

Abstract
Background
β-thalassemia is a rare disease in France, encountered mainly in patients originating from Italy and North Africa. In the setting of the recent French plan for rare diseases, a National Registry for thalassemia has been developed since 2005. Epidemiological and clinical data have been collected on living patients with β-thalassemia major or intermedia, including those who underwent hematopoietic stem cell transplantation.
Design and Methods
A standardized questionnaire was sent to clinicians throughout the national professional networks involved in the management of thalassemic patients and data were updated every 18 months. A cross-sectional study was performed in February 2009.
Results
Data on 378 patients (267 with thalassemia major) with a median age of 20 were recorded. Hematopoietic stem cell transplantation was performed in 52 patients. Stature, rates of parenthood, splenectomy, and cholecystectomy were no different between non-transplanted thalassemia major and thalassemia intermedia patients, after adjustment for age. Among the 215 non-transplanted thalassemia major patients, the median serum ferritin level was 1240 ng/mL and the rates of iron-related complications were 10%, 6%, 10% and 48% for cardiac failure, diabetes, hypothyroidism, and hypogonadism, respectively. From 2005 to 2008, a dramatic switch in chelation treatment, from deferoxamine to deferasirox, was observed.
Conclusions
The rates of complications of iron overload in French thalassemia major patients appeared similar to those reported in other developed countries in which this condition is not endemic. There were no significant differences in height and parenthood rates between patients with the major and the intermedia forms of the disease, underlining the progress in clinical care. Future developments will focus on mortality and morbidity under oral chelation treatment.
Keywords: β-thalassemia, National Registry, France, epidemiology
Introduction
β-thalassemia is encountered in France due to immigration from areas in which the condition is endemic, with the exception of the island of Corsica where 3% of the population are carriers for β-thalassemia trait. Until recently, no national data on mortality, morbidity, or quality of care were available. An epidemiological survey was conducted in 1999 and recorded 362 patients with thalassemia major (TM) or intermedia (TI), of whom most originated from North Africa or Italy.1 In 2004, the French Ministry of Health initiated a National Rare Diseases Plan concerning diseases with a prevalence under 1/2000. Reference centers were created, dedicated to the clinical management of rare diseases, and assigned several missions such as ameliorating global clinical care for patients, improving professional practices and collecting epidemiological data. In this context, a reference center for thalassemia was set up and a National Registry of living β-thalassemia patients was developed as a means to achieve several aspects of the missions. Systematic epidemiological case detection and continuous data collection were performed throughout France for patients with TM, TI, and hemoglobin E/β-thalassemia; data on hematopoietic stem cell transplants in these patients were also recorded.
This article reports data collected in the registry from 2005 to 2008. The main objectives of the study were to provide a description of the demographic and clinical features of β-thalassemia patients living in France and to compare these data according to the type of thalassemia and the treatment options. In addition, the association of age with rates of iron overload complications was evaluated in the subgroup of TM patients and the use of iron chelation therapy over the 4-year period was examined.
Design and Methods
Registry design
Exhaustive identification of French cases was attempted through the distribution of inclusion files to all the physicians who participated in the 1999 national survey. Clinicians were also contacted through two professional networks: the French group for Red Cell and Iron and the French Society of Pediatric Hematology and Immunology. The other sources questioned in order to improve the completeness of case identification were the French database of hematopoietic stem cell transplants, the French network of molecular genetic laboratories involved in the molecular diagnosis of hemoglobinopathies and the four laboratories performing neonatal screening for sickle cell disease.
The French registry was approved by the Commission Nationale Informatique et Libertés and oral informed consent from patients or their parents is required for all inclusion of data. A standardized questionnaire was used to collect data on each patient, including the circumstances of diagnosis, biological and clinical data, markers of iron overload, treatment and social data. Follow-up information was collected after 18 months (range, 12–24 months) and the items recorded were the same as at inclusion plus occurrence of death. For the centers following more than ten patients, a clinical research assistant went to the site to check and complete data. Data were registered by the assistant and then validated by the physician in charge of the patients; data quality control was conducted by the physician responsible for the registry.
