

Abstract
Cystic Fibrosis (CF) is the most common fatal inherited disease of Caucasians, affecting about 1 in 3000 births. Patients with CF have a recessive mutation in the gene encoding the CF transmembrane conductance regulator (CFTR). CFTR is expressed in the epithelium of many organs throughout the exocrine system, however, inflammation and damage of the airways as a result of persistent progressive endobronchial infection is a central feature of CF. The inflammatory response to infection brings about a sustained recruitment of neutrophils to the site of infection. These neutrophils release various pro-inflammatory compounds including proteases, which when expressed at aberrant levels can overcome the endogenous antiprotease defence mechanisms of the lung. Unregulated, these proteases can exacerbate inflammation and result in the degradation of structural proteins and tissue damage leading to bronchiectasis and loss of respiratory function. Other host-derived and bacterial proteases may also contribute to the inflammation and lung destruction observed in the CF lung. Antiprotease strategies to dampen the excessive inflammatory response and concomitant damage to the airways remains an attractive therapeutic option for CF patients.
Keywords: Neutrophil, macrophage, bacteria, proteases, protease inhibitors.
INTRODUCTION
Cystic Fibrosis (CF) lung disease is characterised by progressive chronic infection and inflammation of the airways. The lungs of patients with CF contain elevated levels of neutrophils and proinflammatory cytokines such as tumour necrosis factor-α (TNF-α), interleukin (IL)-6 and IL-8, as well as reduced levels of the anti-inflammatory cytokine IL-10 [1, 2]. This inflammatory response is largely orchestrated by the respiratory epithelium. These cells are part of the first line of defence against pathogens and are responsible for the release of several pro-inflammatory mediators such as IL-8, which acts as a neutrophil chemoattractant [3]. Once recruited to the airways, the neutrophils are primed and activated releasing various compounds such as proteases, oxidants, DNA, and cationic proteins in order to clear the bacteria [4]. When released in excessive amounts, these molecules can cause damage to the respiratory epithelium resulting in yet more pro-inflammatory cytokine release [5-7]. In particular, serine proteases such as neutrophil elastase (NE) enhance the inflammatory response when they are released at the epithelial surface. This results in increased IL-8 expression and further neutrophil influx thereby creating a cycle of inflammation. This cycle of airway inflammation, in conjunction with reduced mucus clearance, is responsible for the mucopurulent plugging and bronchiectasis commonly seen in the diseased CF lung [8].
In order to engage in bacterial phagocytosis neutrophils have to be primed, and in the case of CF, TNF-α and IL-8 are known to play a key role in this activation [9]. When neutrophils are activated in this fashion they release more NE than neutrophils from healthy controls [9]. In addition, these primed and activated CF neutrophils are resistant to the anti-inflammatory affect of IL-10. Once neutrophils have migrated to the site of infection and are primed and activated, they can begin the process of bacterial phagocytosis. The environment of the CF lung however provides several challenges to efficient bacterial phagocytosis. The acidic environment of the CF airway surface due to non-functioning CFTR can result in neutrophil necrosis being favoured over neutrophil apoptosis following bacterial phagocytosis, ultimately resulting in greater damage to the lung [10]. In addition, the hyperviscous mucus found in CF airways severely hampers efficient neutrophil phagocytosis and is thought to increase the incidence of infection by organisms such as by Pseudomonas aeruginosa, Staphylococcus aureus, and Haemophilus influenzae. The predominant organism to colonise the CF lung in the early stages is S. aureus. Although it responds well to antibiotic therapy, S. aureus is thought to leave the lung vulnerable to colonisation by pseudomonads [11]. Unlike S. aureus, P. aeruginosa has a greater potential to resist antibiotic therapies and over time to become the predominant organism in the CF infected lung [11]. When infection with P. aeruginosa in the CF lung becomes persistent the local density of the population increases resulting in the release of quorum-sensing compounds such as N-acyl homoserine lactones. This quorum-sensing leads to a coordinated switch from nonmucoid to mucoid alginate producing P. aeruginosa [12]. The resulting biofilm formation results in the colonising P. aeruginosa becoming even less susceptible to antibiotic therapies and neutrophil clearance. Excessive accumulation of neutrophils at the site of biofilm formation may also result in self damage via release of oxidants and proteolytic enzymes [13].
The main source of protease activity in the CF lung is thought to be activated neutrophils, however, the role of proteases derived from other cell sources, such as mononuclear and epithelial cells, as well as exogenously derived bacterial proteases may also play a vital role in mediating destruction of the lung as seen in CF (as illustrated in Fig. 1). The main emphasis of this review will be to address the role of these endogenous and exogenous proteases and describe past, present and future developments in the search for an efficient antiprotease therapy in CF.
Fig. (1).
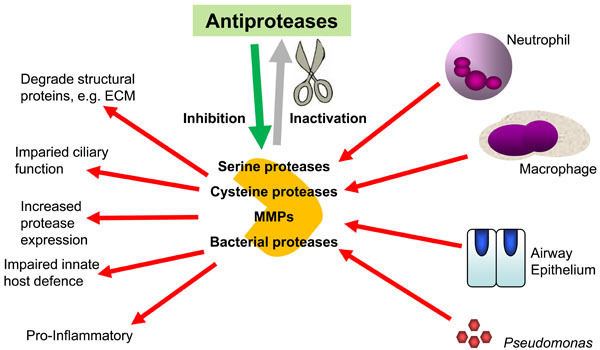
The importance of the balance between proteases and antiproteases in CF airways. The overwhelming of the airway innate antiprotease shield by excessive and dysregulated secretion of host and bacterial proteases in the CF lung leads to exacerbation of the inflammatory response and lung damage.
SERINE PROTEASES
Neutrophil Elastase (NE)
Neutrophils are responsible for the release of proteolytic enzymes such as matrix metalloproteases (MMPs) and the serine protease NE, all of which are capable of destroying the extracellular matrix (ECM). NE plays a key role in the pathophysiology of CF making it one of the major determinants of the pulmonary inflammatory phenotype. NE is a 29 kDa serine protease produced in the neutrophil and stored in primary azurophilic granules for secretion upon activation, phagocytosis and cell death. Although the normal role of NE is to degrade phagocytosed proteins, if it is released from the neutrophil and breaches the lungs antiprotease defences it has the potential to degrade most of the structural proteins of the lung including elastin, fibronectin and other proteins of the ECM [14]. The cleavage of cell surface fibronectin in particular allows improved adherence of P. aeruginosa to the cell facilitating the colonisation of the CF lung. Degradation of opsonic proteins and C3b receptors on the surface of neutrophils has also been shown to significantly reduce the efficacy of antibody mediated phagocytosis [15, 16]. Elevated levels of NE in the CF lung can prevent resolution of inflammation which normally occurs by the removal of apoptotic neutrophils from the airway by macrophages [17].
In the presence of NE, there is reduced apoptotic cell removal as a consequence of NE cleavage of the phosphatidylserine receptor, CD36, and various other apoptotic cell recognition receptors on macrophages [18]. NE can also prolong the inflammatory response by degrading complement resulting in the release of C5a, which acts as a chemoattractant for neutrophils [15]. Increased neutrophil recruitment can also be brought about by the direct effect of NE on the epithelium resulting in the release of IL-8 [19]. The accumulation of neutrophils in the airway can also result in elevated oxygen radical production, airway cell death, and increased leukotriene B4 leading to even greater neutrophil recruitment. NE can also cause a reduction in the ciliary beat frequency of the respiratory epithelium and an increase in serous cell mucus production, which hinder bacterial clearing from the lung [20]. NE is known to exacerbate lung inflammation and injury via its interplay with the potentially damaging MMPs. MMP-9 can be directly activated by NE or indirectly by inactivation of tissue inhibitor metallopeptidase-1 (TIMP-1) the endogenous inhibitor of MMP-9 [21, 22]. The activation of MMP-2 by NE is also possible via a mechanism that requires expression of MMP-14 [23].
Cathepsin G
Cathepsin G is a chymotrypsin-like serine protease stored in primary azurophilic granules within neutrophils and is secreted upon activation. It possesses broad-range specificity for substrates and is capable of degrading the structural proteins of the ECM. It also plays a role in the inhibition of apoptotic cell clearance from CF airways via the removal of cell receptors from the surface of macrophages [24]. Furthermore, cathepsin G along with NE can activate the protease-activated receptor (PAR) family, in particular the PAR-2 receptor. However, cathepsin G and NE can also inactivate PAR-2 preventing its activation by trypsin [25]. Surfactant protein A (SP-A), a pattern recognition molecule that facilitates uptake of microbes by macrophages and neutrophils was shown to be degraded by protease-containing bronchoalveolar lavage fluid (BALF) from CF patients and this degradation could be abrogated by the addition of serine protease inhibitors [26]. The degradation was directly attributed to cathepsin G, NE and proteinase-3 although cathepsin G demonstrated the highest potency against SP-A [26].
Proteinase 3
Proteinase 3, a serine protease secreted by activated neutrophils, also plays a role in the inflammatory response. Proteinase 3 can cleave the mature form of IL-8 to a more active truncated form [27, 28]. This truncated form of IL-8 has a 20-fold increased ability to act as a chemoattractant compared to wild type [27, 28] and also induces increased degranulation and intracellular calcium mobilisation in neutrophils and enhances interaction with the CXC chemokine receptor 1 [28]. Active proteinase 3 is present in CF sputum and its concentration was found to be highly correlated with the concentration of taurine present, which is a reliable marker of airway inflammation and respiratory scores [29]. It is postulated that the unregulated deleterious activities of proteinase 3 could be prevented by treatment with the serine protease inhibitor alpha-1-antitrypsin (AAT) [30].
Endogenous Serine Antiproteases
The release of proteolytic enzymes during the inflammatory response has enormous potential to exacerbate and prolong inflammation causing numerous deleterious effects such as lung tissue destruction and reduced bacterial clearance. To counteract this, the airways are equipped with a highly regulated antiprotease shield to dampen and control excessive proteolytic activity. The protease-antiprotease imbalance hypothesis of chronic neutrophil-mediated lung disease contends that this imbalance, or overwhelming of the natural antiprotease defence mechanisms, determines pulmonary phenotype [31]. Several CF studies have correlated protease lung burden with disease severity and have shown an inverse relationship to the status of the antiprotease defence shield [7, 32-34]. The function of the airway antiprotease defence system is to inhibit the activity of cognate proteases thereby preventing potentially damaging degradation of host tissue (Fig. 1). The primary antiproteases of the airway are AAT, secretory leucoprotease inhibitor (SLPI), elafin, TIMPs and cystatins [31, 35-38]. The primary protease targets of AAT, SLPI and elafin are the serine proteases, NE, proteinase 3 and cathepsin G, all of which are released by activated neutrophils [30, 31, 35, 36].
