

Abstract
Background
Abnormal adhesiveness of red blood cells to endothelium has been implicated in vaso-occlusive crisis of sickle cell disease. The present study examined whether the SAD mouse model exhibits the same abnormalities of red blood cell adhesion as those found in human sickle cell disease.
Design and Methods
The repertoire of adhesive molecules on murine erythrocytes and bEnd.3 microvascular endothelial cells was determined by flow cytometry using monoclonal antibodies or by western blotting. Adhesion was investigated in dynamic conditions and measured at different shear stresses.
Results
CD36, CD47 and intercellular adhesion molecular-4, but not Lutheran blood group antigen/basal cell adhesion molecule, are present on mouse mature erythrocytes. α4β1 are not expressed on SAD and wild type reticulocytes. Endothelial bEnd.3 cells express αVβ3, α4β1, CD47, vascular cell adhesion molecule-1, and Lutheran blood group antigen/basal cell adhesion molecule, but not CD36. Adhesion of SAD red cells is: (i) 2- to 3-fold higher than that of wild type red cells; (ii) further increased on platelet activating factor-activated endothelium; (iii) not stimulated by epinephrine; (iv) inhibited after treating the endothelium with a peptide reproducing one of the binding sequences of mouse intercellular adhesion molecular-4, or with mon-oclonal antibody against murine αv integrin; and (v) inhibited after pretreatment of red blood cells with anti-mouse CD36 monoclonal antibodies. The combination of treatments with intercellular adhesion molecular-4 peptide and anti-CD36 monoclonal antibodies eliminates excess adhesion of SAD red cells. The phosphorylation state of intercellular adhesion molecular-4 and CD36 is probably not involved in the over-adhesiveness of SAD erythrocytes.
Conclusions
Intercellular adhesion molecular-4/αvβ3 and CD36/thrombospondin interactions might contribute to the abnormally high adhesiveness of SAD red cells. The SAD mouse is a valuable animal model for investigating adhesion processes of sickle cell disease.
Keywords: sickle cell disease, mouse, adhesion, ICAM-4, CD36, endothelium, cAMP, phosphorylation
Introduction
The major complications of sickle cell disease are the recurrent painful crises that result from vaso-occlusion processes involving interactions between multiple types of cells (endothelium, leukocytes, red blood cells, platelets). Among these interactions, those between sickle red blood cells (SS RBC) and microvascular endothelium have been highlighted following the initial observation of Hebbel and co-workers, two decades ago, of a significant positive correlation between SS RBC–endothelium adherence and the clinical severity of the disease.1 Indeed, the surface of erythrocytes bears a number of pro-adhesive proteins that are potential receptors for endothelial or subendothelial adhesion molecules. Since the 1980s, a large body of work has shown that the increased propensity of SS RBC to adhere to endothelium depends on the over-expression or the activation of adhesion molecules on the surface of SS RBC and inflamed SS endothelium.2,3 Human mature RBC express CD47, Lutheran blood group antigen/basal cell adhesion molecule (Lu/BCAM), and Landsteiner-Weiner blood group antigen/intercellular adhesion molecule-4 (LW/ICAM-4), with thrombospondin, laminin and αVβ3 integrins as their respective ligands on endothelium. Interestingly, Lu/BCAM- and ICAM-4-mediated RBC adherence to endothelium is enhanced by epinephrine, a stress mediator released during crises.4,5 The signaling pathway involves a rise in intracellular cAMP followed by protein kinase A-dependent phosphorylation and the activation of both adhesive proteins.4–6 Sickle human immature reticulocytes express enhanced levels of CD36 and α4β1 (VLA-4) integrins that interact respectively with thrombospondin, and both thrombospondin and fibronectin, localized on endothelial cells or on exposed subendothelial cell matrix of inflamed endothelium.7,8
The physiological in vivo relevance of such an increased adherence of SS RBC to vascular endothelial cells has essentially been documented by ex vivo or in vivo trans-species approaches. Accordingly, human SS RBC were infused into mouse or rat vasculature (e.g. cremaster muscle, mesocecum) cleared of endogenous blood, and intravital videomicroscopy was conducted to quantify blood flow and cell adhesion to vascular endothelium. Such studies showed that tumor necrosis factor-α (TNFα)-induced retention of SS RBC in the retinal microcirculation could be blocked by an α4β1 antagonist9 and that platelet activating factor (PAF)-induced adhesion of SS RBC to isolated artificially perfused rat mesocecum vasculature could be blocked by monoclonal antibodies to the vitronectin receptor αVβ3 or by peptides based on the αV-binding domains of ICAM-4.10,11 Data from approaches looking at the hemodynamic behavior of human RBC flowing in the vascular system of other species may be flawed because of potential interspecies differences in adhesion molecules and receptors. Additionally, most ex vivo studies are performed in plasma-free medium that cannot represent the complexity of the RBC adhesion process in physiological conditions, in which the critical role of plasma proteins (fibronectin, thrombospondin, von Willebrand factor) and leukocytes is well established. For these reasons, mouse models of SCD could constitute valuable tools to delineate the role of the different erythrocyte and endothelial adhesion molecules in ideally homologous physiological conditions, since the rheologic behavior of labeled RBC can be analyzed in the vasculature of the mouse from which they are collected.