Patients were included in the registry from January 2005 to December 2008. Data were collected once (inclusion) or twice (inclusion and follow-up) for each patient during this time period. A cross-sectional study was performed in February 2009 using the most recent record available for each subject in the registry (last record in 2007 or 2008 for 95% of the cases).
Definition of variables
The two clinical types of thalassemia were defined as follows: TM, symptomatic anemia requiring more than eight transfusions/year (transfusion-dependent) before 4 years of age;2 TI, no or occasional transfusion before 4 years of age. The main complications considered, with their definitions, were hypothyroidism (low free thyroxine or abnormally high levels of thyroid-stimulating hormone), hypogonadism (delayed puberty, arrest of puberty for 2 years, low steroid levels or treatment with testosterone, primary or secondary amenorrhea before the age of 40), heart disease (heart failure or arrhythmia requiring inotropic or anti-arrhythmic therapy), short stature (SD score for height less than or equal to −2, with height and weight converted to SD scores using tables of normal values for the French population) and diabetes (a history of therapy with insulin or oral hypoglycemics).
Statistical analysis
Statistical analyses were performed using the Statistical Package for Social Sciences, version 15.0 (SPSS Inc., Chicago, IL, USA). Data are expressed as median values with ranges for continuous variables and numbers with percentages for categorical variables. Non-parametric tests were used to compare continuous variables (the Mann-Whitney or Kruskall-Wallis test) or categorical variables (Pearson’s χ2 test) across subgroups of patients. These tests were followed by a post hoc Dunn test. P values are two-sided and considered statistically significant when less than 0.05. Multiple logistic regression models were used to model the effect of pertaining to categories of patients, adjusting for major confounding factors (age, gender) on the following outcomes: splenectomy, cholecystectomy, antibodies to hepatitis C virus (HCV), parenthood.
Results
Registry population
From 2005 to 2008, a systematic search to identify all living thalassemia patients resulted in the identification of 378 patients. Patients were recruited from 57 French centers: 31 pediatric centers, mostly Hematology/Oncology departments, and 26 departments of Hematology or Internal Medicine for adult patients. Six centers were following more than 15 patients each, representing 49% of the cohort, and 38 followed no more than five patients (15 centers reported only one patient).
Among the overall cohort, 267 patients were classified as having TM, 110 as having TI and one infant has not yet been classified. Forty-three patients had HbE/thalassemia (24 TI and 19 TM). Seventy percent of the patients (74% of TM patients) were born in France.
The incidence of the disease was estimated in the group of patients under 18 years of age among whom death is rare: among 12,296,400 live births between 1991 and 2005 (source: INSEE), 109 were identified to have TM, leading to an incidence of 1 case for 112,811 live births (or a mean of 7 new cases/ year for TM).
Genetic studies have been carried out for 245 (65%) patients. Analysis of the mutations revealed that the vast majority of the patients are of Mediterranean origin and that the four most common Mediterranean mutations (β 39C>T, IVS1-110G>A, IVS1-6 T>C and IVS1-1G>A) account for 52% of the mutations, whereas 18% were of Asian origin (the most common being HbE and Cd 41/42-CTTT).
Patients’ characteristics
The patients were separated into four categories, according to three criteria: first according to the type of β-thalassemia (TM or TI) and then, for the TM patients only, according to whether they had or had not undergone hematopoietic stem cell transplantation and the outcome of the transplant (Table 1A). Two patients were excluded from analysis: one not yet classified into TM or TI, and one who was transplanted for TI. The median age for all the patients was 20 years and differed significantly between the four categories of patients, with TI patients being older than TM patients (P<0.01).
Table 1A.
Characteristics of patients according to the type of thalassemia and the outcome of hematopoietic stem cell transplantation.