Alpha-1 Antitrypsin (AAT)
The AAT gene is located on chromosome 14 (q31-32.3), spanning 12.2 kb and consisting of seven exons and six introns [39]. The coding exons (II-V) follow three exons (Ia, Ib, Ic), which code for the untranslated region of the AAT gene [40, 41]. The gene is translated into a 418 amino acid protein incorporating a 24 amino acid signal peptide. Glycosylation of AAT occurs at three asparagine (ASN) sites - ASN 46, 83 and 247 - in the endoplasmic reticulum and the protein is packaged in the Golgi apparatus prior to release. The final 52 kDa protein is produced primarily in hepatocytes but has also been shown to be expressed in epithelial cells, macrophages and neutrophils [42-44]. Due to the large number of AAT mRNA transcripts present in the hepatocyte, it is believed that serum AAT is largely derived from the liver from where it diffuses throughout the body and into the lung. In addition to its role in regulating serine protease activity, AAT is also known to be involved in regulating the inflammatory response via several mechanisms unrelated to its antiprotease activity. The increase in cAMP levels brought about by AAT activation of adenylate cyclase and classic downstream activation of cyclic AMP-dependent protein kinase (PKA) by cAMP can inhibit endotoxin-stimulated TNF-α and IL-10 production in human monocytes [45].
AAT is a member of the serpin family of serine protease inhibitors [46]. The tertiary structure of members of this family is similar, as is the exposed reactive centre loop containing the inhibitory active site sequence [47]. AAT inhibits many proteases including trypsin, plasmin, thrombin, factor X and cathepsin G [41]. However, the main cognate protease of AAT is NE. When NE binds to the active site of AAT, the Met358-Ser359 bond, cleavage of this Met-Ser bond results in the generation of a tight, non-covalent interaction between NE and AAT thereby inhibiting NE activity and causing release of the 36-amino acid C-terminal Ser359-Lys394 peptide [48]. The oxidation susceptibility of AAT renders it less active against NE. The association rate constant of AAT drops from 6 x 10-7 M-1s-1 to 3 x 10-4 M-1s-1, a drop of almost 2000-fold [49]. The molecular basis of this oxidation was initially shown to be oxidation of the active site Met358 residue [50]. However, using recombinant-derived AAT, it has since been demonstrated that Met351, another active site residue, is also involved in the AAT-NE interaction [51].
There are nine methionine residues present in the AAT sequence, however, only Met 351 and 358 are fully surface exposed and thereby bind to NE. In the case of porcine pancreatic elastase (PPE), only Met358 appears to be important for binding to, and inhibiting, this protease [52]. The differences in binding of oxidised AAT to NE and PPE can most likely be explained by the previous observation that oxidised AAT is a much less potent inhibitor of NE although it will still bind and inhibit NE albeit at a much slower rate. However, oxidised AAT does not inhibit PPE indicating that the inhibitory mechanisms of AAT for PPE and NE are different [49, 53].
Secretory Leucoprotease Inhibitor (SLPI)
SLPI was initially identified as a serine protease inhibitor present in a number of bodily secretions including nasal, bronchial, salivary, tear and seminal secretions [54-57]. The SLPI gene is 2.65 kb in length and is composed of four exons and three introns [58]. The mature protein is 11.7 kDa in size, consisting of 107 amino acids and comprising two domains [59]. Each domain is homologous to the whey acid protein (WAP) motif, a protein sequence composed of 4 disulfide bridges and approximately 50 amino acids in length [60]. SLPI and elafin are the best characterised of the WAP family of proteins, which currently stands at 15 members, and are situated in a tight cluster on chromosome 20q12-13 [60]. Readers are directed to many excellent reviews on the structural and multifunctional activities of these proteins [61-65]. Expression sites for SLPI are found primarily in mucosal tissue but it can also be produced by inflammatory cells including macrophages and neutrophils [66, 67]. SLPI has been shown to inhibit NE, cathepsin G, chymotrypsin and trypsin [59]. Interestingly, SLPI does not inhibit the other neutrophil serine protease, proteinase 3, in contrast to AAT and elafin [68]. Due to its high association rate constant for NE, SLPI is considered to be the major inhibitor of NE in the upper respiratory tract.
Recent research has shown that SLPI possesses antibacterial properties, which may be important in front-line defence of the upper airways. In addition to its antiprotease and antibacterial properties, it has been shown that SLPI also possesses anti-inflammatory activity [61-66, 69]. In response to LPS, SLPI deficient mice show increased mortality from endotoxin shock [70]. Furthermore, in a mouse model of acute lung injury, prior administration of SLPI decreased lung injury and down-regulated nuclear factor (NF)-κB activation by preventing degradation of the NF-κB inhibitor protein, IκBβ [71, 72]. It has also been shown that SLPI is capable of entering cells and localising in both the cytoplasm and nucleus, where it binds directly to NF-κB binding sites in a site-specific fashion [73]. However the anti-inflammatory and antiprotease properties of SLPI have been found to be greatly reduced when SLPI is oxidised leaving free NE capable of causing lung tissue injury and inflammation [74, 75].
The crystallographic structure of SLPI reveals a boomerang-like shape with an N-terminal domain of residues 1-54 and a C-terminal domain composed of residues 55-107 (Table 1) [76]. The antiprotease active site of SLPI has been located on a loop (residues 67-74) on the C-terminal domain containing the scissile bond Leu72-Met73. As with AAT, the association rate constant of SLPI for NE is very high (10-7 M-1s-1) and the rate of dissociation is low and contains an active site methionine residue (Met73), which is susceptible to oxidative inactivation, rendering it a less effective antiprotease [77]. It has also been shown that the Thr67-Tyr68 bond of the active site loop is susceptible to cleavage by members of the elastolytic cathepsin family, cathepsin B, L and S [78]. Recent studies strongly suggest that cathepsins play a putative role in the cleavage of SLPI in emphysema resulting in decreased SLPI levels and activity [78]. In studies of individuals with community-acquired pneumonia, SLPI levels were found to be increased in the infected and uninvolved lobes, in comparison to lobes in healthy controls [74]. The overall anti-NE activity in infected BALF was low consistent with evidence of SLPI cleavage in infected BALF samples [74]. These studies show that while SLPI levels can increase in certain disease states in response to infection, activity can be compromised by proteolytic cleavage.
Table 1.
Comparison of the Structure and Function of the Serine Protease Inhibitors SLPI and Elafin, Two Closely Related Members of the WAP Family of Proteins
| Structure/Function | SLPI | Elafin |
|---|---|---|
| Size | 11.7 kDa (117 amino acids) | 6 kDa (57 amino acids) |
| WAP domains | 2 | 1 |
| pI | 9.5 | 9.7 |
| Anti-protease activities | Neutrophil elastase, cathepsin G, chymase, trypsin, chymotrypsin | Neutrophil elastase, proteinase 3 |
| Anti-bacterial | E. coli, P. aeruginosa, S. aureus | P. aeruginosa, S. aureus |
| Anti-inflammatory | Inhibits NF-κB activation and cytokine production | Inhibits NF-κB and AP-1 activation and cytokine production |
Elafin
The mature form of elafin is a 6 kDa protein possessing a WAP domain homologous to the WAP domains present in the SLPI protein, and is found in bronchial secretions and in the skin [35, 36, 79, 80] (Table 1). Elafin is expressed by many cell types present in the lung but also by endothelium and alveolar macrophages [36, 81, 82]. The elafin gene is 2.3 kb in length, composed of three exons and two introns, and possesses transcription factor binding sites for AP-1 and NF-κB [36, 83, 84]. Elafin is expressed as a 117 amino acid protein, which includes a 22-residue signal peptide [36]. Both TNF-α and IL-1 have been demonstrated to induce elafin expression in pulmonary epithelial cells although LPS does not [84]. The primary function of elafin is the inhibition of the neutrophil serine proteases NE and proteinase 3 but not cathepsin G [35, 85]. In contrast, SLPI inhibits cathepsin G but not proteinase 3 [68].
As with SLPI, elafin is a potent inhibitor of NE with an association rate constant of 3.6 x 10-6 M-1s-1. Other activities of elafin have been described. For example, overexpression of elafin improves acute lung injury induced by P. aeruginosa in mice although it is not clear if this affect is dependent on the antiprotease activity of elafin or an antibacterial/anti-inflammatory affect [86]. Recently, it has been demonstrated that pre-elafin can protect against LPS-induced lung inflammation in mice by decreasing neutrophil influx into the lung, decreasing gelatinase activity and reducing levels and expression of a number of pro-inflammatory mediators [87]. A study looking at the status of elafin in CF sputum found that elafin was cleaved in sputum from individuals with CF [88]. P. aeruginosa-positive sputum, which contained lower levels of elafin and higher levels of NE, was found to be particularly effective at cleaving recombinant elafin [88]. In vitro incubation of recombinant elafin with excess NE resulted in rapid cleavage of elafin at two cleavage sites at the N-terminal end of elafin [88]. These cleavage products were still able to inhibit NE but they had diminished ability to bind LPS and were less readily immobilised by transglutamination [88].
THERAPEUTIC SERINE ANTIPROTEASE APPROACHES
A large variety of lung diseases are characterised by the presence of multiple unopposed protease activities (serine protease, MMPs and cathepsins). Lung tissue is susceptible to the direct effect of proteolysis in addition to its effect on other soluble host defence proteins present on the respiratory surface. As lung tissue is susceptible to the direct and indirect effects of these proteases it would seem reasonable to hypothesize that antiprotease therapy directed against these activities would be of benefit. Small synthetic inhibitors based on the active sites of many of the naturally occurring antiproteases are currently being tested in animal models [89-92]. However, some of these inhibitors possess certain drawbacks - decreased half-life, increased toxicity and the induction of an immune response - that may limit their use. In addition, many of these inhibitors have not yet been tested in humans. Much of the clinical work in this area has centred on the use of recombinant and plasma-purified versions of the naturally-occurring antiproteases, particularly AAT and SLPI.
Therapeutic Application of AAT
AAT augmentation therapy with plasma-purified AAT has been available for the treatment of AAT deficiency since 1987. Intravenous administration of plasma-purified AAT augments serum and lung levels of AAT and increases anti-elastase capacity in individuals deficient in AAT [93, 94]. A study into the clinical application of AAT in CF demonstrated a fall in NE activity brought about by AAT augmentation therapy, however bacterial loading in the sputum of CF patients was unaltered [95]. A further double-blinded, randomised, placebo-controlled trial demonstrated that aerosolised recombinant AAT (rAAT) was safe and well tolerated by CF patients [96]. However, no significant difference between rAAT and placebo for NE sputum activity was observed, though some secondary efficacy variables such as myeloperoxidase levels were improved [96]. A trial determining the effects of AAT aerosol therapy on airway inflammation in CF also observed no change in sputum NE activity. However levels of sputum taurine - which is known to correlate with CF respiratory exacerbations - did show a progressive decrease during the therapy, suggesting that even in the absence of elastase inhibition, AAT aerosol activity may have a beneficial effect on airway inflammation [97].
