

Abstract
Background
The most frequent form of congenital dyserythropoietic anemia is the type II form. Recently it was shown that the vast majority of patients with congenital dyserythropoietic anemia type II carry mutations in the SEC23B gene. Here we established the molecular basis of 42 cases of congenital dyserythropoietic anemia type II and attempted to define a genotype-phenotype relationship.
Design and Methods
SEC23B gene sequencing analysis was performed to assess the diversity and incidence of each mutation in 42 patients with congenital dyserythropoietic anemia type II (25 described exclusively in this work), from the Italian and the French Registries, and the relationship of these mutations with the clinical presentation. To this purpose, we divided the patients into two groups: (i) patients with two missense mutations and (ii) patients with one nonsense and one missense mutation.
Results
We found 22 mutations of uneven frequency, including seven novel mutations. Compound heterozygosity for a missense and a nonsense mutation tended to produce a more severe clinical presentation, a lower reticulocyte count, a higher serum ferritin level, and, in some cases, more pronounced transfusion needs, than homozygosity or compound heterozygosity for two missense mutations. Homozygosity or compound heterozygosity for two nonsense mutations was never found.
Conclusions
This study allowed us to determine the most frequent mutations in patients with congenital dyserythropoietic anemia type II. Correlations between the mutations and various biological parameters suggested that the association of one missense mutation and one nonsense mutation was significantly more deleterious that the association of two missense mutations. However, there was an overlap between the two categories.
Keywords: CDA II, SEC23B gene, COPII, genotype-phenotype relationship
Introduction
The congenital dyserythropoietic anemias (CDA) are a heterogeneous group of rare genetic disorders marked by ineffective erythropoiesis causing anemia, although a degree of hemolysis is present as well.1,2 CDA type II (CDA II), a recessively inherited condition, is the most common form of CDA. The main European Registries (German, Italian and French) have collated 281 individual or familial cases (unpublished data), although some are not of European descent. CDA II seems more frequent in southern Italy than in northern and central Europe, but has been reported sporadically among Americans, Africans, Indians, and Pakistani.
The vast majority of CDA II patients show a variable degree of anemia, which is mild in most cases (9–10 g/dL). Iron overload develops even in the absence of transfusions and is a major element for prognosis and treatment. CDA II is associated with well-defined cellular and ultrastructural features including binucleated or multinucleated late precursors, flat vesicles of variable length, and a double membrane beneath the cytoplasmic membrane.3,4 The principal biochemical feature is the hypoglycosylation of several proteins, such as transferrin and band 3.5,6 Much research has been devoted to enzymes involved in glycan synthesis. Microsomal N-acetylglucosaminyltransferase II (14q21),7 Golgi α-mannosidase II (5q2.1.2.2),8 as well as other enzymes, were found to be deficient. Originally, the responsible gene was mapped to the long arm of chromosome 20.9 Consistently, the above-cited genes and others were excluded.10 Seven genes in 20q, not all included in glycan synthesis, were further ruled out.11 In a refined contig build (build 36.3), the markers with the highest CDA II logarithmic of odds (LOD) scores overlap the minimal homozygosity region on the short arm of chromosome 20.12 Moreover, because the gene of interest was located next to the centromere, its position was redefined between D20S112-D20S106 markers, encompassing the short arm of chromosome 20. In order to narrow down this region further, we used the information derived from a functional mapping approach. We performed microarray analysis on total RNA from CD34+ and orthochromatic normoblasts (14–21 days after erythroid differentiation induced by erythropoietin) of healthy controls to identify differentially expressed genes during normal erythropoiesis (data not shown). With the assumption that the cis, median and trans N-glycan Golgi processing of erythroblast glycoproteins was impaired, the SEC23B gene became a likely candidate. Schwarz et al.13 independently resorted to a functional approach based on homozygosity mapping. Sequencing analysis in 33 people from 28 unrelated families from the main European Registries13 and 13 CDAII patients from 10 families14 showed a wide spectrum of different mutations in either the compound heterozygous or homozygous state. In Saccharomyces cerevisiae, Sec23 is a component of the COPII complex, which derives from sequential binding of Sar1-GTP, the inner complex proteins Sec23/24 and the outer components Sec13/31 on the endoplasmic reticulum.15,16
The aim of this study was to assess the diversity and incidence of SEC23B gene mutations in a large series of CDA II patients from the Italian and the French Registries, specifically, and the relationship of these mutations to the clinical presentation.
