

Summary
Background
Lipoprotein-associated phospholipase A2 (Lp-PLA2), an inflammatory enzyme expressed in atherosclerotic plaques, is a therapeutic target being assessed in trials of vascular disease prevention. We investigated associations of circulating Lp-PLA2 mass and activity with risk of coronary heart disease, stroke, and mortality under different circumstances.
Methods
With use of individual records from 79 036 participants in 32 prospective studies (yielding 17 722 incident fatal or non-fatal outcomes during 474 976 person-years at risk), we did a meta-analysis of within-study regressions to calculate risk ratios (RRs) per 1 SD higher value of Lp-PLA2 or other risk factor. The primary outcome was coronary heart disease.
Findings
Lp-PLA2 activity and mass were associated with each other (r=0·51, 95% CI 0·47–0·56) and proatherogenic lipids. We noted roughly log-linear associations of Lp-PLA2 activity and mass with risk of coronary heart disease and vascular death. RRs, adjusted for conventional risk factors, were: 1·10 (95% CI 1·05–1·16) with Lp-PLA2 activity and 1·11 (1·07–1·16) with Lp-PLA2 mass for coronary heart disease; 1·08 (0·97–1·20) and 1·14 (1·02–1·27) for ischaemic stroke; 1·16 (1·09–1·24) and 1·13 (1·05–1·22) for vascular mortality; and 1·10 (1·04–1·17) and 1·10 (1·03–1·18) for non-vascular mortality, respectively. RRs with Lp-PLA2 did not differ significantly in people with and without initial stable vascular disease, apart from for vascular death with Lp-PLA2 mass. Adjusted RRs for coronary heart disease were 1·10 (1·02–1·18) with non-HDL cholesterol and 1·10 (1·00–1·21) with systolic blood pressure.
Interpretation
Lp-PLA2 activity and mass each show continuous associations with risk of coronary heart disease, similar in magnitude to that with non-HDL cholesterol or systolic blood pressure in this population. Associations of Lp-PLA2 mass and activity are not exclusive to vascular outcomes, and the vascular associations depend at least partly on lipids.
Funding
UK Medical Research Council, GlaxoSmithKline, and British Heart Foundation.
Introduction
Lipoprotein-associated phospholipase A2 (Lp-PLA2), an enzyme expressed by inflammatory cells in atherosclerotic plaques,1,2 is carried in the circulation bound predominantly to LDL.3 Lp-PLA2 and other human A2 phospholipases (such as secretory phospholipase A2)4 propagate inflammation by producing precursors of arachidonic acid from membrane glycerophospholipids.5 Lp-PLA2 (also called platelet-activating factor acetylhydrolase) hydrolyses oxidised phospholipids to yield pro-inflammatory products that are implicated in endothelial dysfunction, plaque inflammation, and formation of necrotic core in plaque,6,7 and is postulated to link oxidative modification of LDL and development of inflammatory responses in the arterial intima.8,9
Since the initial report in 2000,10 many prospective epidemiological studies have investigated the associations between circulating Lp-PLA2 (assayed either as its enzymatic activity or mass concentration) and subsequent risk of vascular disease outcomes. A meta-analysis of 14 such studies has been reported.11 However, because that review was based on published data, it was unable to provide detailed analyses (eg, separate examination of associations with coronary heart disease and stroke; characterisation of the shape of any dose-response relations) or to adjust consistently for potential confounding factors.
The objective of the Lp-PLA2 Studies Collaboration,12 an analysis of individual data from relevant prospective studies, was to assess the independence, specificity, magnitude, and shape of associations of Lp-PLA2 with coronary heart disease, stroke, and mortality under different circumstances.
Methods
Study design
Details of study selection and data collection have been described previously.12 Information about Lp-PLA2 in relation to major vascular disease morbidity or cause-specific mortality was supplied by 32 prospective studies, 19 of which agreed to participate before their publication. Data were available for 79 036 participants (webappendix pp 2 and 17–20); only two relevant studies (comprising <5% of known incident vascular outcomes) were unable to share data.13,14 Study participants were drawn from three groups (webappendix p 7): (1) 35 945 people with no history of vascular disease at the initial examination (baseline); (2) 35 494 patients with a history of stable vascular disease (ie, diagnosis more than 30 days before baseline of any of myocardial infarction, angina, other coronary heart disease, stroke [including transient ischaemic attack], peripheral vascular disease, or coronary surgery, including revascularisations); and (3) 10 638 patients diagnosed with acute ischaemic events occurring no more than 30 days before baseline. (This final group has been analysed separately because risk factor levels might be more liable to distortion immediately after acute ischaemic events and because these studies had much shorter follow-up than did the other groups.) Baseline information was not available for non-vascular diseases.
Of the 19 studies that measured Lp-PLA2 enzyme activity, eight used radiometric and 11 used colorimetric assays (of which eight used Colorimetric Activity Method [CAM] assays [diaDexus, San Francisco, CA, USA], two used Azwell assays [Azwell, Osaka, Japan], and one used Cayman assays [Cayman Chemical, Ann Arbor, MI, USA]). Of the 25 studies that measured Lp-PLA2 mass concentration, two used in-house enzyme-linked immunoassays and 23 used commercial immunoassays, including three studies that used first generation Phospholipase A2—Cardiovascular (PLAC I) assays, 19 that used second generation (PLAC II), and one study that used third generation (PLAC III; all PLAC assays were manufactured by diaDexus, San Francisco, CA, USA). In registering fatal outcomes, all but one study used international classification of disease codings to at least three digits, and ascertainment was based on death certificates. 24 of the 32 contributing studies were also known to have classified deaths using medical records, autopsy findings, and other supplementary sources. 28 studies used standard definitions of myocardial infarction based on criteria of monitoring trends and determinants in cardiovascular disease.15 21 studies reported diagnosis of stroke subtypes on the basis of typical clinical features and characteristic changes on brain imaging. The study was approved by the Cambridgeshire Ethics Research Committee.
Statistical analyses
The webappendix p 3 provides details of the statistical methods. Because of differences in the mean and SD of concentrations of Lp-PLA2 recorded across studies using different assay methods (webbappendix pp 8 and 18), concentrations were Z transformed to a mean of 0 and an SD of 1 within each study (as well as doing sensitivity analyses with exclusion of studies with outlier values). Cross-sectional associations of Lp-PLA2 with various markers were assessed by calculation of mean Lp-PLA2 concentrations within tenths of these characteristics, with linear mixed models adjusted for age as previously described.16 The primary outcome was coronary heart disease (ie, non-fatal myocardial infarction or fatal coronary heart disease). All participants contributed only either the first non-fatal outcome or death during follow-up recorded at age 20 years or older (ie, deaths preceded by non-fatal coronary heart disease or stroke were not included in the main analyses). Principal analyses used a two-stage approach. Estimates of association were calculated within each study before pooling across studies by random-effects meta-analysis (parallel analyses used fixed-effect models). For cohort studies, hazard ratios were calculated with Cox proportional hazards models stratified by sex and baseline history of vascular disease (and, when appropriate, by trial group). Assumptions of proportionality of hazards were satisfied for both Lp-PLA2 markers. For case-control studies that were nested within prospective cohorts, odds ratios were calculated with either conditional or unconditional logistic regression models, as appropriate. Odds ratios were assumed to approximate hazard ratios and are collectively described as risk ratios (RRs). Studies contributing ten or fewer outcomes to any particular analysis were excluded. When data were missing for covariates, we restricted analyses to subsets of participants with complete information.