A randomised study of CF patients receiving a daily deposition of AAT targeting either the peripheral or bronchial compartment demonstrated increased levels of AAT and decreased levels of NE activity and other endpoints such as neutrophil infiltration, pro-inflammatory cytokines and P. aeruginosa numbers in the lung [98]. Despite the anticipation that peripheral AAT deposition might be superior to bronchial deposition, no significant difference was seen between the two approaches in regards to NE activity and several other parameters [98]. A recent study has looked to improve the efficacy of AAT by addressing some of the key factors of aerosol administration such as reproducibility of delivered dosage both inter- and intra-patient and that the dose be deposited at therapeutic levels in the correct region of the lung despite differing levels of disease severity [99]. Using individually adapted breathing patterns, where both the airflow rate and inhalation were controlled for each subject, they achieved 70% total lung deposition of the AAT filling dose in all three populations studied (healthy, AAT deficient and CF) [99].
Despite this work, aerosolised rAAT administration remains problematic since delivery to the diseased lung via this method is uneven with ‘less diseased’ sites being favoured over ‘diseased’, also it is deposited on the mucus layer rather than at the cell surface [100]. In addition, administration of rAAT is less effective because of the relatively impermeable nature of the epithelium to macromolecules [101]. To overcome these limitations, a more targeted delivery system would prove advantageous. A recent study has shown that it is possible to effectively target AAT to the apical surface of human tracheal xenografts via a fusion protein with a single-chain Fv directed against the extracellular portion of polymeric immunoglobulin receptor (pIgR) [100]. This targeted approach should be amenable to the use of other antiproteases or anti-inflammatory agents.
Therapeutic Application of SLPI and Elafin
Recombinant SLPI (rSLPI) has also been administered to individuals with CF resulting in decreased active NE and decreased IL-8 levels on the epithelial surface of the lung. The latter effect is partly due to the ability of SLPI (and AAT) to inhibit NE-induced up-regulation of IL-8 by the respiratory epithelium [102-104]. Pharmacokinetics of aerosolised rSLPI show that while SLPI levels and anti-elastase capacity are increased in the epithelial lining fluid of CF patients and healthy controls post-aerosolisation, rSLPI did not accumulate on the respiratory epithelial surface [103]. Although administration of SLPI and AAT may prove useful in neutralising serine protease activity in neutrophil-dominated lung diseases, they may have little effect on the other non-serine proteases present on the epithelial surface. Therefore, a combined antiprotease therapeutic strategy using cystatins and TIMPs as well as serine protease inhibitors, may prove to be a most useful way to combat protease-mediated lung destruction. A double-blind, randomised, placebo-controlled Phase I clinical trial of elafin demonstrated that intravenously applied doses were well tolerated in healthy male subjects. Although elafin is in the development pipeline as a therapeutic, no data from CF-related studies has yet been published to our knowledge [105].
Application of Synthetic Protease Inhibitors
DX-890 (also known as EPI-hNE4) has been developed as a NE inhibitor [106]. However, although postulated to be proteolytically stable, this inhibitor has been shown to be only partially effective in neutralising NE activity in Pseudomonas-infected CF sputum probably due to degradation by pepsin [107]. In addition, it has not been tested against NE at higher molar NE:DX-890 ratios (i.e. 2:1 and higher), which would be more representative of in vivo CF lung conditions. Alternatively, many companies have developed low molecular weight synthetic inhibitors of NE. In addition to GSK’s activity in this area, acyl-enzyme inhibitors (ONO-5046, L-694458 and MR-889) and transition-state inhibitors (ONO-6818, AE-3763 and FK-706), all potent NE inhibitors, have been evaluated in clinical trials by Astra Zeneca, Ono and Fujisawa Pharmaceutical to name only a few. Yet, none of these compounds have shown sufficient promise to make it to the market. For instance, Ono had conducted a phase IIa double blind clinical study (low and high doses of ONO-6818 vs placebo) in patients with COPD in Japan since July, 2001, but it voluntarily suspended the study due to abnormal elevation of liver function test values and deferred the commencement of the U.S. phase II study [108]. Crucially, these synthetic compounds, being mechanism-based inhibitors, do not possess any other additional antibacterial and anti-inflammatory properties such as the endogenous inhibitor SLPI.
OTHER HOST-DERIVED PROTEASES
The airway disease in CF is characterised by chronic infection and an inflammatory response dominated by infiltrating neutrophils and therefore much of the work to date has focused on neutrophil serine proteases as key players in mediating lung damage, and corresponding serine protease inhibitors as potential therapeutics, as discussed previously. Although serine proteases and NE in particular predominate in CF [31], it is nevertheless important to recognise that a broad range of extracellular proteases from a variety of cell types also contribute to pulmonary damage seen in CF. Thus, the role and interplay of other protease families as well as the role of their respective inhibitors when contemplating antiprotease therapy must be considered, areas often neglected in terms of antiprotease therapy in CF. Indeed, it is evident from the published work available that a number of complicated cascades of protease activity exists in CF and other chronic infective lung diseases that may further compound the already arduous task of designing efficient antiprotease therapy. Our understanding of the roles of other protease families such as the cysteine cathepsins and metalloproteases is incomplete and perhaps impaired due to degeneracies that exist in protease function and cell to cell variation in specific roles. Thus, further study into the specific roles played by individual proteases in CF is much needed in order to develop efficient antiprotease therapies for CF.
Cysteine Cathepsins
The term cathepsin is given to a number of proteases from various families including cysteine cathepsins (B, C, H, L, S, K, O, F, X, V and W), aspartic proteases (cathepsins D and E), serine proteases (A and G), and metalloproteases (cathepsin III). For the purpose of this section, the potential of cysteine cathepsin inhibitors as therapeutics in CF will be discussed. Evidence is emerging that various members of this family of lysosomal proteases are over-expressed and secreted in the diseased lung by a variety of lung cells and infiltrating inflammatory cells, comprehensively reviewed in [37, 109, 110]. Cathepsins are synthesised as inactive preproenzymes, and while they are directed towards the lysosomal compartment, some cells can secrete significant amounts of mature and procathepsins [109, 110]. Thus, many cathepsins can degrade components of the ECM intracellularly and extracellularly, and cathepsins K, L, and S are among the most potent elastases known [111].
In the lungs, a number of cathepsins are expressed ubiquitously, e.g. cathepsins B, H and L, whereas others are expressed either in macrophages (cathepsin S) or in bronchial epithelial cells (cathepsin K) and type II pneumocytes (cathepsin H) [112-114]. Most cathepsins are unstable at neutral pH and have only weak activity in such environments and instead work at an acidic pH, with the exception of cathepsins S and C, which retain their activity in neutral pH conditions [115, 116]. The pH of epithelial lining fluid in CF is 5.8, decreasing to as low as 5.3 during infective exacerbations [117]. Thus, the acidic environment in the lungs of patients with CF provides optimal conditions for cathepsin activity and highlights the importance of therapeutic interventions directed at regulating this activity. There is also evidence for the interplay between NE and cathepsins in inflammation whereby NE up-regulates expression of cathepsin B and may play an important role in the activation of this enzyme [118, 119]. We and others have shown previously that activities of cathepsins such as B, L, H and S are increased in lung secretions and serum from patients with CF, compared with those in BALF from healthy individuals [119-122].
NE up-regulates expression of cathepsin B in macrophages and increased cathepsin B levels were observed in wild-type mice compared with NE knockout mice challenged with P. aeruginosa [118]. However, like serine proteases, lysosomal cysteine proteases appear to play an important role in regulating the innate immune response. Cathepsin C (dipeptidyl aminopeptidase 1), which is found highly expressed in the lung, was shown to activate the granule-associated serine proteases NE, cathepsin G, granzymes A and B, and mast cell chymase in vitro and in vivo [123-125]. These findings suggest the existence of yet another protease cascade highlighting the intricate relationship that exists between proteases. Cathepsins have been found to contribute significantly to the destruction of a number of host defence proteins which exhibit antimicrobial, antiprotease and immunomodulatory potential. Johnson et al [126] reported the ability of cathepsin L to inactivate AAT, one of the major serine protease inhibitors present in the lung discussed previously. Furthermore, cleavage and inactivation of SLPI and the antimicrobial human β-defensins 2 and 3 and lactoferrin by cathepsins B, L, and S has also been reported [78, 120, 121]. Taken together, these findings indicate the involvement of cathepsins in the diminution of the lung antiprotease and antimicrobial screen possibly leading to lung destruction and favouring conditions for bacterial infection and colonisation.
The activity of cathepsins is of course regulated tightly at different levels, given their exceptional proteolytic activity and the high concentration of cathepsins (up to 1 mM) in lysosomes. Endogenous inhibitors called cystatins regulate their activity. Cystatins are expressed intracellularly (type 1 cystatins or stefins; cystatins A and B) or extracellularly (type 2 cystatins; cystatins C, D, S, SA, SN, E/M and F) [37, 110]. Serine proteases can cleave and inactivate protease inhibitors such as cystatin C, again highlighting interaction between proteases, because serine protease inactivation of cysteine protease inhibitors increases the likelihood of free cathepsin activity [127].
The presence of secreted cathepsins in the lung may be necessary to regulate the activities of these cationic proteins and peptides; however, this process may become dysregulated in diseases such as CF, thus compromising the antiprotease/anti-inflammatory screen and thereby perpetuating inflammation and infection. Although the presence of elevated cathepsin activity has been reported in CF, little or no data on the levels of corresponding endogenous inhibitors is available. Without similar increases in protease inhibitor concentrations, the tight regulation required to keep these powerful proteases in check may contribute to an overall imbalance between proteases and protease inhibitors. Overall, these findings suggest that dysregulated cysteinyl cathepsin activity may play an important role in the pathogenesis and progression of CF lung disease, providing sound evidence for the need of further research into potential antiprotease therapies directed either against these proteases or via increasing levels of endogenous inhibitors, research areas which to date has been severely neglected. A number of cathepsins have been validated as relevant targets for therapies for a diverse range of diseases including cancer and osteoarthritis, and compounds targeting cathepsins S and K are currently in clinical evaluation, whereas others are in experimental phases [128]. As cysteine cathepsins continue to emerge as important components of CF lung disease, there is potential for agents such as these to be applied to CF research in the future.
MATRIX METALLOPROTEASES (MMPs)
In CF lung disease, another major protease group of interest are the MMPs. To date, 24 mammalian MMPs have been described and there appears to be considerable overlap in the substrates that these enzymes can cleave, particularly among the ECM proteins [129, 130]. MMPs are either secreted as latent pro-enzymes or anchored to the cell surface and MMPs can be classified in part on their substrate specificity and in part on their cellular localisation; the collagenases (MMP-1, -8, -13 and -18), gelatinases (MMP-2 and -9), stromelysins (MMP-3, -10 and -11), membrane-type MMPs (MMP-14-17, MMP-24 and -25), matrilysin (MMP-7 and -26), metalloelastase (MMP-12), but there are a number of MMPs that don’t fit into any of these groups.