Design and Methods
Patients
After signed informed consent, blood was obtained for genetic analysis from the probands. Whenever possible, relatives were investigated. Blood from healthy control subjects was obtained after signed consent according to the Declaration of Helsinki. Collection of patients’ data from the International Registry on CDA II, Naples, Italy, and the French Registry on CDA, Le Kremlin-Bicêtre, France, was approved by local university ethical committees. Blood samples were processed within 24 hours.
Forty-two individuals with CDA II from 40 independent families from the Italian and French Registries were included in this study. The mutations of 17 of these patients have already been described,13 however the patients’ clinical presentation have not been detailed. We added 25 patients from the two above Registries in order to broaden the significance of the study. For each of these additional patients, the mutations were elucidated. The diagnosis was based on history, clinical findings, routine laboratory data, analysis of aspirated bone marrow (light microscopy; electron microscopy was resorted to exceptionally), evidence of abnormal band 3 upon polyacrylamide gel electrophoresis in the presence of sodium docecyl sulfate (SDS-PAGE), and/or increased erythrocyte agglutination by anti-i sera. Western blots were used to detect endoplasmic reticulum-specific proteins in most patients. Serum ferritin level, evaluated before iron chelation therapy, was monitored as a key element for prognosis and a guide to the chelation treatment. Taken together, the key elements for the diagnosis were the bone marrow aspirate and the abnormal electrophoretic appearance of band 3. It should be noted that thorough phenotypic data were not available for all patients (Table 1).
Table 1.
SEC23B mutations and laboratory investigations of CDAII patients.

In a number of cases, more than one sibling was affected in a family. Often enough, the second CDA II patient underwent a workup reduced simply to what was needed for the diagnosis and, based on the experience with the elder child, fewer transfusions were administered (Patients 1a and 1b, 35a and 35b, Table 1). SEC23B gene analysis was not performed on a systematic basis in the younger child either. The latter cases (i.e. those for which sequencing was not done) are not included in Table 1.
Sequencing analysis
Genomic DNA was prepared from peripheral blood using a Wizard Genomic DNA purification kit (Promega, Milan, Italy). The mutational search was performed using 20 ng of genomic DNA by direct sequencing. Briefly, all exons, flanking splice junctions, 5′-and 3′-untranslated regions of the SEC23B gene were amplified by polymerase chain reaction (PCR) in a 25 μL volume with Master Mix 2X (Promega). The oligonucleotide primers were designed by Primer3 program (Primer3 v. 0.4.0, freeware online). Sequence primers are available on request ( iolascon@ceinge.unina.it). The PCR products were checked by DNA agarose gel electrophoresis. Direct sequencing was performed using the BigDye® Terminator Cycle Sequencing Kit (Applied Biosystems, Branchberg, NJ, USA) and a 3730 DNA Analyzer (Applied Biosystems).
Statistical methods
In order to analyze the genotype-phenotype correlation, we divided the CDA II patients into two groups: (i) patients with two missense mutations [group A (patients 1a, 1b, 3, 5, 7, 10, 12, 14, 16, 17, 19, 21, 22, 24, 25, 27–34, 36–39), n=27] and (ii) patients with one nonsense and one missense mutation [group B (patients 2, 6, 11, 13, 15, 18, 20, 23, 26, 35a, 35b), n=11]. Patient 23, who exhibited a missense mutation and the splicing mutation c.689+1 G>A, was included in group B. The four patients with only one missense or one nonsense mutation in the heterozygous state (patients 4, 8, 9 and 40) were excluded from the analysis.
A Mann-Whitney test was used to compare differences in quantitative variables between two groups. Qualitative clinical data were compared using the χ2 test. Odds ratios (OR) and 95% confidence intervals (CI) were calculated to assess the relative risk of a more severe phenotype conferred by a specific genotype. A two-sided P value of less than 0.05 was considered statistically significant.
