

Abstract
Cigarette smoking has been associated repeatedly in observational studies with decreased risk of Parkinson's disease (PD), but its relationship to the risk of dementia or Alzheimer's disease (AD) is inconsistent. All of these studies have used clinical diagnoses of disease. We tested the hypothesis that lifetime cigarette use might be associated with reduced risk of neuropathologic changes of Lewy‐related pathology (LRP) in multiple brain regions or with reduced risk of consensus neuropathologic changes of AD in a prospective community‐based study of brain aging and dementia, the Adult Changes in Thought (ACT) study. We observed that heavy lifetime cigarette smoking (>50 pack years) was associated with significantly reduced relative risk (RR) for LRP, but not AD‐type pathologic changes, after correcting for selection bias, and with significantly reduced frequency of LRP in the substantia nigra. These findings are the first of which we are aware to associate reduced LRP in human brain with any exposure, and substantiate observational studies that have associated cigarette smoking with reduced risk of PD. Although cigarette smoking is too toxic to suggest as a treatment, if confirmed, these findings may guide future therapeutic strategies that attempt to suppress LRP in human brain by other means.
Keywords: Alzheimer's disease, Lewy body, smoking, neuropathology
INTRODUCTION
Parkinson's disease (PD) and Alzheimer's disease (AD) are two common age‐related neurodegenerative diseases. Each disease has consensus clinical criteria for a “probable” diagnosis; however, definitive diagnosis of either requires neuropathologic examination 6, 15. PD is prominently a movement disorder defined pathologically by regionally ordered accumulation of Lewy bodies and related structures referred to as Lewy‐related pathology (LRP) (3), whereas AD is a form of dementia characterized pathologically by density of neuritic plaques (NPs) and regionally ordered accumulation of neurofibrillary tangles (NFTs) (1). The clinical situation is further complicated by a condition called dementia with Lewy bodies (DLB) that most commonly is characterized by the presence of LRP with or without typical AD pathologic changes (12). Importantly, by current clinical criteria, it is difficult to distinguish AD from DLB with high specificity (23). This means that research cohorts of clinically defined probable AD are varyingly “contaminated” to an unknown extent by patients with LRP.
A concerted research effort is under way to identify therapeutic interventions that suppress accumulation of NPs, NFTs or LRP by blocking formation or promoting clearance from brain. One clue to an effective treatment may reside in smokers. Indeed, at least a dozen case‐control or cohort studies from across the globe have reported that cigarette smoking is inversely associated with PD 20, 21, 22. A pooled analysis of 11 of these observational studies (2816 cases and 8993 controls) confirmed inverse associations between clinically diagnosed PD and smoking that were stronger in current than former smokers, stronger for Caucasian and Asian than Hispanic and African American patients, and inversely associated with pack years smoked at every age of onset except those older than 75 years, but not related to sex (20). More recently, the Cancer Prevention Study II Nutrition Cohort (143 325 participants followed for 9 years) investigated the relationship between cigarette smoking and 413 cases of incident PD (22). Consonant with previous observations, participants who smoked into older age had the lowest risk of incident PD that did not differ between men and women; a 30% to 60% reduction in risk for incident PD was observed for those who smoked up to 24 years prior to diagnosis of PD. These observations have inspired experimentation and speculation on the role of nicotine as a modulator of dopaminergic neurodegeneration (19). Despite these many clinical and experimental studies, we are unaware of any neuropathologic study that has pursued the structural correlates of the apparent protective effect of smoking in patients with PD. Thus, it remains unknown if smoking is associated with reduced LRP in parallel with decreased risk for PD.
In contrast to PD, outcomes from a similar number of observational studies of smoking and the risk of clinically diagnosed AD or all‐cause dementia have been highly inconsistent [reviewed in (7)]. As mentioned, the common “contamination” of clinically defined AD cohorts with DLB and its associated LRP confounds interpretation of these observational studies. As with PD, we are unaware of any neuropathologic study that has investigated the associations between smoking and the burden of structural changes observed in dementia. Thus, it remains unknown whether smoking is associated with reduced pathologic changes of AD, LRP, both or neither. In this study we addressed this gap in knowledge by determining the association between lifetime cigarette use and pathologic changes of AD (NPs and NFTs) or LRP in a prospective cohort study of dementia.
