

Abstract
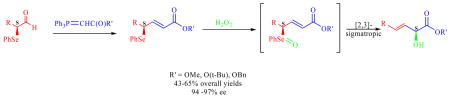
An efficient and enantiocontrolled 3-step synthesis of α-hydroxy-(E)-β,γ-unsaturated esters is reported. Enantioenriched α-selenyl aldehydes, prepared in one step by asymmetric, organocatalytic α-selenylation of aldehydes, were directly subjected to a Wittig reaction followed by allylic selenide to selenoxide oxidation and final spontaneous [2,3]-sigmatropic rearrangement to yield the target compounds in 43–65 % overall yield and in 94–97 % ee.
Asymmetric synthesis utilizing organoselenium compounds has become increasingly popular in recent years.1 Selenium incorporation can be done in either a nucleophilic or an electrophilic fashion.2 Once selenium is introduced into the molecule, many different chemical transformations can occur including oxidations leading to either syn-elimination3 or, in the case of an allylic selenide, [2,3]-sigmatropic rearrangement to give an allylic alcohol.4 Pursuing our recently published work preparing chiral non-racemic γ-hydroxy-(E)-α,β-unsaturated sulfones and esters,5 we have developed a complementary asymmetric synthetic method producing α-hydroxy-(E)-β,γ-unsaturated esters.
α-Hydroxy esters and their corresponding acids are key structural units of valuable synthetic intermediates as well as natural products.1,6 Based on the utility of these structural units, we set out to design a simple, asymmetric general strategy for their synthesis. Suprisingly, most reports of selenium oxidation and [2,3]-sigmatroptic rearrangements have been done utilizing chiral oxidants as opposed to installing chirality prior to oxidation.7 There have been a few reports involving diastereoselective oxidations of selenides containing chiral moieties,8 but the scope is quite limited, with low to moderate yields and only modest diastereomeric excess (de). Other methods have also been reported using conformationally locked ring systems to induce stereocontrol.9
Recent reports on the organocatalytic, asymmetric α-selenenylation of aldehydes in high yields (> 85 %) and high enantiomeric excess (> 95 %)10,11 gave an excellent method for controlling the absolute stereochemistry of our α-hydroxy-(E)-β,γ-unsaturated target systems.
We chose a variety of aldehydes (3-phenylpropanal, 2-cyclohexylethanal, N-Boc-4-piperidineethanal, hexadecanal, and 6-benzyloxyhexanal) as well as three different ester functional groups (OMe, O-t-butyl, and O-benzyl) to determine the generality of this reaction. α-Selenyl aldehydes 3a-e, prepared according to an Italian protocol,11 were immediately subjected in situ to a Wittig reaction to give the enantiomerically enriched compounds 5–11 (Scheme 1, A) in 48–83 % yields. Oxidation of the γ-seleneyl-(E)-α,β-unsaturated esters to the selenoxide using hydrogen peroxide caused spontaneous [2,3]-sigmatropic rearrangement to give the final enantioenriched α-hydroxy-(E)-β,γ-unsaturated esters (Scheme 1, B) in 63–90 % yields and excellent ee’s (≥94 %). The exclusive (E)-geometry of the carbon-carbon double bond in intermediates 5–11 as well as the final products 12–18 was confirmed by 1H NMR spectroscopy (J = 14–16 Hz). It is important to note that when α-hydroxy ester final products 12, 13 and 14 were dissolved in a variety of solvents (e. g. THF, CHCl3, CH3CN, and hexanes) at room temperature, they were stable to racemization (as determined via chiral HPLC) for at least 24 hours. We also used mCPBA as an oxidant of selenium, and we obtained a comparable yield and ee when compared to hydrogen peroxide (system tested was hexadecanal). The preferred method of oxidation remained hydrogen peroxide due to the convenience of the reaction conditions (non-anhydrous).
Scheme 1.