To assess shapes of association, study-specific RRs calculated within fifths of baseline Lp-PLA2 values were pooled on the log scale by multivariate random-effects meta-analysis and plotted against the mean levels in each fifth. We estimated 95% CIs from floated variances that correspond to the amount of information underlying each group (including the reference group).17 Since associations were roughly log-linear, we calculated regression coefficients to estimate the RR associated with one Z score higher Lp-PLA2, equivalent to a 1 SD higher Lp-PLA2. RRs were adjusted progressively for conventional risk factors. Because directly measured LDL cholesterol values were available in only a subset of participants, non-HDL cholesterol was used as the principal marker of cholesterol content in pro-atherogenic lipoproteins, avoiding potential biases of use of LDL cholesterol estimated by the Friedewald formula (webappendix p 6). The Wald χ2 statistic indicated the evidence of association. Heterogeneity was assessed by the I2 statistic.18 Diversity in study characteristics was investigated by grouping studies by recorded characteristics and by meta-regression. We investigated effect modification by formal tests of interaction, with main emphasis on age, sex, and lipid-related variables. Analyses related to prediction of vascular risk were not attempted, principally owing to the briefness of follow-up (eg, median follow-up of <6 years) and missing information about relevant risk factors.19 We used Stata (version 11.0) for analyses.
Role of the funding source
The independent academic coordinating centre, based at the University of Cambridge and University of Oxford, designed the study, did data collection and management, did statistical analysis, and wrote the report. GlaxoSmithKline was represented on the study's Operations Group. The study was undertaken independently from the funders. AT and JD had full access to all the data in the study and had final responsibility for the decision to submit for publication.
Results
Mean age at entry of the 79 036 participants was 64 years (SD 10). 50 290 (64%) were men, 46 418 (59%) were from western Europe, and 20 663 (26%) were from North America (webappendix pp 17–18). 7639 incident coronary heart disease outcomes, 2547 ischaemic strokes, 198 haemorrhagic strokes, 1191 unclassified strokes, and 1490 deaths from other vascular diseases, 4424 deaths from non-vascular diseases, and 233 from unknown causes were recorded during at least 474 976 person-years at risk. In the 71 439 participants who were initially healthy or had a history of stable vascular disease at baseline, Lp-PLA2 activity was available in 57 931 participants from 18 studies and Lp-PLA2 mass in 58 224 participants from 21 studies (table 1 and webappendix p 7).
Table 1.
Summary of data available and associations with Lp-PLA2 activity and mass at baseline survey
|
Lp-PLA2activity (up to 57 931 participants from 18 studies) |
Lp-PLA2mass (up to 58 224 participants from 21 studies) |
||||||
|---|---|---|---|---|---|---|---|
| n | Mean (SD) or % | Correlation* (95% CI) | n | Mean (SD) or % | Correlation* (95% CI) | ||
| Anthropometric markers | |||||||
| Age at survey (years) | 57 931 | 64 (8) | 0·02 (−0·00 to 0·05) | 58 224 | 64 (8) | 0·06 (0·04 to 0·08) | |
| Body-mass index (kg/m2) | 46 278 | 27 (4) | 0·04 (0·02 to 0·05) | 48 366 | 27 (5) | −0·01 (−0·03 to 0·01) | |
| Systolic blood pressure (mm Hg) | 47 019 | 138 (22) | 0·02 (−0·00 to 0·03) | 48 316 | 137 (21) | 0·02 (0·01 to 0·04) | |
| Lipid markers | |||||||
| Total cholesterol (mmol/L) | 57 681 | 5·5 (1·0) | 0·41 (0·37 to 0·45) | 57 550 | 5·5 (1·0) | 0·28 (0·25 to 0·31) | |
| Non-HDL cholesterol (mmol/L) | 56 749 | 4·26 (1·02) | 0·49 (0·45 to 0·52) | 53 572 | 4·26 (1·00) | 0·30 (0·27 to 0·34) | |
| HDL cholesterol (mmol/L) | 56 838 | 1·23 (0·35) | −0·24 (−0·29 to −0·19) | 53 639 | 1·24 (0·36) | −0·07 (−0·12 to −0·02) | |
| Loge triglycerides (mmol/L) | 55 649 | 0·40 (0·51) | 0·22 (0·19 to 0·26) | 52 595 | 0·38 (0·51) | 0·07 (0·04 to 0·11) | |
| LDL cholesterol (mmol/L)† | 28 006 | 3·09 (0·81) | 0·48 (0·41 to 0·55) | 29 114 | 3·44 (0·80) | 0·28 (0·22 to 0·34) | |
| Apolipoprotein B (g/L) | 36 399 | 1·10 (0·24) | 0·45 (0·38 to 0·51) | 28 778 | 1·05 (0·23) | 0·24 (0·17 to 0·30) | |
| Apolipoprotein AI (g/L) | 33 790 | 1·45 (0·23) | −0·15 (−0·23 to −0·05) | 28 797 | 1·41 (0·22) | −0·07 (−0·13 to 0·00) | |
| Inflammatory markers | |||||||
| Loge C-reactive protein (mg/L) | 52 443 | 0·87 (1·10) | 0·03 (0·01 to 0·05) | 47 674 | 0·83 (1·08) | 0·08 (0·04 to 0·11) | |
| Fibrinogen (μmol/L) | 17 533 | 10·04 (2·17) | 0·00 (−0·02 to 0·02) | 13 169 | 11·12 (2·17) | 0·05 (0·03 to 0·07) | |
| Loge leucocyte count (×109/L) | 12 388 | 1·87 (0·27) | 0·03 (0·01 to 0·05) | 10 731 | 1·84 (0·28) | 0·07 (0·04 to 0·10) | |
| Categorical variables | |||||||
| Sex | |||||||
| Men | 36 222 | 63% | Ref | 36 857 | 63% | Ref | |
| Women | 21 709 | 37% | −0·21 (−0·25 to −0·17) | 21 367 | 37% | −0·10 (−0·13 to −0·07) | |
| Ethnic origin | |||||||
| White | 50 922 | 96% | Ref | 47 376 | 96% | Ref | |
| Non-white | 1906 | 4% | −0·07 (−0·11 to −0·03) | 2083 | 4% | −0·08 (−0·12 to −0·03) | |
| Smoking status | |||||||
| Other | 44 576 | 86% | Ref | 45 871 | 86% | Ref | |
| Current | 7268 | 14% | 0·03 (0·01 to 0·05) | 7595 | 14% | 0·08 (0·06 to 0·11) | |
| History of diabetes | |||||||
| No | 46 741 | 82% | Ref | 46 824 | 82% | Ref | |
| Yes | 9934 | 18% | 0·00 (−0·02 to 0·02) | 10 209 | 18% | −0·03 (−0·05 to −0·02) | |
Data are shown for the 71 439 participants who were initially healthy or had a history of stable vascular disease at baseline only. Data for the 10 638 participants with recent acute ischaemic events are shown in webappendix p 24. Mean Lp-PLA2 activity and mass by assay method are shown in webappendix p 8. 44 716 participants had information about both Lp-PLA2 activity and mass. Lp-PLA2=lipoprotein-associated phospholipase A2. Ref=reference category.