METHODS
Participants
The Adult Changes in Thought (ACT) study is a community‐based prospective cohort study that enrolled participants from the Seattle‐area members of the Group Health Cooperative (GHC), the largest health maintenance organization based in the Puget Sound region. The base population for the ACT study was GHC members who were 65 years or older. Cohort enrollment occurred between 1994 and 1996. A simple random sample (n = 6782) was drawn from the study base. Initial review excluded potential subjects with existing diagnosis of dementia or those who were in a skilled nursing facility. Of the 5422 eligible subjects, 2841 (52%) refused to participate in the longitudinal ACT study because of a variety of medical, personal and other reasons. Refusals prior to enrollment were somewhat more common among subjects in the oldest age group and among women, although the numbers enrolled in these groups were still sufficient to provide relatively stable incidence estimates. There were no exclusions for PD or other movement disorders, as long as the participants were cognitively normal at enrollment. Of the eligible subjects, 2581 provided informed consent and were enrolled in the ACT cohort. These participants provided demographic information, medical history, family history, psychosocial history and known or suspected dementia risk factors during an in‐person interview. As of June 2007, 1167 participants in the ACT cohort had died; of these, 250 had undergone autopsy and 238 had complete neuropathologic assessment.
Detailed ACT study design and methods have been described (11).
Exposure measurement
We administered questionnaires and acquired neuropsychological measurements using the cognitive assessment screening instrument (CASI) on all ACT participants every 2 years. The questionnaire data includes overall self‐rated health status, smoking history and alcohol history. Smoking habits were assessed at baseline and reassessed at each biennial visit when the number of cigarettes smoked per day and the number of pipes or cigars smoked per week were estimated. Lifetime pack years of smoking was calculated from the information obtained in the biennial questionnaires, and was defined as the average number of packs of cigarettes smoked per day multiplied by the number of years that the individual smoked.
Outcome measurement
All brain autopsies were performed following appropriate informed consent and approval of IRBs. We performed a standard neuropathologic workup blinded to clinical measures or smoking status that included gross and microscopic examination of all autopsied subjects. Histological evaluations included hematoxylin–eosin (HE), modified Bielschowsky and Congo Red staining. In addition, we performed alpha‐synuclein (SNCA) immunohistochemical analysis (antibody LB509, dilution 1:400; generous gift from John Q. Trojanowski) in the medulla, substantia nigra, hippocampus, entorhinal cortex/parahippocampal gyrus, amygdala, cingulate gyrus and frontal cortex exactly as previously described (12). We followed the consensus criteria that included both classic Lewy bodies (as seen with HE histological stain) and abnormal SNCA deposition in Lewy inclusions and neurites; in aggregate, these are referred to as LRP 12, 14. Braak stage for NFT distribution (2) and Consortium to Establish a Registry for Alzheimer's Disease (CERAD) NP score (17) were determined for each case. Cases with a Braak NFT stage of IV, V or VI, and CERAD NP score of B or C were judged to have met neuropathologic criteria (NC) for AD and are referred to as AD neuropathologic change (ADNC) (10).
Statistical analysis
The primary neuropathologic outcomes considered were LRP and ADNC. The exposure of interest was lifetime pack years of smoking calculated from information obtained in the biennial questionnaires as described earlier. Because of the potential for measurement error in self‐recalled smoking, we elected to base our primary analyses on three categories: non‐/light smokers (0–5 pack years); moderate smokers (>5–50 pack years); and heavy smokers (>50 pack years). Analyses were also carried out using continuous pack‐year data and several other categorizations to establish robustness of the primary analyses. In addition, possible non‐linearity of exposure–response between neuropathologic changes and pack years was explored using cubic splines (4).
Differences in demographic and autopsy measures across smoking groups were assessed using anova for continuous measures and Fisher's exact test for categorical measures. Relative risk (RR) of neuropathologic changes by smoking status was estimated using a generalized linear model (GLM) with a log link, Gaussian error and robust estimates of the standard errors of the model coefficients 13, 16. We used moderate smokers (>5–50 pack years) as the referent. RRs were adjusted for age at death and gender, which we believe to be the primary potential confounders based on previous studies that showed that older age and male gender are associated with smoking status and PD (9).