MMP synthesis and function are tightly controlled by a family of endogenous inhibitors called TIMPs. To date, 4 have been identified, TIMP-1, -2 and -4, are secreted, while TIMP-3 is anchored in the ECM. Many cell types produce MMPs and TIMPs in the lung, including epithelial (MMP-1, -2, -9, TIMP-1 and -2), smooth muscle cells (MMP-2, -3, -7, MMP-14/MT1-MMP, TIMP-1 and -2), neutrophils (MMP-8, -9 and TIMP-1), alveolar macrophages (MMP-1, -9, -12 and TIMP-1), and mast cells (MMP-9) [38]. Typically, MMPs are not expressed in normal healthy tissues. However, while increased levels of MMP-2, -7, -8 and -9 in CF BALF and serum have been documented, conflicting data on the levels of TIMP-1 in CF lung secretions has been reported [122, 131-135]. As previously mentioned, evidence exists for the interplay between NE and MMPs, as NE activates MMP-9 directly but also indirectly by inactivating TIMP-1, the naturally occurring inhibitor of MMP-9 [22, 136]. Furthermore, NE may activate MMP-2 through a mechanism that requires MMP-14 expression or by directly affecting gene expression [23, 118].
Similar to what has been found with serine and cysteine protease families, MMPs regulate the turnover of the ECM, but also act broadly in inflammation to regulate barrier function, innate immunity, and inflammatory cytokine and chemokine activity [17, 28, 137]. Similar to members of the serine and elastolytic cathepsin families, a number of MMPs are capable of degrading a wide variety of soluble proteins present on the respiratory tract. Serine proteases can cleave and inactivate protease inhibitors such as TIMP-1 increasing the likelihood of free MMP activity [20]. Likewise, a large number of MMPs (MMP-1, -3, -7, -8, -9, and -12) can cleave and inactivate AAT, thereby allowing NE, cathepsin G, and proteinase 3 to remain active [138-142]. In addition, Ramadas et al. [143] recently reported the ability of recombinant MMP-12 to cleave recombinant SLPI, however, no evidence for this event in vivo was reported. In contrast, both SLPI and elafin are resistant to proteolytic inactivation by MMP-2, -8 and -12 [143, 144].
Another related group of proteases that may play a role in CF is the ADAM ("a disintegrin and metalloprotease") family of proteases that includes proteins containing disintegrin-like and metalloprotease-like domains. ADAMs are typically distributed over the bronchial epithelium and in inflammatory cells suggesting a localised function [145]. As most ADAMs are involved in release of inflammatory mediators, growth factors and mucin, they may play an important role in the first line of defence and in initiation of repair events in the airways [145]. However, little or no information exists on the status and integrity of these proteases in the CF lung. Two of the most prominent ADAMs are TNF-α converting enzyme (TACE, ADAM17) and ADAM10. Again, interplay between NE and this family of proteases exists as NE can activate TACE thereby facilitating the induction of MUC5AC mucin expression [146]. Mucus hypersecretion is a prominent manifestation in patients with CF and contributes significantly to the morbidity and mortality of CF patients by plugging airways and causing recurrent infections. MUC1 plays a critical role in protecting mucosal epithelia from microbial and enzymatic attack and MUC1 shedding is controlled by TACE and MMP-14 [147, 148]. MUC1 has also been shown to act as an adhesion site for Pseudomonas, an interaction that is abolished by proteolytic cleavage by NE, suggesting a possible role for this protein in binding and removal of bacteria [149].
PROTEASOME-RELATED PROTEASES
Recently other protease-mediated pathways which are implicated in CF pathogenesis have come to light. The most common disease causing mutation of CF, F508del, causes mutant CFTR to be rapidly destroyed by endoplasmic reticulum (ER)-associated degradation (ERAD), which acts as a quality system for protein folding [150]. The ERAD system is responsible for the ubiquitination and subsequent destruction of misfolded proteins via the ubiquitin-proteasome system. The deletion of a phenyalanine at position 508 of the CFTR results in a temperature-sensitive folding defect and premature degradation via the ERAD system [151]. The proteasome, which is responsible for the proteolytic degradation of ubiquitinated proteins, can be inhibited by the drug bortezomib which is a potent, reversible and selective inhibitor of chymotryptic threonine protease activity [152]. Recent work has shown that bortezomib can rescue CFTR from ERAD, resulting in the appearance of mature CFTR and functional chloride channels [150]. A reduction of IL-8 expression levels via proteasomal inhibition was also demonstrated [150]. However, this favourable anti-inflammatory effect may come at the expense of a reduction in normal proteasomal degradation.
BACTERIAL PROTEASES
The secretion and control of host proteolytic species has been shown to be pivotal in the pathology of the diseased CF lung. Initially bacterial proteases were thought of as merely secreted digestive enzymes for the liberation of nutrients from the host tissue. However, it became evident that bacterial proteolytic enzymes were acting in various ways to disrupt key host processes thereby aiding colonisation of hostile host tissue. They achieve this by several means such as activation of cascade pathways, disruption of cytokine signalling and inactivation of cell receptors and crucially via inactivation of host protease inhibitors [153]. During colonisation of the CF lung bacteria secrete numerous virulence factors including proteases which can regulate the inflammatory process. The opportunistic pathogen P. aeruginosa also produces numerous virulence factors, several of which including the metalloelastase (LasB), are secreted outside the cell [154]. LasB has been shown to dampen the inflammatory response through the alteration of key membrane receptors such as the urokinase-type plasminogen activator receptor (uPAR) [155]. The proteolytic cleavage of uPAR by LasB results in a drastic reduction in the capacity of the cells to bind urokinase and loss of the ability of uPAR to interact with the matrix adhesive protein vitronectin thus altering the function of inflammatory cells during infection. P. aeruginosa LasB is also capable of cleaving similar substrates to those of NE and is known to cause tissue damage in the lung.
Another key virulence factor of P. aeruginosa is alginate exopolysaccaride which aids in the persistence of the organism in the CF lung. Alginate production is switched on primarily in the environment found in the CF lung and is only rarely encountered in other infections [156]. The importance of alginate as a virulence factor is highlighted by the correlation between alginate production and poor prognosis and high mortality of CF patients [11]. It has been shown that LasB is secreted in relatively high amounts by nonmucoid P. aeruginosa and conversely that it is reduced in mucoid alginate producing P. aeruginosa. This is thought to be in response to alginate production which requires intracellular LasB cleavage of nucleoside diphoshate kinase (NdK) from its 16 kDa from to a 12 kDa form for the generation of GTP which itself is required for the synthesis of alginate [154]. A LasB-specific inhibitor could target both the damaging effects of secreted LasB and crucially intracellular LasB thereby inhibiting biofilm formation. Therapies such as this in conjunction with antibiotics could lead to more effective clearance of colonising bacteria from the CF lung. The use of an antiprotease therapy to combat biofilm formation is especially attractive. Antiprotease therapy, unlike traditional antibiotics which target fundamental processes such as cell wall synthesis thereby creating enormous selection pressures, should be well tolerated and less likely to result in the generation of resistant pathogens [153].
CONCLUSIONS
In conclusion, evidence is accumulating to show that each protease family has a multitude of regulatory functions and may, either individually or as a component of one or more protease cascades, be of pivotal importance in CF. However, there is a need for further clarification as to the status, activity and integrity of such proteases and their endogenous inhibitors in CF lung disease. In CF, dysregulated protease activity results in upregulation of proinflammatory mediators, increased recruitment of inflammatory cells to the lung, impaired phagocytosis, increased mucin production, and inactivation of important innate and antimicrobial proteins resulting in sustained inflammation and predisposition to infection. One way to treat such protease-mediated events in chronic infective lung disease is with antiprotease therapy. Developing rational therapies, however, requires further identification of specific protease substrates and characterisation of the downstream consequences of protease activity in CF. In addition the identification and targeting of bacterial proteases involved in chronic infection processes such as the regulation of biofilm production would be a useful addition to CF therapeutic armoury.
Future research will also have to address the reasons for prolonged extracellular protease activity in CF compared with those conditions characterised by a shorter duration of protease activity, e.g. pneumonia. Because of the multiple protease activities present in CF, it may be important to identify definitively whether there is a key protease or proteases central to direct tissue destruction or activation of other proteases in the diseased lung, as suggested by a number of papers. Under these circumstances, neutralisation of one such protease with a specific antiprotease may be sufficient to lessen the overall protease burden in CF without the need for inhibition of all proteases.
ACKNOWLEDGEMENTS
We wish to acknowledge funding from the Northern Ireland Chest Heart and Stroke Association and the Alpha One Foundation.