Results
Description of the patients’ phenotype
The clinical features of the 42 affected individuals, i.e. 38 unrelated individuals and two pairs of siblings in two unrelated families (patients 1a and 1b, and 35a and 35b), are presented in Table 1. The mean age of the patients at diagnosis was 15.1 years (range, from birth to 52 years). The male to female ratio was 16:26. The mean hemoglobin concentration was 9.8 g/dL (range, 6.0 to 13.7 g/dL) before splenectomy (hemoglobin values were post-splenectomy in patients 1a and 15). In two cases consanguinity was demonstrated (patients 1a and 1b, and patient 3). The mean ferritin value was 358.1 μg/L (range, 15 to 2500 μg/L) and the mean absolute reticulocyte count at diagnosis was 90474×106/L (ranging, 10000×106/L to 180000×106/L).
Patients 2, 18, 20, 29 and 39 had a severe, transfusion-dependent condition. Among these, patient 20 underwent splenectomy in May 2008; however, she died soon afterward (December 2008) at the age of 21, from heart failure due to massive iron overload. Patient 29, a 9-year old boy, has required transfusions since infancy. He was splenectomized at the age of 6 and this reduced his transfusion needs. For the past 4 years he has received regular iron-chelation therapy. Patient 39 also received transfusions. She was splenectomized at the age of 4 years. Patient 1a has a younger sister (1b) showing roughly the same phenotype. Yet, she was not splenectomized. Patient 8 has a younger brother (not shown), in whom the diagnosis of anemia was established first due to a rather severe presentation (the nature of the aggravating factor remained elusive). Splenectomy was performed on the basis of an uncertain diagnosis and it was only later that CDA II was identified. This patient also presented behavioral disorders. Patient 18 has an elder brother (not shown); both require monthly transfusions and both will undergo subtotal splenectomy in due course. Patient 27 has a twin sister (not shown) with the same moderate phenotype. Patient 35a has a younger sister (35b) showing a milder phenotype.
The vast majority of the patients displayed a mild to moderate phenotype. Nevertheless, 15 of 40 patients (patients 1b, 3, 5, 6, 9, 13, 15, 20, 22, 26, 29, 30, 33, 38 and 39) (37.5%) underwent splenectomy (mean age 17.9 years, range from 4 to 44 years), sometimes based on the erroneous diagnosis of hereditary spherocytosis (Iolascon et al., unpublished data).
We collected information about neonatal signs in 23 patients. During this period jaundice and anemia sometimes required transfusion support in four of these 23 patients (17.4%).
Sequencing analysis of SEC23B
The SEC23B gene spans approximately 54 kb on human chromosome 20p11.23 and has 19 coding exons, 3’ and 5’ untranslated regions. Multiple alternatively spliced transcript variants encoding the same protein (767 amino acids) have been found. We referred to transcript variant ENST00000377475 of Ensembl Genome Browser (http://www.ensembl.org/index.html) for sequence analysis. We found 22 mutations lying all along the SEC23B gene. There were seven stop mutations (2 frameshift and 5 nonsense mutations) and 14 missense mutations (Figure 1, Online Supplementary Table S1). All involved conserved residues (Online Supplementary Figure S1). We also found one splice site mutation (patient 23). Five missense mutations (R503Q, R530Q, Y562C, F656L, K723Q) and one nonsense and one frameshift mutation (R550X and T654TfsX13, respectively) have not described in previous articles.13,14 Analysis of 120 control chromosomes showed that these changes were not present in normal subjects.
Figure 1.

Site of all mutations in the SEC23B gene and protein. Top. Exons and introns are shown (distance in scale). Above the physical map, missense mutations are shown in straight characters. single nucleotide polymorphisms are presented in italics. Below the map, nonsense (direct stop codons and frameshift mutations) and splicing mutations are shown. The mutations first described are marked in bold. Bottom. Domains of the SEC23B component are shown and delineated with numbered amino acids. ZNF: zinc finger; BS:β-sheet. The extent of the site for binding other components of the COPII complex, inferred from a yeast model, is shown below.