Participants who underwent autopsy were a self‐selected sample of deceased ACT participants. Full neuropathologic assessment was not possible to complete on 12 autopsies because of inadequate tissue sampling; for simplicity, we will henceforth refer to “autopsied” subjects when we mean “autopsied with complete neuropathologic assessment”. Differences in demographic and clinical measures between those deceased members of the ACT cohort for whom an autopsy was and was not performed were assessed using Student's t‐test for continuous measures and Fisher's exact test for categorical measures. Potential selection bias was addressed by inverse probability weighting to adjust the composition of the autopsied group to reflect the characteristics of the deceased group as a whole (24). This method essentially creates a pseudo sample, in which the autopsied subjects are weighted by the inverse of the probability of being autopsied. In this way, the autopsied subjects account in the analysis for those deceased subjects with similar characteristics who were not autopsied. It was assumed that—given information on age at death, gender, ethnicity (Caucasian vs. other), marital status, baseline CASI score, follow‐up time and clinical diagnosis of dementia during follow‐up—neuropathologic results of deceased subjects could be regarded as missing at random. Using information on these covariates for all deceased subjects (n = 1167), logistic regression was used to estimate the probability that a deceased participant underwent autopsy. The inverses of these probabilities were used as weights in the GLM models described earlier to obtain estimates of RR of neuropathologic changes by smoking status that were adjusted for selection bias. Ten thousand bootstrap replications were applied to this GLM analysis, and 95% confidence intervals (CIs) for the RRs were constructed using the bias‐corrected accelerated (BCa) method (5). Statistical significance was set at α = 0.05.
RESULTS
Relative to those deceased subjects who were not autopsied, participants who were autopsied were significantly older at time of death, had greater time elapsed between study entry and death, were more likely to be Caucasian and had higher education levels, higher baseline CASI scores, lower rates of self‐reported hypertension and coronary artery disease and higher rates of dementia diagnosed during the ACT study (Table 1). The autopsied and not autopsied ACT participants did not significantly differ in marital status, body mass index (BMI), diagnosis of diabetes mellitus (DM), diagnosis of cerebrovascular accident (CVA), frequency of APOEε4 allele or smoking status.
Table 1.
Characteristics of 1167 ACT participants who died as of June 1, 2007. Continuous variables expressed as mean [standard deviation (SD), range] and categorical variables expressed as frequency (%). Abbreviations: BMI = body mass index; CAD = coronary artery disease; CASI = cognitive assessment screening instrument; CVA = cerebrovascular accident; DM = diabetes mellitus.
| Not autopsied (n = 929) | Autopsied (n = 238) | P * | |
|---|---|---|---|
| Mean (SD, range) | |||
| Age at baseline (years) | 78 (7, 65–101) | 79 (7, 65–96) | 0.018 |
| Age at death (years) | 84 (7, 66–105) | 86 (7, 68–101) | <0.0001 |
| Time from study entry to death (years) | 6 (3, 0–13) | 7 (3, 0–13) | <0.0001 |
| CASI score at baseline† | 91 (6, 18–100) | 93 (5, 70–100) | 0.0002 |
| BMI at baseline‡ | 27 (5, 16–49) | 27 (5, 16–49) | 0.2 |
| Frequency (%) | |||
| Male | 450 (48) | 103 (43) | 0.2 |
| Caucasian | 849 (91) | 230 (97) | 0.0055 |
| Completing college or higher | 265 (29) | 86 (36) | 0.026 |
| Married at study entry | 457 (49) | 132 (55) | 0.098 |
| APOEε4 allele§ | 168 (25) | 58 (27) | 0.6 |
| Baseline hypertension¶ | 432 (47) | 90 (38) | 0.019 |
| Baseline DM** | 129 (14) | 31 (13) | 0.8 |
| Baseline CAD†† | 261 (28) | 50 (21) | 0.027 |
| Baseline CVA‡‡ | 155 (17) | 33 (14) | 0.3 |
| Clinical dementia | 218 (23) | 82 (34) | 0.0008 |
| Smoked at least 100 cigarettes in lifetime | 537 (58) | 140 (59) | 0.8 |
Student's t‐test for differences in means and Fisher's exact test for difference in proportions.
17 subjects with missing data.
28 subjects with missing data.
290 subjects with missing data.
5 subjects with missing data.