Preparation of α-hydroxy-(E)-β,γ-unsaturated esters
To illustrate the utility of this method for natural product synthesis, we completed a formal total synthesis of (+)-symbioramide (20), a naturally occurring bioactive ceramide composed of D-erythro-dihydrosphingosine and (2R,3E)-2-hydroxy-3-octadecenoic acid.12 We prepared key intermediate (−)-19 that had been used in a recent synthesis13 of this biologically active ceramide (Scheme 2). Intermediate (−)-19 had been prepared in 7 steps and 11.8 % overall yield from L-serine.13a Using the new method described here, we prepared this intermediate mono-protected diol (−)-19 in only 5 steps and in 28 % overall yield and 95% ee (Scheme 3). It is important to note that, in order to achieve the natural stereochemistry at the chiral 2-position, we employed the opposite proline-derived catalyst in the asymmetric α-selenenylation of hexadecanal. This formal total synthesis of (+)-symbioramide (20) allowed us to confirm the absolute stereochemistry of our final α-hydroxy carbonyl products. Previously published characterization of mono-protected diol (−)-19 indicates that the absolute stereochemistry at the 2 position is R, with [α]D = −77°, matching our experimental data for intermediate (−)-19 prepared via Scheme 3.
Scheme 2.

Previously Published Synthesis of (+)-symbioramide (20) Using Key Intermediate (−)-19
Scheme 3.

Formal Total Synthesis of (+)-symbioramide (20)
In one final example, we prepared also a vitamin D3 chiron that incorporated a side chain with the structural motif generated via this synthetic method. An SN2 displacement of iodide 2214 using allylmagnesium bromide yielded terminal olefin 23. Oxidative cleavage afforded aldehyde 24. Using the method described here (Scheme 4), we were able to prepare α-hydroxy-(E)-β,γ-unsaturated benzyl ester 26 as a single diastereomer. This fragment can be easily modified in order to prepare final C,D-ring side-chain modified vitamin D analogs.
Scheme 4.