Complications according to the type of thalassemia and outcome of hematopoietic stem cell transplantation
Table 1A also shows the complications occurring in the overall cohort and the four categories of patients. Height and weight did not differ significantly according to category (Table 1A). In the whole cohort, the percentages of patients with SD below or equal to −2 were 20.1% for height and 16.2% for weight. These proportions were similar in the four categories of patients. The final SD for height, available for 168 patients over 20 years old, did not differ statistically according to gender (P=0.58) or disease category (P=0.19).
Approximately 50% of all the patients underwent splenectomy at a median age of 9 years (range, 1.5 to 36 years), with no statistical differences between the four groups of patients.
Results of HCV antibody testing were available for 342 patients, not available for eight patients and considered as negative for 26 others (11 TI patients who had never been transfused and 15 transfused children born in France after the implementation of systematic HCV antibody testing in blood donors in 1990). Sixty-four patients (17%) were positive for HCV antibodies, 26 of whom were HCV-RNA positive. The prevalence of positivity for HCV antibodies was higher among patients with TM than among those with TI, with the difference being more pronounced after adjustment for age (P<0.001, Table 1B). Of note, 26 patients have been treated with antiviral therapy for their HCV infection: 13 became HCV-RNA negative and 13 remained with a positive viremia. One patient showed serological evidence of infection by human immunodeficiency virus and three others were positive for the HBs antigen, indicating chronic hepatitis B virus infection.
Table 1B.
Characteristics of patients according to the type of thalassemia and the outcome of hematopoietic stem cell transplantation outcome.

Serum ferritin levels tested within the previous year differed statistically between the four groups of patients (Table 1A), with TI patients having the lowest values and patients in whom hematopoietic stem cell transplantation had failed having the highest (non-transplanted TM versus TI: P<0.01, non-transplanted TM versus successfully transplanted TM: P<0.05).
Seventeen male and 33 female patients had at least one child, parenthood being more frequently encountered in TI than in TM. Nonetheless, when age, gender and disease category were taken into account, only age remained significantly associated with the fact of having children in adult patients (Table 1B). The percentage of patients who had used artificial fertilization was not available.
Treatment
Nearly all TM patients and 16% of TI patients received regular transfusions. As regards the TI patients, regular transfusion therapy was started in late childhood in four patients, during adolescence in three and during adulthood in ten. Chelation therapy was administered to 95% of TM and 44% of TI patients. During the 4-year period of analysis, a total of 428 records (including both inclusion and follow-up forms) were entered into the registry for the 263 patients under chelation therapy. The use of the different iron chelators and the change in iron chelation therapy over time are presented in Figure 1. Whereas deferoxamine was the most commonly used therapeutic chelator in 2005–2006 (62%), since 2007, deferasirox has become the most frequent treatment (71%). In 2008, 84% of the patients were receiving oral chelation therapy.
Figure 1.
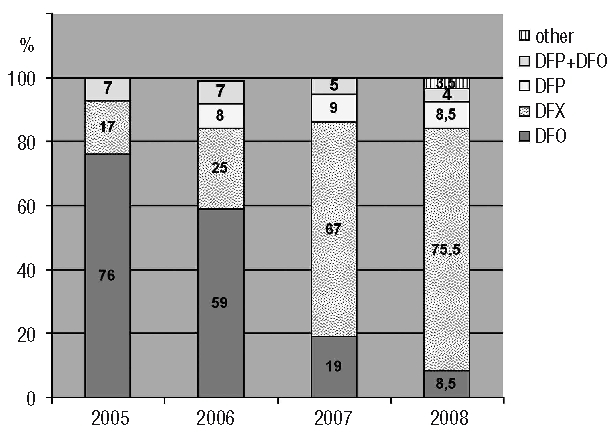
Change in iron chelation therapy over the last 4 years. Results are expressed as percentage of the forms (both inclusion and follow-up forms) received each year for the 263 patients under chelation therapy (2005: n=29; 2006: n=111; 2007: n=150; 2008: n=138). DFO: deferoxamine; DFX: deferasirox; DFP: deferiprone; other: other combinations.