2 subjects with missing data.
1 subject with missing data.
3 subjects with missing data.
Information on smoking history was available for all 238 autopsied subjects. Demographic and clinical characteristics of autopsied participants by pack years of smoking are presented in Table 2. Compared with moderate and heavy smokers, non‐/light smokers died at an older age and were more commonly female. Table 3 shows neuropathologic changes stratified by smoking history. Despite being more commonly male, heavy smokers tended to have less LRP across all brain regions: 19% of heavy smokers; 31% of moderate smokers; and 35% non‐/light smokers. This pattern was statistically significant only in the substantia nigra (P = 0.04, Fisher's exact test), where LRP was found in 5% of heavy smokers vs. 21% or 18% in moderate or non‐/light smokers, respectively. No other significant association was observed between neuropathologic endpoints and smoking status. For those with LRP, classification of LRP predominance (12) are as follows: amygdala (n = 15); brainstem (n = 23); limbic (n = 15); neocortical (n = 13); unclassified (n = 6). We defined a summary index of ADNC as Braak NFT stage IV, V or VI, and CERAD NP score of B or C per NIA/Reagan criteria. As expected, LRP was more commonly co‐morbid with ADNC (43%) than without ADNC (26%; P = 0.03, Fisher's exact test). Individuals with LRP vs. those without LRP were similar for mean [standard deviation (SD)] age at death [87 (7) years vs. 86 (7) years] and more commonly male (50% vs. 40%), but these differences were not statistically significant.
Table 2.
Demographic and clinical characteristics by lifetime pack years for 238 participants with autopsy. Continuous variables expressed as mean [standard deviation (SD), range] and categorical variables expressed as frequency (%), except where otherwise noted. Abbreviations: BMI = body mass index; CAD = coronary artery disease; CASI = cognitive assessment screening instrument; CVA = cerebrovascular accident; DM = diabetes mellitus.
| 0–5 pack years (n = 114) | >5–50 pack years (n = 82) | >50 pack years (n = 42) | P * | |
|---|---|---|---|---|
| Median (range) pack year | 0 (0–5) | 27 (5.4–50) | 66 (50.4–223) | |
| Mean (SD, range) | ||||
| Age at baseline | 80 (7, 67–96) | 78 (6, 65–88) | 78 (7, 66–90) | 0.011 |
| Age at death | 88 (7, 72–101) | 85 (7, 68–97) | 85 (7, 72–100) | 0.0029 |
| CASI score at baseline† | 92 (5, 72–100) | 94 (5, 70–100) | 94 (4, 80–98) | 0.2 |
| BMI at baseline‡ | 26 (5, 17–49) | 27 (5, 16–38) | 27 (4, 18–36) | 0.3 |
| Frequency (%) | ||||
| Male | 32 (28) | 48 (59) | 23 (55) | <0.0001 |
| Caucasian | 110 (96) | 80 (98) | 40 (95) | 0.8 |
| Completing college or higher | 44 (39) | 27 (33) | 15 (36) | 0.7 |
| APOEε4 allele§ | 29 (29) | 20 (27) | 9 (24) | 0.8 |
| Baseline hypertension | 44 (39) | 37 (45) | 9 (21) | 0.033 |
| Baseline DM | 12 (11) | 9 (11) | 10 (24) | 0.086 |
| Baseline CAD | 26 (23) | 11 (13) | 13 (31) | 0.059 |
| Baseline CVA | 18 (16) | 11 (13) | 4 (10) | 0.7 |
| Clinical dementia | 43 (38) | 26 (32) | 13 (31) | 0.6 |
anova for differences in means and Fisher's exact test for categorical variables.
Two subjects with missing data.
Three subjects with missing data.
Twenty‐five subjects with missing data.
Table 3.