Several strains of transgenic mouse expressing human sickle β-globin chains have been generated.12 Some models, such as the Berkeley mouse, exclusively express human globin chains and exhibit a very severe phenotype entailing a very low percentage of viable pups, very short lifespan, and extreme fragility during manipulation.13 Other models, such as the SAD mouse, have combined murine and human globin chains.14 The SAD1 (SAD) Hbβ single/single hemizygous mice used in this work harbor a recombinant hβ-globin gene construct expressing human hemoglobin SAD (α2β2SAD) which contains two mutations [Antilles (β231) and D-Punjab (β121Q)] in addition to the βs6v mutation. These two additional mutations result in increased polymer formation in the presence of mouse hemoglobin. The SAD mouse has mild sickle cell disease with shortened survival, priapism, and kidney defects typical of sickle cell disease.15 The strain is bred on the C57Bl/6J genetic background (more than 12 backcrosses). Their RBC contain 19% human Hb SAD. Their sickle phenotype is mild in normoxic conditions, but becomes severe during hypoxia.14
The purpose of the current study was to determine whether SAD1 sickle transgenic mice display abnormalities of RBC adhesion to endothelial cells, similar to those of human sickle RBC, and could, therefore, serve as a useful animal model for evaluating the in vivo efficacy of potential anti-adhesogens.
Design and Methods
Details on animals, reagents, antibodies, cell culture, and RBC purification and membrane preparation are provided in the Online Supplementary Appendix.
Treatment of the bEnd.3 murine microvascular endothelial cell line
As cerebrovascular complications occur recurrently in sickle cell disease, we chose the mouse endothelioma cell line bEnd.3 as the endothelial system (ATCC, Manassas, VA, USA). Endothelial cell monolayers were activated by either 5 ng/mL of mouse TNFα (BD Biosciences) or 0.2 ng/mL of PAF. In adhesion experiments, the monolayers were treated with 250 μM peptides or 10 μg/mL antibodies for 30 min.
Flow cytometry analysis of surface antigens of erythrocytes and bEnd.3 endothelial cells
RBC were collected from SAD and wild-type mice before and after a 48 h-period of 10% O2 hypoxia. Endothelial cells were grown until 80% confluence before adding TNFα or PAF to the culture medium. The flow cytometry procedure is detailed in the Online Supplementary Appendix.
Immunoprecipitation of phosphorylated proteins from red cell membranes
RBC membranes (200 μg protein) were solubilized in 1 mL immunoprecipitation buffer (20 mM Tris pH 7.5, 150 mM NaCl, 1 mM EDTA, protease and phosphatase inhibitors, 1% Triton X100 and 0.2% IGEPAL-CA630) for 1 h at 4°C. After centrifugation at high speed, phosphorylated proteins of the supernatant were immunoprecipitated overnight at 4°C with 30 μL agarose-conjugated monoclonal antibodies against either phosphorylated threonine or phosphorylated tyrosine. The agarose beads were pelleted and washed five times with immunoprecipitation buffer. The beads were resuspended in 10 μL of 3X Laemmli buffer contaning 3% β-mercaptoethanol, and boiled for 5 min. The supernatants obtained after a final centrifugation were analyzed by immunoblotting. The immunoblotting procedure is detailed in the Online Supplementary Appendix.
Erythrocyte cyclic AMP assay
Freshly collected RBC (2×108) were placed in 360 μL Hank’s balanced salt solution and were stimulated by epinephrine (50 nM final, 2 min at 37°C). Cells were lysed by addition of 40 μL of a 2.5% solution of dodecyltrimethylammonium bromide, followed by vigorous agitation and incubation at room temperature for 10 min to achieve complete cell lysis. Cyclic AMP was measured in 40 μL of lysate by an enzyme immunoassay using cAMP-acetylcholine esterase conjugate as tracer, after acetylation of both samples and standards, according to the manufacturer’s instructions.
Erythrocyte adhesion assay under flow conditions
Endothelial bEnd.3 cells were cultured to confluence in flat glass capillaries. As SAD and wild-type RBC were injected together into the same capillary, they were discriminated by different fluorescent labels (PKH67 and PKH26, respectively). Their adhesion to the endothelial monolayer was measured at four different shear stresses, according to a previously described method.16 The adhesion assay procedure is detailed in the Online Supplementary Appendix.
Results
Adhesion molecules on mouse microvascular bEnd.3 endothelial cells
Levels of expression are depicted in Online Supplementary Figure S1. Vascular cell adhesion molecule-1 (VCAM-1), ICAM-1, E-selectin, and P-selectin were weakly expressed on resting bEnd.3 cells, and were over-expressed after activation by TNFα for 4 or 16 h, confirming the endothelial phenotype of bEnd.3 cells (Online Supplementary Figure S1A). αvβ3 and α4β1 integrins, as well as CD44 and CD47 were significantly expressed. On the other hand, CD36 was very moderately expressed. These levels of expression were unchanged by PAF activation for 16 h (Online Supplementary Figure S1B). The presence of Lu/BCAM in bEnd.3 cells was confirmed by immunoblotting (Online Supplementary Figure S1C).
Adhesion molecules on mouse red blood cells
Flow cytometry confirmed the previously reported finding of a significantly higher percentage of reticulocytes in the RBC population from SAD mice than from wild-type mice (3.9±0.2% versus 2.9±0.3%, n=4). These percentages were unchanged after a 48-h period of hypoxic stress (10% oxygen). Flow cytometry also revealed that CD36, CD47, and CD147 were expressed at higher levels on mouse reticulocytes than on mature RBC (Figure 1A). The level of CD36 on SAD RBC was half that on wild-type RBC whereas CD47 and CD147 levels were similar on RBC of the two types of mice (Figure 1A). The lower expression of CD36 on SAD RBC was also revealed by western blot analysis (Figure 1C). Hypoxia did not change the expression of CD36 and CD47, but significantly reduced that of CD147 on both SAD and wild-type RBC and reticulocytes (Figure 1A). α4β1 integrins were not detectable on either mature RBC or reticulocytes from both SAD and wild-type mice, in normoxia as well as after hypoxia (data not shown). Our polyclonal antibody identified ICAM-4 in mouse RBC membranes as a 37 KD glycoprotein and a 27 KD deglycosylated protein (Figure 1B, upper panel). The antibody specificity was validated since the protein bands were abated when peptide-saturated antiserum or pre-immune serum was used instead of immune serum (Figure 1B, upper panel). Western blot analysis showed that expression of ICAM-4 was lower in three different SAD RBC membrane preparations than in three different wild-type RBC membrane preparations (Figure 1B, lower panel).
