

Abstract
The ERYTHROPOIETIN (EPO) gene is regulated by the transcription factor Hypoxia Inducible Factor-α (HIF-α). In this pathway, Prolyl Hydroxylase Domain protein 2 (PHD2) hydroxylates two prolyl residues in HIF-α, which in turn promotes HIF-α degradation by the von Hippel Lindau (VHL) protein. Evidence that HIF-2α is the important isoform for EPO regulation in humans comes from the recent observation that mutations in the HIF2A gene are associated with cases of erythrocytosis. We report here a new erythrocytosis-associated mutation, p.Asp539Glu, in the HIF2A gene. Similar to all reported cases, the affected residue is in close vicinity and C-terminal to the primary hydroxylation site in HIF-2α, Pro531. This mutation, however, is notable in producing a rather subtle amino acid substitution. Nonetheless, we find that this mutation compromises binding of HIF-2α to both PHD2 and VHL, and we propose that this mutation is the cause of erythrocytosis in this individual.
Keywords: erythrocytosis, Hypoxia Inducible Factor, HIF-2α, erythropoietin
Introduction
The glycoprotein erythropoietin (EPO) is the central regulator of red blood cell mass, and EPO is regulated primarily at the transcriptional level.1,2 The key transcription factor controlling the EPO gene is Hypoxia Inducible Factor, which is composed of an oxygen-sensitive α subunit (HIF-α) and an oxygen-insensitive β subunit.3 There are three α subunit isoforms, HIF-1α, HIF-2α, and HIF-3α. Under normoxic conditions, a family of three enzymes known as Prolyl Hydroxylase Domain proteins (PHDs; also known as HIF Prolyl Hydroxylases and Egg Laying Defective Nine proteins) site-specifically hydroxylates HIF-α on two prolyl residues.4 This modification provides a recognition motif for the von Hippel Lindau tumor suppressor protein (VHL), a component of an E3 ubiquitin ligase complex that targets HIF-α for degradation by the ubiquitin-proteasome pathway.5 This mechanism allows HIF-α levels to be highly responsive to changes in oxygen concentration, since the hydroxylation reaction utilizes molecular oxygen as a substrate and is also inhibited by reactive oxygen species generated under hypoxic conditions. Thus, under hypoxia, HIF-α is hypohydroxylated and stabilized, allowing it to heterodimerize with HIF-β and transactivate a broad range of genes involved in cellular and systemic responses to hypoxia, of which the EPO gene is a cardinal example.3,6
Idiopathic erythrocytosis is an uncommon disorder characterized by abnormally elevated red cell mass.7,8 Investigations have revealed that some cases previously diagnosed as idiopathic erythrocytosis can now be attributed to mutations in proteins of the oxygen-sensing pathway that regulates HIF.9, 10 Thus, certain cases are due to homozygous or compound heterozygous missense mutations in the VHL gene (OMIM 263400) that diminish the ability of VHL to promote the degradation of HIF-α,11 while others are due to heterozygous mutations in the PHD2 gene (OMIM 609820) that either impair the ability of PHD2 to hydroxylate HIF-α (in the case of missense mutations) or lead to truncated proteins (in the case of frameshift and nonsense mutations).12 Recent studies have shown that mutations in the HIF2A gene (OMIM 611783) comprise a third cause of erythrocytosis attributable to defects in the oxygen-sensing pathway, and these studies implicate HIF-2α as the central HIF-α isoform regulating EPO in human adults.13 All reported mutations to date are heterozygous missense mutations that affect residues that are C-terminal to the primary hydroxylation site in HIF-2α, Pro531, and that variably affect the interaction with PHD2 and/or VHL.13–17 Collectively, these mutations, while affecting different proteins in the oxygen-sensing pathway, all lead to aberrant upregulation of HIF-2α.
In this report, we describe a patient with a novel HIF2A mutation associated with erythrocytosis that is predicted to result in a p.Asp539Glu amino acid change. Similar to the mutations previously described, it is a heterozygous mutation affecting a residue C-terminal to the hydroxylacceptor Pro531. Notably, though, this is perhaps the most subtle mutation thus far described, switching one acidic residue with another. As a point of reference, the first HIF-2α mutation described (p.Gly537Trp) changes the smallest amino acid for the bulkiest.13 Nonetheless, the present mutation impairs interaction of HIF-2α with both PHD2 and VHL, thereby underscoring the exquisite sensitivity of the pathway to what otherwise might be considered a seemingly conservative substitution.