Neuropathologic characteristics by lifetime pack years for 238 participants with autopsy. Values are n (%). Abbreviations: CERAD = Consortium to Establish a Registry for Alzheimer's Disease; LRP = Lewy‐related pathology; NP = neuritic plaque.
| 0–5 pack years (n = 114) | >5–50 pack years (n = 82) | >50 pack years (n = 42) | |
|---|---|---|---|
| LRP in any region | 35 (31) | 29 (35) | 8 (19) |
| LRP in substantia nigra*, † | 24 (21) | 15 (18) | 2 (5) |
| LRP in medulla‡ | 27 (24) | 20 (25) | 5 (12) |
| LRP in amygdala* | 27 (24) | 19 (23) | 6 (14) |
| LRP in cingulate gyrus§ | 17 (16) | 7 (9) | 2 (5) |
| LRP in frontal cortex¶ | 12 (11) | 6 (8) | 1 (3) |
| Braak stage** | |||
| None, I | 30 (26) | 15 (18) | 13 (31) |
| II, III | 43 (38) | 41 (50) | 19 (45) |
| IV–VI | 41 (36) | 26 (32) | 10 (24) |
| CERAD NP score** | |||
| None | 25 (22) | 27 (33) | 14 (33) |
| A | 44 (39) | 22 (27) | 17 (40) |
| B or C | 45 (39) | 33 (40) | 11 (26) |
One subject with missing data.
P < 0.05 by Fisher's exact test.
Two subjects with missing data.
Seventeen subjects with missing data.
Five subjects with missing data.
See (1).
Table 4 presents the estimated RR of LRP by pack‐year category with the moderate smokers as the reference group. Although the association, adjusted for age at death and gender, was not statistically significant, heavy smokers had lower risk of LRP than moderate smokers [RR (95% CI) 0.55 (0.28, 1.07)]. Figure 1 shows a cubic spline smooth estimate of the (unadjusted) relationship between pack years and risk of LRP using a GLM. Estimates adjusted for covariates and for selection bias were similar in structure. The smoothed association indicated declining risk of LRP beyond about 40 pack years of smoking, but with extremely wide pointwise confidence bounds. To assess sensitivity to outliers with very heavy smoking, the GLM analysis was repeated, omitting those who smoked more than 80 pack years (n = 10). Similar GLM models using continuous pack years and different categorizations of pack years also were assessed. All analyses consistently yielded results that were suggestive, but not conclusive, of a decrease in risk of LRP for the heaviest smokers.
Table 4.
Relative risk (RR) and 95% confidence interval (CI) for the relationship between neuropathologic changes and smoking. RR and CI are modeled as three categories: 0–5; >5–50; and >50 pack years, with >5–50 pack years as referent. Abbreviations: ADNC = Alzheimer's disease neuropathologic change; LRP = Lewy‐related pathology.
| >50 pack years vs. >5–50 pack years | 0–5 pack years vs. >5–50 pack years | |
|---|---|---|
| LRP changes | ||
| RR adjusted for age at death and gender | 0.55 (0.28, 1.07) | 0.82 (0.54, 1.25) |
| RR after selection bias adjustment | 0.43 (0.15, 0.90)* | 0.67 (0.38, 1.14) |
| ADNCs | ||
| RR adjusted for age at death and gender | 0.43 (0.15, 1.22) | 1.10 (0.65, 1.88) |
| RR after selection bias adjustment | 0.65 (0.17, 2.25) | 1.16 (0.55, 2.81) |
P < 0.05.
Figure 1.
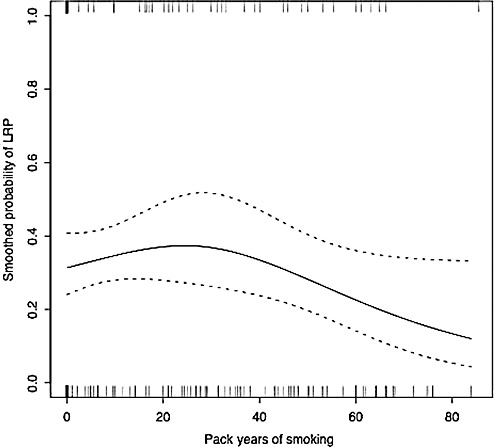
Cubic spline estimate and associated pointwise 95% confidence bounds of the association between the probability of Lewy‐related pathology (LRP) and smoking pack years. Observed pack years of smoking by LRP status is indicated below (absence of LRP) and above (presence of LRP) the plot. For ease of visualization, eight subjects with >85.5 pack years of smoking, all of whom were without LRP, are omitted from the plot (but not from estimation of the smoothed association).