Partial correlation coefficient (or for categorical variables, the difference in standardised Lp-PLA2 compared with the reference category) adjusted for age, sex, baseline history of diabetes, and baseline history of vascular disease (as appropriate).
Directly measured LDL cholesterol.
For Lp-PLA2 activity, much of the variation in mean values across studies was explained by differences in the assay methods used (webappendix p 8). In studies using the CAM assay, the mean was 151 nmol/min/mL (SD 32), whereas it was 42 nmol/min/mL (14) in studies using radiometric assays. For mass, apart from the two studies that used in-house ELISA, mean concentrations were broadly similar across studies, with a mean of 312 μg/L (SD 95) in studies that used the PLAC II assay. Lp-PLA2 activity and mass were roughly linearly associated with each other (partial correlation coefficient r=0·51, 95% CI 0·47–0·56; figure 1). Lp-PLA2 activity was higher in men than in women (table 1) and positively correlated with non-HDL cholesterol (r=0·49, 0·45–0·52), directly measured LDL cholesterol (r=0·48, 0·41–0·55), apolipoprotein B (r=0·45, 0·38–0·51), and loge triglycerides (r=0·22, 0·19–0·26), and inversely correlated with HDL cholesterol (r=−0·24, −0·29 to −0·19) and apolipoprotein AI (r=−0·15, −0·23 to −0·05; figure 1). Lp-PLA2 activity was only weakly or non-significantly associated with age, systolic blood pressure, body-mass index, smoking, loge C-reactive protein (CRP), and fibrinogen or leucocyte count (table 1 and figure 1). Associations of Lp-PLA2 mass followed similar patterns, although mass was more strongly associated with smoking and less strongly associated with lipids than was activity (table 1 and webappendix p 9). A combined estimate of the within-person variability of Lp-PLA2 could not be made reliably because results from different studies were widely divergent.20 Furthermore, only some of the sources of heterogeneity could be identified (eg, studies that used CAM assays tended to have higher reproducibility values than did those that used radiometric activity assays; webappendix p 10).
Figure 1.
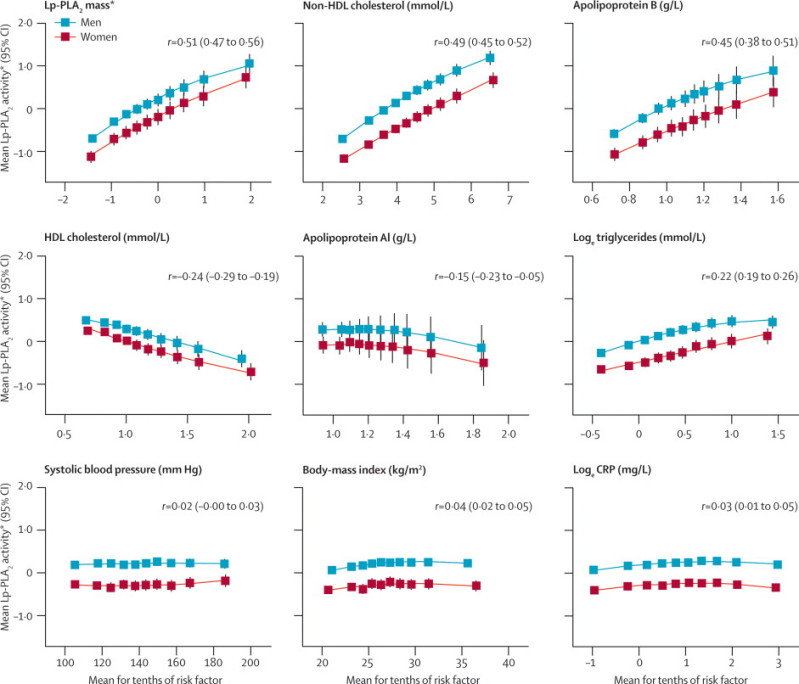
Cross-sectional associations of Lp-PLA2 activity
Table 1 shows number of participants included in each analysis. Webappendix p 9 shows cross-sectional associations of Lp-PLA2 mass. Error bars represent 95% CIs. r=Pearson's partial correlation coefficient (95% CI) adjusted for age, sex, history of diabetes, and baseline history of vascular disease. Lp-PLA2=lipoprotein-associated phospholipase A2. *Lp-PLA2 activity and mass were standardised to a mean of 0·00 (SD 1·00) in each study.
We noted roughly log-linear associations of Lp-PLA2 activity with risk of coronary heart disease and all vascular mortality, and less distinct associations with ischaemic stroke and the aggregate of non-vascular mortality (figure 2 and webappendix p 11). Because RRs did not differ significantly in initially healthy participants and in patients with stable vascular disease, we combined them to improve precision (figure 3). The RR for coronary heart disease with 1 SD higher Lp-PLA2 activity was reduced from 1·16 (95% CI 1·10–1·21) in minimally adjusted analyses to 1·10 (1·05–1·16) after further adjustment for conventional risk factors (the Wald χ reduced from 33 to 14, most of which was due to adjustment for lipids; table 2). We recorded no clear evidence of heterogeneity (I2=20%, 95% CI 0–59) nor of effect modification (webappendix pp 12–13). In subsets of participants with relevant information, RRs for coronary heart disease were: 1·10 (95% CI 1·02–1·18) after adjustment for apolipoproteins AI and B (instead of HDL cholesterol and non-HDL cholesterol, respectively); 1·12 (1·07–1·18) after adjustment for several conventional risk factors plus cholesterol concentrations and apolipoprotein B; 1·13 (1·08–1·19) after adjustment for directly measured LDL and HDL cholesterol (instead of non-HDL and HDL cholesterol); and 1·07 (1·00–1·14) after adjustment for several conventional risk factors plus Lp-PLA2 mass (webappendix p 21). The RR for ischaemic stroke after adjustment for conventional risk factors was 1·08 (0·97–1·20; table 2). Adjusted RRs were 0·97 (0·79–1·19) for haemorrhagic stroke, 1·02 (0·93–1·12) for unclassified stroke, and 1·16 (1·09–1·24) for all vascular mortality (figure 3 and webappendix p 14). The RR for the aggregate of non-vascular mortality was 1·10 (1·04–1·17) after adjustment for several risk factors (figure 3), with an RR for cancer death of 1·05 (0·97–1·14), and 1·18 (1·07–1·30) for non-vascular mortality not attributed to cancer (webappendix p 22). There were too few outcomes to attempt detailed subdivisions of non-vascular deaths (eg, by cancer site).
Figure 2.