Design and Methods
Patient and genetic studies
The patient is a 17-year old girl of North African ancestry. She has a history of an atrial septal defect, type II, that was closed 15 years ago. At that time, an elevated hemoglobin level had already been observed. Currently, hemoglobin levels are consistently around 17.7 g/dL with red blood cell counts of 6.3×1012/L (normal range 3.7 −5×1012/L). The patient’s EPO levels are marginally elevated (25 IU/L, normal range < 20 IU/L). Methemoglobin levels, as well as other blood counts, are within the normal range.
Both her height and her weight are −3 SD. She has mild mental retardation.
She has several complaints that can partly be attributed to her high level of hemoglobin: headaches, dizziness, dyspnoe d’effort, tiredness and a mild Raynaud phenomenon. Pulmonary, cardiac, and renal investigations showed no abnormalities. In particular, the patient does not suffer from chronic obstructive pulmonary disease, right-to-left cardiac shunts, or EPO-producing tumors.
Since the patient suffered from dizziness and headaches, the efficacy of lowering her hemoglobin to ameliorate her subjective well-being was addressed by repeated phlebotomy. This proved quite successful and could be tapered over time with maintenance of lower hemoglobin levels (in the range of 14.5–16.1 g/dL).
The inappropriately high level of EPO, together with the absence of any pulmonary, cardiac, or renal abnormalities, suggested a hereditary cause of secondary erythrocytosis in this patient.18,19 Therefore, the coding region of genes encoding hemoglobin (HBA and HBB), bisphosphoglyceratemutase (BPGM), PHD2 (PHD2), VHL (VHL), and HIF-2α (HIF2A) were investigated by PCR and subsequent DNA sequence analysis (primer sequences available on request). Informed consent was obtained, and procedures were performed in agreement with the Helsinki Declaration.
In vitro assays
pcDNA3-GAL4-HIF-2α (516–549) has been described previously.13 pcDNA3-GAL4-HIF-2α (516–549) D539E was constructed by subcloning into the Eco RI/Xba I site of pcDNA3-GAL4 an oligonucleotide duplex comprised of the following sequences: 5′-AAT TCA GTA CCC AGA CGG ATT TCA ATG AGC TGG ACT TGG AGA CAC TGG CAC CCT ATA TCC CCA TGG ACG GGG AAG AGT CC AAT TGA GCC CCA TCT GCC CCG AGG AGT AG-3′ and 5′-CTA GCT ACT CCT CGG GGC AGA TGG GGC TCA ATT GGA ACT CTT CCC CGT CCA TGG GGA TAT AGG GTG CCA GTG TCT CCA AGT CCA GCT CAT TGA AAT CCG TCT GGG TAC TG-3′. Authenticity of the construct was verified by sequencing. PHD2 binding assays were performed employing 35S-labeled in vitro translated GAL4-HIF-2α (516–549) fusion proteins (prepared using wheat germ extracts) and recombinant baculovirus-expressed (His)6FlagPHD220 as described previously.13
Hydroxyproline-531 (Hyp-531)-substituted wild-type or D539E HIF-2α (527–542) peptides, as well as N-terminal biotinylated Hyp-564 HIF-1α (556–574) peptide, were obtained from Genscript. VHL binding assays were performed by assessing the capacity of Hyp-531 HIF-2α peptide to compete with immobilized Hyp-564 HIF-1α (556–574) binding for 35S-labeled in vitro translated VHL (prepared using rabbit reticulocyte lysates) as described previously.13
Results shown are representative of two independent experiments.
Results and Discussion
DNA sequence analysis of HBA, HBB, BPGM, PHD2, and VHL showed no abnormalities. However, in HIF2A, a novel heterozygous C>G substitution was identified at nt c.1617, predicting the substitution of aspartic acid at residue 539 by glutamic acid (Figure 1). This single base change was not detected among 106 normal control alleles when screened by BbsI digestion.
Figure 1.