REFERENCES
- 1.Greally PM, Hussein MJ, Cook AJ, Sampson AP, Piper PJ, Price JF. Sputum tumour necrosis factor-alpha and leukotriene concentrations in cystic fibrosis. Arch Dis Child. 1993;68:389–92. doi: 10.1136/adc.68.3.389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bonfield TL, Panuska JR, Konstan MW, et al. Inflammatory cytokines in cystic fibrosis lungs. Am J Respir Crit Care Med. 1995;152:2111–18. doi: 10.1164/ajrccm.152.6.8520783. [DOI] [PubMed] [Google Scholar]
- 3.Strieter RM. Interleukin-8: a very important chemokine of the human airway epithelium. Am J Physiol. 2002;283:L688–L9. doi: 10.1152/ajplung.00146.2002. [DOI] [PubMed] [Google Scholar]
- 4.Birrer P. Consequences of unbalanced protease in the lung: protease involvement in destruction and local defence mechanisms of the lung. Agents Actions Suppl. 1993;40:3–12. doi: 10.1007/978-3-0348-7385-7_1. [DOI] [PubMed] [Google Scholar]
- 5.Sommerhoff CP, Nadel JA, Basbaum CB, Caughey GH. Neutrophil elastase and cathepsin G stimulate secretion from bovine airway gland serous cells. J Clin Invest. 1990;85:682–89. doi: 10.1172/JCI114492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Breuer R, Christensen TG, Lucey EC, Bolbochan G, Stone PJ, Snider GL. Elastase causes secretory discharge in bronchi of hamsters with elastase-induced secretory cell metaplasia. Exp Lung Res. 1993;19:273–82. doi: 10.3109/01902149309031724. [DOI] [PubMed] [Google Scholar]
- 7.O'Connor CM, Gaffney K, Keane J, et al. α1-Proteinase inhibitor, elastase activity, and lung disease severity in cystic fibrosis. Am Rev Respir Dis. 1993;148:1665–70. doi: 10.1164/ajrccm/148.6_Pt_1.1665. [DOI] [PubMed] [Google Scholar]
- 8.Jacquot J, Tabary O, Le Rouzic P, Clement P. Airway epithelial cell inflammatory signaling in cystic fibrosis. Int J Biochem Cell Biol. 2008;40:1703–15. doi: 10.1016/j.biocel.2008.02.002. [DOI] [PubMed] [Google Scholar]
- 9.Taggart C, Coakely RJ, Greally P, Canny G, O'Neill SJ, McElvaney NG. Increased elastase release by CF neutrophils is mediated by tumor necrosis factor-alpha and interleukin-8. Am J Physiol Lung Cell Mol Physiol. 2000;278:L33–L41. doi: 10.1152/ajplung.2000.278.1.L33. [DOI] [PubMed] [Google Scholar]
- 10.Coakley RJ, Taggart C, McElvaney NG, O'Neill SJ. Cytosolic pH and the inflammatory microenvironment modulate cell death in human neutrophils after phagocytosis. Blood. 2002;100:3383–91. doi: 10.1182/blood.V100.9.3383. [DOI] [PubMed] [Google Scholar]
- 11.May TB, Shinabarger D, Maharaj R, et al. Alginate synthesis by Pseudomonas aeruginosa: a key pathogenic factor in chronic pulmonary infections of cystic fibrosis patients. Clin Microbiol Rev. 1991;4:191–206. doi: 10.1128/cmr.4.2.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Singh PK, Schaefer AL, Paresk MR, Moninger TO, Welsh MJ, Greenberg EP. Quorum sensing signals indicate that cystic fibrosis lungs are infected with bacterial biofilms. Nature. 2000;407:762–4. doi: 10.1038/35037627. [DOI] [PubMed] [Google Scholar]
- 13.Bass DA, DeChatlet LR, Burk RF, Shirley P, Szedja P. Polymorphonuclear leukocyte bactericidal activity and oxidative metabolism during glutathione peroxidase deficiency. Infect Immun. 1977;18:78–84. doi: 10.1128/iai.18.1.78-84.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Doring G. The role of neutrophil elastase in chronic inflammation. Am J Respir Crit Care Med. 1994;150:S114–117. doi: 10.1164/ajrccm/150.6_Pt_2.S114. [DOI] [PubMed] [Google Scholar]
- 15.Fick PA, Robins RA, Squier SU, Schoderbek WE, Russ WD. Complement activation in cystic fibrosis respiratory fluids: in vivo and in vitro generation of C5a and chemotatic activity. Pediatr Res. 1986;20:1258–68. doi: 10.1203/00006450-198612000-00014. [DOI] [PubMed] [Google Scholar]
- 16.Berger M, Soerensen RJ, Tosi MF, Dearborn DG, Doring G. Complement receptor expression on neutrophils at an inflammatory site, the Pseudomonas infected lung in cystic fibrosis. J Clin Invest. 1989;84:1302–13. doi: 10.1172/JCI114298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Taggart CC, Greene CM, Carroll TP, O'Neill SJ, McElvaney NG. Elastolytic proteases: Inflammation resolution and dysregulation in chronic infective lung disease. Am J Respir Crit Care Med. 2005;171:1070–6. doi: 10.1164/rccm.200407-881PP. [DOI] [PubMed] [Google Scholar]
- 18.Vandivier RW, Fadok VA, Hoffmann PR, et al. Elastase-mediated phosphatidylserine receptor cleavage impairs apopotic cell clearance in cystic fibrosis and bronchiectasis. J Clin Invest. 2002;109:661–70. doi: 10.1172/JCI13572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Walsh DE, Greene CM, Carroll TP, et al. Interleukin-8 up-regulation by neutrophil elastase is mediated by MyD88/IRAK/TRAF-6 in human bronchial epithelium. J Biol Chem. 2001;276:35494–9. doi: 10.1074/jbc.M103543200. [DOI] [PubMed] [Google Scholar]
- 20.Fahy JV, Schuster A, Ueki I, Boushey HA, Nadel JA. Mucus hypersecretion in bronchiestasis: the role of neutrophil proteases. Am Rev Respir Dis. 1992;146:1430–3. doi: 10.1164/ajrccm/146.6.1430. [DOI] [PubMed] [Google Scholar]
- 21.Ferry G, Longchampt M, Pennel L, de Nanteuil G, Canet E, Tucker GC. Activation of MMP-9 by neutrophil elastase in an in vivo model of acute lung injury. FEBS Lett. 1997;402:111–15. doi: 10.1016/s0014-5793(96)01508-6. [DOI] [PubMed] [Google Scholar]
- 22.Itoh Y, Nagase H. Preferential inactivation of tissue inhibitor of metalloproteinases-1 that is bound to the precursor of matrix metalloproteinase 9 (progelatinase B) by human neutrophil elastase. J Biol Chem. 1995;270:16518–21. doi: 10.1074/jbc.270.28.16518. [DOI] [PubMed] [Google Scholar]
- 23.Shamamian P, Schwartz JD, Pocock BJ, et al. Activation of progelatinase A (MMP-2) by neutrophil elastase, cathepsin G, and proteinase-3: a role for inflammatory cells in tumor invasion and angiogenesis. J Cell Physiol. 2001;189:197–206. doi: 10.1002/jcp.10014. [DOI] [PubMed] [Google Scholar]
- 24.Elizur A, Cannon CL, Ferkol TW. Airway inflammation in cystic fibrosis. Chest. 2008;133:489–95. doi: 10.1378/chest.07-1631. [DOI] [PubMed] [Google Scholar]
- 25.Dulon S, Cande C, Bunnett NW, Hollenberg MD, Chignard M, Pidard D. Proteinase-activated receptor-2 and human lung epithelial cells: disarming by neutrophil serine proteinases. Am J Respir Cell Mol Biol. 2003;28:339–46. doi: 10.1165/rcmb.4908. [DOI] [PubMed] [Google Scholar]
- 26.Rubio F, Cooley J, Accurso FJ, Remold-O'Donnell E. Linkage of neutrophil serine proteases and decreased surfactant protein-A (SP-A) levels in inflammatory lung disease. Thorax. 2004;59:318–23. doi: 10.1136/thx.2003.014902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Padrines M, Wolf M, Walz A, Baggiolini M. Interleukin-8 processing by neutrophil elastase, cathepsin G and proteinase-3. FEBS Lett. 1994;352:231–5. doi: 10.1016/0014-5793(94)00952-x. [DOI] [PubMed] [Google Scholar]
- 28.Van den Steen PE, Proost P, Wuyts A, Van Damme J, Opdenakker G. Neutrophil gelatinase B potentiates interleukin-8 tenfold by aminoterminal processing, whereas it degrades CTAP-III, PF-4, and GRO-alpha and leaves RANTES and MCP-2 intact. Blood. 2000;96:2673–81. [PubMed] [Google Scholar]
- 29.Witko-Sarsat V, Halbwachs-Mecarelli L, Schuster A, et al. Proteinase 3, a potent secretagogue in airways, is present in cystic fibrosis sputum. Am J Respir Cell Mol Biol. 1999;20:729–36. doi: 10.1165/ajrcmb.20.4.3371. [DOI] [PubMed] [Google Scholar]
- 30.Duranton J, Bieth JG. Inhibition of proteinase 3 by [alpha] 1-antitrypsin in vitro predicts very fast inhibition in vivo. Am J Respir Cell Mol Biol. 2003;1:57–61. doi: 10.1165/rcmb.2002-0258OC. [DOI] [PubMed] [Google Scholar]
- 31.Balfour-Lynn IM. The protease-antiprotease battle in the cystic fibrosis lung. J R Soc Med. 1999;92(Suppl 37):23–30. doi: 10.1177/014107689909237s05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Birrer P, McElvaney N, Rüderberg A, et al. Protease-antiprotease imbalance in the lungs of children with cystic fibrosis. Am J Respir Crit Care Med. 1994;150:207–213. doi: 10.1164/ajrccm.150.1.7912987. [DOI] [PubMed] [Google Scholar]
- 33.Suter S, Schaad UB, Tegner H, Ohlsson K, Desgrandchamps D, Waldvogel FA. Levels of free granulocyte elastase in bronchial secretions from patients with cystic fibrosis: effect of antimicrobial treatment against Pseudomonas aeruginosa. J Infect Dis. 1986;153:902–9. doi: 10.1093/infdis/153.5.902. [DOI] [PubMed] [Google Scholar]
- 34.Suter S. Imbalance between polymorphonuclear leukocyte proteases and antiproteases in chronic pyogenic infections and its relation to the proteolysis of complement component C3. Complement. 1986;3:1–24. doi: 10.1159/000467874. [DOI] [PubMed] [Google Scholar]
- 35.Ying QL, Simon SR. Kinetics of the inhibition of human leukocyte elastase by elafin, a 6-kilodalton elastase-specific inhibitor from human skin. Biochemistry. 1993;32:1866–74. doi: 10.1021/bi00058a021. [DOI] [PubMed] [Google Scholar]