Synthesis of a 23-(E)-ene-25(S)-hydroxy Vitamin D3 Chiron
In summary, we report an efficient, generalized, asymmetric, organocatalytic procedure for synthesis of α-hydroxy-(E)-β,γ-unsaturated esters in high enantiomeric purities. This three-step synthesis works with a variety of substrates, in good overall yields (46–65 %), and with excellent control of absolute stereochemistry (ee values ≥94 %). We have proven the utility of this method through a formal total synthesis of natural product (+)-symbioramide (20) as well as through the preparation of a 23-(E)-ene-25(S)-hydroxy building block useful for synthesis of new vitamin D side-chain analogs.15
Supplementary Material
Table 1.
Reaction Results from Scheme 1.
| compound (A,B) | R | R′ | A yield, % | B yield, % | ee (%)a |
|---|---|---|---|---|---|
| 5, 12 | Bn (1a) | OMe (4a) | 73 | 87 | 95 |
| 6, 13 | Bn (1a) | O(t-Bu) (4b) | 73 | 63 | 95 |
| 7, 14 | Bn (1a) | OBn (4c) | 81 | 70 | 95 |
| 8, 15 | cyclohexyl (1b) | OBn (4c) | 79 | 67 | 95 |
| 9, 16 | N-Boc-piperidine (1c) | OBn (4c) | 83 | 67 | 94 |
| 10, 17 | BnO(CH2)4 (1d) | OBn (4c) | 48 | 90 | 97 |
| 11, 18b | CH3(CH2)13(1e) | OBn (4c) | 77 | 83 | 95 |
ee values determined using chiral HPLC and racemic standards;
catalyst (2R)-2 was used.
Acknowledgments
thank the NIH (CA 93547) for support.
Footnotes
Supporting Information Available: Experimental procedures and full spectroscopic data for all new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Wirth T. Tetrahedron. 1999;55:1. [Google Scholar]
- 2.(a) Sharpless KB, Lauer RF. J Am Chem Soc. 1973;95:2697. [Google Scholar]; (b) Sharpless KB, Young MW. J Org Chem. 1975;40:947. [Google Scholar]; (c) Grieco PA, Gilman S, Nishizawa M. J Org Chem. 1976;41:1485. [Google Scholar]; (d) Tomoda S, Nomura Y. Chem Lett. 1981:1069. [Google Scholar]
- 3.(a) Clark RD, Heathcock CH. J Org Chem. 1976;41:1396. doi: 10.1021/jo00870a023. [DOI] [PubMed] [Google Scholar]; (b) Nicalaou KC, Lysenko Z. J Am Chem Soc. 1977;99:3185. [Google Scholar]; (c) Friedrich LE, Lam PYS. J Org Chem. 1981;46:306. [Google Scholar]; (d) Scarborough RM, Smith AB. Tetrahedron Lett. 1977;18:4361. [Google Scholar]; (e) Liotta D, Markiewicz W, Santiesteban H. Tetrahedron Lett. 1977;18:4365. [Google Scholar]
- 4.(a) Reich HJ. J Org Chem. 1975;40:2570. [Google Scholar]; (b) Clive DL, Chittattu G, Curtis NJ, Menchen SM. J Chem Soc, Chem Commun. 1978:770. [Google Scholar]
- 5.Petersen KS, Posner GH. Org Lett. 2008;10:4685. doi: 10.1021/ol8020513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.(b) Tamura Y, Yakura T, Haruta JI, Kita Y. Tetrahedron Lett. 1985;26:3837. [Google Scholar]; (b) Tamura Y, Annoura H, Yamamoto H, Kondo H, Kita Y, Fujioka H. Tetrahedron Lett. 1987;28:5709. [Google Scholar]; (c) Takanami T, Tokoro H, Kato DI, Nishiyama S, Sugai T. Tetrahedron Lett. 2005;46:3291. [Google Scholar]; (d) Hong YT, Cho CW, Skucas E, Krische M. Org Lett. 2007;9:3745. doi: 10.1021/ol7015548. [DOI] [PubMed] [Google Scholar]; (e) Faulkner DJ, Petersen MR. J Am Chem Soc. 1973;95:553. doi: 10.1021/ja00783a040. [DOI] [PubMed] [Google Scholar]; (f) Masamune TI, Ono M, Matsue H. Bull Chem Soc Jpn. 1975 [Google Scholar]; (g) Coppola GM, Schuster HF. α-Hydroxy Acids in Enantioselective Synthesis. VCH; Weinheim: 1997. [Google Scholar]
- 7.(a) Davis FA, Stringer OD, McCauley JP. Tetrahedron. 1985;41:4747. [Google Scholar]; (b) Reich HJ, Yelm KE. J Org Chem. 1992;57:5672. [Google Scholar]; (c) Davis FA, Reddy RT. J Org Chem. 1992;57:2599. [Google Scholar]; (d) Komatsu N, Nishibayashi Y, Sugita T, Uemura SJ. J Chem Soc, Chem Commun. 1992:46. [Google Scholar]; (e) Komatsu N, Matsunaga S, Sugita T, Uemura S. J Org Chem. 1993;58:3697. [Google Scholar]; (f) Komatsu N, Nishibayashi Y, Sugita T, Uemura S. J Am Chem Soc. 1993;115:5847. [Google Scholar]; (g) Komatsu N, Nishibayashi Y, Uemura S. Tetrahedron Lett. 1993;34:2339. [Google Scholar]
- 8.(a) Nishibayashi Y, Singh JD, Fukuzawa S, Uemura S. J Org Chem. 1995;60:4114. [Google Scholar]; (b) Davoren JE, Harcken C, Martin SF. J Org Chem. 2008;73:391. doi: 10.1021/jo701739v. [DOI] [PubMed] [Google Scholar]
- 9.(a) Zoretic PA, Chambers RJ, Marbury GD, Riebiro AA. J Org Chem. 1985;50:2981. [Google Scholar]; (b) Koprowski M, Krawczyk E, Skowronska A, McParlin M, Choi N, Radojevic S. Tetrahedron. 2001;57:1105. [Google Scholar]
- 10.Sunden H, Rios R, Cordova A. Tetrahedron Lett. 2007;48:7865. [Google Scholar]
- 11.Tiecco M, Carlone A, Sternativo S, Marini F, Bartoli G, Melchiorre P. Angew Chem, Int Ed. 2007;46:6882. doi: 10.1002/anie.200702318. [DOI] [PubMed] [Google Scholar]
- 12.Kobayashi J. J Nat Prod. 1989;52:225. doi: 10.1021/np50065a047. [DOI] [PubMed] [Google Scholar]
- 13.(a) Azuma H, Takao R, Niiro H, Shikata K, Tamagaki S, Tachibana T, Ogino K. J Org Chem. 2003;68:2790. doi: 10.1021/jo0206824. [DOI] [PubMed] [Google Scholar]; (b) Yoshida J, Masako N, Seki H, Hino J. J Chem Soc, Perkin Trans 1. 1992;3:343. [Google Scholar]
- 14.Posner GH, Lee JK, White MC, Hutchings RH, Dai H, Kachinski JL, Dolan P, Kensler TW. J Org Chem. 1997;62:3299. doi: 10.1021/jo970049w. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.