Fifty-two patients underwent hematopoietic stem cell transplantation, 42 of whom became transfusion-independent. The records for transplanted patients were not exhaustive given that, according to the National Hematopoietic Stem Cell Transplantation database, as of December 2008, an additional 39 patients were alive after a successful hematopoietic stem cell transplant, often performed more than 10 years ago.
Complications in thalassemia major patients
In an analysis restricted to patients with TM who did not undergo hematopoietic stem cell transplantation, patients were divided into four age groups (Table 2). The frequency of organ damage related to iron overload increased with age, with the occurrence of complications being rare in childhood. The median height, expressed as SD, was also less impaired in the youngest patients. Hypogonadism was the most frequent complication encountered (66 cases, 54 receiving hormone replacement therapy). Among the 215 patients, 9.7% had experienced heart failure. Of note, at the end of 2008, only 40% of TM patients aged 10 years or more had undergone T2* cardiac magnetic resonance assessment in the preceding 18 months.
Table 2.
Complications in patients with thalassemia major.

A significant difference was observed between the four age groups in the rate of all complications related to iron overload while the serum ferritin levels did not differ statistically (Table 2). Serum ferritin levels were not significantly higher in patients who had experienced iron overload-related complications, e.g. hypothyroidism, heart failure, and diabetes (data not shown). Interestingly, similar serum ferritin levels were observed irrespective of the size of the centers: the median serum ferritin of the TM patients treated in the six largest centers was 1259 ng/mL compared with 1193 ng/mL for the patients followed in the other centers.
Discussion
The exhaustiveness of the case collection in the Registry was determined in various ways. For TM particularly, it is assumed that the case collection is almost complete, given that transfusion treatment requires frequent hospital visits and hence the incidence of TM can be estimated. With an incidence of approximately 1 case for 100,000 population, β-thalassemia is a very rare disease in France. This incidence is slightly lower than that recorded in the UK2 and similar to that reported in Germany and in Belgium.3,4
In this study we provide, for the first time, a detailed profile of the state of health of thalassemia patients living in France. These data reflect our current practice as 70% of the patients were born in France and can benefit from recent progress in treatments early in life.
The choice of recording data from successfully engrafted thalassemia patients (who represent around 20% of the TM patients included) was taken in order to enable future studies comparing long-term results of standard treatment and hematopoietic stem cell transplantation. The patients receiving standard transfusion treatment after a failed transplant have been analyzed as a separate group considering that the conditioning for transplantation may have had an impact on their morbidity.
We showed that height is nowadays only moderately affected in thalassemia patients, with the proportion of TM patients with short stature being around 20%, which is lower but quite similar to findings in other recent studies.5–9 In our study, in agreement with the findings of Vogiatzi et al., the type of thalassemia (TM versus TI) does not significantly influence final height or weight although one would expect better final height in patients with TI.10 Furthermore, although significant differences were seen in the prevalences of splenectomy, cholecystectomy and parenthood between non-transplanted TM and TI patients, these differences did not remain statistically significant after adjustment for age. These findings reflect the fact that the registered TI patients have a quite severe condition, with 16% being regularly transfused and 42% receiving occasional transfusions.
Moreover, in France as in North America, splenectomies performed to reduce transfusion requirements were, until recently, very frequently performed in TM.9 Finally, the similar rate of parenthood observed in TI and TM patients could be explained by a global improvement in the clinical care of TM patients including the use of assisted reproduction in TM, which affects fertility.5,8,9 In contrast, the prevalence of HCV infection and serum ferritin levels remained significantly different between the patients with the two types of thalassemia after adjustment for age (P<0.001) as a consequence of the high rate of transfusion in TM.