Figure 1.

Adhesion molecules on erythrocytes of wild type and SAD mice. (A) Flow cytometry analysis of CD36, CD47, and CD147 on erythrocytes from mice bled before and after undergoing 10% O2 hypoxia for 48 h. Results are expressed as mean fluorescence intensity (MFI). Columns represent the mean results from three means of three mice (white columns: wild-type mice; black columns: SAD mice). Bars represent SEM. **P< 0.01; ***P<0.001 by ANOVA. (B) Western blot analysis of ICAM-4 in RBC membranes. Upper panel: Mouse ICAM-4 is revealed as a 37 KD glycoprotein and a 27 KD deglycosylated protein, after digestion by N-glycosidase F (PNGase F). These bands are specific inasmuch as they were abated when the antibody was pre-incubated overnight at 4°C with a saturating concentration of the immunizing peptide or when the rabbit preimmune serum was used. Lower panel: Semi-quantitative western blot analysis of ICAM-4 expression in RBC membrane of three different SAD and wild-type C57Bl/6 mice. Equal protein loading was demonstrated by Ponceau red staining. (C) Immunoprecipitation of phosphorylated proteins from RBC membrane, followed by immunoblotting.
Phosphorylation of intercellular adhesion molecule-4 and CD36
The amino acid sequence of the cytoplasmic domain of mouse ICAM-4 contains three potential phosphorylation sites: a carboxy-terminal threonine residue (position 264) and two tyrosine residues (positions 255 and 259).17 It has been reported that CD36 is constitutively phosphorylated on threonine 92 of the thrombospondin binding sequence in the ectodomain in human resting platelets and in a megakaryocytic cell line.18 Immunoprecipitation assays did not reveal phosphorylated ICAM-4 on either the threonine or the tyrosine residues (Figure 1C). Likewise, no phosphorylation of CD36 was detected (Figure 1C). As a control, tyrosine phosphorylation of band 3 was determined and found to be stronger on SAD than on wild-type membranes, as was previously reported for human sickle cells19 (Figure 1C).
Higher adhesion to endothelium of sickle cell SAD red blood cells
In basal conditions, the number of SAD RBC adherent to the bEnd.3 monolayer was 2.5-fold higher than that of wild-type RBC, independently of shear stress levels ranging from 0.04 to 0.4 Pa (Figure 2A).
Figure 2.
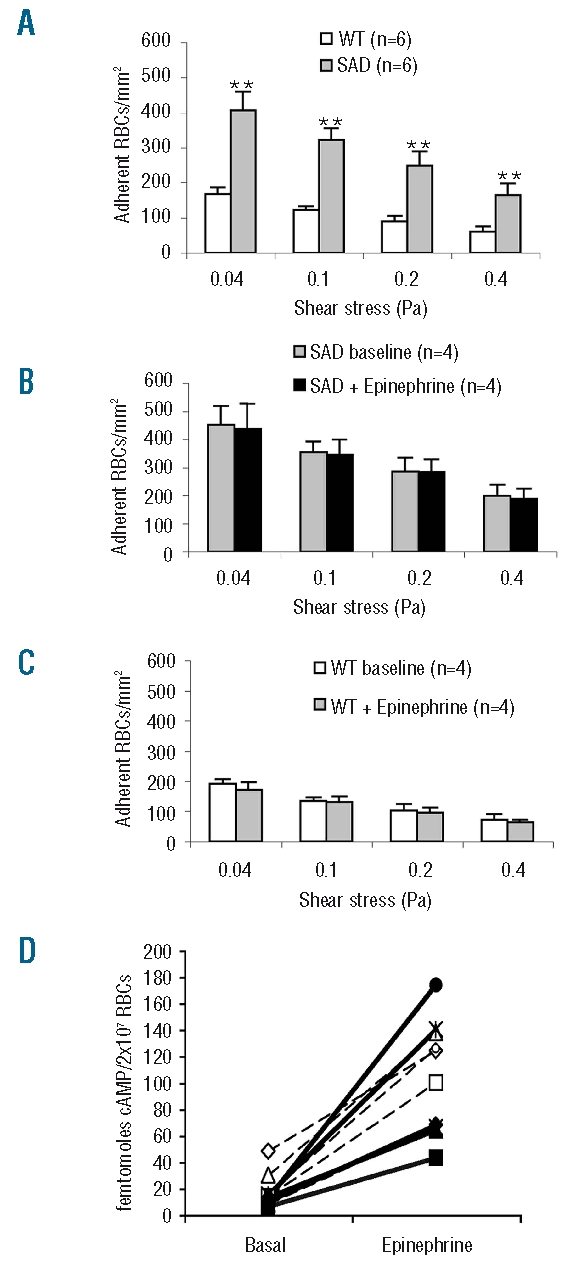
The adhesion of SAD RBC to an endothelial monolayer is greater than that of wild-type (WT) RBC, and not altered by epinephrine (50 nM) in spite of stimulation of cAMP synthesis. (A) Adherence to endothelium in basal condition. (B) Adhesion of epinephrine-treated SAD RBC. (C) Adhesion of epinephrine-treated WT RBC. (D) Effect of epinephrine on cyclic AMP content of RBC from five SAD (black symbols and solid lines) and five WT mice (white symbols and dashed lines). Columns denote means and bars denote SEM. **P<0.01 between SAD and WT by ANOVA.