Figure 2 shows the frequency of the mutations. The most represented missense mutations were E109K (34%) and R14W (14%), lying close to the zinc finger domain. Among nonsense mutations, the most frequent were R79X (4%), situated in the zinc finger domain, and R550X (3%), located in the helical domain (Figures 1 and 2). Four patients (patients 4, 8, 9 and 40) of the 42 studied showed only one mutation in the heterozygous state in association with one or two non-synonymous single nucleotide polymorphisms (Table 1). These single nucleotide polymorphisms were also recorded in some patients carrying two mutations. We assessed minor allele frequency for all three single nucleotide polymorphisms (rs41309927, rs17807673, rs2273526) by using 50 healthy controls (Online Supplementary Figure S2). We could exclude a dominant negative effect because the involved changes were detected also in one unaffected parent. The presence of a polymorphism in the heterozygous state excluded a deletion at least within the corresponding region of the gene.
Figure 2.
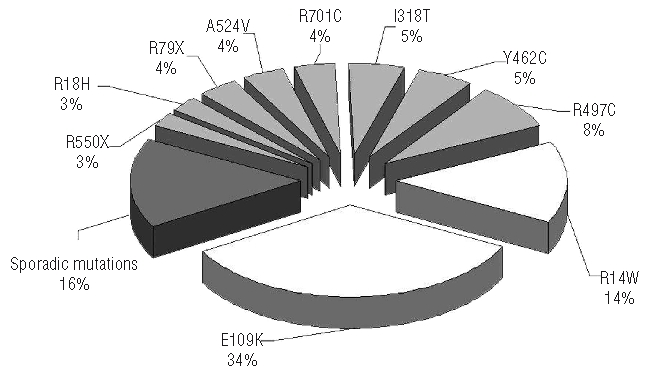
Allelic distribution of SEC23B mutations. This pie chart shows the incidence of the most frequent mutations (white), the less frequent mutations (light gray) and mutations found in one kindred only (dark gray). These last include the following mutations: three nonsense (R217X, R264X, R401X), two frameshift (D355IfsX8, T654TfsX13), one splicing (c.689+1 G>A), and six missense (Q386R, R503Q, R530Q, Y562C, F656L, K723Q).
Genotype-phenotype correlations
Patients were divided into two groups: group A (27 patients with two missense mutations) and group B (11 patients with one missense and one nonsense mutation). The four patients with only one mutation currently found in the SEC23B gene were not included in the statistics. For the purpose of comparing parameters, however, it is important to note that not all patients from each group were taken into account. Patients for whom the data were incomplete were not included (Table 2, Figures 3a, 3b, 3c and 3s). Thus, the series for every set of comparisons were smaller than the entire groups A and B.
Table 2.
Clinical parameters of CDA II patients*.
Figure 3.
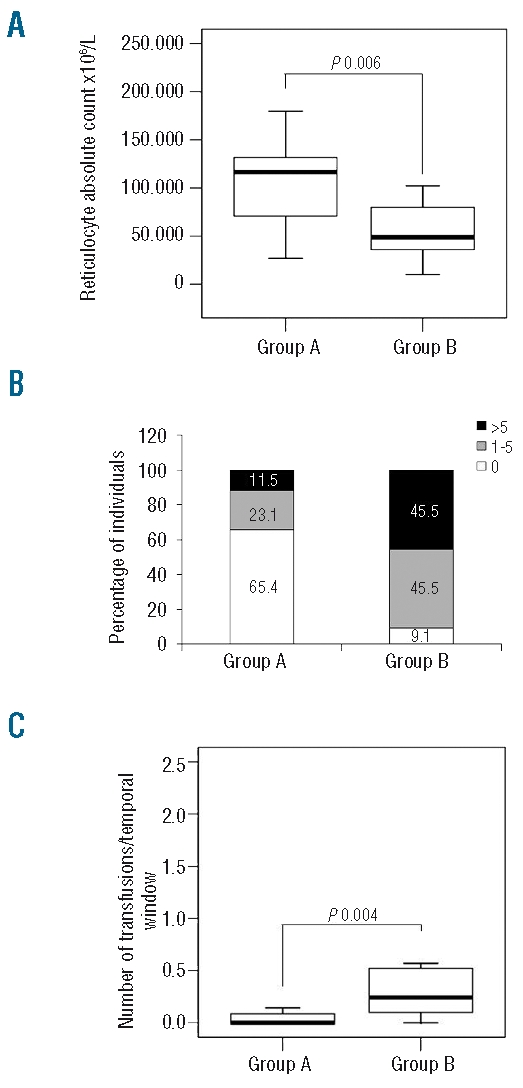
Genotype-phenotype correlation. Patients for whom the data were incomplete were not included. (A) Reticulocyte count. Patients with one nonsense + one missense mutation have a lower reticulocyte count [Group B, n=9: 48895; 54512 ± 32633 (x106/L)] when compared with patients with two missense mutations [Group A, n=24: 116600; 104115 ± 41986 (x106/L)] (P=0.006, Mann-Whitney test). (B) Patients were subdivided into three categories on the basis of number of transfusions: (i) TD (transfusion-dependent) + more than five transfusions (intermittent transfusions), (ii) one to five transfusions (occasional transfusions, due to intervening infections, doctor in charge’s anxiety or lack of knowledge about CDA II) and (iii) no transfusion. The percentage of individuals in each category is provided in ordinates. Patients with one nonsense + one missense mutation (group B, n = 11) were more transfused compared with patients with two missense mutations (group A, n= 26) (P=0.005, χ2 test). (C) Ratio of number of transfusions/time window. Patients with one nonsense + one missense mutation (group B, n=11: 0.2; 0.7 ± 1.2) had a higher ratio compared to patients with two missense mutations (group A, n=23: 0.0; 0.2±0.6) (P=0.004, Mann-Whitney test). Data are presented as median; mean ± SD.
