- Griffith HR, den Hollander JA, Stewart CC, Evanochko WT, Buchthal SD, Harrell LE, Zamrini EY, Brockington JC, Marson DC. Elevated brain scyllo-inositol concentrations in patients with Alzheimer's disease. NMR Biomed. 2007;20:709–716. doi: 10.1002/nbm.1132. [DOI] [PubMed] [Google Scholar]
- Lannfelt L, Blennow K, Zetterberg H, Batsman S, Ames D, Harrison J, Masters CL, Targum S, Bush AI, Murdoch R, Wilson J, Ritchie CW. Safety, efficacy, and biomarker findings of PBT2 in targeting Abeta as a modifying therapy for Alzheimer's disease: a phase IIa, double-blind, randomised, placebo-controlled trial. Lancet Neurol. 2008;7:779–786. doi: 10.1016/S1474-4422(08)70167-4. [DOI] [PubMed] [Google Scholar]
- Bush AI, Tanzi RE. The galvanization of beta-amyloid in Alzheimer's disease. Proc Natl Acad Sci U S A. 2002;99:7317–7319. doi: 10.1073/pnas.122249699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adlard PA, Cherny RA, Finkelstein DI, Gautier E, Robb E, Cortes M, Volitakis I, Liu X, Smith JP, Perez K, Laughton K, Li QX, Charman SA, Nicolazzo JA, Wilkins S, Deleva K, Lynch T, Kok G, Ritchie CW, Tanzi RE, Cappai R, Masters CL, Barnham KJ, Bush AI. Rapid restoration of cognition in Alzheimer's transgenic mice with 8-hydroxy quinoline analogs is associated with decreased interstitial Abeta. Neuron. 2008;59:43–55. doi: 10.1016/j.neuron.2008.06.018. [DOI] [PubMed] [Google Scholar]
- Bush AI, Tanzi RE. Therapeutics for Alzheimer's disease based on the metal hypothesis. Neurotherapeutics. 2008;5:421–432. doi: 10.1016/j.nurt.2008.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levites Y, Smithson LA, Price RW, Dakin RS, Yuan B, Sierks MR, Kim J, McGowan E, Reed DK, Rosenberry TL, Das P, Golde TE. Insights into the mechanisms of action of anti-Abeta antibodies in Alzheimer's disease mouse models. Faseb J. 2006;20:2576–2578. doi: 10.1096/fj.06-6463fje. [DOI] [PubMed] [Google Scholar]
- Leissring MA. Proteolytic degradation of the amyloid beta-protein: the forgotten side of Alzheimer's disease. Curr Alzheimer Res. 2006;3:431–435. doi: 10.2174/156720506779025206. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ. Clearing the brain's amyloid cobwebs. Neuron. 2001b;32:177–180. doi: 10.1016/s0896-6273(01)00475-5. [DOI] [PubMed] [Google Scholar]
- Melchor JP, Pawlak R, Strickland S. The tissue plasminogen activator-plasminogen proteolytic cascade accelerates amyloid-beta (Abeta) degradation and inhibits Abeta-induced neurodegeneration. J Neurosci. 2003;23:8867–8871. doi: 10.1523/JNEUROSCI.23-26-08867.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobsen JS, Comery TA, Martone RL, Elokdah H, Crandall DL, Oganesian A, Aschmies S, Kirksey Y, Gonzales C, Xu J, Zhou H, Atchison K, Wagner E, Zaleska MM, Das I, Arias RL, Bard J, Riddell D, Gardell SJ, Abou-Gharbia M, Robichaud A, Magolda R, Vlasuk GP, Bjornsson T, Reinhart PH, Pangalos MN. Enhanced clearance of Abeta in brain by sustaining the plasmin proteolysis cascade. Proc Natl Acad Sci U S A. 2008;105:8754–8759. doi: 10.1073/pnas.0710823105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabbro S, Seeds NW. Plasminogen activator activity is inhibited while neuroserpin is up-regulated in the Alzheimer disease brain. J Neurochem. 2009;109:303–315. doi: 10.1111/j.1471-4159.2009.05894.x. [DOI] [PubMed] [Google Scholar]
- Vardy ER, Catto AJ, Hooper NM. Proteolytic mechanisms in amyloid-beta metabolism: therapeutic implications for Alzheimer's disease. Trends Mol Med. 2005;11:464–472. doi: 10.1016/j.molmed.2005.08.004. [DOI] [PubMed] [Google Scholar]
- Gandy S, Greengard P. Regulated cleavage of the Alzheimer amyloid precursor protein: molecular and cellular basis. Biochimie. 1994;76:300–303. doi: 10.1016/0300-9084(94)90162-7. Review. [DOI] [PubMed] [Google Scholar]
- Koo EH. Phorbol esters affect multiple steps in beta-amyloid precursor protein trafficking and amyloid beta-protein production. Mol Med. 1997;3:204–211. [PMC free article] [PubMed] [Google Scholar]