Incubation of SAD red blood cells with epinephrine did not increase their adhesion to endothelium
Neither SAD nor wild-type RBC were responsive to epinephrine (50 nM) in terms of adhesion to endothelial cells (Figure 2B and 2C). This absence of effect did not result from unresponsiveness of the β2-adrenergic-receptors of mouse RBC to epinephrine, since the cAMP content of SAD RBC, which was similar to that of wild-type RBC in basal conditions, was equally and strongly increased in both types of erythrocytes after 2 min incubation with the same concentration of hormone (Figure 2D).
Involvement of intercellular adhesion molecule-4/integrin αV chain interaction in the increased propensity of SAD red blood cells to adhere to endothelium
The contribution of ICAM-4 to over-adhesiveness of SAD RBC was assessed with the octapeptide T-8-I, which mimics one of the binding sites of human ICAM-4 to αV integrin.10 The adhesion of SAD RBC to the ICAM-4 peptide (T-8-I)-treated bEnd.3 monolayer was half that recorded on control peptide (A-8-C)-treated endothelium. This difference was maintained, irrespectively of the wall shear stresses applied, and was statistically significant (P<0.001, ANOVA) (Figure 3A). Likewise, the adhesion of SAD RBC to endothelium treated with anti-αV monoclonal antibody was significantly reduced, by half, in comparison to that recorded on endothelium treated with rat IgG (Figure 3C). Wild-type RBC also adhered less to bEnd.3 cells pretreated with either T-8-I or anti-αV integrin monoclonal antibody (Figure 3B and D). However, the decrease in the number of adhered wild-type RBC was much less than that of SAD RBC. Moreover, it is of note that the inhibition achieved by T-8-I peptide and anti- αV monoclonal antibody was of same magnitude.
Figure 3.

Inhibition of SAD RBC adhesion on bEnd.3 endothelial cells by ICAM-4 peptide and by anti-αV integrin monoclonal antibody. Endothelial cell monolayers on microslides were treated by 250 μM T-8-I (ICAM-4 peptide) or A-8-C (control peptide), or 10 μg/mL anti-αV integrin antibody or control rat IgG, for 30 min before injection of RBC. (A) Adherence of SAD RBC on T-8-I peptide-treated endothelial cells. (B) Adherence of wild-type (WT) RBC on T-8-I treated endothelial cells. (C) Adherence of SAD RBC on endothelial cells treated with αV integrin antibodies. (D) Adherence of WT RBC on endothelial cells treated with αV integrin antibodies. ANOVA indicates that the differences observed for SAD RBC are highly significant (P< 0.001).
Contribution of red blood cell CD36 to the exaggerated adherence of SAD red blood cells
When function blocking anti-CD36 antibody was applied to SAD RBC prior to the adhesion assay, the number of adherent RBC was reduced by 25%, as compared to that of SAD RBC treated with control mouse IgG (Figure 4A). Although moderate, the difference was statistically significant. No such effect was found for wild-type RBC treated with the antibody (Figure 4B).
Figure 4.

Additive contributions of ICAM-4 and CD36 to increased adhesion of SAD RBC to endothelium. A and B: RBC were incubated in the presence 10 μg/mL monoclonal antibody anti-mouse CD36 or control mouse IgG, for 30 min. Anti-CD36 antibody significantly reduced the adhesion of SAD RBC (P<0.05) (A), but not that of wild-type (WT) RBC (B). C and D: RBC were treated with anti-CD36 mono-clonal antibody or control IgG, and bEnd.3 cell monolayers with 250 μM T-8-I or A-8-C peptide, for 30 min. The double treatment resulted in a highly significant fall of SAD RBC adhesion (C) and a moderate fall of WT RBC adhesion (D) (P<0.001 by ANOVA). Columns represent mean results from four different mice, and bars represent SEM.
We next determined the respective proportions of cells adherent through ICAM-4 and cells adherent through CD36 among the total over-adherent SAD RBC population: we did this by measuring adhesion of CD36-blocked SAD RBC on T-8-I pretreated endothelial cells. The adhesion of SAD RBC in these conditions fell greatly (Figure 4C), to a level close to that recorded for wild-type RBC in basal conditions (see Figure 2A). It is noteworthy that for wild-type RBC, the combined treatments also produced greater inhibition than individual treatments (Figure 4D).
Increased adhesion of SAD red blood cells to endothelium treated with platelet activating factor
Since the level of PAF in the blood has been reported to be 2-fold higher in patients with sickle cell disease20 and to enhance sickle cell adhesion in an ex vivo microcirculatory preparation,10 we investigated SAD RBC adhesion on a PAF-treated bEnd.3 monolayer. We found that PAF significantly increased the adhesion of SAD RBC (Figure 5A), but not that of wild-type RBC (Figure 5B).
Figure 5.