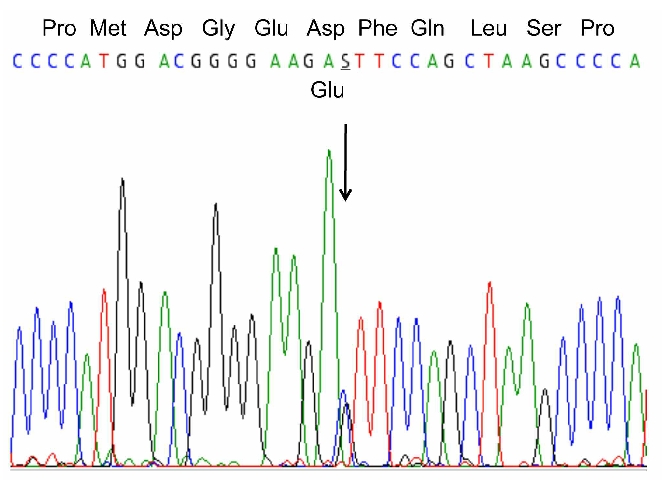
Genetic studies. DNA sequence analysis of nts c.1599–1633 of HIF2A. At nt c.1617 a heterozygous C>G substitution was identified. This novel missense mutation predicts the subsitution of aspartic acid by glutamic acid at residue 539 of HIF-2α.
We assessed the functional consequences of this mutation as follows. First, we incubated an 35S-labeled version of HIF-2α (residues 516–549 fused to GAL4) with recombinant PHD2. We immunoprecipitated the latter and determined the recovery of the HIF-2α fusion protein. Wild-type HIF-2α (516–549) binds with easily detectable affinity to PHD2, appropriate for an enzyme:substrate interaction (Figure 2A, lane 3). Importantly, we find that the p.Asp539Glu mutation weakens this interaction (Figure 2A, compare lanes 3 and 6).
Figure 2.
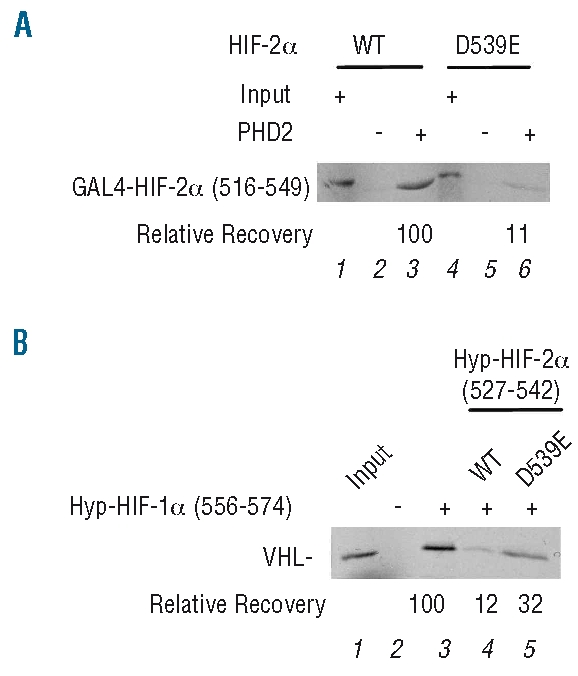
In vitro studies. (A) PHD2 binding assay. Anti-Flag (M2) agarose with or without immobilized recombinant (His)6FlagPHD2 was incubated with 35S labeled, in vitro translated wild-type or D539E GAL4-HIF-2α (516–549), washed, eluted, and the eluates subjected to SDS-PAGE. Input represents 10% of the total amount of wild-type or mutant GAL4-HIF-2α (516–549). Relative recovery is as indicated. (B) VHL binding assay. Streptavidin-agarose with or without biotinylated Hyp-564 HIF-1α (556–574) was incubated with 35S labeled, in vitro translated VHL in absence or presence of 50 nM of wild-type or D539E Hyp-531 HIF-2α (527–542) peptide. Resins were washed, eluted, and the eluates subjected to SDS-PAGE. Input represent 10% of the total amount of VHL employed in each reaction. The relative recovery of VHL is as indicated.
We next assessed the effect of this mutation on recognition by VHL in a competition assay in which 35S-labeled VHL was incubated with immobilized Hyp-564 HIF-1α (556–574) in the absence or presence of wild-type or Asp539Glu Hyp-531 HIF-2α (527–542) peptide. In this experiment, VHL binds with high affinity to Hyp-HIF-1α (556–574), as expected (Figure 2B, lane 3), and wild-type Hyp-HIF-2α (527–542) acts as an effective competitor for VHL binding (Figure 2B, compare lanes 3 and 4). We find that the p.Asp539Glu mutation diminishes the capacity of the Hyp-HIF-2α peptide to compete for VHL binding (Figure 2B, compare lanes 4 and 5), implying that this mutation impairs interaction of VHL. We conclude that the p.Asp539Glu mutation impairs the interaction of HIF-2α with both PHD2 and VHL. However, these are in vitro results, and it is conceivable that the interaction with and subsequent post-translational modification of HIF-2α by either PHD2 or VHL may not be rate limiting in vivo. That being said, the existence of both heterozygous PHD2 mutations and hypomorphic VHL mutations associated with erythrocytosis suggests that both interactions may, in fact, be important in this pathway.