- Skovronsky DM, Moore DB, Milla ME, Doms RW, Lee VM. Protein kinase C-dependent alpha-secretase competes with beta-secretase for cleavage of amyloid-beta precursor protein in the trans-golgi network. J Biol Chem. 2000;275:2568–2575. doi: 10.1074/jbc.275.4.2568. [DOI] [PubMed] [Google Scholar]
- Rossner S, Beck M, Stahl T, Mendla K, Schliebs R, Bigl V. Constitutive overactivation of protein kinase C in guinea pig brain increases alpha-secretory APP processing without decreasing beta-amyloid generation. Eur J Neurosci. 2000;12:3191–3200. doi: 10.1046/j.1460-9568.2000.00211.x. [DOI] [PubMed] [Google Scholar]
- Leiser SC, Bowlby MR, Comery TA, Dunlop J. A cog in cognition: How the alpha7 nicotinic acetylcholine receptor is geared towards improving cognitive deficits. Pharmacol Ther. 2009 doi: 10.1016/j.pharmthera.2009.03.009. [DOI] [PubMed] [Google Scholar]
- Qi XL, Nordberg A, Xiu J, Guan ZZ. The consequences of reducing expression of the alpha7 nicotinic receptor by RNA interference and of stimulating its activity with an alpha7 agonist in SH-SY5Y cells indicate that this receptor plays a neuroprotective role in connection with the pathogenesis of Alzheimer's disease. Neurochem Int. 2007;51:377–383. doi: 10.1016/j.neuint.2007.04.002. [DOI] [PubMed] [Google Scholar]
- Mousavi M, Hellstrom-Lindahl E. Nicotinic receptor agonists and antagonists increase sAPPalpha secretion and decrease Abeta levels in vitro. Neurochem Int. 2009;54:237–244. doi: 10.1016/j.neuint.2008.12.001. [DOI] [PubMed] [Google Scholar]
- Shimohama S. Nicotinic receptor-mediated neuroprotection in neurodegenerative disease models. Biol Pharm Bull. 2009;32:332–336. doi: 10.1248/bpb.32.332. [DOI] [PubMed] [Google Scholar]
- de Jonge WJ, Ulloa L. The alpha7 nicotinic acetylcholine receptor as a pharmacological target for inflammation. Br J Pharmacol. 2007;151:915–929. doi: 10.1038/sj.bjp.0707264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang HY, Lee DH, D'Andrea MR, Peterson PA, Shank RP, Reitz AB. beta-Amyloid(1-42) binds to alpha7 nicotinic acetylcholine receptor with high affinity. Implications for Alzheimer's disease pathology. J Biol Chem. 2000;275:5626–5632. doi: 10.1074/jbc.275.8.5626. [DOI] [PubMed] [Google Scholar]
- Small DH, Maksel D, Kerr ML, Ng J, Hou X, Chu C, Mehrani H, Unabia S, Azari MF, Loiacono R, Aguilar MI, Chebib M. The beta-amyloid protein of Alzheimer's disease binds to membrane lipids but does not bind to the alpha7 nicotinic acetylcholine receptor. J Neurochem. 2007;101:1527–1538. doi: 10.1111/j.1471-4159.2006.04444.x. [DOI] [PubMed] [Google Scholar]
- Strobel G. Keystone Drug News: Agonists for M1, Serotonin Receptors Prime Cholinergic Pump. 2008d avaiable online at http://www.alzforum.org/new/detail.asp?id=1791.
- Marcade M, Bourdin J, Loiseau N, Peillon H, Rayer A, Drouin D, Schweighoffer F, Desire L. Etazolate, a neuroprotective drug linking GABA(A) receptor pharmacology to amyloid precursor protein processing. J Neurochem. 2008;106:392–404. doi: 10.1111/j.1471-4159.2008.05396.x. [DOI] [PubMed] [Google Scholar]
- Greig NH, Sambamurti K, Yu QS, Brossi A, Bruinsma GB, Lahiri DK. An overview of phenserine tartrate, a novel acetylcholinesterase inhibitor for the treatment of Alzheimer's disease. Curr Alzheimer Res. 2005;2:281–290. doi: 10.2174/1567205054367829. [DOI] [PubMed] [Google Scholar]
- Utsuki T, Yu QS, Davidson D, Chen D, Holloway H, Brossi A, Sambamurti K, Lahiri D, Greig NH, Giordano T. Identification of Novel Small Molecule Inhibitors of APP Protein Synthesis as a Route to Lower Alzheimer's disease Amyloid-{beta} peptide. J Pharmacol Exp Ther. 2006 doi: 10.1124/jpet.106.103309. [DOI] [PubMed] [Google Scholar]
- Klunk WE, Mathis CA. The future of amyloid-beta imaging: a tale of radionuclides and tracer proliferation. Curr Opin Neurol. 2008;21:683–687. doi: 10.1097/WCO.0b013e3283168e1a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klunk WE, Engler H, Nordberg A, Wang Y, Blomqvist G, Holt DP, Bergstrom M, Savitcheva I, Huang GF, Estrada S, Ausen B, Debnath ML, Barletta J, Price JC, Sandell J, Lopresti BJ, Wall A, Koivisto P, Antoni G, Mathis CA, Langstrom B. Imaging brain amyloid in Alzheimer's disease with Pittsburgh Compound-B. Ann Neurol. 2004;55:306–319. doi: 10.1002/ana.20009. [DOI] [PubMed] [Google Scholar]