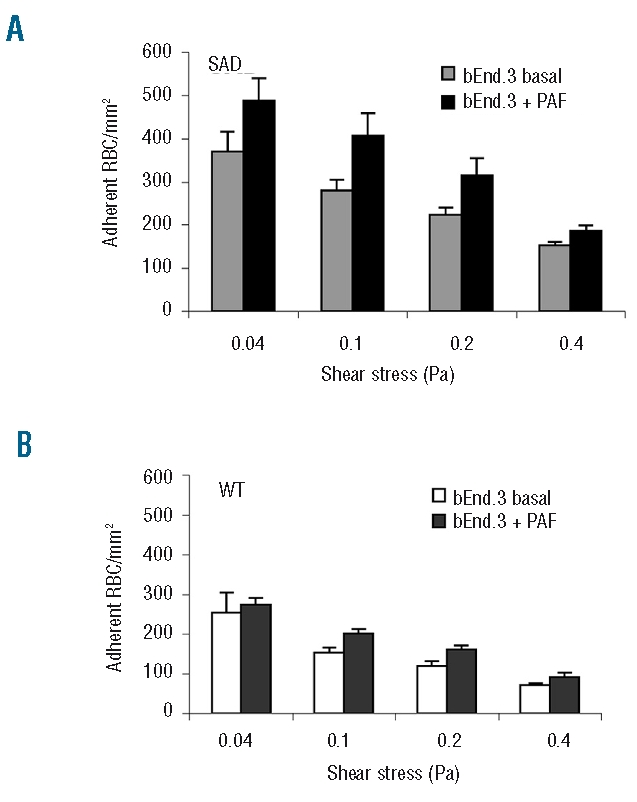
The adhesion of SAD RBC on PAF-activated endothelium is increased. Confluent endothelial monolayers on microslides were treated with PAF (0.2 ng/mL) for 10 min before injection of RBC. (A) At all four wall shear stresses, SAD RBC adhered in higher numbers to PAF-treated than to untreated endothelial cells (P<0.05 by ANOVA). (B) The adherence of wild-type (WT) RBC on PAF-treated endothelium was equivalent to that on non-treated endothelium. Columns represent the mean results from six different mice and bars represent SEM.
Discussion
The SAD mouse model of sickle cell disease offers the opportunity to work in a perfectly homologous system for evaluating the in vivo efficacy of potential anti-adhesogens. We examined whether RBC from these mice exhibit the abnormally high adhesiveness to endothelial cells reported for human sickle RBC.
We first identified the adhesion molecules expressed on mouse RBC and on the mouse microvascular endothelium cell line bEnd.3, which was used in this study because brain microcirculation complications are important features of sickle cell disease. We found that the expression of these molecules differs somewhat from that on the human counterparts.
CD36 expression was found to be significant on both mature and young mouse RBC, but lower on SAD than on wild-type RBC, and not altered by hypoxia. These findings differ from the previously reported faint and exclusive expression of CD36 on human reticulocytes, increased during sickle cell anemia.8,21 Given its significant expression on both young and mature SAD RBC in association with hyper-reticulocytosis, and in spite of its weaker expression, CD36 may contribute significantly to RBC adhesiveness by binding to endothelial thrombospondin.22 Thrombospondin is also a ligand of CD47 which is highly expressed on mouse RBC, as it is on human RBC.8 CD47 levels on SAD and wild-type RBC are similar and not altered by hypoxia.
CD147 expression is 10-fold lower than CD47 expression, on both young and mature mouse erythrocytes, paralleling findings for human RBC.8 The level of CD147 expression on both SAD and wild-type RBC is the same and significantly lowered by hypoxia. If CD147 plays a role in the recirculation of mature erythrocytes from the spleen into the general circulation, as was recently suggested,23 the reduction in CD147 expression may reflect spleen sequestration of deoxygenated RBC. In humans, α4β1 integrins are not present on mature RBC and only very faintly expressed on reticulocytes. However, the expression of α4β1 integrins is increased on reticulocytes of subjects with sickle cell disease.8,24 We could not detect α4β1 integrins on either mature or young mouse RBC (data not shown). ICAM-4 is an erythrocyte-specific inter-cellular adhesion molecule which binds to αVβ3 integrins on endothelial cells. Like Lu/BCAM, which is not present on mouse RBC,25 it can be activated by signal transduction pathways. We show here that ICAM-4 is indeed present in mouse as it is in human RBC membranes, and that its protein level is lower in SAD RBC membranes than in wild-type ones.
bEnd.3 cells have maintained a typical endothelial phenotype as indicated by their higher expression of VCAM-1, ICAM-1, E-selectin and P-selectin following TNFα activation. Like human endothelial cells, such as human umbilical vein endothelial cells, the cell type most widely used in previous studies, b.End.3 cells express αVβ3 integrins, the ICAM-4 counter-receptor. Lu/BCAM has been found on bEnd.3 cells as it was on the renal vascular endothelium of mice.25 Low levels of CD36 and α4 integrin, and a high level of β1 integrin are found on bEnd.3 cells as on human microvascular endothelial cells.26–28
It has been reported that resting SS and normal human RBC do not adhere much to a human umbilical vein endothelial cell monolayer, and that brief epinephrine treatment hugely increases the adhesion of SS but not normal RBC.5 Such a selective effect was attributed to the activation of ICAM-4 on SS RBC through a signaling pathway involving increased cAMP synthesis and phosphorylation of ICAM-4 by protein kinase A. However, this effect of epinephrine was observed on RBC collected from only 50% of the cohort of patients.
Our findings in a mouse model of sickle cell disease are somewhat at variance with these reported for humans. We found that, in basal conditions, SAD RBC are much more adherent to endothelium than are wild-type RBC. We assume that reticulocytes, which are more abundant and express more adhesion molecules in SAD mice, may contribute to the hyper-adhesiveness. Moreover, SAD RBC do not adhere more after epinephrine treatment in spite of an elevation of intracellular cAMP.