The possible impact of selection bias was assessed by a weighted GLM analysis, with the observation for each autopsied subject weighted by the inverse of his or her estimated probability of being autopsied. These estimated probabilities ranged from approximately 3% to 60% and were based on a logistic regression analysis using the following covariates: age at death; gender; ethnicity (Caucasian vs. other); marital status (married, unmarried or other); baseline CASI score; follow‐up time; and clinical diagnosis of dementia during follow‐up. After adjustment for selection bias, the estimated RR of LRP for heavy smokers compared with moderate smokers was 0.43 (95%CI 0.15, 0.90), similar to the estimated RR without adjustment for selection bias, but stronger and statistically significant. However, the statistical significance was sensitive to a few influential subjects who were given relatively large weight because their characteristics were underrepresented in the autopsy sample.
For comparison, Table 4 presents the estimated RR of ADNC by pack‐year category with the moderate smokers again as the reference group. There was no evidence of an association between pack years of smoking and risk of developing ADNC, adjusting for age at death and gender, regardless of whether or not a selection bias adjustment was included. The interactions between pack‐year category and ADNC in a GLM for LRP, adjusting for age at death and gender, were not statistically significant either with or without selection bias adjustment.
DISCUSSION
In this community‐based study, we observed that cigarette smoking was associated with significantly reduced RR for LRP after correcting for selection bias and with significantly reduced frequency of LRP in the substantia nigra. This apparent protective effect was observed only in heavy smokers, suggesting the possibility of an exposure–response relationship. These findings are of interest because they are the first, of which we are aware, to associate reduced LRP in the human brain with any exposure. Moreover, they suggest that substantia nigra neurons, a major target of LRP‐associated neurodegeneration, may be especially receptive to this form of neuroprotection. Although cigarette smoking is too toxic to suggest as a treatment, these findings, if confirmed, may guide future therapeutic strategies that attempt to suppress LRP by other means (18).
Our study has several strengths. One is that our community‐based sample was derived from a typical urban/suburban US population. Furthermore, our subjects were a prospectively followed cohort of initially non‐demented community‐dwelling elderly individuals. This prospective study design limits the type of biases common in case‐control studies (eg, recall bias, as information on smoking was collected at enrollment prior to the development of any cognitive impairment, as well as from biennial questionnaires). Finally, neuropathologic data were collected by investigators who were blinded to exposure data and clinical information.
Several shortcomings and caveats exist that limit the generalization of our findings. First, individuals must have survived to age 65 years without dementia in order to enroll in ACT. Therefore, individuals who died younger than 65 years of age from the carcinogenic, cardiovascular or pulmonary complications of cigarette smoking were not included. This might be a source of bias as one could speculate that smokers who survive to age 65 years may represent an especially metabolically vigorous group that is resistant to the damaging effects not only of smoking but also processes that lead to the development of LRP. From this perspective, it is at least interesting to note that smokers in our study did not have significantly altered levels of AD pathologic changes, perhaps undermining this speculation. Regardless, we must acknowledge the possibility that the apparent impact on LRP is not causally related to smoking status, but rather is a consequence of other, unmeasured, factors that are associated with heavy smoking in subjects who survive to old age.
Although this is one of the largest samples of autopsied individuals followed in a prospective cohort study, another shortcoming is interpreting null findings when sample sizes are small. This may have contributed to the apparently regionally restricted statistically significant exposure–response relationships, where our results consistently indicated reduced LRP in heavy smokers, but the small sample size in this group (n = 42) limited our analytical power. We also recognized a trend for association with AD neuropathologic changes but note that this was less consistent than the association of reduced LRP in heaviest smokers. Thus, although the LRP‐smoking association is likely strongest among standard neuropathologic measures of neurodegeneration, further study and larger samples will be needed to confirm not only our positive but also our negative findings.
We investigated LRP but did not further subclassify cases into clinicopathological groups such as PD or DLB. This is a strength if LRP represents a disease entity that has varied clinical presentations, the view to which we subscribe. Regardless, we lack sufficient number of cases to meaningfully stratify our sample into clinicopathologic groups, and so we must await larger studies to determine if our association of heavy cigarette smoking with reduced LRP is restricted to certain clinical presentations of LRP. One speculation—as the association observed was strongest for LRP in the substantia nigra—is that heavy cigarette smoking might be more strongly related with reduced risk of parkinsonism from LRP than of cognitive impairment from LRP; this resonates with observational studies of smoking and clinical diagnosis of PD or AD 7, 20, 21, 22.