Among basal hematologic parameters we evaluated the hemoglobin concentration, the absolute reticulocyte count, and the serum ferritin concentration in both groups.
Hemoglobin concentration tended to be lower in group B patients (n= 11; 8.9±1.4 g/dL) than in group A patients (n = 25; 9.9±2 g/dL), although the difference was not statistically significant (P=0.12) (Table 2).
The absolute reticulocyte count was lower in group B patients (n=9) than in group A patients (n=24) (P=0.006) (Figure 3a). To define the role of genotype in CDA II severity better, we performed the same comparison of absolute reticulocyte count excluding the five transfusion-dependent patients [2 from group A (patients 29 and 39) and 3 from group B (patients 2, 18 and 20)], obtaining the same results (data not shown).
Serum ferritin concentration tended to be higher in group B patients (n=10; 751±875 μg/L) than in group A patients (n=19; 196±100 μg/L), although the difference was not statistically significant (P=0.12) (Table 2). Since the serum ferritin level depends on age, we calculated the ferritin level/dosage age ratio and divided it into two ‘high’ and ‘low’ ratio categories on the basis of the median value (14.2). We then analyzed the distribution of these two categories between patients from groups A and B. Patients from group B (n=10) had a significantly higher ferritin level/dosage age ratio than patients from group A (n=19) (P=0.03) (Online Supplementary Figure S3). Using the number of transfusions as an index of disease severity, we compared the number of transfusions before splenectomy between the two groups. Patients from group B (n=11) had a higher risk of receiving a larger number of transfusions when compared to patients from group A (n=26) (0 versus 1–5 transfusions, OR=14.2, 95% CI: 1.4–147.1, P=0.01; 0 versus >5 transfusions, OR=28.3, 95% CI: 2.4–336.0, P=0.001) (Table 2, Figure 3b). Likewise, patients from group B had a higher risk of transfusion dependence than group A patients, although the difference was not statistically significant (OR=4.5, 95% CI: 0.6–31.9, P=0.11). We also analyzed the number of transfusions in a particular time window (corresponding to the current age for not splenectomized patients and the age before splenectomy for those splenectomized), i.e. the number of transfusions in relationship to this time interval. The ratio of number of transfusions/time window was higher in patients from group B (n = 11) than in patients from group A (n = 23) (P=0.004) (Figure 3c).
Bioinformatic analysis of protein structure
The SEC23B component is a member of the SEC23 subfamily that also includes the SEC23A component (see below). The SEC23B component has similarity (66.4%) with the Sec23 component of COPII in yeast, which is the coat protein complex responsible for vesicle budding from the endoplasmic reticulum. We assessed the amino-acid conservation of all missense mutations in 13 species that were aligned with the Homo sapiens SEC23B component (Online Supplementary Figure S1). Among the investigated species, we used the needle program (http://www.ebi.ac.uk/Tools/emboss/align/) to evaluate the human SEC23B component versus its orthologs in the mouse, monkey and zebrafish (98.6%, 84.1% and 93% of similarity, respectively). Mutated positions were conserved in most species from Homo sapiens to Aspergillus clavatus.