- Agdeppa ED, Kepe V, Petri A, Satyamurthy N, Liu J, Huang SC, Small GW, Cole GM, Barrio JR. In vitro detection of (S)-naproxen and ibuprofen binding to plaques in the Alzheimer's brain using the positron emission tomography molecular imaging probe 2-(1-[6-[(2-[(18)F]fluoroethyl)(methyl)amino]-2-naphthyl]ethylidene)malono nitrile. Neuroscience. 2003;117:723–730. doi: 10.1016/s0306-4522(02)00907-7. [DOI] [PubMed] [Google Scholar]
- Fagan AM, Mintun MA, Mach RH, Lee SY, Dence CS, Shah AR, Larossa GN, Spinner ML, Klunk WE, Mathis CA, Dekosky ST, Morris JC, Holtzman DM. Inverse relation between in vivo amyloid imaging load and cerebrospinal fluid Abeta(42) in humans. Ann Neurol. 2005 doi: 10.1002/ana.20730. [DOI] [PubMed] [Google Scholar]
- Reiman EM, Chen K, Liu X, Bandy D, Yu M, Lee W, Ayutyanont N, Keppler J, Reeder SA, Langbaum JB, Alexander GE, Klunk WE, Mathis CA, Price JC, Aizenstein HJ, Dekosky ST, Caselli RJ. Fibrillar amyloid-{beta} burden in cognitively normal people at 3 levels of genetic risk for Alzheimer's disease. Proc Natl Acad Sci U S A. 2009 doi: 10.1073/pnas.0900345106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack CR, Jr, Lowe VJ, Weigand SD, Wiste HJ, Senjem ML, Knopman DS, Shiung MM, Gunter JL, Boeve BF, Kemp BJ, Weiner M, Petersen RC. Serial PIB and MRI in normal, mild cognitive impairment and Alzheimer's disease: implications for sequence of pathological events in Alzheimer's disease. Brain. 2009 doi: 10.1093/brain/awp062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mormino EC, Kluth JT, Madison CM, Rabinovici GD, Baker SL, Miller BL, Koeppe RA, Mathis CA, Weiner MW, Jagust WJ. Episodic memory loss is related to hippocampal-mediated {beta}-amyloid deposition in elderly subjects. Brain. 2008 doi: 10.1093/brain/awn320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aizenstein HJ, Nebes RD, Saxton JA, Price JC, Mathis CA, Tsopelas ND, Ziolko SK, James JA, Snitz BE, Houck PR, Bi W, Cohen AD, Lopresti BJ, DeKosky ST, Halligan EM, Klunk WE. Frequent amyloid deposition without significant cognitive impairment among the elderly. Arch Neurol. 2008;65:1509–1517. doi: 10.1001/archneur.65.11.1509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee VMY, Trojanowski JQ. The disordered neuronal cytoskeleton in Alzheimer's disease. Current Opinion in Neurobiology. 1992;2:653–656. doi: 10.1016/0959-4388(92)90034-i. [DOI] [PubMed] [Google Scholar]
- Hutton M. Molecular genetics of chromosome 17 tauopathies. Ann N Y Acad Sci. 2000;920:63–73. doi: 10.1111/j.1749-6632.2000.tb06906.x. [DOI] [PubMed] [Google Scholar]
- Hutton M, Lendon CL, Rizzu P, Baker M, Froelich S, Houlden H, Pickering-Brown S, Chakraverty S, Isaacs A, Grover A, Hackett J, Adamson J, Lincoln S, Dickson D, Davies P, Petersen RC, Stevens M, de Graaff E, Wauters E, van Baren J, Hillebrand M, Joosse M, Kwon JM, Nowotny P, Heutink P, et al. Association of missense and 5′-splice-site mutations in tau with the inherited dementia FTDP-17. Nature. 1998;393:702–705. doi: 10.1038/31508. [DOI] [PubMed] [Google Scholar]
- Spillantini MG, Murrell JR, Goedert M, Farlow MR, Klug A, Ghetti B. Mutation in the tau gene in familial multiple system tauopathy with presenile dementia. Proc Natl Acad Sci U S A. 1998;95:7737–7741. doi: 10.1073/pnas.95.13.7737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee VM, Goedert M, Trojanowski JQ. Neurodegenerative tauopathies. Annu Rev Neurosci. 2001;24:1121–1159. doi: 10.1146/annurev.neuro.24.1.1121. [DOI] [PubMed] [Google Scholar]
- Boeve BF, Hutton M. Refining frontotemporal dementia with parkinsonism linked to chromosome 17: introducing FTDP-17 (MAPT) and FTDP-17 (PGRN) Arch Neurol. 2008;65:460–464. doi: 10.1001/archneur.65.4.460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goedert M, Jakes R. Mutations causing neurodegenerative tauopathies. Biochim Biophys Acta. 2005;1739:240–250. doi: 10.1016/j.bbadis.2004.08.007. [DOI] [PubMed] [Google Scholar]