The present findings are notablefor a number of reasons. First, they support the notion that HIF-2α is the critical isoform regulating EPO in human adults. Second, they indicate that a seemingly conservative substitution is sufficient to disrupt this pathway. Glutamic acid differs from aspartic acid by only a single methylene group, yet this substitution results in impaired interaction with both PHD2 and VHL. Impairment of both interactions is consistent with the observation of direct contacts between the corresponding residue in HIF-1α, Asp571, and PHD2 and VHL in the crystal structures of the respective complexes.21–23 In fact, the interaction of HIF-1α Asp571 with PHD2 and VHL is quite similar, involving salt bridge formation and hydrogen bonding with an arginine residue: Arg396 of PHD2 (Figure 3A), and Arg107 of VHL (Figure 3B). The longer side chain of glutamic acid, as compared to aspartic acid, is likely to disrupt these interactions when incorporated at position 571 of HIF-1α (Figure 3).
Figure 3.
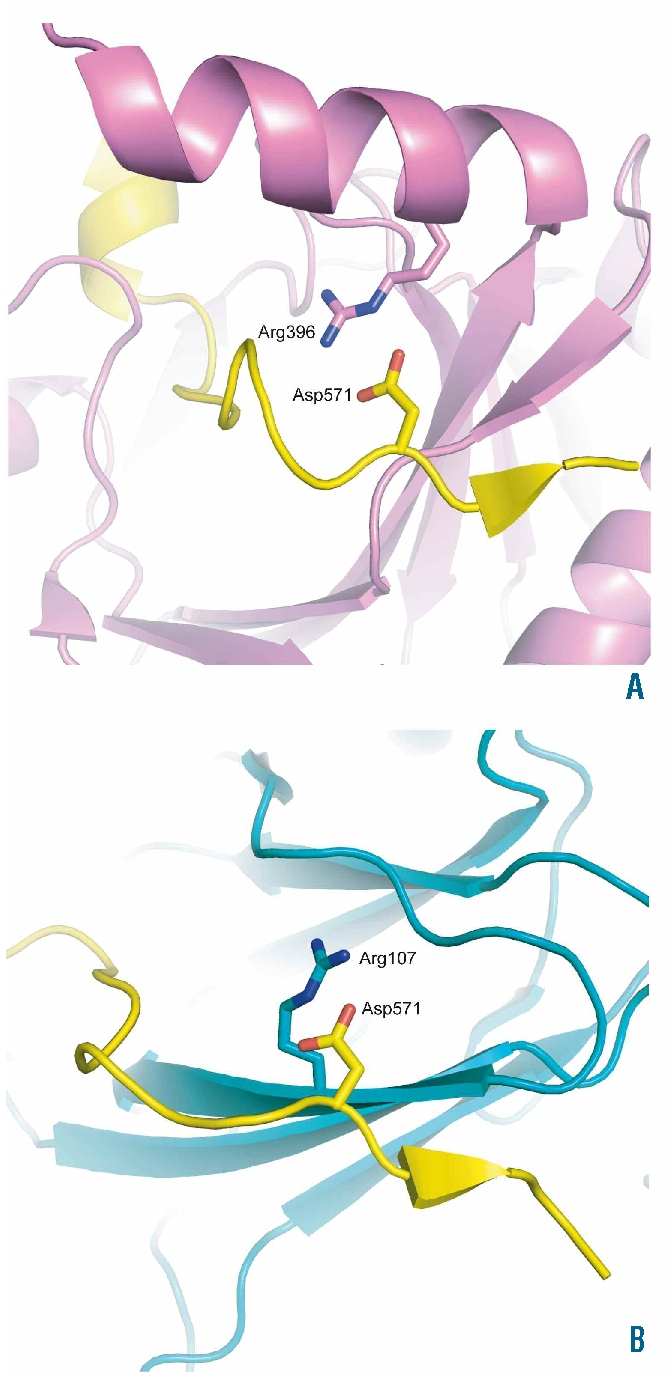
Structural effects. Images showing the position of the HIF-1α residue, Asp571, that is equivalent to HIF-2α Asp539 in the cocrystal structures of the CODD domain of HIF-1α with PHD2 (PDB ID 3HQR),21 and VHL (PDB ID 1LM8)22 (Panels A and B, respectively). HIF-1α is colored yellow, PHD2 violet, and VHL teal. HIF-1α residue Asp571 and selected residues are shown in stick representation. One of the contacts involved in HIF-1α binding to PHD2 and VHL involves the formation of salt bridges and hydrogen bonds with Arg396 of PHD2 (Panel A), and Arg107 of VHL (Panel B). In both cases, incorporation of the longer side chain of glutamic acid at position 571 in HIF-1α is likely to disrupt these interactions. The figure was generated using the program PyMOL (DeLano, W.L. The PyMOL Molecular Graphics System (2002) on World Wide Web http://www.pymol.org).