Discussion
Two recent studies showed that CDA II is caused in the vast majority of cases by mutations in the SEC23B gene.13,14 We examined a large series of CDA II patients from the Italian and French Registries. In keeping with current knowledge on CDA II, a characteristic clinical finding was low effective red cell production, inadequate to maintain normal hemoglobin levels. In general, anemia was mild to moderate with a mean hemoglobin concentration of 9.8 g/dL. However, there were a few very severe cases. The vast majority of patients had two mutations (in the homozygous or compound heterozygous state), in accordance with the pattern of autosomal recessive inheritance. Missense mutations were the most frequent events (85.5% allele frequency), whereas nonsense mutations were observed in eight chromosomes (10.5%). Two frameshift mutations and one splice site mutation were also observed. We did not check the cDNA in this last case (patient 23). In no case was homozygosity or compound heterozygosity for nonsense mutation(s) found, a situation likely to be lethal. Studies in knock-out animal models should confirm this hypothesis. This study identified seven additional mutations, localized in the β-sheet, the helical and the gelsolin-like domains. The other 15 mutations have already been described by Schwarz et al.13 and Bianchi et al.14 (Online Supplementary Table S1). Affected individuals carried missense mutations at conserved residues in SEC23B homologs from humans to Aspergillus clavatus. None of the disease-associated variants was present in the NCBI and Ensembl SNP databases or in 120 control chromosomes. These new data further support that mutations in the SEC23B gene cause CDA II. We found that four mutations R14W, E109K, R497C, and I318T account for more than 50% of all mutations in the SEC23B gene in CDA II. To further validate this finding we added to the 25 new cases described here to the 33 and 13 CDA II cases presented before.13,14 In this total of 71 cases from 63 families, 33% of the mutations were E109K, 17% were R14W and 6% were R497C. These data substantially confirm our distribution of mutations (Figure 2). Such information could be useful in directing the search for the causative mutation in new patients. The most frequent mutations, E109K and R14W, were present in people from different ethnic origins. They occurred in codons containing a complete or overlapping CpG dinucleotide (gccGAAttg and gaaCGGgat, respectively), a ‘hot spot’ for mutations. As in many rare genetic diseases, the mutation spectrum must be quite homogeneous all over the world (with a few recurrent mutations in the case of CDA II), with none producing a selective advantage. Only genetic isolates are likely to promote a given mutation through endogamy.
We observed the presence of several polymorphisms, in cis or in trans to the causative mutations: rs41309927, rs17807673, rs2273526. Among normal individuals, their frequency was 6%, 12% and 7% for the minor allele, respectively. The possible effects of these polymorphisms on the expression of the SEC23B gene are under investigation.
We looked for possible relationships between phenotype and genotype. One way of doing this was to divide the patients with two missense mutations (group A) from those with one missense and one nonsense mutation (group B). Hemoglobin concentration did not differ significantly between the two groups. In contrast, the reticulocyte count was significantly lower and the transfusion needs significantly higher in group B patients. These results could suggest that nonsense mutations impair erythrocyte production more severely and confirm our data on erythroid maturation: silencing of the SEC23B gene causes an increase in G2 phase block.13
In group B, there was three patients heavily dependent on transfusions (patients 2, 18, 20) and, to some extent, two other patients (patients 29 and 39) who stood apart. Otherwise, groups A and B overlapped. While patients 26 and 35a, who received more than five transfusions, belonged to group B, patients 29 and 39, who were transfusion-dependent, unexpectedly belonged to group A. Of the 11 patients who had needed fewer than five transfusions, six and five belonged to groups A and B, respectively.
Pre-splenectomy serum ferritin levels appeared higher in group B when related to age. Using our approach, trends could be perceived between groups A and B.