Iron overload complications were analyzed only in the homogeneous group of non-transplanted TM patients. Median serum ferritin levels were around 1000 ng/mL in all age groups, thus being in the lower range of values reported for other registries,9,11 suggesting that the management of iron overload and the patients’ compliance to the treatment were satisfactory. Moreover, serum ferritin levels did not differ between large centers and those following fewer than 15 patients, indicating that iron overload, as assessed by serum ferritin levels, is homogenously managed in the different centers.
Serum ferritin levels did not differ according to age or the occurrence of iron-related complications. However, the collection of serum ferritin levels in the year preceding the study was the only marker used to assess iron overload and the cross-sectional type of the analysis does not allow us to determine the implication of chronic iron exposure in the rate of related complications. As regards clinical complications, the rates of cardiac disease, hypogonadism, diabetes, growth retardation and hypothyroidism were also similar to those reported in other registries and cohorts.5,6,7,9,11 Hypogonadism, observed in nearly 50% of patients aged over 15, remained the most common endocrinopathy encountered in French TM patients. Rates of growth failure were higher after 15 years as a consequence of hypogonadism and absence of the pubertal growth spurt.
The registry also monitored data on treatments and clinical investigations used. Although it had been active for 4 years only, it already showed new trends in these fields.
The type of iron chelation treatment has changed dramatically over the past 4 years: in 2005 and 2006, deferoxamine was still the most frequent treatment but was progressively replaced by deferasirox which was introduced onto the market in France at the end of 2006. The prescription of deferiprone (available since 1999), alone or in combination with deferoxamine, remained stable during the period concerned, accounting for about 15% of the iron chelation therapy. Within this 4-year period, the median serum ferritin levels did not change significantly among the patients (data not shown).
Recent progress in TM treatments that may improve life expectancy include hematopoietic stem cell transplantation,13–14 the use of cardiac T2* measurements and chelation regimens targeted on cardiac iron overload.15–18 While hematopoietic stem cell transplantation is widely used in France, cardiac magnetic resonance studies were performed in only 40% of the adult TM patients in 2007 and 2008. Although the number of centers performing cardiac magnetic resonance studies is increasing regularly, there is a delay in the spread of this technique when compared to its use in other countries such as the UK or Italy. Consequently, intensification of chelation treatment with combination therapy probably remains under-used.
In conclusion, thalassemia is a very rare disease in France. Patients are spread all over the country and treated in a considerable number of centers. Although the mortality rate is not known, the global state of health of French TM patients appears very similar to that reported in North America and Europe, especially concerning median serum ferritin levels and rates of iron overload-related complications.
The existence of the registry has already encouraged the centers to collect data, to complete patients’ investigations, for example by endocrinological studies and cardiac magnetic resonance evaluation, and has strengthened the relations between the national reference center and clinicians. Further developments will include mortality studies for both transplanted and non-transplanted patients and an evaluation of the impact of the registry on patients’ management.
Acknowledgments
the authors would like to thank Danielle Lena, Centre d’Enseignement et de Recherche en Génétique Médicale for initiating the registry and for her continuous support, Christelle Grangier, Centre de Reference “Thalassémie”, for data collection and recording, Michel Sanz, Service d’Informatique Médicale for technical support on the database, all clinicians participating in the registry and thalassemia patients and their families for cooperation.