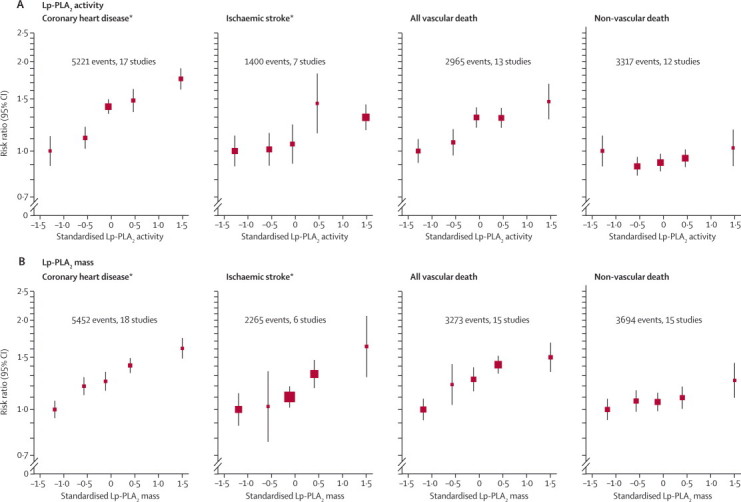
Minimally adjusted risk ratios for coronary heart disease, ischaemic stroke, and death due to vascular and non-vascular causes by fifths of Lp-PLA2 activity or mass at baseline
Risk ratios were adjusted for age, sex, baseline history of vascular disease, history of diabetes, and trial group (as appropriate). The webappendix p 11 shows more fully adjusted risk ratios. Data are shown for the 71 439 participants who were initially healthy or had a history of stable vascular disease at baseline only. One unit on the standardised scale is equal to 1 SD on the untransformed scale. Error bars represent 95% CIs. The sizes of the boxes are proportional to the inverse of the variance of the risk ratios. Lp-PLA2=lipoprotein-associated phospholipase A2. *Fatal and non-fatal events.
Figure 3.
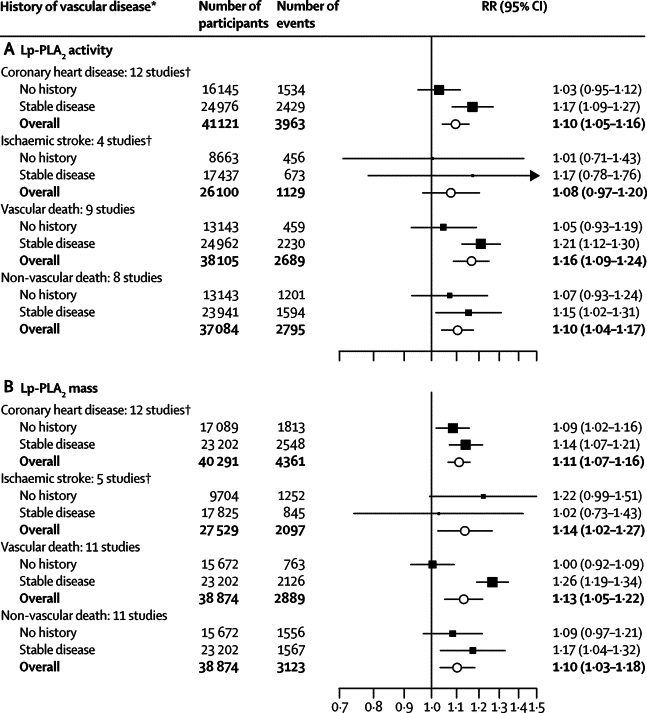
Risk ratios for coronary heart disease, ischaemic stroke, and vascular and non-vascular mortality per 1 SD higher Lp-PLA2 activity or mass at baseline, adjusted for several risk factors
Risk ratios were adjusted for the non-lipid and lipid risk factors described in table 2. We noted no significant differences in risk ratios between people with and without a history of stable vascular disease at baseline, apart from for vascular death with Lp-PLA2 mass (p=0·007). Data for patients with recent acute ischaemic events are shown in webappendix p 16. Error bars represent 95% CIs. The sizes of the boxes are proportional to the inverse of the variance of the RRs. Lp-PLA2=lipoprotein-associated phospholipase A2. RR=risk ratio. *Diagnosis more than 30 days before baseline of myocardial infarction, angina, other coronary heart disease, stroke (including transient ischaemic attack), peripheral vascular disease, or coronary surgery (including revascularisations). †Fatal and non-fatal events.
Table 2.
Risk ratios for coronary heart disease and ischaemic stroke per 1 SD higher Lp-PLA2 activity or mass at baseline, with progressive adjustment for baseline levels of potential confounders
|
Lp-PLA2activity |
Lp-PLA2mass |
|||||
|---|---|---|---|---|---|---|
| RR (95% CI) | Wald χ21 | I2 (95% CI) | RR (95% CI) | Wald χ21 | I2 (95% CI) | |
| Coronary heart disease* | ||||||
| Minimally adjusted† | 1·16 (1·10–1·21) | 33 | 34 (0–67) | 1·15 (1·11–1·19) | 55 | 19 (0–58) |
| Plus lipid-lowering drug use | 1·16 (1·10–1·22) | 32 | 37 (0–68) | 1·15 (1·11–1·19) | 56 | 19 (0–58) |
| Plus systolic blood pressure | 1·16 (1·10–1·22) | 32 | 36 (0–67) | 1·14 (1·10–1·19) | 55 | 18 (0–57) |
| Plus body-mass index | 1·15 (1·10–1·21) | 31 | 36 (0–68) | 1·15 (1·11–1·19) | 56 | 18 (0–57) |
| Plus smoking status | 1·15 (1·09–1·21) | 29 | 36 (0–68) | 1·14 (1·10–1·18) | 52 | 16 (0–56) |
| Plus non-HDL cholesterol | 1·12 (1·07–1·17) | 28 | 10 (0–49) | 1·11 (1·07–1·16) | 32 | 17 (0–56) |
| Plus HDL cholesterol | 1·11 (1·06–1·16) | 20 | 11 (0–51) | 1·11 (1·07–1·16) | 25 | 29 (0–64) |
| Plus loge triglyceride | 1·10 (1·05–1·16) | 14 | 20 (0–59) | 1·11 (1·07–1·16) | 25 | 26 (0–62) |
| Ischaemic stroke‡ | ||||||
| Minimally adjusted† | 1·08 (1·02–1·15) | 7 | 0 (0–85) | 1·18 (1·07–1·30) | 11 | 71 (25–88) |
| Plus lipid-lowering drug use | 1·08 (1·02–1·15) | 7 | 0 (0–85) | 1·18 (1·07–1·30) | 10 | 71 (27–89) |
| Plus systolic blood pressure | 1·08 (1·02–1·15) | 7 | 0 (0–85) | 1·16 (1·05–1·28) | 9 | 71 (26–89) |
| Plus body-mass index | 1·09 (1·02–1·15) | 7 | 0 (0–85) | 1·16 (1·05–1·28) | 9 | 70 (25–88) |
| Plus smoking status | 1·08 (1·01–1·14) | 6 | 0 (0–85) | 1·14 (1·04–1·26) | 7 | 69 (20–88) |
| Plus non-HDL cholesterol | 1·07 (0·98–1·16) | 2 | 22 (0–88) | 1·13 (1·02–1·26) | 5 | 70 (24–88) |
| Plus HDL cholesterol | 1·07 (0·97–1·19) | 2 | 39 (0–79) | 1·13 (1·02–1·26) | 5 | 70 (24–88) |
| Plus loge triglyceride | 1·08 (0·97–1·20) | 2 | 41 (0–80) | 1·14 (1·02–1·27) | 6 | 70 (22–88) |
Analyses were restricted to participants with complete information. The Wald χ21 statistic indicates the significance of the accompanying RR. The I2 statistic estimates the percentage of heterogeneity in the study-specific RRs that can be accounted for by between-study differences and not chance. RRs for other outcomes are shown in webappendix p 22. Lp-PLA2=lipoprotein-associated phospholipase A2. RR=risk ratio.