- Lewis J, McGowan E, Rockwood J, Melrose H, Nacharaju P, Van Slegtenhorst M, Gwinn-Hardy K, Paul Murphy M, Baker M, Yu X, Duff K, Hardy J, Corral A, Lin WL, Yen SH, Dickson DW, Davies P, Hutton M. Neurofibrillary tangles, amyotrophy and progressive motor disturbance in mice expressing mutant (P301L) tau protein. Nat Genet. 2000;25:402–405. doi: 10.1038/78078. [DOI] [PubMed] [Google Scholar]
- Gotz J, Chen F, Barmettler R, Nitsch RM. Tau filament formation in transgenic mice expressing P301L tau. J Biol Chem. 2001;276:529–534. doi: 10.1074/jbc.M006531200. [DOI] [PubMed] [Google Scholar]
- Seabrook GR, Ray WJ, Shearman M, Hutton M. Beyond amyloid: the next generation of Alzheimer's disease therapeutics. Mol Interv. 2007;7:261–270. doi: 10.1124/mi.7.5.8. [DOI] [PubMed] [Google Scholar]
- Small SA, Duff K. Linking Abeta and tau in late-onset Alzheimer's disease: a dual pathway hypothesis. Neuron. 2008;60:534–542. doi: 10.1016/j.neuron.2008.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kauwe JS, Cruchaga C, Mayo K, Fenoglio C, Bertelsen S, Nowotny P, Galimberti D, Scarpini E, Morris JC, Fagan AM, Holtzman DM, Goate AM. Variation in MAPT is associated with cerebrospinal fluid tau levels in the presence of amyloid-beta deposition. Proc Natl Acad Sci U S A. 2008;105:8050–8054. doi: 10.1073/pnas.0801227105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukherjee O, Kauwe JS, Mayo K, Morris JC, Goate AM. Haplotype-based association analysis of the MAPT locus in late onset Alzheimer's disease. BMC Genet. 2007;8:3. doi: 10.1186/1471-2156-8-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rademakers R, Melquist S, Cruts M, Theuns J, Del-Favero J, Poorkaj P, Baker M, Sleegers K, Crook R, De Pooter T, Bel Kacem S, Adamson J, Van den Bossche D, Van den Broeck M, Gass J, Corsmit E, De Rijk P, Thomas N, Engelborghs S, Heckman M, Litvan I, Crook J, De Deyn PP, Dickson D, Schellenberg GD, Van Broeckhoven C, Hutton ML. High-density SNP haplotyping suggests altered regulation of tau gene expression in progressive supranuclear palsy. Hum Mol Genet. 2005;14:3281–3292. doi: 10.1093/hmg/ddi361. [DOI] [PubMed] [Google Scholar]
- Santacruz K, Lewis J, Spires T, Paulson J, Kotilinek L, Ingelsson M, Guimaraes A, DeTure M, Ramsden M, McGowan E, Forster C, Yue M, Orne J, Janus C, Mariash A, Kuskowski M, Hyman B, Hutton M, Ashe KH. Tau suppression in a neurodegenerative mouse model improves memory function. Science. 2005;309:476–481. doi: 10.1126/science.1113694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harada A, Oguchi K, Okabe S, Kuno J, Terada S, Ohshima T, Sato-Yoshitake R, Takei Y, Noda T, Hirokawa N. Altered microtubule organization in small-calibre axons of mice lacking tau protein. Nature. 1994;369:488–491. doi: 10.1038/369488a0. [DOI] [PubMed] [Google Scholar]
- Takei Y, Teng J, Harada A, Hirokawa N. Defects in axonal elongation and neuronal migration in mice with disrupted tau and map1b genes. J Cell Biol. 2000;150:989–1000. doi: 10.1083/jcb.150.5.989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikegami S, Harada A, Hirokawa N. Muscle weakness, hyperactivity, and impairment in fear conditioning in tau-deficient mice. Neurosci Lett. 2000;279:129–132. doi: 10.1016/s0304-3940(99)00964-7. [DOI] [PubMed] [Google Scholar]
- Roberson ED, Scearce-Levie K, Palop JJ, Yan F, Cheng IH, Wu T, Gerstein H, Yu GQ, Mucke L. Reducing endogenous tau ameliorates amyloid beta-induced deficits in an Alzheimer's disease mouse model. Science. 2007;316:750–754. doi: 10.1126/science.1141736. [DOI] [PubMed] [Google Scholar]
- Donahue CP, Muratore C, Wu JY, Kosik KS, Wolfe MS. Stabilization of the tau exon 10 stem loop alters pre-mRNA splicing. J Biol Chem. 2006;281:23302–23306. doi: 10.1074/jbc.C600143200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donahue CP, Ni J, Rozners E, Glicksman MA, Wolfe MS. Identification of tau stem loop RNA stabilizers. J Biomol Screen. 2007;12:789–799. doi: 10.1177/1087057107302676. [DOI] [PubMed] [Google Scholar]
- Gamblin TC, Berry RW, Binder LI. Modeling tau polymerization in vitro: a review and synthesis. Biochemistry. 2003;42:15009–15017. doi: 10.1021/bi035722s. [DOI] [PubMed] [Google Scholar]