As estimates based on autopsied subjects alone may be subject to selection bias, we attempted to assess the strength of our findings by carrying out weighted analyses that effectively created a “virtual” sample in which all deceased ACT subjects had been autopsied. This approach assumes that—conditional on the specified covariates—neuropathologic data lacking from individuals who did not undergo autopsy were missing at random, or, in other words, that—within covariate strata—the distribution of neuropathologic changes was the same in those who were and who were not autopsied. Although this seems reasonable, it is not possible to test this assumption. Adjusting from the autopsy group to all deceased members of ACT is appropriate because of known important differences between these two groups (eg, prevalence of dementia). Moreover, adjusting our results to account for these known differences between the autopsy group and all deceased members of the cohort enhances our ability to generalize our conclusions to those individuals who met eligibility criteria for ACT and who were followed until death within a given time period. Further selection bias adjustments of our analyses to reflect the entire ACT cohort (living and deceased) are theoretically possible, but require knowledge of potential systematic differences between the survivors and the deceased. Such knowledge is limited, and in some cases not measurable. Thus, although recognizing the theoretical advantages of adjusting for selection bias between the autopsied and the entire cohort, we have elected not to attempt further adjustments of our findings to reflect the entire ACT cohort because of the potential inaccuracy of these extrapolations.
Finally, cigarette smoking may act directly to inhibit SNCA fibrillation. Compounds such as nicotine and hydroquinone in cigarettes might cause such inhibition (8). Alternatively, cigarette smoking may serve as a surrogate for factors that might reduce the risk for the development of PD or LRP. For example, behavioral traits that may increase the risk for cigarette smoking may be associated with the clinical and/or pathological manifestation of PD. Additional molecular biological studies are necessary to determine whether smoking directly affects the neurochemical aspects of LRP or alternatively, whether smoking indirectly reduces the risk for developing PD via other mechanisms. In summary, we observed an association between cigarette smoking and reduced risk of developing LRP, but not AD pathologic changes, in the heaviest smokers. These findings are consonant with earlier studies of diminished risk of clinically assessed PD with cigarette smoking, suggest one explanation to partially account for widely varying results of observational studies of smoking and dementia, and may provide new insights into developing alternative therapeutics to suppress the development of LRP in the elderly.
ACKNOWLEDGMENTS
This work was supported by grants from the National Institutes of Health (AG05136, AG06781, AG023801 and AG000258) and the Nancy and Buster Alvord Endowment. This material is also the result of work supported by resources from the VA Puget Sound Health Care System, Seattle, Washington.
The authors declare that they have no conflict of interest with this work.
REFERENCES
- 1. Anonymous (1997) Consensus recommendations for the postmortem diagnosis of Alzheimer's disease. The National Institute on Aging, and Reagan Institute Working Group on Diagnostic Criteria for The Neuropathological Assessment of Alzheimer's disease. Neurobiol Aging 18:S1–S2. [PubMed] [Google Scholar]
- 2. Braak H, Braak E (1991) Neuropathological stageing of Alzheimer‐related changes. Acta Neuropathol 82:239–259. [DOI] [PubMed] [Google Scholar]
- 3. Braak H, Ghebremedhin E, Rub U, Bratzke H, Del Tredici K (2004) Stages in the development of Parkinson's disease‐related pathology. Cell Tissue Res 318:121–134. [DOI] [PubMed] [Google Scholar]
- 4. De Boor C (1978) A Practical Guide to Splines. Springer Verlag: Berlin. [Google Scholar]
- 5. Efron B, Tibshirani R (1986) Bootstrap methods for standard errors, confidence intervals, and other measured of statistical accuracy. Stat Sci 1:54–75. [Google Scholar]