Appendix
In addition to authors, the following investigators contributed to the French Registry (listed in alphabetic order): Atmani Sai, MD, Hôpital Laennec, Creil; Belgodere Danielle, MD, Centre Hospitalier de Bastia; Benichou Jean-Jacques, MD, Hôpital Bicêtre, Le Kremlin Bicêtre; Benkerrou Malika, MD, Hôpital Robert Debré, Paris; Bernard Fréderic, MD, Hôpital Arnaud de Villeneuve, Montpellier; Bertrand Yves, MD, Institut d’Hématologie et d’Oncologie Pédiatrique, Lyon; Blanc Michel, MD, Centre Hospitalier de Chambéry; Blot Nathalie, MD, Hôpitaux du Pays du Mont Blanc, Sallanches; Brousse Valentine, MD, Hôpital Necker, Paris; Carreiro Miguel, MD, Centre Hospitalier de Montauban; Castex Marie-Pierre, MD, Hôpital des Enfants, Toulouse; Christian Bernard, MD, Hôpital Notre Dame de Bon Secours, Metz; Dore Eric, MD, Institut d’Hématologie et d’Oncologie Pédiatrique, Lyon; Dorvaux Véronique, MD, Hôpital Notre Dame de Bon Secours, Metz; Driss Françoise, MD, Hôpital Bicêtre, Le Kremlin Bicêtre; Dulieu Fabienne, MD, Hôpital Lenval, Nice; Dumesnil Cécile, MD, Hôpital Charles Nicolle, Rouen; Fieschi Jean-Baptiste, MD, Hôpital de la Miséricorde, Ajaccio; Fimbel Béatrice, MD, Centre Hospitalier de Tours; Gestas, MD, Clinique Cordella, Tahiti; Gouraud François, MD, Centre Hospitalier de Meaux; Guichard Henriette, MD, Hôpital Notre Dame de Bon Secours, Metz; Harle Jean-Robert, MD, Hôpital de la Conception, Marseille; Ithier Ghislaine, MD, Hôpital Robert Debré, Paris; Jaubert Jérôme, MD, Hôpital Nord, St Etienne; Karsenti Jean-Michel, MD, Hôpital l’Archet, Nice; Kebaili Kamila, MD, Institut d’Hématologie et d’Oncologie Pédiatrique, Lyon; Kurtz François, MD, Hôpital de Hautepierre, Strasbourg; Lahary Agnès, MD, Hôpital Charles Nicolle, Rouen; Lalande Muriel, MD, Hôpital Arnaud de Villeneuve, Montpellier; Lambliotte Anne, MD, Hôpital Jeanne de Flandre, Lille; Lehnert Agnès, MD, CHAM de Montargis; Leverger Guy, MD, Hôpital Trousseau, Paris; Lionnet François, MD, Hôpital Tenon, Paris; Loko Gylna, MD, Hôpital du Lamentin, Martinique; Lucchini Marie-Josée, MD, Hôpital de la Miséricorde, Ajaccio; Maakaroun Abdallah, MD, Hôpital Jacques-Cœur, Bourges; Macey Nathalie, MD, Centre Hospitalier d’Annonay; Mathey Cathy, MD, Centre Hospitalier d’Aix en Provence; Mesples Bettina, MD, Hôpital Louis Mourier, Colombes; Mialou Valérie, MD, Institut d’Hématologie et d’Oncologie Pédiatrique, Lyon; Michalet Mauricette, MD, Hôpital Edouard Herriot, Lyon; Michel Gérard, MD, Hôpital de la Timone, Marseille; Monpoux Fabrice, MD, Hôpital l’Archet, Nice; Munzer Martine, MD, Centre Hospitalier de Reims; Navarro Robert, MD, Hôpital Lapeyronie, Montpellier; Nezri Meyer, MD, Centre Hospitalier de Martigues; Odievre Marie-Helene, MD, Hôpital Louis Mourier, Colombes; Orus Didier, MD, Hôpital St Joseph-Marseille; Oziol Eric, MD, Centre Hospitalier de Béziers; Paillard Catherine, MD, Hôpital Hôtel Dieu, Clermont-Ferrand; Pautard Brigitte, MD, Hôpital Nord, Amiens; Piguet Christophe, MD, Hôpital Dupuytren, Limoges; Pincemaille Olivier, MD, Centre Hospitalier de Bastia; Rakotoarijaona Andriamiarina, MD, Centre Hospitalier de Longjumeau; Ribeil Antoine, MD, Hôpital