For Lp-PLA2 activity: 12 studies, 41 121 participants, and 3963 events; for Lp-PLA2 mass: 12 studies, 40 291 participants, and 4361 events.
Adjusted for age and history of diabetes, and stratified by sex, baseline history of vascular disease, and trial group (as appropriate).
For Lp-PLA2 activity: four studies, 26 100 participants, and 1129 events; for Lp-PLA2 mass: five studies, 27 529 participants, and 2097 events
We recorded roughly log-linear associations of Lp-PLA2 mass with vascular and non-vascular outcomes (figure 2 and webappendix p 11). Because RRs did not differ significantly in initially healthy participants and in those with stable vascular disease (apart from for the outcome of vascular death; webappendix p 23), they were combined (figure 3). The RR for coronary heart disease with 1 SD higher Lp-PLA2 mass reduced from 1·15 (1·11–1·19) to 1·11 (1·07–1·16) after adjustment for several risk factors (the Wald χ2 reduced from 55 to 25; table 2). Again, we noted no clear evidence of heterogeneity (I2=26%, 95% CI 0–62) nor of effect modification (webappendix pp 12–13). The adjusted RR for coronary heart disease with Lp-PLA2 mass was 1·08 (1·04–1·12) after further adjustment for Lp-PLA2 activity (further reducing the Wald χ2 from 33 to 14; webappendix p 21). Adjusted RRs for other outcomes were: 1·14 (1·02–1·27) for ischaemic stroke (table 2); 1·13 (1·05–1·22) for all vascular mortality; 1·10 (1·03–1·18) for the aggregate of non-vascular mortality; 1·08 (0·98–1·18) for cancer death; and 1·13 (1·04–1·23) for non-vascular mortality not attributed to cancer (webappendix p 22).
Adjusted RRs for coronary heart disease with Lp-PLA2 mass and activity were broadly similar to those with non-HDL cholesterol and systolic blood pressure (figure 4). For both Lp-PLA2 markers, we recorded qualitatively similar results to those reported in sensitivity analyses that: adjusted RRs further for CRP or fibrinogen (webappendix p 21); used fixed-effect models (webappendix p 15); omitted any individual study to assess its relative effect on the overall result; and included fatal outcomes without censoring previous non-fatal outcomes (data available on request).
Figure 4.
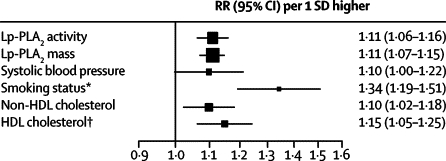
Adjusted risk ratios for coronary heart disease per 1 SD higher baseline Lp-PLA2 activity, mass, and several conventional risk factors in a common set of participants
Analyses were restricted to participants with complete information (3278 events in 34 762 participants who were initially healthy or had a history of stable vascular disease at baseline from seven studies). RRs were adjusted for the non-lipid and lipid risk factors described in table 2. Error bars represent 95% CIs. The sizes of the boxes are proportional to the inverse of the variance of the risk ratios. Lp-PLA2=lipoprotein-associated phospholipase A2. RR=risk ratio. *Current smoker versus other (never or ex-smoker). †To aid comparison with the other risk factors, the RR with HDL cholesterol is shown per 1 SD lower baseline levels.
The 10 638 patients diagnosed with recent acute ischaemic events had much briefer median follow-up than did other participants (1·1 [IQR 0·6–2·3] vs 5·8 [4·0–8·4] years). The cross-sectional correlations of Lp-PLA2 in these patients were broadly similar to those described above (webappendix p 24). RRs for recurrent vascular outcomes in these patients were essentially null, albeit with wide confidence intervals (webappendix p 16).
Discussion
Our analysis of 79 036 participants has shown that Lp-PLA2 activity and mass are associated with each other, proatherogenic lipids, and vascular risk. Lp-PLA2 activity was more strongly associated with various lipid markers than was Lp-PLA2 mass, which could indicate their varying distributions across lipoprotein classes, differences in measurement precision, or both.21–23 By contrast with previous suggestions of risk thresholds,24 our analysis shows roughly log-linear associations of Lp-PLA2 with risk of coronary heart disease and total vascular mortality. The shape of relations of circulating Lp-PLA2 with ischaemic stroke and with the aggregate of non-vascular mortality are less clear than is that with coronary heart disease, perhaps as a result of the fewer outcomes recorded. As has been reported previously for CRP and fibrinogen,25,26 Lp-PLA2 is associated with risk of both major vascular and non-vascular outcomes. However, because Lp-PLA2 mass and activity are not materially correlated with these circulating inflammatory markers, Lp-PLA2 measurements could have the potential to provide distinct insight into the relation between inflammation and atherothrombosis.
The strength of association for coronary heart disease with Lp-PLA2 was reduced after adjustment for baseline concentrations of lipids and apolipoproteins. Since Lp-PLA2 is physically linked (through apolipoprotein B) with LDL, however, the validity of statistical attempts to distinguish the effects of Lp-PLA2 on risk of coronary heart disease from those of proatherogenic lipids remains uncertain. A practical approach, as used in this study, is to present RRs with and without statistical adjustment for such lipid markers, and across participants with different lipid concentrations at baseline. Even in adjusted analyses, however, substantial residual confounding might persist because lipids (and other risk factors) are measured with some error,23 and because detailed information about some potential confounding factors (eg, medication for vascular diseases) was not uniformly available from the contributing studies.
The adjusted RR of baseline Lp-PLA2 with risk of coronary heart disease was similar to those for concentration of non-HDL cholesterol and systolic blood pressure. The RR with each of these conventional risk factors was, however, fairly moderate in magnitude—ie, about 10–15% higher risk per 1 SD higher value of the risk factor, or about a third as strong as in previous reports in which mean age at baseline survey was about 10 years lower than that reported in this study.27 Our findings of such weaker than expected RRs with conventional risk factors could be explained by the older mean age of participants in this study, since RRs with vascular risk factors tend to decrease with age (by contrast with absolute risk).27–29 Furthermore, the high percentage of participants who had prevalent vascular disease at entry could have disrupted natural relations between risk factors and subsequent coronary heart disease. These effects could, therefore, have blunted RRs with Lp-PLA2.