- 36.Sallenave JM, Silva A. Characterization and gene sequence of the precursor of elafin, an elastase-specific inhibitor in bronchial secretions. Am J Respir Cell Mol Biol. 1993;8:439–45. doi: 10.1165/ajrcmb/8.4.439. [DOI] [PubMed] [Google Scholar]
- 37.Burden RE, Scott CJ. Cysteine cathepsins in pulmonary diseases. In: Taggart CC, Greene CM, editors. Mechanisms of pulmonary innate immunity. India: Research Signpost; 2008. pp. 135–56. [Google Scholar]
- 38.Gueders MM, Foidart JM, Noel A, Cataldo DD. Matrix metalloproteinases (MMPs) and tissue inhibitors of MMPs in the respiratory tract: potential implications in asthma and other lung diseases. Eur J Pharmacol. 2006;533:133–44. doi: 10.1016/j.ejphar.2005.12.082. [DOI] [PubMed] [Google Scholar]
- 39.Long GL, Chandra T, Woo SL, Davie EW, Kurachi K. Complete sequence of the cDNA for human alpha 1-antitrypsin and the gene for the S variant. Biochemistry. 1984;23:4828–37. doi: 10.1021/bi00316a003. [DOI] [PubMed] [Google Scholar]
- 40.Brantly M, Nukiwa T, Crystal RG. Molecular basis of alpha-1-antitrypsin deficiency. Am J Med. 1988;84:13–31. doi: 10.1016/0002-9343(88)90154-4. [DOI] [PubMed] [Google Scholar]
- 41.Crystal RG, Brantly ML, Hubbard RC, Curiel DT, States DJ, Holmes MD. The alpha 1-antitrypsin gene and its mutations. Clinical consequences and strategies for therapy. Chest. 1989;95:196–208. doi: 10.1378/chest.95.1.196. [DOI] [PubMed] [Google Scholar]
- 42.Travis J, Salvesen GS. Human plasma proteinase inhibitors. Annu Rev Biochem. 1983;52:655–709. doi: 10.1146/annurev.bi.52.070183.003255. [DOI] [PubMed] [Google Scholar]
- 43.Mornex JF, Chytil-Weir A, Martinet Y, Courtney M, LeCocq JP, Crystal RG. Expression of the alpha-1-antitrypsin gene in mononuclear phagocytes of normal and alpha-1-antitrypsin-deficient individuals. J Clin Invest. 1986;77:1952–61. doi: 10.1172/JCI112524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.du Bois RM, Bernaudin JF, Paakko P, et al. Human neutrophils express the alpha 1-antitrypsin gene and produce alpha 1-antitrypsin. Blood. 1991;77:2724–30. [PubMed] [Google Scholar]
- 45.Janciauskiene SM, Stevens T. Alpha1-antitrypsin, old dog, new tricks. Alpha1-antitrypsin exerts in vitro anti-inflammatory activity in human monocytes by elevating cAMP. J Biol Chem. 2007;282:8573–82. doi: 10.1074/jbc.M607976200. [DOI] [PubMed] [Google Scholar]
- 46.Lomas DA, Carrell RW. Serpinopathies and the conformational dementias. Nat Rev Genet. 2002;3:759–68. doi: 10.1038/nrg907. [DOI] [PubMed] [Google Scholar]
- 47.Whisstock J, Skinner R, Lesk AM. An atlas of serpin conformations. Trends Biochem Sci. 1998;23:63–7. doi: 10.1016/s0968-0004(97)01172-9. [DOI] [PubMed] [Google Scholar]
- 48.Banda MJ, Rice AG, Griffin GL, Senior RM. The inhibitory complex of human alpha 1-proteinase inhibitor and human leukocyte elastase is a neutrophil chemoattractant. J Exp Med. 1988;167:1608–15. doi: 10.1084/jem.167.5.1608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Beatty K, Bieth J, Travis J. Kinetics of association of serine proteinases with native and oxidized alpha-1-proteinase inhibitor and alpha-1-antichymotrypsin. J Biol Chem. 1980;255:3931–4. [PubMed] [Google Scholar]
- 50.Johnson D, Travis J. The oxidative inactivation of human alpha-1-proteinase inhibitor. Further evidence for methionine at the reactive center. J Biol Chem. 1979;254:4022–6. [PubMed] [Google Scholar]
- 51.Taggart C, Cervantes-Laurean D, Kim G, et al. Oxidation of either methionine 351 or methionine 358 in alpha 1-antitrypsin causes loss of anti-neutrophil elastase activity. J Biol Chem. 2000;275:27258–65. doi: 10.1074/jbc.M004850200. [DOI] [PubMed] [Google Scholar]
- 52.Griffiths SW, Cooney CL. Relationship between protein structure and methionine oxidation in recombinant human alpha 1-antitrypsin. Biochemistry. 2002;41:6245–52. doi: 10.1021/bi025599p. [DOI] [PubMed] [Google Scholar]
- 53.Padrines M, Schneider-Pozzer M, Bieth JG. Inhibition of neutrophil elastase by alpha-1-proteinase inhibitor oxidized by activated neutrophils. Am Rev Respir Dis. 1989;139:783–90. doi: 10.1164/ajrccm/139.3.783. [DOI] [PubMed] [Google Scholar]
- 54.Appelhans B, Ender B, Sachse G, Nikiforov T, Appelhans H, Ebert W. Secretion of antileucoprotease from a human lung tumor cell line. FEBS Lett. 1987;224:14–18. doi: 10.1016/0014-5793(87)80413-1. [DOI] [PubMed] [Google Scholar]
- 55.Franken C, Meijer CJ, Dijkman JH. Tissue distribution of antileukoprotease and lysozyme in humans. J Histochem Cytochem. 1989;37:493–8. doi: 10.1177/37.4.2926127. [DOI] [PubMed] [Google Scholar]
- 56.Heinzel R, Appelhans H, Gassen G, et al. Molecular cloning and expression of cDNA for human antileukoprotease from cervix uterus. Eur J Biochem. 1986;160:61–7. doi: 10.1111/j.1432-1033.1986.tb09940.x. [DOI] [PubMed] [Google Scholar]
- 57.Lee CH, Igarashi Y, Hohman RJ, Kaulbach H, White MV, Kaliner MA. Distribution of secretory leukoprotease inhibitor in the human nasal airway. Am Rev Respir Dis. 1993;147:710–16. doi: 10.1164/ajrccm/147.3.710. [DOI] [PubMed] [Google Scholar]
- 58.Stetler G, Brewer MT, Thompson RC. Isolation and sequence of a human gene encoding a potent inhibitor of leukocyte proteases. Nucleic Acids Res. 1986;14:7883–96. doi: 10.1093/nar/14.20.7883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Thompson RC, Ohlsson K. Isolation, properties, and complete amino acid sequence of human secretory leukocyte protease inhibitor, a potent inhibitor of leukocyte elastase. Proc Natl Acad Sci USA. 1986;83:6692–6. doi: 10.1073/pnas.83.18.6692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Clauss A, Lilja H, Lundwall A. A locus on human chromosome 20 contains several genes expressing protease inhibitor domains with homology to whey acidic protein. Biochem J. 2002;368:233–42. doi: 10.1042/BJ20020869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Weldon S, McGarry N, Taggart CC. SLPI and elafin: multifunctional components of the respiratory tract. In: Taggart CC, Greene CM, editors. Mechanisms of pulmonary innate immunity. India: Research Signpost; 2008. p. 201. [Google Scholar]
- 62.Bingle CD, Vyakarnam A. Novel innate immune functions of the whey acidic protein family. Trends Immunol. 2008;29:444–53. doi: 10.1016/j.it.2008.07.001. [DOI] [PubMed] [Google Scholar]
- 63.Moreau T, Baranger K, Dadé S, Dallet-Choisy S, Guyot N, Zani ML. Multifaceted roles of human elafin and secretory leukocyte proteinase inhibitor (SLPI), two serine protease inhibitors of the chelonianin family. Biochimie. 2008;90:284–95. doi: 10.1016/j.biochi.2007.09.007. [DOI] [PubMed] [Google Scholar]
- 64.Fitch PM, Roghanian A, Howie SE, Sallenave JM. Human neutrophil elastase inhibitors in innate and adaptive immunity. Biochem Soc Trans. 2006;34:279–82. doi: 10.1042/BST20060279. [DOI] [PubMed] [Google Scholar]
- 65.Williams SE, Brown TI, Roghanian A, Sallenave JM. SLPI and elafin: one glove, many fingers. Clin Sci (Lond) 2006;110:21–35. doi: 10.1042/CS20050115. [DOI] [PubMed] [Google Scholar]
- 66.Jin FY, Nathan C, Radzioch D, Ding A. Secretory leukocyte protease inhibitor: a macrophage product induced by and antagonistic to bacterial lipopolysaccharide. Cell. 1997;88:417–26. doi: 10.1016/s0092-8674(00)81880-2. [DOI] [PubMed] [Google Scholar]
- 67.Sallenave JM, Si-Ta Har M, Cox G, Chignard M, Gauldie J. Secretory leukocyte proteinase inhibitor is a major leukocyte elastase inhibitor in human neutrophils. J Leukoc Biol. 1997;61:695–702. doi: 10.1002/jlb.61.6.695. [DOI] [PubMed] [Google Scholar]
- 68.Rao NV, Marshall BC, Gray BH, Hoidal JR. Interaction of secretory leukocyte protease inhibitor with proteinase-3. Am J Respir Cell Mol Biol. 1993;8:612–16. doi: 10.1165/ajrcmb/8.6.612. [DOI] [PubMed] [Google Scholar]
- 69.Rogan MP, Geraghty P, Greene CM, O'Neill SJ, Taggart CC, McElvaney NG. Antimicrobial proteins and polypeptides in pulmonary innate defence. Respir Res. 2006;7:29. doi: 10.1186/1465-9921-7-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Nakamura A, Mori Y, Hagiwara K, et al. Increased susceptibility to LPS-induced endotoxin shock in secretory leukoprotease inhibitor (SLPI)-deficient mice. J Exp Med. 2003;197:669–74. doi: 10.1084/jem.20021824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lentsch AB, Jordan JA, Czermak BJ, et al. Inhibition of NF-kappaB activation and augmentation of IkappaBbeta by secretory leukocyte protease inhibitor during lung inflammation. Am J Pathol. 1999;154:239–47. doi: 10.1016/s0002-9440(10)65270-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Mulligan MS, Lentsch AB, Huber-Lang M, et al. Anti-inflammatory effects of mutant forms of secretory leukocyte protease inhibitor. Am J Pathol. 2000;156:1033–9. doi: 10.1016/S0002-9440(10)64971-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Taggart CC, Cryan SA, Weldon S, et al. Secretory leukocyte protease inhibitor binds to NF-{kappa}B binding sites in monocytes and inhibits p65 binding. J Exp Med. 2005;202:1659–68. doi: 10.1084/jem.20050768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Greene C, Taggart C, Lowe G, Gallagher P, McElvaney N, O'Neill S. Local impairment of anti-neutrophil elastase capacity in community acquired pneumonia. J Infect Dis. 2003;188:769–76. doi: 10.1086/377238. [DOI] [PubMed] [Google Scholar]