The corresponding structure of VHL bound to HIF-2α, or of PHD2 bound to HIF-2α, has yet to be reported. Nonetheless, the impaired interaction both with PHD2 and with VHL that is observed with the p.Asp539Glu mutation is also seen with most HIF-2α mutations studied to date.13,16 An exception is the p.Met535Val mutation, which selectively impairs interaction with PHD2 but not with VHL.16
Footnotes
Funding: this work was supported by NIH grant R01-CA090261 (FSL) and a University of Pennsylvania Research Foundation grant (FSL).
FSL received research grant support from GlaxoSmithKline.
Authorship and Disclosures
RvW designed and performed the genetic research, performed structural analysis, interpreted data, wrote the manuscript, approved the final version for publication. SS performed the functional studies, interpreted data, wrote the manuscript, approved the final version for publication. ACWW performed the genetic research, collected, analyzed, and interpreted data, approved the final version for publication. EGH performed structural analysis, wrote the manuscript, approved the final version for publication. MJP designed the genetic research, approved the final version for publication. MB wrote the manuscript, approved the final version for publication. FSL designed the functional studies, interpreted data, wrote the manuscript, approved the final version for publication.
The other authors report no potential conflicts of interest.
References
- 1.Jelkmann W. Erythropoietin after a century of research: younger than ever. Eur J Haematol. 2007;78(3):183–205. doi: 10.1111/j.1600-0609.2007.00818.x. [DOI] [PubMed] [Google Scholar]
- 2.Fandrey J. Oxygen-dependent and tissue-specific regulation of erythropoietin gene expression. Am J Physiol Regul Integr Comp Physiol. 2004;286(6):R977–88. doi: 10.1152/ajpregu.00577.2003. [DOI] [PubMed] [Google Scholar]
- 3.Semenza GL. Life with oxygen. Science. 2007;318(5847):62–4. doi: 10.1126/science.1147949. [DOI] [PubMed] [Google Scholar]
- 4.Schofield CJ, Ratcliffe PJ. Oxygen sensing by HIF hydroxylases. Nat Rev Mol Cell Biol. 2004;5(5):343–54. doi: 10.1038/nrm1366. [DOI] [PubMed] [Google Scholar]
- 5.Kaelin WG. Von Hippel-Lindau disease. Annu Rev Pathol. 2007;2:145–73. doi: 10.1146/annurev.pathol.2.010506.092049. [DOI] [PubMed] [Google Scholar]
- 6.Gordan JD, Simon MC. Hypoxia-inducible factors: central regulators of the tumor phenotype. Curr Opin Genet Dev. 2007;17(1):71–7. doi: 10.1016/j.gde.2006.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.McMullin MF, Bareford D, Campbell P, Green AR, Harrison C, Hunt B, et al. Guidelines for the diagnosis, investigation and management of polycythaemia/erythrocytosis. Br J Haematol. 2005;130(2):174–95. doi: 10.1111/j.1365-2141.2005.05535.x. [DOI] [PubMed] [Google Scholar]
- 8.Hodges VM, Rainey S, Lappin TR, Maxwell AP. Pathophysiology of anemia and erythrocytosis. Crit Rev Oncol Hematol. 2007;64(2):139–58. doi: 10.1016/j.critrevonc.2007.06.006. [DOI] [PubMed] [Google Scholar]
- 9.Percy MJ, Lee FS. Familial erythrocytosis: molecular links to red blood cell control. Haematologica. 2008;93(7):963–7. doi: 10.3324/haematol.13250. [DOI] [PubMed] [Google Scholar]