The strength and potential limitations of this investigation merit consideration. It is a large and comprehensive study, encompassing more than 95% of the relevant available data. Use of individual records allowed detailed analysis and a consistent approach to adjustment for several potential confounders. Because most contributing studies agreed to participate before publication of their data, the effect of selective reporting should be reduced. However, because data for serial Lp-PLA2 measurements were sparse and apparently divergent, we could not reliably correct for regression dilution.23 If, for example, the true correlation of Lp-PLA2 concentrations taken a few years apart in the same people is about 0·5, then the degree of underestimation of RRs could be as large as two-fold. Furthermore, the median follow-up duration in this study was about 6 years, which is too brief to enable informative study of the incremental value of Lp-PLA2 measurement in standard 10-year prediction of vascular disease risk. These limitations draw attention to the need for large studies of first-ever coronary heart disease with serial measurements and extended follow-up. Furthermore, more detailed studies are needed of non-vascular outcomes, especially because recorded associations of Lp-PLA2 with risk of non-cancer, non-vascular deaths might be attributed, at least partly, to confounding by comorbidity at baseline. Nevertheless, a potential limitation of any observational studies of circulating Lp-PLA2 is that the enzyme in the blood could be an imperfect indicator of its relevance to atherosclerotic plaques.
Loss-of-function mutations in the PLA2G7 gene, which are common in east-Asian populations, effectively abolish Lp-PLA2 activity (or, in heterozygotes, substantially reduce activity).30 Vascular risk is, however, not clearly lower in people carrying such mutations,31,32 although available studies might have been limited by heterogeneous outcomes and possible pleiotropic effects.8 Because known Lp-PLA2-related genotypes that are common in people of European continental ancestry have only weak effects on Lp-PLA2 activity,33 their study would need very large numbers of patients with coronary heart disease. Randomised trials of potent reversible pharmacological inhibitors of Lp-PLA2 activity should help to establish whether modification of Lp-PLA2 can reverse vascular risk.34–37
Acknowledgments
Acknowledgments
The independent academic Lp-PLA2 Studies Collaboration coordinating centre has been supported by specific grants from the UK Medical Research Council (G0601284) and GlaxoSmithKline, and is underpinned by a programme grant from the British Heart Foundation (RG/08/014). Alexander Thompson and Emanuele Di Angelantonio were supported by UK Medical Research Council doctoral training grants. A variety of sources have supported recruitment, follow-up, and laboratory measurements in the 32 studies contributing to the Lp-PLA2 Studies Collaboration. Investigators of several of these studies have contributed to a list naming some of these funding sources, which can be found at http://www.phpc.cam.ac.uk/ceu/lsc. Pierre Jacob and Clément Pravin provided statistical support. Hannah Sneath, Angela Harper, and Karina Prasad provided administrative support. Mary Cushman provided limited tabular data on Lp-PLA2 reproducibility on behalf of the Multi-Ethnic Study of Atherosclerosis (MESA) investigators.
Contributors
Alexander Thompson and John Danesh drafted the report. Pei Gao and Lia Orfei did the analyses. All members of the writing committee provided critical revisions. All investigators shared individual data and had opportunities to contribute to the interpretation of the results and critical revision of the report. Members of the operations group monitored the study's progress. The data management team undertook data collation and harmonisation. All members of the coordinating centre contributed to the collection, harmonisation, analysis, and interpretation of the data.
The Lp-PLA2 Studies Collaboration
Writing committee Alexander Thompson, Pei Gao*, Lia Orfei*, Sarah Watson, Emanuele Di Angelantonio, Stephen Kaptoge, University of Cambridge, Cambridge, UK; Christie Ballantyne, Baylor College of Medicine, Houston, TX, USA; Christopher P Cannon, Brigham and Women's Hospital, Boston, MA, USA; Michael Criqui, University of California, San Diego, La Jolla, CA, USA; Mary Cushman, University of Vermont, Burlington, VT, USA; Albert Hofman, Erasmus Medical Centre, Rotterdam, Netherlands; Chris Packard, University of Glasgow, Glasgow, UK; Simon G Thompson, MRC Biostatistics Unit, Cambridge, UK; Rory Collins, University of Oxford, Oxford, UK; John Danesh, University of Cambridge, Cambridge, UK. *These authors contributed equally.
Investigators (study acronyms are defined in webappendix p 2) ARIC: Christie Ballantyne; Bruneck: Johann Willeit, Stefan Kiechl, Christian Wiedermann; CHS: Mary Cushman, Bruce Psaty, Curt Furberg; EPIC-Norfolk: Kay-Tee Khaw, Manjinder Sandhu; FHS Offspring: Emelia J Benjamin, Ramachandran S Vasan, Renate B Schnabel; FRISC II: Jonas Oldgren; GENICA: Gian Paolo Rossi, Maurizio Cesari, Livia Lenzini, Mario Zanchetta; GUSTO IV: Stefan K James; HPFS: Eric Rimm, Ida Hatoum; HPS: Rory Collins; IHCS: Jeffrey L Anderson, Heidi T May, Benjamin D Horne, John F Carlquist, Joseph B Muhlestein; KAROLA: Wolfgang Koenig, Hermann Brenner, Dietrich Rothenbacher; LURIC: Winfried März, Bernhard Böhm, Bernhard R Winkelmann, Karl Winkler; MDCS: Goran Berglund, Margaretha Persson; MAYO CLINIC (Olmsted County): Veronique Roger, Yariv Gerber; MAYO CLINIC (Referral practice): Peter B Berger, Emmanouil S Brilakis, Joseph P McConnell; MONICA-KORA: Wolfgang Koenig, Christa Meisinger; NHS: Eric Rimm, Ida Hatoum; NOMAS: Ralph Sacco, Mitchell Elkind; NPHS II: Philippa J Talmud; OPUS-TIMI 16: Michelle O'Donoghue; PEACE: Marc S Sabatine, David A Morrow; PROSPER: Chris Packard; Muriel Caslake; PROVEIT-TIMI 22: Eugene Braunwald, Christopher P Cannon; Rancho Bernardo: Elizabeth Barrett-Connor, Lori B Daniels, Gail A Laughlin; Rotterdam Study: Albert Hofman, Isabella Kardys, Jacqueline C M Witteman; SDVC: Michael Criqui; THROMBO: James P Corsetti, David L Rainwater, Arthur J Moss; WHI-HaBPS: Sylvia Wassertheil-Smoller; WHS: Paul Ridker; WOSCOPS: Chris Packard.
Operations group Christie Ballantyne, Christopher P Cannon, Rory Collins, Michael Criqui, Mary Cushman, John Danesh, Albert Hofman, Jeanenne J Nelson, Chris Packard, Simon G Thompson, Nevine Zariffa, Andrew Zalewski.
Data Management Team Sarah Watson, Mat Walker.
Coordinating Centre Alexander Thompson (coordinator), Lia Orfei, Pei Gao, Sarah Watson, Emanuele Di Angelantonio, Mat Walker, Philip Perry, Stephen Kaptoge, Angela Wood, Simon G Thompson, Rory Collins, John Danesh (principal investigator).