- Gamblin TC, King ME, Dawson H, Vitek MP, Kuret J, Berry RW, Binder LI. In vitro polymerization of tau protein monitored by laser light scattering: method and application to the study of FTDP-17 mutants. Biochemistry. 2000;39:6136–6144. doi: 10.1021/bi000201f. [DOI] [PubMed] [Google Scholar]
- Nacharaju P, Lewis J, Easson C, Yen S, Hackett J, Hutton M, Yen SH. Accelerated filament formation from tau protein with specific FTDP-17 missense mutations. FEBS Lett. 1999;447:195–199. doi: 10.1016/s0014-5793(99)00294-x. [DOI] [PubMed] [Google Scholar]
- Wischik CM, Edwards PC, Lai RY, Roth M, Harrington CR. Selective inhibition of Alzheimer disease-like tau aggregation by phenothiazines. Proc Natl Acad Sci U S A. 1996;93:11213–11218. doi: 10.1073/pnas.93.20.11213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khlistunova I, Biernat J, Wang Y, Pickhardt M, von Bergen M, Gazova Z, Mandelkow E, Mandelkow EM. Inducible expression of Tau repeat domain in cell models of tauopathy: aggregation is toxic to cells but can be reversed by inhibitor drugs. J Biol Chem. 2006;281:1205–1214. doi: 10.1074/jbc.M507753200. [DOI] [PubMed] [Google Scholar]
- Masuda M, Suzuki N, Taniguchi S, Oikawa T, Nonaka T, Iwatsubo T, Hisanaga S, Goedert M, Hasegawa M. Small molecule inhibitors of alpha-synuclein filament assembly. Biochemistry. 2006;45:6085–6094. doi: 10.1021/bi0600749. [DOI] [PubMed] [Google Scholar]
- Gustke N, Steiner B, Mandelkow EM, Biernat J, Meyer HE, Goedert M, Mandelkow E. The Alzheimer-like phosphorylation of tau protein reduces microtubule binding and involves Ser-Pro and Thr-Pro motifs. FEBS Lett. 1992;307:199–205. doi: 10.1016/0014-5793(92)80767-b. [DOI] [PubMed] [Google Scholar]
- Goedert M, Sisodia SS, Price DL. Tau proteins of Alzheimer paired helical filaments: Abnormal phosphorylation of all six brain isoforms. Neuron. 1992;8:159–168. doi: 10.1016/0896-6273(92)90117-v. [DOI] [PubMed] [Google Scholar]
- Geschwind DH. Tau phosphorylation, tangles, and neurodegeneration: the chicken or the egg? Neuron. 2003;40:457–460. doi: 10.1016/s0896-6273(03)00681-0. [DOI] [PubMed] [Google Scholar]
- Friedhoff P, von Bergen M, Mandelkow EM, Mandelkow E. Structure of tau protein and assembly into paired helical filaments. Biochim Biophys Acta. 2000;1502:122–132. doi: 10.1016/s0925-4439(00)00038-7. [DOI] [PubMed] [Google Scholar]
- Le Corre S, Klafki HW, Plesnila N, Hubinger G, Obermeier A, Sahagun H, Monse B, Seneci P, Lewis J, Eriksen J, Zehr C, Yue M, McGowan E, Dickson DW, Hutton M, Roder HM. An inhibitor of tau hyperphosphorylation prevents severe motor impairments in tau transgenic mice. Proc Natl Acad Sci U S A. 2006;103:9673–9678. doi: 10.1073/pnas.0602913103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakashima H, Ishihara T, Suguimoto P, Yokota O, Oshima E, Kugo A, Terada S, Hamamura T, Trojanowski JQ, Lee VM, Kuroda S. Chronic lithium treatment decreases tau lesions by promoting ubiquitination in a mouse model of tauopathies. Acta Neuropathol (Berl) 2005;110:547–556. doi: 10.1007/s00401-005-1087-4. [DOI] [PubMed] [Google Scholar]
- Noble W, Planel E, Zehr C, Olm V, Meyerson J, Suleman F, Gaynor K, Wang L, LaFrancois J, Feinstein B, Burns M, Krishnamurthy P, Wen Y, Bhat R, Lewis J, Dickson D, Duff K. Inhibition of glycogen synthase kinase-3 by lithium correlates with reduced tauopathy and degeneration in vivo. Proc Natl Acad Sci U S A. 2005;102:6990–6995. doi: 10.1073/pnas.0500466102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phiel CJ, Wilson CA, Lee VM, Klein PS. GSK-3alpha regulates production of Alzheimer's disease amyloid-beta peptides. Nature. 2003;423:435–439. doi: 10.1038/nature01640. [DOI] [PubMed] [Google Scholar]
- Feyt C, Kienlen-Campard P, Leroy K, N'Kuli F, Courtoy PJ, Brion JP, Octave JN. Lithium chloride increases the production of amyloid-beta peptide independently from its inhibition of glycogen synthase kinase 3. J Biol Chem. 2005;280:33220–33227. doi: 10.1074/jbc.M501610200. [DOI] [PubMed] [Google Scholar]
- Macdonald A, Briggs K, Poppe M, Higgins A, Velayudhan L, Lovestone S. A feasibility and tolerability study of lithium in Alzheimer's disease. Int J Geriatr Psychiatry. 2008;23:704–711. doi: 10.1002/gps.1964. [DOI] [PubMed] [Google Scholar]