Necker, Paris; Robert Alain, MD, Hôpital des Enfants, Toulouse; Rohrlich Pierre Simon, MD, Hôpital St Jacques, Besançon; Sanderson Frederick, MD, Hôpital l’Archet, Nice; Schneider Jean-Marc, MD, Hôpital Bel-Air, Thionville; Silva Muriel, MD, Hôpital Jacques Monod, Le Havre; Ssi-Yan-Kai Raymond, MD, Fondation Albert Barbot, La Réunion; Stankovic Katia, MD, Hôpital Tenon, Paris; Stephan Jean-Louis, MD, Hôpital Nord, St Etienne; Swiader Laure, MD, Hôpital de la Timone, Marseille; Traulle Catherine, MD, Centre Hospialier Lyon Sud, Lyon; Troncy Jacques, MD, Hôpital Edouard Herriot, Lyon; Vaillant Willy, MD, Hôpital Purpan, Toulouse; Vannier Jean-Pierre, MD, Hôpital Charles Nicolle, Rouen; Verrot Denis, MD, Hôpital Saint Joseph, Marseille; Veyssier Catherine, MD, Centre Hospitalier Poissy St Germain, St Germain en Laye; Vigne Gérard, MD, Centre Hospitalier d’Arles.
Footnotes
Funding: the Centre de Référence Thalassémie is supported by The French Ministry of Health (DHOS), The National Registry for Thalassemia receives support from the Institut National de la Santé et la Recherche Médicale (INSERM) and the Institut National de Veille Sanitaire (INVS).
Authorship and Disclosures
IT and CB were the principal investigators, designed the study, take primary responsibility for the paper and wrote it. IT collected and analyzed the data. AL and MCS performed the statistical analyses; all authors contributed to the critical revision and approved the version to be published.
The authors reported no potential conflicts of interest
References
- 1.Badens C, North ML, Lena-Russo D. Beta-thalassemia in metropolitan France. Presse Med. 2003;32(22):1016–21. [PubMed] [Google Scholar]
- 2.Modell B, Khan M, Darlison M, King A, Layton M, Old J, et al. Survival in beta-thalassemia major in the UK: data from the UK thalassemia register. Lancet. 2000;355 (9220):2051–2. doi: 10.1016/S0140-6736(00)02357-6. [DOI] [PubMed] [Google Scholar]
- 3.ickerhoff R, Genzel-Boroviczeny O, Kohne E. Haemoglobinopathies and newborn haemoglobinopathy screening in Germany. J Clin Pathol. 2009;62(1):34. doi: 10.1136/jcp.2008.058909. [DOI] [PubMed] [Google Scholar]
- 4.Gulbis B, Cotton F, Ferster A, Ketelslegers O, Dresse MF, Rongé-Collard E, et al. Neonatal haemoglobinopathy screening in Belgium. J Clin Pathol. 2009;62(1):49–52. doi: 10.1136/jcp.2008.060517. [DOI] [PubMed] [Google Scholar]
- 5.Vogiatzi MG, Macklin EA, Trachtenberg FL, Fung EB, Cheung AM, Vichinsky E, et al. Thalassemia Clinical Research Network. Differences in the prevalence of growth, endocrine and vitamin D abnormalities among the various thalassaemia syndromes in North America. Br J Haematol. 2009;146(5):546–56. doi: 10.1111/j.1365-2141.2009.07793.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.De Sanctis V, Eleftheriou A, Malaventura C. Thalassaemia International Federation Study Group on Growth and Endocrine Complications in Thalassaemia. Prevalence of endocrine complications and short stature in patients with thalassaemia major: a multicenter study by the Thalassaemia International Federation (TIF) Pediatr Endocrinol Rev. 2004;2 (Suppl 2):249–55. [PubMed] [Google Scholar]