- 6. Gibb WR, Lees AJ (1988) The relevance of the Lewy body to the pathogenesis of idiopathic Parkinson's disease. J Neurol Neurosurg Psychiatry 51:745–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hernan MA, Alonso A, Logroscino G (2008) Cigarette smoking and dementia: potential selection bias in the elderly. Epidemiology 19:448–450. [DOI] [PubMed] [Google Scholar]
- 8. Hong DP, Fink AL, Uversky VN (2009) Smoking and Parkinson's disease: does nicotine affect alpha‐synuclein fibrillation? Biochim Biophys Acta 1794:282–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kasten M, Chade A, Tanner CM (2007) Epidemiology of Parkinson's disease. Handb Clin Neurol 83:129–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Knopman DS, Parisi JE, Salviati A, Floriach‐Robert M, Boeve BF, Ivnik RJ et al (2003) Neuropathology of cognitively normal elderly. J Neuropathol Exp Neurol 62:1087–1095. [DOI] [PubMed] [Google Scholar]
- 11. Kukull WA, Higdon R, Bowen JD, McCormick WC, Teri L, Schellenberg GD et al (2002) Dementia and Alzheimer disease incidence: a prospective cohort study. Arch Neurol 59:1737–1746. [DOI] [PubMed] [Google Scholar]
- 12. Leverenz JB, Hamilton R, Tsuang DW, Schantz A, Vavrek D, Larson EB et al (2008) Empiric refinement of the pathologic assessment of Lewy‐related pathology in the dementia patient. Brain Pathol 18:220–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lumley T, Kronmal R, Ma S (2006) Relative Risk Regression in Medical Research: Models, Contrasts, Estimators, and Algorithms. University of Washington: Seattle, WA. [Google Scholar]
- 14. McKeith IG, Galasko D, Kosaka K, Perry EK, Dickson DW, Hansen LA et al (1996) Consensus guidelines for the clinical and pathologic diagnosis of dementia with Lewy bodies (DLB): report of the consortium on DLB international workshop. Neurology 47:1113–1124. [DOI] [PubMed] [Google Scholar]
- 15. McKhann G, Drachman D, Folstein M, Katzman R, Price D, Stadlan EM (1984) Clinical diagnosis of Alzheimer's disease: report of the NINCDS‐ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer's Disease. Neurology 34:939–944. [DOI] [PubMed] [Google Scholar]
- 16. McNutt LA, Wu C, Xue X, Hafner JP (2003) Estimating the relative risk in cohort studies and clinical trials of common outcomes. Am J Epidemiol 157:940–943. [DOI] [PubMed] [Google Scholar]
- 17. Mirra SS, Heyman A, McKeel D, Sumi SM, Crain BJ, Brownlee LM et al (1991) The Consortium to Establish a Registry for Alzheimer's Disease (CERAD). Part II. Standardization of the neuropathologic assessment of Alzheimer's disease. Neurology 41:479–486. [DOI] [PubMed] [Google Scholar]
- 18. Ono K, Hirohata M, Yamada M (2007) Anti‐fibrillogenic and fibril‐destabilizing activity of nicotine in vitro: implications for the prevention and therapeutics of Lewy body diseases. Exp Neurol 205:414–424. [DOI] [PubMed] [Google Scholar]
- 19. Quik M, O'Neill M, Perez XA (2007) Nicotine neuroprotection against nigrostriatal damage: importance of the animal model. Trends Pharmacol Sci 28:229–235. [DOI] [PubMed] [Google Scholar]
- 20. Ritz B, Ascherio A, Checkoway H, Marder KS, Nelson LM, Rocca WA et al (2007) Pooled analysis of tobacco use and risk of Parkinson disease. Arch Neurol 64:990–997. [DOI] [PubMed] [Google Scholar]
- 21. Tan LC, Koh WP, Yuan JM, Wang R, Au WL, Tan JH et al (2008) Differential effects of black versus green tea on risk of Parkinson's disease in the Singapore Chinese Health Study. Am J Epidemiol 167:553–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Thacker EL, O'Reilly EJ, Weisskopf MG, Chen H, Schwarzschild MA, McCullough ML et al (2007) Temporal relationship between cigarette smoking and risk of Parkinson disease. Neurology 68:764–768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Tsuang D, Simpson K, Larson EB, Peskind E, Kukull W, Bowen JB et al (2006) Predicting lewy body pathology in a community‐based sample with clinical diagnosis of Alzheimer's disease. J Geriatr Psychiatry Neurol 19:195–201. [DOI] [PubMed] [Google Scholar]
- 24. Zhao LP, Lipsitz S (1992) Designs and analysis of two‐stage studies. Stat Med 11:769–782. [DOI] [PubMed] [Google Scholar]