- Gendron TF, Petrucelli L. The role of tau in neurodegeneration. Mol Neurodegener. 2009;4:13. doi: 10.1186/1750-1326-4-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu PJ, Wulf G, Zhou XZ, Davies P, Lu KP. The prolyl isomerase Pin1 restores the function of Alzheimer-associated phosphorylated tau protein. Nature. 1999;399:784–788. doi: 10.1038/21650. [DOI] [PubMed] [Google Scholar]
- Zhou XZ, Kops O, Werner A, Lu PJ, Shen M, Stoller G, Kullertz G, Stark M, Fischer G, Lu KP. Pin1-dependent prolyl isomerization regulates dephosphorylation of Cdc25C and tau proteins. Mol Cell. 2000;6:873–883. doi: 10.1016/s1097-2765(05)00083-3. [DOI] [PubMed] [Google Scholar]
- Pastorino L, Sun A, Lu PJ, Zhou XZ, Balastik M, Finn G, Wulf G, Lim J, Li SH, Li X, Xia W, Nicholson LK, Lu KP. The prolyl isomerase Pin1 regulates amyloid precursor protein processing and amyloid-beta production. Nature. 2006;440:528–534. doi: 10.1038/nature04543. [DOI] [PubMed] [Google Scholar]
- Dickey CA, Yue M, Lin WL, Dickson DW, Dunmore JH, Lee WC, Zehr C, West G, Cao S, Clark AM, Caldwell GA, Caldwell KA, Eckman C, Patterson C, Hutton M, Petrucelli L. Deletion of the ubiquitin ligase CHIP leads to the accumulation, but not the aggregation, of both endogenous phospho- and caspase-3-cleaved tau species. J Neurosci. 2006a;26:6985–6996. doi: 10.1523/JNEUROSCI.0746-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimura H, Schwartz D, Gygi SP, Kosik KS. CHIP-Hsc70 complex ubiquitinates phosphorylated tau and enhances cell survival. J Biol Chem. 2004;279:4869–4876. doi: 10.1074/jbc.M305838200. [DOI] [PubMed] [Google Scholar]
- Dickey CA, Dunmore J, Lu B, Wang JW, Lee WC, Kamal A, Burrows F, Eckman C, Hutton M, Petrucelli L. HSP induction mediates selective clearance of tau phosphorylated at proline-directed Ser/Thr sites but not KXGS (MARK) sites. Faseb J. 2006b;20:753–755. doi: 10.1096/fj.05-5343fje. [DOI] [PubMed] [Google Scholar]
- Dickey CA, Kamal A, Lundgren K, Klosak N, Bailey RM, Dunmore J, Ash P, Shoraka S, Zlatkovic J, Eckman CB, Patterson C, Dickson DW, Nahman NS, Jr, Hutton M, Burrows F, Petrucelli L. The high-affinity HSP90-CHIP complex recognizes and selectively degrades phosphorylated tau client proteins. J Clin Invest. 2007;117:648–658. doi: 10.1172/JCI29715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo W, Dou F, Rodina A, Chip S, Kim J, Zhao Q, Moulick K, Aguirre J, Wu N, Greengard P, Chiosis G. Roles of heat-shock protein 90 in maintaining and facilitating the neurodegenerative phenotype in tauopathies. Proc Natl Acad Sci U S A. 2007;104:9511–9516. doi: 10.1073/pnas.0701055104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrucelli L, Dickson D, Kehoe K, Taylor J, Snyder H, Grover A, De Lucia M, McGowan E, Lewis J, Prihar G, Kim J, Dillmann WH, Browne SE, Hall A, Voellmy R, Tsuboi Y, Dawson TM, Wolozin B, Hardy J, Hutton M. CHIP and Hsp70 regulate tau ubiquitination, degradation and aggregation. Hum Mol Genet. 2004;13:703–714. doi: 10.1093/hmg/ddh083. [DOI] [PubMed] [Google Scholar]
- Dou F, Netzer WJ, Tanemura K, Li F, Hartl FU, Takashima A, Gouras GK, Greengard P, Xu H. Chaperones increase association of tau protein with microtubules. Proc Natl Acad Sci U S A. 2003;100:721–726. doi: 10.1073/pnas.242720499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B, Maiti A, Shively S, Lakhani F, McDonald-Jones G, Bruce J, Lee EB, Xie SX, Joyce S, Li C, Toleikis PM, Lee VM, Trojanowski JQ. Microtubule-binding drugs offset tau sequestration by stabilizing microtubules and reversing fast axonal transport deficits in a tauopathy model. Proc Natl Acad Sci U S A. 2005;102:227–231. doi: 10.1073/pnas.0406361102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wartmann M, Altmann KH. The biology and medicinal chemistry of epothilones. Curr Med Chem Anticancer Agents. 2002;2:123–148. doi: 10.2174/1568011023354489. [DOI] [PubMed] [Google Scholar]
- Landhuis E. Chicago: Brain-Penetrant Microtubule Stabilizer in Tauopathy Mice. 2008 avaiable online at http://www.alzforum.org/new/detail.asp?id=1903.