Conflicts of interest
Alexander Thompson's institution has received research funding from the British Heart Foundation, GlaxoSmithKline, and UK Medical Research Council. He has received honoraria and reimbursement of costs for speaking at scientific meetings from GlaxoSmithKline. Pei Gao, Lia Orfei, Sarah Watson, Emanuele Di Angelantonio, and Stephen Kaptoge's institution has received research funding from the British Heart Foundation, GlaxoSmithKline, and UK Medical Research Council. Christie Ballantyne's institution has received research funding from diaDexus and GlaxoSmithKline, and he has received consultancy fees from GlaxoSmithKline. Christopher P Cannon has received research funding from Bristol-Myers Squibb, Sanofi-Aventis, Intekrin, Novartis, Takeda, and GlaxoSmithKline. His institution has received research funding from Accumetrics, AstraZeneca, and Merck. He has received honoraria from Bristol-Myers Squibb and Sanofi-Aventis, and reimbursement of costs for attending scientific meetings from AstraZeneca, GlaxoSmithKline, and Merck. He is a member of Data Safety Monitoring Boards for GlaxoSmithKline and Merck. He has received consultancy payments from, and holds an equity interest in, Automedics Medical Systems. He has received payment for expert testimony from the University of Michigan. Michael Criqui has received honoraria and reimbursement of costs for attending scientific meetings from GlaxoSmithKline. Mary Cushman's institution has received research funding from GlaxoSmithKline. She has received honoraria from GlaxoSmithKline and the US National Institutes of Health, and reimbursement of costs for attending scientific meetings from GlaxoSmithKline. Albert Hofman declares that he has no conflicts of interest. Chris Packard's institution has received research funding from GlaxoSmithKline and he has received honoraria and reimbursement of costs for attending scientific meetings from GlaxoSmithKline. Simon G Thompson has received honoraria and reimbursement of costs for attending scientific meetings from GlaxoSmithKline. Rory Collins is paid by the British Heart Foundation, National Health Service, and UK Biobank, and has received research funding and reimbursement of costs for attending scientific meetings (but no honoraria or consultancy payments) from AstraZeneca, Bayer, Bristol-Myers Squibb, British Heart Foundation, Cancer Research UK, European Union, GlaxoSmithKline, Kadoorie Trust, Medical Research Council, Merck, Roche, Sanofi, Schering, Solvay, and UK Biobank. John Danesh has received research funding from the British Heart Foundation, BUPA Foundation, Denka, diaDexus, European Union, Evelyn Trust, Fogarty International Centre, GlaxoSmithKline, Medical Research Council, Merck, National Heart, Lung, and Blood Institute, National Institute of Neurological Disorders and Stroke, Novartis, Pfizer, Roche, the Wellcome Trust, and UK Biobank. He has also received honoraria and reimbursement of costs for speaking at scientific meetings from GlaxoSmithKline and Novartis. He is a member of the Merck Sharp and Dohme UK Atherosclerosis Advisory Board and Novartis Cardiovascular and Metabolic Advisory Board.
Web Extra Material
References
- 1.Kolodgie FD, Burke AP, Skorija KS. Lipoprotein-associated phospholipase A2 protein expression in the natural progression of human coronary atherosclerosis. Arterioscler Thromb Vasc Biol. 2006;26:2523–2529. doi: 10.1161/01.ATV.0000244681.72738.bc. [DOI] [PubMed] [Google Scholar]
- 2.Mannheim D, Herrmann J, Versari D. Enhanced expression of Lp-PLA2 and lysophosphatidylcholine in symptomatic carotid atherosclerotic plaques. Stroke. 2008;39:1448–1455. doi: 10.1161/STROKEAHA.107.503193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Stafforini DM, Tjoelker LW, McCormick SP. Molecular basis of the interaction between plasma platelet-activating factor acetylhydrolase and low density lipoprotein. J Biol Chem. 1999;274:7018–7024. doi: 10.1074/jbc.274.11.7018. [DOI] [PubMed] [Google Scholar]
- 4.Rosenson RS, Hislop C, McConnell D, for the PLASMA Investigators Effects of 1-H-indole-3-glyoxamide (A-002) on concentration of secretory phospholipase A2 (PLASMA study): a phase II double-blind, randomised, placebo-controlled trial. Lancet. 2009;373:649–658. doi: 10.1016/S0140-6736(09)60403-7. [DOI] [PubMed] [Google Scholar]
- 5.Corson MA. Phospholipase A2 inhibitors in atherosclerosis: the race is on. Lancet. 2009;373:608–610. doi: 10.1016/S0140-6736(09)60378-0. [DOI] [PubMed] [Google Scholar]
- 6.Karabina SA, Elisaf M, Bairaktari E, Tzallas C, Siamopoulos KC, Tselepis AD. Increased activity of platelet-activating factor acetylhydrolase in low-density lipoprotein subfractions induces enhanced lysophosphatidylcholine production during oxidation in patients with heterozygous familial hypercholesterolaemia. Eur J Clin Invest. 1997;27:595–602. doi: 10.1046/j.1365-2362.1997.1570706.x. [DOI] [PubMed] [Google Scholar]
- 7.Macphee CH, Moores KE, Boyd HF. Lipoprotein-associated phospholipase A2, platelet-activating factor acetylhydrolase, generates two bioactive products during the oxidation of low-density lipoprotein: use of a novel inhibitor. Biochem J. 1999;338:479–487. [PMC free article] [PubMed] [Google Scholar]
- 8.Zalewski A, Macphee C. Role of lipoprotein-associated phospholipase A2 in atherosclerosis: biology, epidemiology, and possible therapeutic target. Arterioscler Thromb Vasc Biol. 2005;25:923–931. doi: 10.1161/01.ATV.0000160551.21962.a7. [DOI] [PubMed] [Google Scholar]
- 9.Caslake MJ, Packard CJ. Lipoprotein-associated phospholipase A2 (platelet-activating factor acetylhydrolase) and cardiovascular disease. Curr Opin Lipidol. 2003;14:347–352. doi: 10.1097/00041433-200308000-00002. [DOI] [PubMed] [Google Scholar]
- 10.Packard CJ, O'Reilly DS, Caslake MJ. Lipoprotein-associated phospholipase A2 as an independent predictor of coronary heart disease. West of Scotland Coronary Prevention Study Group. N Engl J Med. 2000;343:1148–1155. doi: 10.1056/NEJM200010193431603. [DOI] [PubMed] [Google Scholar]
- 11.Garza CA, Montori VM, McConnell JP, Somers VK, Kullo IJ, Lopez-Jimenez F. Association between lipoprotein-associated phospholipase A2 and cardiovascular disease: a systematic review. Mayo Clin Proc. 2007;82:159–165. doi: 10.4065/82.2.159. [DOI] [PubMed] [Google Scholar]
- 12.The Lp-PLA2 Studies Collaboration Collaborative meta-analysis of individual participant data from observational studies of Lp-PLA2 and cardiovascular diseases. Eur J Cardiovasc Prev Rehabil. 2007;14:3–11. doi: 10.1097/01.hjr.0000239464.18509.f1. [DOI] [PubMed] [Google Scholar]