Patient 39 had a more severe phenotype than patient 19. However, both patients 19 and 39, originating from the same Polynesian Island, were compound heterozygotes for the same mutations. We assume that patient 39 carried an aggravating mutation. We did not have a chance to investigate this possibility since we lost sight of this patient long ago. In a few cases (patients 18 and 20), CDA II is as severe as β-thalassemia major with patients having heavy transfusion requirements and major iron overload. Such patients should be offered genetic counseling. It is not possible to exclude the coinheritance of genetic factors causing iron overload, such as HFE mutants.
Four patients (patients 4, 8, 9 and 40) showed only one mutation and this remains an unsolved issue. We did not look for large DNA deletions, but the heterozygous state for several polymorphisms seems to exclude the possibility of such deletions. The sequence of the promoter region was not investigated in detail, so it is possible that we missed mutations in this region. Nevertheless, we assume that the elusive mutation lies in SEC23B, because compound heterozygosity is more likely than two simple heterozygosities, supposing that the second mutation is in a different locus.
In summary, we identified an array of mutations in the SEC23B gene within a cohort of 42 unrelated CDA II patients originating mostly from Italy and from France, but also from other parts of the world. Correlation between the mutations and various biological parameters showed that addition of one missense mutation and one nonsense mutation tended to produce a more severe presentation then the association of two missense mutations; or rather, there is a trend for patients carrying two missense mutations to be more mildly affected. Nevertheless, excluding five patients whose phenotype was extremely severe or fatal and who stood widely apart, there was an obvious overlap between the two groups. Homozygosity for two nonsense mutations was never encountered and must be fatal. Three mutations accounted for more than 50% of mutated alleles, which is information potentially useful for post-natal and pre-natal diagnosis and a guide for rational molecular diagnosis.
Acknowledgments
we thank C. Barro and B. Pégourié-Bandelier (Grenoble), P. Boivin (Clichy), A. Boutet, F. Méchinaud and C. Thomas (Nantes), M.-P. Chauveheid, G. Leverger, G. Tchernia and L. Garçon (Paris), S Daliphard and M. Munzer (Reims), P. Fenaux, C. Gardin and G. Leroux (Bobigny), G.L. Forni (Genova), F. Gauthier, C. Guitton, Ms.I. Marie and M. A. Proust (Le Kremlin-Bicêtre), A. Jaccard (Limoges), M.-J. King (Bristol), J. Pasché and L. Roda (Papeete), E. Plouvier (Besançon), P. Revell (Stafford), U. Ramenghi (Turin), D. Steschenko (Nancy), P. Teira (Toulouse), S. N. Wickramasinghe (London), and the AIEOP (Italian Association for Pediatric Hematology and Oncology) for referring their patients to us or helping us in the diagnosis in one way or another.
Footnotes
Funding: support was provided by the Italian Ministero dell’Università e della Ricerca, by Telethon (Italy) project GGP09044 to A.I., by grants MUR-PS 35-126/Ind and by grants from Regione Campania (DGRC2362/07).
The Online version of this article has a Supplementary Appendix.
Authorship and Disclosures
CP, LG and SP cared for the patients and collected clinical data. RR and MRE worked on the molecular studies. MRE and RA performed bioinformatic analyses. RR performed statistical analyses. MFT did the routine laboratory tests. AI and JD were responsible for the CDA II registries, supervised the criteria for inclusion of patients, and contributed to writing the manuscript.
The authors reported no potential conflicts of interest.