- Sigurdsson EM. Immunotherapy targeting pathological tau protein in Alzheimer's disease and related tauopathies. J Alzheimers Dis. 2008;15:157–168. doi: 10.3233/jad-2008-15202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boimel M, Grigoriadis N, Lourbopoulos A, Touloumi O, Rosenmann D, Abramsky O, Rosenmann H. Statins reduce the neurofibrillary tangle burden in a mouse model of tauopathy. J Neuropathol Exp Neurol. 2009;68:314–325. doi: 10.1097/NEN.0b013e31819ac3cb. [DOI] [PubMed] [Google Scholar]
- Fassbender K, Simons M, Bergmann C, Stroick M, Lutjohann D, Keller P, Runz H, Kuhl S, Bertsch T, von Bergmann K, Hennerici M, Beyreuther K, Hartmann T. Simvastatin strongly reduces levels of Alzheimer's disease beta - amyloid peptides Abeta 42 and Abeta 40 in vitro and in vivo. Proc Natl Acad Sci U S A. 2001;98:5856–5861. doi: 10.1073/pnas.081620098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Refolo LM, Pappolla MA, LaFrancois J, Malester B, Schmidt SD, Thomas-Bryant T, Tint GS, Wang R, Mercken M, Petanceska SS, Duff KE. A cholesterol-lowering drug reduces beta-amyloid pathology in a transgenic mouse model of Alzheimer's disease. Neurobiol Dis. 2001;8:890–899. doi: 10.1006/nbdi.2001.0422. [DOI] [PubMed] [Google Scholar]
- Sparks DL, Sabbagh MN, Connor DJ, Lopez J, Launer LJ, Petanceska S, Browne P, Wassar D, Johnson-Traver S, Lochhead J, Ziolkowski C. Atorvastatin therapy lowers circulating cholesterol but not free radical activity in advance of identifiable clinical benefit in the treatment of mild-to-moderate AD. Curr Alzheimer Res. 2005;2:343–353. doi: 10.2174/1567205054367900. [DOI] [PubMed] [Google Scholar]
- Caccamo A, Oddo S, Billings LM, Green KN, Martinez-Coria H, Fisher A, LaFerla FM. M1 receptors play a central role in modulating AD-like pathology in transgenic mice. Neuron. 2006;49:671–682. doi: 10.1016/j.neuron.2006.01.020. [DOI] [PubMed] [Google Scholar]
- McKee AC, Carreras I, Hossain L, Ryu H, Klein WL, Oddo S, LaFerla FM, Jenkins BG, Kowall NW, Dedeoglu A. Ibuprofen reduces Abeta, hyperphosphorylated tau and memory deficits in Alzheimer mice. Brain Res. 2008;1207:225–236. doi: 10.1016/j.brainres.2008.01.095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oddo S, Billings L, Kesslak JP, Cribbs DH, LaFerla FM. Abeta immunotherapy leads to clearance of early, but not late, hyperphosphorylated tau aggregates via the proteasome. Neuron. 2004;43:321–332. doi: 10.1016/j.neuron.2004.07.003. [DOI] [PubMed] [Google Scholar]
- O'Collins VE, Macleod MR, Donnan GA, Horky LL, van der Worp BH, Howells DW. 1,026 experimental treatments in acute stroke. Ann Neurol. 2006;59:467–477. doi: 10.1002/ana.20741. [DOI] [PubMed] [Google Scholar]
- Waldmeier P, Bozyczko-Coyne D, Williams M, Vaught JL. Recent clinical failures in Parkinson's disease with apoptosis inhibitors underline the need for a paradigm shift in drug discovery for neurodegenerative diseases. Biochem Pharmacol. 2006;72:1197–1206. doi: 10.1016/j.bcp.2006.06.031. [DOI] [PubMed] [Google Scholar]
- Aggarwal S, Cudkowicz M. ALS drug development: reflections from the past and a way forward. Neurotherapeutics. 2008;5:516–527. doi: 10.1016/j.nurt.2008.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price DL, Sisodia SS. Mutant genes in familial Alzheimer's disease and transgenic models. Annual Review of Neuroscience. 1998;21:479–505. doi: 10.1146/annurev.neuro.21.1.479. [DOI] [PubMed] [Google Scholar]
- Games D, Buttini M, Kobayashi D, Schenk D, Seubert P. Mice as models: transgenic approaches and Alzheimer's disease. J Alzheimers Dis. 2006;9:133–149. doi: 10.3233/jad-2006-9s316. [DOI] [PubMed] [Google Scholar]
- Hsiao K. Transgenic mice expressing Alzheimer amyloid precursor proteins. Exp Gerontol. 1998;33:883–889. doi: 10.1016/s0531-5565(98)00045-x. [DOI] [PubMed] [Google Scholar]
- Spires TL, Hyman BT. Transgenic models of Alzheimer's disease: learning from animals. NeuroRx. 2005;2:423–437. doi: 10.1602/neurorx.2.3.423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutton M, Lewis J, Dickson D, Yen SH, McGowan E. Analysis of tauopathies with transgenic mice. Trends Mol Med. 2001;7:467–470. doi: 10.1016/s1471-4914(01)02123-2. [DOI] [PubMed] [Google Scholar]