- 7.Shalitin S, Carmi D, Weintrob N, Phillip M, Miskin H, Kornreich L, et al. Serum ferritin level as a predictor of impaired growth and puberty in thalassemia major patients. Eur J Haematol. 2005;74(2):93–100. doi: 10.1111/j.1600-0609.2004.00371.x. [DOI] [PubMed] [Google Scholar]
- 8.Skordis N, Petrikkos L, Toumba M, Hadjigavriel M, Sitarou M, Kolnakou A, et al. Update on fertility in thalassaemia major. Pediatr Endocrinol Rev. 2004;2 (Suppl 2):296–302. [PubMed] [Google Scholar]
- 9.Cunningham MJ, Macklin EA, Neufeld EJ, Cohen AR Thalassemia Clinical Research Network. Complications of beta-thalassemia major in North America. Blood. 2004;104(1):34–9. doi: 10.1182/blood-2003-09-3167. [DOI] [PubMed] [Google Scholar]
- 10.Borgna-Pignatti C. Modern treatment of thalassaemia intermedia. Br J Haematol. 2007;138(3):291–304. doi: 10.1111/j.1365-2141.2007.06654.x. [DOI] [PubMed] [Google Scholar]
- 11.Cario H, Stahnke K, Sander S, Kohne E. Epidemiological situation and treatment of patients with thalassemia major in Germany: results of the German multicenter beta-thalassemia study. Ann Hematol. 2000;79(1):7–12. doi: 10.1007/s002770050002. [DOI] [PubMed] [Google Scholar]
- 12.Borgna-Pignatti C, Cappellini MD, De Stefano P, Del Vecchio GC, Forni GL, Gamberini MR, et al. Cardiac morbidity and mortality in deferoxamine- or deferiprone-treated patients with thalassemia major. Blood. 2006;107(9):3733–7. doi: 10.1182/blood-2005-07-2933. [DOI] [PubMed] [Google Scholar]
- 13.Lucarelli G, Galimberti M, Polchi P, Angelucci E, Baronciani D, Giardini C, et al. Bone marrow transplantation in patients with thalassemia. N Engl J Med. 1990;322(7):417–21. doi: 10.1056/NEJM199002153220701. [DOI] [PubMed] [Google Scholar]
- 14.Locatelli F, Rocha V, Reed W, Bernaudin F, Ertem M, Grafakos S, et al. Related umbilical cord blood transplantation in patients with thalassemia and sickle cell disease. Blood. 2003;101(6):2137–43. doi: 10.1182/blood-2002-07-2090. [DOI] [PubMed] [Google Scholar]
- 15.Modell B, Khan M, Darlison M, Westwood MA, Ingram D, Pennell DJ. Improved survival of thalassaemia major in the UK and relation to T2* cardiovascular magnetic resonance. J Cardiovasc Magn Reson. 2008;10(1):42. doi: 10.1186/1532-429X-10-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Anderson LJ, Wonke B, Prescott E, Holden S, Walker JM, Pennell DJ. Comparison of effects of oral deferriprone and subcutaneous desferrioxamine on myocardial iron concentrations and ventricular function in beta-thalassaemia. Lancet. 2002;360(9332):516–20. doi: 10.1016/s0140-6736(02)09740-4. [DOI] [PubMed] [Google Scholar]
- 17.Pennell DJ, Berdoukas V, Karagiorga M, Ladis V, Piga A, Aessopos A, et al. Randomized controlled trial of deferiprone or deferoxamine in beta-thalassemia major patients with asymptomatic myocardial siderosis. Blood. 2006;107(9):3738–44. doi: 10.1182/blood-2005-07-2948. [DOI] [PubMed] [Google Scholar]
- 18.Telfer P, Coen PG, Christou S, Hadjigavriel M, Kolnakou A, Pangalou E, et al. Survival of medically treated thalassemia patients in Cyprus. Trends and risk factors over the period 1980–2004. Haematologica. 2006;91(9):1187–92. [PubMed] [Google Scholar]