References
- 1.Wickramasinghe SN. Congenital dyserythropoietic anemias. Curr Opin Hematol. 2000;7(2):71–8. doi: 10.1097/00062752-200003000-00001. [DOI] [PubMed] [Google Scholar]
- 2.Wickramasinghe SN, Wood WG. Advances in the understanding of the congenital dyserythropoietic anemias. Br J Haematol. 2005;131(4):431–46. doi: 10.1111/j.1365-2141.2005.05757.x. [DOI] [PubMed] [Google Scholar]
- 3.Crookston JH, Crookston MC, Burnie KL, Francombe WH, Dacie JV, Davis JA, et al. Hereditary erythroblastic multinuclearity associated with a positive acidified-serum test: a type of congenital dyserythropoietic anaemia. Br J Haematol. 1969;17(1):11–26. doi: 10.1111/j.1365-2141.1969.tb05660.x. [DOI] [PubMed] [Google Scholar]
- 4.Iolascon A, D’Agostaro G, Perrotta S, Izzo P, Tavano R, Miraglia del Giudice B. Congenital dyserythropoietic anemia type II: molecular basis and clinical aspects. Haematologica. 1996;81(6):543–59. [PubMed] [Google Scholar]
- 5.Alloisio N, Texier P, Denoroy L, Berger C, Miraglia del Giudice E, Perrotta S, et al. The cisternae decorating the red blood cell membrane in congenital dyserythropoietic anemia (type II) originate from the endoplasmic reticulum. Blood. 1996;87(10):4433–9. [PubMed] [Google Scholar]
- 6.Anselstetter V, Horstmann HJ, Heimpel H. Congenital dyserythropoietic anaemia, types I and II: aberrant pattern of erythrocyte membrane proteins in CDAII, as revealed by two-dimensional polyacrylamide gel electrophoresis. Br J Haematol. 1977;35(2):209–15. doi: 10.1111/j.1365-2141.1977.tb00577.x. [DOI] [PubMed] [Google Scholar]
- 7.Fukuda MN, Dell A, Scartezzini P. Primary defect of congenital dyserythropoietic anemia type II. Failure in glycosylation of erythrocyte lactosaminoglycan proteins caused by lowered N-acetylglucosaminyltransferase II. J Biol Chem. 1987;262(15):7195–206. [PubMed] [Google Scholar]
- 8.Fukuda MN, Masri KA, Dell A, Luzzatto L, Moremen KW. Incomplete synthesis of N-glycans in congenital dyserythropoietic anemia type II caused by a defect in the gene encoding a-mannosidase II. Proc Natl Acad Sci USA. 1990;87(19):7443–7. doi: 10.1073/pnas.87.19.7443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gasparini P, Miraglia del Giudice E, Delaunay J, Totaro A, Granatiero M, Melchionda S, et al. Localization of the congenital dyserythropoietic anemia II locus to chromosome 20q11.2 by genomewide search. Am J Hum Genet. 1997;61(5):1112–6. doi: 10.1086/301609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Iolascon A, Miraglia del Giudice E, Perrotta S, Granatiero M, Zelante L, Gasparini P. Exclusion of three candidate genes as determinants of congenital dyserythropoietic anemia type II (CDA II) Blood. 1997;90(10):4197–200. [PubMed] [Google Scholar]
- 11.Lanzara C, Ficarella R, Totaro A, Chen X, Roberto R, Perrotta S, et al. Congenital dyserythropoietic anemia type II: exclusion of seven candidate genes. Blood Cells Mol Dis. 2003;30(1):22–9. doi: 10.1016/s1079-9796(03)00009-3. [DOI] [PubMed] [Google Scholar]
- 12.Denecke J, Marquardt T. Congenital dyserythropoietic anemia type II (CDAII/HEMPAS): where are we now? Biochim Biophys Acta. 2009;1792(9):915–20. doi: 10.1016/j.bbadis.2008.12.005. [DOI] [PubMed] [Google Scholar]
- 13.Schwarz K, Iolascon A, Verissimo F, Trede Nikolaus S, Horsley Wyatt, Chen Wen, et al. Mutations in the human secretory COPII coat component SEC23B cause congenital dyserythropoietic anemia type II (CDA II) Nat Gen. 2009;41(8):936–40. doi: 10.1038/ng.405. [DOI] [PubMed] [Google Scholar]
- 14.Bianchi P, Fermo E, Vercellati C, Boschetti C, Barcellini W, Iurlo A, et al. Congenital dyserythropoietic anemia type II (CDAII) is caused by mutations in the SEC23B gene. Hum Mutat. 2009;30(9):1292–8. doi: 10.1002/humu.21077. [DOI] [PubMed] [Google Scholar]
- 15.Bi X, Corpina RA, Goldberg J. Structure of the Sec 23/24-Sar1 pre-budding complex of the COPII vesicle coat. Nature. 2002;419(6904):271–7. doi: 10.1038/nature01040. [DOI] [PubMed] [Google Scholar]
- 16.Bi X, Mancias JD, Goldberg J. Insights into COPII coat nucleation from the structure of Sec23. Sar1 complexed with the active fragment of Sec31. Dev Cell. 2007;13(5):635–45. doi: 10.1016/j.devcel.2007.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]