- Gotz J, Deters N, Doldissen A, Bokhari L, Ke Y, Wiesner A, Schonrock N, Ittner LM. A decade of tau transgenic animal models and beyond. Brain Pathol. 2007;17:91–103. doi: 10.1111/j.1750-3639.2007.00051.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graff-Radford NR, Crook JE, Lucas J, Boeve BF, Knopman DS, Ivnik RJ, Smith GE, Younkin LH, Petersen RC, Younkin SG. Association of low plasma Abeta42/Abeta40 ratios with increased imminent risk for mild cognitive impairment and Alzheimer disease. Arch Neurol. 2007;64:354–362. doi: 10.1001/archneur.64.3.354. [DOI] [PubMed] [Google Scholar]
- Khachaturian ZS, Snyder PJ, Doody R, Aisen P, Comer M, Dwyer J, Frank RA, Holzapfel A, Khachaturian AS, Korczyn AD, Roses A, Simpkins JW, Schneider LS, Albert MS, Egge R, Deves A, Ferris S, Greenberg BD, Johnson C, Kukull WA, Poirier J, Schenk D, Thies W, Gauthier S, Gilman S, Bernick C, Cummings JL, Fillit H, Grundman M, Kaye J, Mucke L, Reisberg B, Sano M, Pickeral O, Petersen RC, Mohs RC, Carrillo M, Corey-Bloom JP, Foster NL, Jacobsen S, Lee V, Potter WZ, Sabbagh MN, Salmon D, Trojanowski JQ, Wexler N, Bain LJ. A roadmap for the prevention of dementia II: Leon Thal Symposium 2008. Alzheimers Dement. 2009;5:85–92. doi: 10.1016/j.jalz.2009.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golde TE. The therapeutic importance of understanding mechanisms of neuronal cell death in neurodegenerative disease. Mol Neurodegener. 2009;4:8. doi: 10.1186/1750-1326-4-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blennow K. Cerebrospinal fluid protein biomarkers for Alzheimer's disease. NeuroRx. 2004;1:213–225. doi: 10.1602/neurorx.1.2.213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox NC, Crum WR, Scahill RI, Stevens JM, Janssen JC, Rossor MN. Imaging of onset and progression of Alzheimer's disease with voxel- compression mapping of serial magnetic resonance images. Lancet. 2001;358:201–205. doi: 10.1016/S0140-6736(01)05408-3. [DOI] [PubMed] [Google Scholar]
- Jack CR, Jr, Dickson DW, Parisi JE, Xu YC, Cha RH, O'Brien PC, Edland SD, Smith GE, Boeve BF, Tangalos EG, Kokmen E, Petersen RC. Antemortem MRI findings correlate with hippocampal neuropathology in typical aging and dementia. Neurology. 2002;58:750–757. doi: 10.1212/wnl.58.5.750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller SG, Weiner MW, Thal LJ, Petersen RC, Jack CR, Jagust W, Trojanowski JQ, Toga AW, Beckett L. Ways toward an early diagnosis in Alzheimer's disease: The Alzheimer's Disease Neuroimaging Initiative (ADNI) Alzheimers Dement. 2005;1:55–66. doi: 10.1016/j.jalz.2005.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leow AD, Yanovsky I, Parikshak N, Hua X, Lee S, Toga AW, Jack CR, Jr, Bernstein MA, Britson PJ, Gunter JL, Ward CP, Borowski B, Shaw LM, Trojanowski JQ, Fleisher AS, Harvey D, Kornak J, Schuff N, Alexander GE, Weiner MW, Thompson PM. Alzheimer's disease neuroimaging initiative: a one-year follow up study using tensor-based morphometry correlating degenerative rates, biomarkers and cognition. Neuroimage. 2009;45:645–655. doi: 10.1016/j.neuroimage.2009.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw LM, Vanderstichele H, Knapik-Czajka M, Clark CM, Aisen PS, Petersen RC, Blennow K, Soares H, Simon A, Lewczuk P, Dean R, Siemers E, Potter W, Lee VM, Trojanowski JQ. Cerebrospinal fluid biomarker signature in Alzheimer's disease neuroimaging initiative subjects. Ann Neurol. 2009;65:403–413. doi: 10.1002/ana.21610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider LS, Lahiri DK. The perils of Alzheimer's drug development. Curr Alzheimer Res. 2009;6:77–78. doi: 10.2174/156720509787313871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardy JA, Higgins GA. Alzheimer's disease: the amyloid cascade hypothesis. Science. 1992;256:184–185. doi: 10.1126/science.1566067. [DOI] [PubMed] [Google Scholar]