- 75.Taggart CC, Greene CM, McElvaney NG, O'Neill S. Secretory leucoprotease inhibitor prevents lipopolysaccharide-induced IkappaBalpha degradation without affecting phosphorylation or ubiquitination. J Biol Chem. 2002;277:33648–53. doi: 10.1074/jbc.M203710200. [DOI] [PubMed] [Google Scholar]
- 76.Grutter MG, Fendrich G, Huber R, Bode W. The 2.5 A X-ray crystal structure of the acid-stable proteinase inhibitor from human mucus secretions analysed in its complex with bovine alphachymotrypsin. EMBO J. 1988;7:345–51. doi: 10.1002/j.1460-2075.1988.tb02819.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Heinzel-Wieland R, Steffens GJ, Flohe L. Inhibitory characteristics and oxidant resistance of site specific variants of recombinant human antileukoproteinase (ALP) Biomed Biochim Acta. 1991;50:677–81. [PubMed] [Google Scholar]
- 78.Taggart CC, Lowe GJ, Greene CM, et al. Cathepsin B, L, and S cleave and inactivate secretory leucoprotease inhibitor. J Biol Chem. 2001;276:33345–52. doi: 10.1074/jbc.M103220200. [DOI] [PubMed] [Google Scholar]
- 79.Sallenave JM, Ryle AP. Purification and characterization of elastase-specific inhibitor. Sequence homology with mucus proteinase inhibitor. Biol Chem Hoppe Seyler. 1991;372:13–21. doi: 10.1515/bchm3.1991.372.1.13. [DOI] [PubMed] [Google Scholar]
- 80.Molhuizen HO, Alkemade HA, Zeeuwen PL, de Jongh GJ, Wieringa B, Schalkwijk J. SKALP/elafin: an elastase inhibitor from cultured human keratinocytes. Purification, cDNA sequence, and evidence for transglutaminase cross-linking. J Biol Chem. 1993;268:12028–32. [PubMed] [Google Scholar]
- 81.Sumi Y, Inoue N, Azumi H, et al. Expression of tissue transglutaminase and elafin in human coronary artery: implication for plaque instability. Atherosclerosis. 2002;160:31–9. doi: 10.1016/s0021-9150(01)00542-1. [DOI] [PubMed] [Google Scholar]
- 82.Mihaila A, Tremblay GM. Human alveolar macrophages express elafin and secretory leukocyte protease inhibitor. Z Naturforsch [C] 2001;56:291–7. doi: 10.1515/znc-2001-3-420. [DOI] [PubMed] [Google Scholar]
- 83.Zhang M, Magit D, Pardee AB, Sager R. Re-expression of elafin in 21MT2 breast carcinomas by phorbol 12-myristate 13-acetate is mediated by the Ap1 site in the elafin promoter. Cancer Res. 1997;57:4631–6. [PubMed] [Google Scholar]
- 84.Bingle L, Tetley TD, Bingle CD. Cytokine-mediated induction of the human elafin gene in pulmonary epithelial cells is regulated by nuclear factor-kappaB. Am J Respir Cell Mol Biol. 2001;25:84–91. doi: 10.1165/ajrcmb.25.1.4341. [DOI] [PubMed] [Google Scholar]
- 85.Ying QL, Simon SR. Kinetics of the inhibition of proteinase 3 by elafin. Am J Respir Cell Mol Biol. 2001;24:83–9. doi: 10.1165/ajrcmb.24.1.4300. [DOI] [PubMed] [Google Scholar]
- 86.Simpson AJ, Wallace WA, Marsden ME, et al. Adenoviral augmentation of elafin protects the lung against acute injury mediated by activated neutrophils and bacterial infection. J Immunol. 2001;167:1778–86. doi: 10.4049/jimmunol.167.3.1778. [DOI] [PubMed] [Google Scholar]
- 87.Vachon E, Bourbonnais Y, Bingle CD, Rowe SJ, Janelle MF, Tremblay GM. Anti-inflammatory effect of pre-elafin in lipopolysaccharide-induced acute lung inflammation. Biol Chem. 2002;383:1249–56. doi: 10.1515/BC.2002.138. [DOI] [PubMed] [Google Scholar]
- 88.Guyot N, Butler MW, McNally P, et al. Elafin, an elastase-specific inhibitor, is cleaved by its cognate enzyme neutrophil elastase in sputum from individuals with cystic fibrosis. J Biol Chem. 2008;283:32377–85. doi: 10.1074/jbc.M803707200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Wright JL, Farmer SG, Churg A. Synthetic serine elastase inhibitor reduces cigarette smoke-induced emphysema in guinea pigs. Am J Respir Crit Care Med. 2002;166:954–60. doi: 10.1164/rccm.200202-098OC. [DOI] [PubMed] [Google Scholar]
- 90.Shinguh Y, Yamazaki A, Inamura N, et al. Biochemical and pharmacological characterization of FR134043, a novel elastase inhibitor. Eur J Pharmacol. 1998;345:299–308. doi: 10.1016/s0014-2999(98)00028-4. [DOI] [PubMed] [Google Scholar]
- 91.Williams JC, Stein RL, Giles RE, Krell RD. Biochemistry and pharmacology of ICI 200, 880, a synthetic peptide inhibitor of human neutrophil elastase. Ann N Y Acad Sci. 1991;624:230–43. doi: 10.1111/j.1749-6632.1991.tb17022.x. [DOI] [PubMed] [Google Scholar]
- 92.Tarjan E, Peto L, Appel J, Tolnay P. Protective effect of specific elastase inhibitor in experimental pulmonary emphysema. Schweiz Med Wochenschr. 1984;114:918–20. [PubMed] [Google Scholar]
- 93.Wewers MD, Casolaro MA, Crystal RG. Comparison of alpha-1-antitrypsin levels and antineutrophil elastase capacity of blood and lung in a patient with the alpha-1-antitrypsin phenotype null-null before and during alpha-1-antitrypsin augmentation therapy. Am Rev Respir Dis. 1987;135:539–43. doi: 10.1164/arrd.1987.135.3.539. [DOI] [PubMed] [Google Scholar]
- 94.Hubbard RC, Sellers S, Czerski D, Stephens L, Crystal RG. Biochemical efficacy and safety of monthly augmentation therapy for alpha 1-antitrypsin deficiency. JAMA. 1988;260:1259–64. [PubMed] [Google Scholar]
- 95.Moore JE, Shaw A, Millar BC, et al. Reduction in neutrophil elastase concentration by recombinant alphal-antitrypsin (recAAT) does not alter bacterial loading in the sputum of cystic fibrosis patients. Br J Biomed Sci. 2004;61(3):146–7. doi: 10.1080/09674845.2004.11732661. [DOI] [PubMed] [Google Scholar]
- 96.Martin SL, Downey D, Bilton D, Keogan MT, Edgar J, Elborn JS. Safety and efficacy of recombinant alpha(1)-antitrypsin therapy in cystic fibrosis. Pediatr Pulmonol. 2006;2:177–83. doi: 10.1002/ppul.20345. [DOI] [PubMed] [Google Scholar]
- 97.Cantin AM, Berthiaume Y, Cloutier D, Martel M. Prolastin aerosol therapy and sputum taurine in cystic fibrosis. Clin Invest Med. 2006;4:201–7. [PubMed] [Google Scholar]
- 98.Griese M, Latzin P, Kappler M, et al. alpha1-Antitrypsin inhalation reduces airway inflammation in cystic fibrosis patients. Eur Respir J. 2007;2:240–50. doi: 10.1183/09031936.00047306. [DOI] [PubMed] [Google Scholar]
- 99.Brand P, Schulte M, Wencker M, et al. Lung deposition of inhaled alpha1-proteinase inhibitor in cystic fibrosis and alpha1-antitrypsin deficiency. Eur Respir J. 2009;34(2):354–60. doi: 10.1183/09031936.00118408. [DOI] [PubMed] [Google Scholar]
- 100.Ferkol T, Cohn LA, Phillips TE, Smith A, Davis PB. Targeted delivery of antiprotease to the epithelial surface of human tracheal xenografts. Am J Respir Crit Care Med. 2003;167:1374–9. doi: 10.1164/rccm.200209-1119OC. [DOI] [PubMed] [Google Scholar]
- 101.Cereijido M, Ponce A, Gonzalez-Marical J. Tight junctions and apical basolateral polarity. J Membr Biol. 1989;110:1–9. [Google Scholar]
- 102.McElvaney NG, Nakamura H, Birrer P, et al. Modulation of airway inflammation in cystic fibrosis. In vivo suppression of interleukin-8 levels on the respiratory epithelial surface by aerosolization of recombinant secretory leukoprotease inhibitor. J Clin Invest. 1992;90:1296–301. doi: 10.1172/JCI115994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.McElvaney NG, Doujaiji B, Moan MJ, Burnham MR, Wu MC, Crystal RG. Pharmacokinetics of recombinant secretory leukoprotease inhibitor aerosolized to normals and individuals with cystic fibrosis. Am Rev Respir Dis. 1993;148:1056–60. doi: 10.1164/ajrccm/148.4_Pt_1.1056. [DOI] [PubMed] [Google Scholar]
- 104.Gillissen A, Birrer P, McElvaney NG, et al. Recombinant secretory leukoprotease inhibitor augments glutathione levels in lung epithelial lining fluid. J Appl Physiol. 1993;75:825–32. doi: 10.1152/jappl.1993.75.2.825. [DOI] [PubMed] [Google Scholar]
- 105.Proteo Biotech AG. Available from: www.proteo.de/framesets/r-and-d.php?lang=en . [Accessed: October 26, 2009].
- 106.Grimbert D, Vecellio L, Delépine P, et al. Characteristics of EPI-hNE4 aerosol: a new elastase inhibitor for treatment of cystic fibrosis. J Aerosol Med. 2003;16(2):121–9. doi: 10.1089/089426803321919889. [DOI] [PubMed] [Google Scholar]
- 107.Attucci S, Gauthier A, Korkmaz B, et al. EPI-hNE4, a proteolysis-resistant inhibitor of human neutrophil elastase and potential anti-inflammatory drug for treating cystic fibrosis. J Pharmacol Exp Ther. 2006;318(2):803–9. doi: 10.1124/jpet.106.103440. [DOI] [PubMed] [Google Scholar]
- 108.Ono Pharmaceutical Co., Ltd. Available from: http://www.ono.co.jp/eng/default. htm . [Accessed: October 26, 2009].