Our work shows that ICAM-4 and CD36 are responsible for all the over-adhesiveness of resting SAD RBC, since simultaneous blocking of these two ligands lowers the adhesion of SAD RBC to the level of that of wild-type RBC. The involvement of reticulocyte CD36 in the adhesion process of human SS RBC to endothelium was demonstrated by inhibition with anti-CD36 antibodies.29 However, the contribution of CD36 has been called into question by the finding that sickle cell patients who have a CD36 deficiency of reticulocytes and mature RBC can have a normal clinical course.30 Our results suggest that in the mouse model, the participation of CD36 in in vivo adhesive processes might be of significance, given that CD36 is expressed at high levels on both reticulocytes and mature RBC.
We also observed that, like human SS RBC, which exhibit increased adhesiveness in the blood vessels of PAF-treated mesocecum of the rat,11 mouse SAD RBC are more adherent to mouse endothelium pretreated with PAF. An increased expression of endothelial adhesion molecules does not account for this effect.
The intriguing finding of this work is the mediation of the increase in adhesion of SAD RBC by under-expressed adhesion molecules, i.e., ICAM-4 and CD36. Such dissociation between adhesiveness and adhesion molecule level is not without precedent. For instance, Lu/BCAM was found to be over-expressed on RBC of sickle cell patients treated with hydroxyurea8 when adhesive properties of sickle cells were reduced.24 It is, therefore, conceivable that ICAM-4 and CD36 of SAD RBC, although present in reduced amounts, might be activated by a state of abnormal constitutive phosphorylation. Under our experimental conditions, we did not detect phosphorylation on tyrosine or threonine residues of ICAM-4, either from SAD or wild-type mice. A possible hypothesis concerning CD36 was that it is constitutively ectophosphorylated on wild-type RBC, and dephosphorylated on SAD RBC, resulting in increased thrombospondin binding, as reported for platelets and megakaryocytes.18 This mechanism can be ruled out since we did not detect phosphorylation of CD36 in RBC membranes of either SAD or wild-type mice (Figure 2C). Alternatively, ICAM-4 molecules on SAD RBC could be gathered as clusters, enabling stronger binding to αVβ3 integrin clusters on endothelial cells.31,32 Likewise, an increased clustering of CD36 on SAD RBC, as observed by electronic microscopy in platelet membranes,33 may strengthen the interaction between erythrocyte CD36 and endothelial thrombospondin.
Why are ICAM-4 and CD36 less expressed in the SAD RBC membrane? The low membrane expression of ICAM-4 might result from the release of a soluble isoform into the plasma: this has been reported to occur in the mouse, with the postulated function of modulating the binding of membrane-associated ICAM-4 for erythroblast release from erythroblastic islands.17 By competing with RBC membrane-associated ICAM-4 for binding to endothelial αVβ3 integrins, soluble ICAM-4 might reduce red cell/endothelium interactions thereby moderating an otherwise exaggerated adhesiveness of SS RBC to the vascular wall. The reduced level of CD36 on SAD RBC could result from the high plasma level of endothelin-1 found in both SS patients and SAD mice,34,35 which decreases CD36 protein expression, an effect recently reported for vascular smooth muscle cells.36
In conclusion, the present work shows an over-adhesiveness to endothelium of SAD mouse RBC, with this phenomenon depending on two adhesion proteins, ICAM-4 and CD36, as previously reported for human SS RBC. Thus, the transgenic SAD mouse could serve as a useful model for investigating the individual contributions of the different erythrocyte and endothelial adhesion molecules to vaso-occlusive events, and for evaluating the therapeutic efficacy of blocking peptides or monoclonal antibodies. However, unlike human SS RBC, mouse SS RBC are unresponsive, in terms of endothelial adhesion, to β2-adrenergic stimulation.
Acknowledgments
the authors would like to thank Yves Beuzard, Pierre-Louis Tharaux, Nathalie Saaba, and Jin Huang, INSERM U689 for providing samples of blood, and Dominique Goossens for reading the manuscript.
Footnotes
Funding: this investigation was supported by a grant from “Agence Nationale de la Recherche” (SCADHESION 2007) and a grant from “Region Ile-de-France” (SESAME 2007 n° F-08-1104/R).
The Online version of this article has a Supplementary Appendix.
Authorship and Disclosures
All authors met the criteria for being contributing authors. MMTTT and JPC contributed equally to the design of the research and analysis of the results. MMTTT was the principal investigator and wrote the paper. CVL performed adhesion assays and JP flow cytometry analysis. The adhesion system was set up by MPW.
The authors report no potential conflicts of interest.