- 10.Lee FS. Genetic causes of erythrocytosis and the oxygen-sensing pathway. Blood Rev. 2008;22:321–32. doi: 10.1016/j.blre.2008.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ang SO, Chen H, Hirota K, Gordeuk VR, Jelinek J, Guan Y, et al. Disruption of oxygen homeostasis underlies congenital Chuvash polycythemia. Nat Genet. 2002;32(4):614–21. doi: 10.1038/ng1019. [DOI] [PubMed] [Google Scholar]
- 12.Percy MJ, Zhao Q, Flores A, Harrison C, Lappin TR, Maxwell PH, et al. A family with erythrocytosis establishes a role for prolyl hydroxylase domain protein 2 in oxygen homeostasis. Proc Natl Acad Sci USA. 2006;103(3):654–9. doi: 10.1073/pnas.0508423103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Percy MJ, Furlow PW, Lucas GS, Li X, Lappin TR, McMullin MF, et al. A gain-of-function mutation in the HIF2A gene in familial erythrocytosis. N Engl J Med. 2008;358(2):162–8. doi: 10.1056/NEJMoa073123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Martini M, Teofili L, Cenci T, Giona F, Torti L, Rea M, et al. A novel heterozygous HIF2AM535I mutation reinforces the role of oxygen sensing pathway disturbances in the pathogenesis of familial erythrocytosis. Haematologica. 2008;93(7):1068–71. doi: 10.3324/haematol.13210. [DOI] [PubMed] [Google Scholar]
- 15.Gale DP, Harten SK, Reid CD, Tuddenham EG, Maxwell PH. Autosomal dominant erythrocytosis and pulmonary arterial hypertension associated with an activating HIF2 alpha mutation. Blood. 2008;112(3):919–21. doi: 10.1182/blood-2008-04-153718. [DOI] [PubMed] [Google Scholar]
- 16.Furlow PW, Percy MJ, Sutherland S, Bierl C, McMullin MF, Master SR, et al. Erythrocytosis-associated HIF-2alpha mutations demonstrate a critical role for residues C-terminal to the hydroxylacceptor proline. Biol Chem. 2009;284(14):9050–8. doi: 10.1074/jbc.M808737200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Percy MJ, Beer PA, Campbell G, Dekker AW, Green AR, Oscier D, et al. Novel exon 12 mutations in the HIF2A gene associated with erythrocytosis. Blood. 2008;111(11):5400–2. doi: 10.1182/blood-2008-02-137703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.McMullin MF. The classification and diagnosis of erythrocytosis. Int J Lab Hematol. 2008;30(6):447–59. doi: 10.1111/j.1751-553X.2008.01102.x. [DOI] [PubMed] [Google Scholar]
- 19.Gordeuk VR, Stockton DW, Prchal JT. Congenital polycythemias/erythrocytoses. Haematologica. 2005;90(1):109–16. [PubMed] [Google Scholar]
- 20.Huang J, Zhao Q, Mooney SM, Lee FS. Sequence determinants in hypoxia-inducible factor-1alpha for hydroxylation by the prolyl hydroxylases PHD1, PHD2, and PHD3. J Biol Chem. 2002;277(42):39792–800. doi: 10.1074/jbc.M206955200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chowdhury R, McDonough MA, Mecinovic J, Loenarz C, Flashman E, Hewitson KS, et al. Structural basis for binding of hypoxia-inducible factor to the oxygen-sensing prolyl hydroxylases. Structure. 2009;17(7):981–9. doi: 10.1016/j.str.2009.06.002. [DOI] [PubMed] [Google Scholar]
- 22.Min JH, Yang H, Ivan M, Gertler F, Kaelin WG, Jr, Pavletich NP. Structure of an HIF-1alpha -pVHL complex: hydroxyproline recognition in signaling. Science. 2002;296(5574):1886–9. doi: 10.1126/science.1073440. [DOI] [PubMed] [Google Scholar]
- 23.Hon WC, Wilson MI, Harlos K, Claridge TD, Schofield CJ, Pugh CW, et al. Structural basis for the recognition of hydroxyproline in HIF-1alpha by pVHL. Nature. 2002;417(6892):975–8. doi: 10.1038/nature00767. [DOI] [PubMed] [Google Scholar]