- 109.Wolters PJ, Chapman HA. Importance of lysosomal cysteine proteases in lung disease. Respir Res. 2001;1:170–7. doi: 10.1186/rr29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Bühling F, Waldburg N, Reisenauer A, Heimburg A, Golpon H, Welte T. Lysosomal cysteine proteases in the lung: role in protein processing and immunoregulation. Eur Respir J. 2004;23:620–8. doi: 10.1183/09031936.04.00105304. [DOI] [PubMed] [Google Scholar]
- 111.Chapman HA, Jr, Shi GP. Protease injury in the development of COPD: Thomas A. Neff Lecture. Chest. 2000;117:295S–99S. doi: 10.1378/chest.117.5_suppl_1.295s. [DOI] [PubMed] [Google Scholar]
- 112.Bühling F, Gerber A, Häckel C, et al. Expression of cathepsin K in lung epithelial cells. Am J Respir Cell Mol Biol. 1999;20:612–19. doi: 10.1165/ajrcmb.20.4.3405. [DOI] [PubMed] [Google Scholar]
- 113.Shi GP, Munger JS, Meara JP, Rich DH, Chapman HA. Molecular cloning and expression of human alveolar macrophage cathepsin S, an elastinolytic cysteine protease. J Biol Chem. 1992;267:7258–62. [PubMed] [Google Scholar]
- 114.Brasch F, Ten Brinke A, Johnen G, et al. Involvement of cathepsin H in the processing of the hydrophobic surfactant-associated protein C in type II pneumocytes. Am J Respir Cell Mol Biol. 2002;26:659–70. doi: 10.1165/ajrcmb.26.6.4744. [DOI] [PubMed] [Google Scholar]
- 115.Kirschke H, Wiederanders B. Cathepsin S and related lysosomal endopeptidases. Methods Enzymol. 1994;244:500–11. doi: 10.1016/0076-6879(94)44036-0. [DOI] [PubMed] [Google Scholar]
- 116.Wolters PJ, Raymond WW, Blount JL, Caughey GH. Regulated expression, processing, and secretion of dog mast cell dipeptidyl peptidase I. J Biol Chem. 1998;273:15514–20. doi: 10.1074/jbc.273.25.15514. [DOI] [PubMed] [Google Scholar]
- 117.Tate S, MacGregor G, Davis M, Innes JA, Greening AP. Airways in cystic fibrosis are acidified: detection by exhaled breath condensate. Thorax. 2002;57:926–9. doi: 10.1136/thorax.57.11.926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Geraghty P, Rogan MP, Greene CM, et al. Neutrophil elastase up-regulates cathepsin B and matrix metalloprotease-2 expression. J Immunol. 2007;178:5871–8. doi: 10.4049/jimmunol.178.9.5871. [DOI] [PubMed] [Google Scholar]
- 119.Burnett D, Abrahamson M, Devalia JL, Sapsford RJ, Davies RJ, Buttle DJ. Synthesis and secretion of procathepsin B and cystatin C by human bronchial epithelial cells in vitro: modulation of cathepsin B activity by neutrophil elastase. Arch Biochem. Biophys. 1995;317:305–10. doi: 10.1006/abbi.1995.1167. [DOI] [PubMed] [Google Scholar]
- 120.Taggart CC, Greene CM, Smith SG, et al. Inactivation of human β-defensins 2 and 3 by elastolytic cathepsins. J Immunol. 2003;171:931–7. doi: 10.4049/jimmunol.171.2.931. [DOI] [PubMed] [Google Scholar]
- 121.Rogan MP, Taggart CC, Greene CM, Murphy PG, O'Neill SJ, McElvaney NG. Loss of microbicidal activity and increased formation of biofilm due to decreased lactoferrin activity in patients with cystic fibrosis. J Infect Dis. 2004;190:1245–53. doi: 10.1086/423821. [DOI] [PubMed] [Google Scholar]
- 122.László A, Sohár I, Gyurkovits K. Activity of the lysosomal cysteine proteinases (cathepsin B,H,L) and a metalloproteinase (MMP-7-ase) in the serum of cystic fibrosis homozygous children. Acta Paediatr Hung. 1987;28:175–8. [PubMed] [Google Scholar]
- 123.McGuire MJ, Lipsky PE, Thiele DL. Generation of active myeloid and lymphoid granule serine proteases requires processing by the granule thiol protease dipeptidyl peptidase I. J Biol Chem. 1993;268:2458–67. [PubMed] [Google Scholar]
- 124.Murakami M, Karnik SS, Husain A. Human prochymase activation. A novel role for heparin in zymogen processing. J Biol Chem. 1995;270:2218–23. [PubMed] [Google Scholar]
- 125.Pham CT, Ley TJ. Dipeptidyl peptidase I is required for the processing and activation of granzymes A and B in vivo. Proc Natl Acad Sci USA. 1999;96:8627–32. doi: 10.1073/pnas.96.15.8627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Johnson DA, Barrett AJ, Mason RW. Cathepsin L inactivates alpha 1-proteinase inhibitor by cleavage in the reactive site region. J Biol Chem. 1986;261:14748–51. [PubMed] [Google Scholar]
- 127.Abrahamson M, Buttle DJ, Mason RW, et al. Regulation of cystatin C activity by serine proteinases. Biomed Biochim Acta. 1991;50:587–93. [PubMed] [Google Scholar]
- 128.Vasiljeva O, Reinheckel T, Peters C, Turk D, Turk V, Turk B. Emerging roles of cysteine cathepsins in disease and their potential as drug targets. Curr Pharm Des. 2007;13:387–40. doi: 10.2174/138161207780162962. [DOI] [PubMed] [Google Scholar]
- 129.Sternlicht MD, Werb Z. How matrix metalloproteinases regulate cell behavior. Annu Rev Cell Dev Biol. 2001;17:463–516. doi: 10.1146/annurev.cellbio.17.1.463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Parks WC, Wilson CL, Lopez-Boado YS. Matrix metalloproteinases as modulators of inflammation and innate immunity. Nat Rev Immunol. 2004;4:617–29. doi: 10.1038/nri1418. [DOI] [PubMed] [Google Scholar]
- 131.Peterson-Carmichael SL, Harris WT, Goel R, et al. Association of lower airway inflammation with physiologic findings in young children with cystic fibrosis. Pediatr Pulmonol. 2009;44:503–11. doi: 10.1002/ppul.21044. [DOI] [PubMed] [Google Scholar]
- 132.Ratjen F, Hartog CM, Paul K, Wermelt J, Braun J. Matrix metalloproteases in BAL fluid of patients with cystic fibrosis and their modulation by treatment with dornase alpha. Thorax. 2002;57:930–4. doi: 10.1136/thorax.57.11.930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Hilliard TN, Regamey N, Shute JK, et al. Airway remodelling in children with cystic fibrosis. Thorax. 2007;62:1074–80. doi: 10.1136/thx.2006.074641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Gaggar A, Li Y, Weathington N, et al. Matrix metalloprotease-9 dysregulation in lower airway secretions of cystic fibrosis patients. Am J Physiol Lung Cell Mol Physiol. 2007;293:L96–L104. doi: 10.1152/ajplung.00492.2006. [DOI] [PubMed] [Google Scholar]
- 135.Sagel SD, Kapsner RK, Osberg I. Induced sputum matrix metalloproteinase-9 correlates with lung function and airway inflammation in children with cystic fibrosis. Pediatr Pulmonol. 2005;39:224–32. doi: 10.1002/ppul.20165. [DOI] [PubMed] [Google Scholar]
- 136.Ferry G, Lonchampt M, Pennel L, de Nanteuil G, Canet E, Tucker GC. Activation of MMP-9 by neutrophil elastase in an in vivo model of acute lung injury. FEBS Lett. 1997;402:111–15. doi: 10.1016/s0014-5793(96)01508-6. [DOI] [PubMed] [Google Scholar]
- 137.Manicone AM, McGuire JK. Matrix metalloproteinases as modulators of inflammation. Semin Cell Dev Biol. 2008;19:34–41. doi: 10.1016/j.semcdb.2007.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Desrochers PE, Jeffrey JJ, Weiss SJ. Interstitial collagenase (matrix metalloproteinase-1) expresses serpinase activity. J Clin Invest. 1991;87:2258–65. doi: 10.1172/JCI115262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Zhang Z, Winyard PG, Chidwick K, et al. Proteolysis of human native and oxidised alpha 1-proteinase inhibitor by matrilysin and stromelysin. Biochim Biophys Acta. 1994;1199:224–8. doi: 10.1016/0304-4165(94)90119-8. [DOI] [PubMed] [Google Scholar]
- 140.Michaelis J, Vissers MC, Winterbourn CC. Cleavage of alpha 1-antitrypsin by human neutrophil collagenase. Matrix Suppl. 1992;1:80–1. [PubMed] [Google Scholar]
- 141.Liu Z, Zhou X, Shapiro SD, et al. The serpin alpha1-proteinase inhibitor is a critical substrate for gelatinase B/MMP-9 S. Cell. 2000;102:647–55. doi: 10.1016/s0092-8674(00)00087-8. [DOI] [PubMed] [Google Scholar]
- 142.Shapiro SD, Goldstein NM, Houghton AM, Kobayashi DK, Kelley D, Belaaouaj A. Neutrophil elastase contributes to cigarette smoke-induced emphysema in mice. Am J Pathol. 2003;163:2329–35. doi: 10.1016/S0002-9440(10)63589-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Ramadas RA, Wu L, LeVine AM. Surfactant protein A enhances production of secretory leukoprotease inhibitor and protects it from cleavage by matrix metalloproteinases. J Immunol. 2009;182:1560–7. doi: 10.4049/jimmunol.182.3.1560. [DOI] [PubMed] [Google Scholar]
- 144.Henry MT, McMahon K, Costello C, Fitzgerald MX, O'Connor CM. Secretory leukocyte proteinase inhibitor and elafin are resistant to degradation by MMP-8. Exp Lung Res. 2002;28:85–97. doi: 10.1080/019021402753462022. [DOI] [PubMed] [Google Scholar]
- 145.Dijkstra A, Postma DS, Noordhoek JA, et al. Expression of ADAMs ("a disintegrin and metalloprotease") in the human lung. Virchows Arch. 2009;454:441–9. doi: 10.1007/s00428-009-0748-4. [DOI] [PubMed] [Google Scholar]
- 146.Shao MX, Nadel JA. Neutrophil elastase induces MUC5AC mucin production in human airway epithelial cells via a cascade involving protein kinase C, reactive oxygen species, and TNF-alpha-converting enzyme. J Immunol. 2005;175:4009–16. doi: 10.4049/jimmunol.175.6.4009. [DOI] [PubMed] [Google Scholar]
- 147.Thathiah A, Blobel CP, Carson DD. Tumor necrosis factor-alpha converting enzyme/ADAM 17 mediates MUC1 shedding. J Biol Chem. 2003;278:3386–94. doi: 10.1074/jbc.M208326200. [DOI] [PubMed] [Google Scholar]
- 148.Thathiah A, Carson DD. MT1-MMP mediates MUC1 shedding independently of TACE/ADAM17. Biochem J. 2004;362:363–73. doi: 10.1042/BJ20040513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Lillehoj EP, Hyun SW, Kim BT, et al. Muc1 mucins on the cell surface are adhesion sites for Pseudomonas aeruginosa. Am J Physiol Lung Cell Mol Physiol. 2001;280:L181–L187. doi: 10.1152/ajplung.2001.280.1.L181. [DOI] [PubMed] [Google Scholar]
- 150.Vij N, Fang S, Zeitlin PL. Selective inhibition of endoplasmic reticulum-associated degradation rescues F508-cystic fibrosis transmembrane regulator and suppresses interleukin-8 levels. J Biol Chem. 2006;281:17369–78. doi: 10.1074/jbc.M600509200. [DOI] [PubMed] [Google Scholar]
- 151.Ward CL, Omura S, Kopito RR. Degradation of CFTR by the ubiquitin-proteasome pathway. Cell. 1995;83:121–7. doi: 10.1016/0092-8674(95)90240-6. [DOI] [PubMed] [Google Scholar]
- 152.Mitchell BS. A phase 2 study of bortezomib in relapsed refractory myeloma. N Eng J Med. 2003;348:2597–8. doi: 10.1056/NEJMoa030288. [DOI] [PubMed] [Google Scholar]
- 153.Travis J, Potempa J. Bacterial proteinases as targets for the development of second-generation antibiotics. Biochimica Biophysica Acta. 2000;1477:35–50. doi: 10.1016/s0167-4838(99)00278-2. [DOI] [PubMed] [Google Scholar]
- 154.Kamath S, Kapatral V, Chakrabarty AM. Cellular function of elastase in Pseudomonas aeruginosa: role in the cleavage of nucleoside disphosphate kinase and in alginate synthesis. Mol Microbiol. 1998;30:933–41. doi: 10.1046/j.1365-2958.1998.01121.x. [DOI] [PubMed] [Google Scholar]
- 155.Leduc D, Beaufort N, de Bentzmann S, et al. The Pseudomonas aeruginosa LasB metalloproteinase regulates the human urokinase-type plasminogen activator receptor through domain-specific endoproteolysis. Infect Immun. 2007;75(8):3848–58. doi: 10.1128/IAI.00015-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Govan JRW, Deretic V. Microbial pathogenesis in cystic fibrosis: mucoid Pseudomonas aeruginosa and Burkholderia cepacia. Microbiol Rev. 1996;660:539–74. doi: 10.1128/mr.60.3.539-574.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]