References
- 1.Hebbel RP, Boogaerts MA, Eaton JW, Steinberg MH. Erythrocyte adherence to endothelium in sickle-cell anemia. A possible determinant of disease severity. N Engl J Med. 1980;1;302(18):992–5. doi: 10.1056/NEJM198005013021803. [DOI] [PubMed] [Google Scholar]
- 2.Hebbel RP, Osarogiagbon R, Kaul D. The endothelial biology of sickle cell disease: inflammation and a chronic vasculopathy. Microcirculation. 2004;11(2):129–51. [PubMed] [Google Scholar]
- 3.Frenette PS. Sickle cell vasoocclusion: heterotypic, multicellular aggregations driven by leukocyte adhesion. Microcirculation. 2004;11(2):167–77. [PubMed] [Google Scholar]
- 4.Hines PC, Zen Q, Burney SN, Shea DA, Ataga KI, Orringer EP, et al. Novel epinephrine and cyclic AMP-mediated activation of BCAM/Lu-dependent sickle (SS) RBC adhesion. Blood. 2003;101(8):3281–7. doi: 10.1182/blood-2001-12-0289. [DOI] [PubMed] [Google Scholar]
- 5.Zennadi R, Hines PC, De Castro LM, Cartron JP, Parise LV, Telen MJ. Epinephrine acts through erythroid signaling pathways to activate sickle cell adhesion to endothelium via LW-alphavbeta3 interactions. Blood. 2004;104(12):3774–81. doi: 10.1182/blood-2004-01-0042. [DOI] [PubMed] [Google Scholar]
- 6.Gauthier E, Rahuel C, Wautier MP, El Nemer W, Gane P, Wautier JL, et al. Protein kinase A-dependent phosphorylation of Lutheran/basal cell adhesion molecule glycoprotein regulates cell adhesion to laminin alpha5. J Biol Chem. 2005;280(34):30055–62. doi: 10.1074/jbc.M503293200. [DOI] [PubMed] [Google Scholar]
- 7.Swerlick RA, Eckman JR, Kumar A, Jeitler M, Wick TM. Alpha 4 beta 1-integrin expression on sickle reticulocytes: vascular cell adhesion molecule-1-dependent binding to endothelium. Blood. 1993;82(6):1891–9. [PubMed] [Google Scholar]
- 8.Odievre MH, Bony V, Benkerrou M, Lapoumeroulie C, Alberti C, Ducrocq R, et al. Modulation of erythroid adhesion receptor expression by hydroxyurea in children with sickle cell disease. Haematologica. 2008;93(4):502–10. doi: 10.3324/haematol.12070. [DOI] [PubMed] [Google Scholar]
- 9.Lutty GA, Taomoto M, Cao J, McLeod DS, Vanderslice P, McIntyre BW, et al. Inhibition of TNF-alpha-induced sickle RBC retention in retina by a VLA-4 antagonist. Invest Ophthalmol Vis Sci. 2001;42(6):1349–55. [PubMed] [Google Scholar]
- 10.Kaul DK, Liu XD, Zhang X, Mankelow T, Parsons S, Spring F, et al. Peptides based on alphaV-binding domains of erythrocyte ICAM-4 inhibit sickle red cell-endothelial interactions and vaso-occlusion in the microcirculation. Am J Physiol Cell Physiol. 2006;291(5):C922–30. doi: 10.1152/ajpcell.00639.2005. [DOI] [PubMed] [Google Scholar]
- 11.Kaul DK, Tsai HM, Liu XD, Nakada MT, Nagel RL, Coller BS. Monoclonal antibodies to alphaVbeta3 (7E3 and LM609) inhibit sickle red blood cell-endothelium interactions induced by platelet-activating factor. Blood. 2000;95(2):368–74. [PubMed] [Google Scholar]
- 12.Nagel RL, Fabry ME. The panoply of animal models for sickle cell anaemia. Br J Haematol. 2001;112(1):19–25. doi: 10.1046/j.1365-2141.2001.02286.x. [DOI] [PubMed] [Google Scholar]
- 13.Paszty C, Brion CM, Manci E, Witkowska HE, Stevens ME, Mohandas N, et al. Transgenic knockout mice with exclusively human sickle hemoglobin and sickle cell disease. Science. 1997;278(5339):876–8. doi: 10.1126/science.278.5339.876. [DOI] [PubMed] [Google Scholar]
- 14.Trudel M, De Paepe ME, Chretien N, Saadane N, Jacmain J, Sorette M, et al. Sickle cell disease of transgenic SAD mice. Blood. 1994;84(9):3189–97. [PubMed] [Google Scholar]
- 15.De Paepe ME, Trudel M. The transgenic SAD mouse: a model of human sickle cell glomerulopathy. Kidney Int. 1994;46(5):1337–45. doi: 10.1038/ki.1994.403. [DOI] [PubMed] [Google Scholar]
- 16.Cooke BM, Usami S, Perry I, Nash GB. A simplified method for culture of endothelial cells and analysis of adhesion of blood cells under conditions of flow. Microvasc Res. 1993;45(1):33–45. doi: 10.1006/mvre.1993.1004. [DOI] [PubMed] [Google Scholar]
- 17.Lee G, Spring FA, Parsons SF, Mankelow TJ, Peters LL, Koury MJ, et al. Novel secreted isoform of adhesion molecule ICAM-4: potential regulator of membrane-associated ICAM-4 interactions. Blood. 2003;101(5):1790–7. doi: 10.1182/blood-2002-08-2529. [DOI] [PubMed] [Google Scholar]
- 18.Asch AS, Liu I, Briccetti FM, Barnwell JW, Kwakye-Berko F, Dokun A, et al. Analysis of CD36 binding domains: ligand specificity controlled by dephosphorylation of an ectodomain. Science. 1993;262(5138):1436–40. doi: 10.1126/science.7504322. [DOI] [PubMed] [Google Scholar]