- 13.Robins SJ, Collins D, Nelson JJ, Bloomfield HE, Asztalos BF. Cardiovascular events with increased lipoprotein-associated phospholipase A2 and low high-density lipoprotein-cholesterol: the Veterans Affairs HDL Intervention Trial. Arterioscler Thromb Vasc Biol. 2008;28:1172–1178. doi: 10.1161/ATVBAHA.107.160739. [DOI] [PubMed] [Google Scholar]
- 14.Jackson LA. Description and status of the azithromycin and coronary events study (ACES) J Infect Dis. 2000;181:S579–S581. doi: 10.1086/315628. [DOI] [PubMed] [Google Scholar]
- 15.Tunstall-Pedoe H, Kuulasmaa K, Amouyel P, Arveiler D, Rajakangas AM, Pajak A. Myocardial infarction and coronary deaths in the World Health Organization MONICA Project. Registration procedures, event rates, and case-fatality rates in 38 populations from 21 countries in four continents. Circulation. 1994;90:583–612. doi: 10.1161/01.cir.90.1.583. [DOI] [PubMed] [Google Scholar]
- 16.The Fibrinogen Studies Collaboration Associations of plasma fibrinogen levels with established cardiovascular disease risk factors, inflammatory markers, and other characteristics: individual participant meta-analysis of 154,211 adults in 31 prospective studies: the fibrinogen studies collaboration. Am J Epidemiol. 2007;166:867–879. doi: 10.1093/aje/kwm191. [DOI] [PubMed] [Google Scholar]
- 17.Easton DF, Peto J, Babiker AG. Floating absolute risk: an alternative to relative risk in survival and case-control analysis avoiding an arbitrary reference group. Stat Med. 1991;10:1025–1035. doi: 10.1002/sim.4780100703. [DOI] [PubMed] [Google Scholar]
- 18.Higgins JP, Thompson SG. Quantifying heterogeneity in a meta-analysis. Stat Med. 2002;21:1539–1558. doi: 10.1002/sim.1186. [DOI] [PubMed] [Google Scholar]
- 19.The Fibrinogen Studies Collaboration Measures to assess the prognostic ability of the stratified Cox proportional hazards model. Stat Med. 2009;28:389–411. doi: 10.1002/sim.3378. [DOI] [PubMed] [Google Scholar]
- 20.The Fibrinogen Studies Collaboration Regression dilution methods for meta-analysis: assessing long-term variability in plasma fibrinogen among 27,247 adults in 15 prospective studies. Int J Epidemiol. 2006;35:1570–1578. doi: 10.1093/ije/dyl233. [DOI] [PubMed] [Google Scholar]
- 21.Caslake MJ, Packard CJ, Suckling KE, Holmes SD, Chamberlain P, Macphee CH. Lipoprotein-associated phospholipase A2, platelet-activating factor acetylhydrolase: a potential new risk factor for coronary artery disease. Atherosclerosis. 2000;150:413–419. doi: 10.1016/s0021-9150(99)00406-2. [DOI] [PubMed] [Google Scholar]
- 22.Gazi I, Lourida ES, Filippatos T, Tsimihodimos V, Elisaf M, Tselepis AD. Lipoprotein-associated phospholipase A2 activity is a marker of small, dense LDL particles in human plasma. Clin Chem. 2005;51:2264–2273. doi: 10.1373/clinchem.2005.058404. [DOI] [PubMed] [Google Scholar]
- 23.Clarke R, Shipley M, Lewington S. Underestimation of risk associations due to regression dilution in long-term follow-up of prospective studies. Am J Epidemiol. 1999;150:341–353. doi: 10.1093/oxfordjournals.aje.a010013. [DOI] [PubMed] [Google Scholar]
- 24.Davidson MH, Corson MA, Alberts MJ. Consensus panel recommendation for incorporating lipoprotein-associated phospholipase A2 testing into cardiovascular disease risk assessment guidelines. Am J Cardiol. 2008;101:51F–57F. doi: 10.1016/j.amjcard.2008.04.019. [DOI] [PubMed] [Google Scholar]
- 25.The Fibrinogen Studies Collaboration Plasma fibrinogen level and the risk of major cardiovascular diseases and nonvascular mortality: an individual participant meta-analysis. JAMA. 2005;294:1799–1809. doi: 10.1001/jama.294.14.1799. [DOI] [PubMed] [Google Scholar]
- 26.The Emerging Risk Factors Collaboration C-reactive protein concentration and risk of coronary heart disease, stroke, and mortality: an individual participant meta-analysis. Lancet. 2010;375:132–140. doi: 10.1016/S0140-6736(09)61717-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.The Emerging Risk Factors Collaboration Major lipids, apolipoproteins, and risk of vascular disease. JAMA. 2009;302:1993–2000. doi: 10.1001/jama.2009.1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Manolio TA, Pearson TA, Wenger NK, Barrett-Connor E, Payne GH, Harlan WR. Cholesterol and heart disease in older persons and women. Review of an NHLBI workshop. Ann Epidemiol. 1992;2:161–176. doi: 10.1016/1047-2797(92)90051-q. [DOI] [PubMed] [Google Scholar]
- 29.Prospective Studies Collaboration Blood cholesterol and vascular mortality by age, sex, and blood pressure: a meta-analysis of individual data from 61 prospective studies with 55 000 vascular deaths. Lancet. 2007;370:1829–1839. doi: 10.1016/S0140-6736(07)61778-4. [DOI] [PubMed] [Google Scholar]
- 30.Stafforini DM, Satoh K, Atkinson DL. Platelet-activating factor acetylhydrolase deficiency. A missense mutation near the active site of an anti-inflammatory phospholipase. J Clin Invest. 1996;97:2784–2791. doi: 10.1172/JCI118733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yamada Y, Matsuo H, Segawa T. Assessment of genetic risk for myocardial infarction. Thromb Haemost. 2006;96:220–227. [PubMed] [Google Scholar]
- 32.Hou L, Chen S, Yu H. Associations of PLA2G7 gene polymorphisms with plasma lipoprotein-associated phospholipase A2 activity and coronary heart disease in a Chinese Han population: the Beijing atherosclerosis study. Hum Genet. 2009;125:11–20. doi: 10.1007/s00439-008-0587-4. [DOI] [PubMed] [Google Scholar]
- 33.Hoffmann MM, Winkler K, Renner W. Genetic variants and haplotypes of lipoprotein associated phospholipase A2 and their influence on cardiovascular disease (The Ludwigshafen Risk and Cardiovascular Health Study) J Thromb Haemost. 2009;7:41–48. doi: 10.1111/j.1538-7836.2008.03216.x. [DOI] [PubMed] [Google Scholar]
- 34.Mohler ER, 3rd, Ballantyne CM, Davidson MH. The effect of darapladib on plasma lipoprotein-associated phospholipase A2 activity and cardiovascular biomarkers in patients with stable coronary heart disease or coronary heart disease risk equivalent: the results of a multicenter, randomized, double-blind, placebo-controlled study. J Am Coll Cardiol. 2008;51:1632–1641. doi: 10.1016/j.jacc.2007.11.079. [DOI] [PubMed] [Google Scholar]
- 35.Wilensky RL, Shi Y, Mohler ER., 3rd Inhibition of lipoprotein-associated phospholipase A2 reduces complex coronary atherosclerotic plaque development. Nat Med. 2008;14:1059–1066. doi: 10.1038/nm.1870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.The Stabilization of Atherosclerotic Plaque by Initiation of Darapladib Therapy Trial (STABILITY) http://clinicaltrials.gov/ct2/show/NCT00799903 (accessed April 6, 2010).
- 37.The Stabilization Of pLaques usIng Darapladib-Thrombolysis In Myocardial Infarction 52 Trial (SOLID-TIMI 52) http://clinicaltrials.gov/ct2/show/NCT0100072 (accessed April 6, 2010).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.