- 19.Terra HT, Saad MJ, Carvalho CR, Vicentin DL, Costa FF, Saad ST. Increased tyrosine phosphorylation of band 3 in hemoglobinopathies. Am J Hematol. 1998;58(3):224–30. doi: 10.1002/(sici)1096-8652(199807)58:3<224::aid-ajh11>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- 20.Oh SO, Ibe BO, Johnson C, Kurantsin-Mills J, Raj JU. Platelet-activating factor in plasma of patients with sickle cell disease in steady state. J Lab Clin Med. 1997;130(2):191–6. doi: 10.1016/s0022-2143(97)90095-0. [DOI] [PubMed] [Google Scholar]
- 21.Joneckis CC, Ackley RL, Orringer EP, Wayner EA, Parise LV. Integrin alpha 4 beta 1 and glycoprotein IV (CD36) are expressed on circulating reticulocytes in sickle cell anemia. Blood. 1993;82(12):3548–55. [PubMed] [Google Scholar]
- 22.Dawson DW, Pearce SF, Zhong R, Silverstein RL, Frazier WA, Bouck NP. CD36 mediates the In vitro inhibitory effects of thrombospondin-1 on endothelial cells. J Cell Biol. 1997;138(3):707–17. doi: 10.1083/jcb.138.3.707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Coste I, Gauchat JF, Wilson A, Izui S, Jeannin P, Delneste Y, et al. Unavailability of CD147 leads to selective erythrocyte trapping in the spleen. Blood. 2001;97(12):3984–8. doi: 10.1182/blood.v97.12.3984. [DOI] [PubMed] [Google Scholar]
- 24.Gambero S, Canalli AA, Traina F, Albuquerque DM, Saad ST, Costa FF, et al. Therapy with hydroxyurea is associated with reduced adhesion molecule gene and protein expression in sickle red cells with a concomitant reduction in adhesive properties. Eur J Haematol. 2007;78(2):144–51. doi: 10.1111/j.1600-0609.2006.00788.x. [DOI] [PubMed] [Google Scholar]
- 25.Rahuel C, Filipe A, Ritie L, El Nemer W, Patey-Mariaud N, Eladari D, et al. Genetic inactivation of the laminin alpha5 chain receptor Lu/BCAM leads to kidney and intestinal abnormalities in the mouse. Am J Physiol Renal Physiol. 2008;294(2):F393–406. doi: 10.1152/ajprenal.00315.2007. [DOI] [PubMed] [Google Scholar]
- 26.McCormick CJ, Craig A, Roberts D, Newbold CI, Berendt AR. Intercellular adhesion molecule-1 and CD36 synergize to mediate adherence of Plasmodium falciparum-infected erythrocytes to cultured human microvascular endothelial cells. J Clin Invest. 1997;100(10):2521–9. doi: 10.1172/JCI119794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Swerlick RA, Lee KH, Wick TM, Lawley TJ. Human dermal microvascular endothelial but not human umbilical vein endothelial cells express CD36 in vivo and in vitro. J Immunol. 1992;148(1):78–83. [PubMed] [Google Scholar]
- 28.Xu Y, Swerlick RA, Sepp N, Bosse D, Ades EW, Lawley TJ. Characterization of expression and modulation of cell adhesion molecules on an immortalized human dermal microvascular endothelial cell line (HMEC-1) J Invest Dermatol. 1994;102(6):833–7. doi: 10.1111/1523-1747.ep12382086. [DOI] [PubMed] [Google Scholar]
- 29.Sugihara K, Sugihara T, Mohandas N, Hebbel RP. Thrombospondin mediates adherence of CD36+ sickle reticulocytes to endothelial cells. Blood. 1992;80(10):2634–42. [PubMed] [Google Scholar]
- 30.Lee K, Gane P, Roudot-Thoraval F, Godeau B, Bachir D, Bernaudin F, et al. The nonexpression of CD36 on reticulocytes and mature red blood cells does not modify the clinical course of patients with sickle cell anemia. Blood. 2001;98(4):966–71. doi: 10.1182/blood.v98.4.966. [DOI] [PubMed] [Google Scholar]
- 31.Schaffner-Reckinger E, Gouon V, Melchior C, Plancon S, Kieffer N. Distinct involvement of beta3 integrin cytoplasmic domain tyrosine residues 747 and 759 in integrinmediated cytoskeletal assembly and phosphotyrosine signaling. J Biol Chem. 1998;273(20):12623–32. doi: 10.1074/jbc.273.20.12623. [DOI] [PubMed] [Google Scholar]
- 32.Metral S, Machnicka B, Bigot S, Colin Y, Dhermy D, Lecomte MC. AlphaII-spectrin is critical for cell adhesion and cell cycle. J Biol Chem. 2009;284(4):2409–18. doi: 10.1074/jbc.M801324200. [DOI] [PubMed] [Google Scholar]
- 33.Berger G, Caen JP, Berndt MC, Cramer EM. Ultrastructural demonstration of CD36 in the alpha-granule membrane of human platelets and megakaryocytes. Blood. 1993;82(10):3034–44. [PubMed] [Google Scholar]
- 34.Tharaux PL, Hagege I, Placier S, Vayssairat M, Kanfer A, Girot R, et al. Urinary endothelin-1 as a marker of renal damage in sickle cell disease. Nephrol Dial Transplant. 2005;20(11):2408–13. doi: 10.1093/ndt/gfi111. [DOI] [PubMed] [Google Scholar]
- 35.Sabaa N, de Franceschi L, Bonnin P, Castier Y, Malpeli G, Debbabi H, et al. Endothelin receptor antagonism prevents hypoxia-induced mortality and morbidity in a mouse model of sickle-cell disease. J Clin Invest. 2008;118(5):1924–33. doi: 10.1172/JCI33308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kwok CF, Juan CC, Ho LT. Endothelin-1 decreases CD36 protein expression in vascular smooth muscle cells. Am J Physiol Endocrinol Metab. 2007;292(2):E648–52. doi: 10.1152/ajpendo.00084.2006. [DOI] [PubMed] [Google Scholar]