

Abstract
Effective diagnosis of inflammation and cancer by molecular imaging is challenging because of interference from non-selective accumulation of the contrast agents in normal tissues. Here we report a series of novel fluorescence imaging agents that efficiently target cyclooxygenase-2 (COX-2), which is normally absent from cells, but is found at high levels in inflammatory lesions, and in many premalignant and malignant tumors. After either intraperitoneal or intravenous injection, these reagents become highly enriched in inflamed or tumor tissue compared to normal tissue and this accumulation provides sufficient signal for in vivo fluorescence imaging. Further, we show that only the intact parent compound is found in the region of interest. COX-2-specific delivery was unambiguously confirmed using animals bearing targeted deletions of COX-2 and by blocking the COX-2 active site with high affinity inhibitors in both in vitro and in vivo models. Because of their high specificity, contrast, and detectability, these COX-2 beacons are ideal candidates for detection of inflammatory lesions or early-stage COX-2-expressing human cancers, such as those in the esophagus, oropharynx, and colon.
Keywords: Cyclooxygenase-2, Molecular Imaging, Targeted, Fluorescence, Inflammation, Premalignancy
Molecular imaging presents exciting opportunities for selective detection of specific cell populations, such as those bearing markers of disease (1, 2). Cyclooxygenase-2 (COX-2) is an attractive target for molecular imaging because it is expressed in only a few normal tissues and is greatly up-regulated in inflamed tissues as well as many premalignant and malignant tumors (3, 4). COX-2 is an important contributor to the etiology of inflammation and cancer as illustrated by the efficacy of COX-2-selective inhibitors as anti-inflammatory agents, cancer preventive agents, and adjuvant cancer therapeutic agents (5). The importance of COX-2 in tumor progression has been thoroughly documented in the esophagus and colon where COX-2 is detected in premalignant lesions and its levels appear to increase during tumor progression (6–8). The importance of COX-2 in survival and response to therapy has been elegantly demonstrated by Edelman et al who reported that non-small cell lung cancer patients expressing high levels of COX-2 in their tumors have reduced survival compared to patients expressing low levels of COX-2 (9). Patients with high tumor expression of COX-2 benefit from the combination of carboplatin and gemcitabine plus the COX-2 inhibitor, celecoxib, whereas patients with low expression exhibit a poorer response to carboplatin/gemcitabine/ celecoxib than to carboplatin/gemcitabine alone (9).
PET or SPECT imaging agents (18F-, 11C-, or 123I-labeled COX-2 inhibitors) have been described for nuclear imaging (10–17). These have all been based on the diarylheterocycle structural class analogous to celecoxib and rofecoxib. Although selective uptake into macrophages or tumor cells expressing COX-2 has been demonstrated in vitro for some compounds, such selectivity has not been rigorously demonstrated in vivo and significant non-specific binding has been observed. (18). Thus, despite recognition of the potential of COX-2-targeted imaging agents, in vivo proof-of-concept for this strategy is lacking.
Fluorescent COX-2 inhibitors are attractive candidates as targeted imaging agents. Such compounds have the advantage that each molecule bears the fluorescent tag and the compounds are non-radioactive and stable. Thus, they can be used conveniently for cellular imaging, animal imaging, and clinical imaging of tissues where topical or endoluminal illumination is possible (e.g., esophagus, colon, and upper airway via endoscopy, colonoscopy, and bronchoscopy, respectively). Prior work from our laboratory demonstrated that fluorescent COX-2 inhibitors can be useful biochemical probes of protein binding but these earlier compounds were neither potent inhibitors of COX-2 nor did they possess appropriate fluorescence properties to be useful for cellular or in vivo imaging (19). Thus, we initiated a program to design and synthesize a series of fluorescent COX-2 inhibitors that could be used for these applications. The design strategy for candidate development was based on our prior discovery that amide derivatives of the non-selective COX inhibitor, indomethacin, are selective COX-2 inhibitors. Many compounds were synthesized and screened for COX-2 inhibition in vitro and in intact cells. Then the most promising compounds were evaluated as imaging agents in intact cells and in animal models of inflammation and cancer. We describe herein the optimized candidates, their selective uptake by COX-2-expressing cells and tumors, and genetic and pharmacological validation that their in vivo target is COX-2.
Materials and Methods
Synthesis and characterization of all compounds is described in Supplemental Data.
Inhibition assay using purified COX-1 and COX-2
Cyclooxygenase activity of ovine COX-1 or human COX-2 was assayed by a method that quantifies conversion of [1-14C]arachidonic acid to [1-14C]prostaglandin products. Reaction mixtures of 200 µL consisted of hematin-reconstituted protein in 100 mM Tris-HCl, pH 8.0, 500 µM phenol, and [1-14C]arachidonic acid (50 µM, ~55–57 mCi/mmol, Perkin Elmer). For the time-dependent inhibition assay, hematin-reconstituted COX-1 (44 nM) or COX-2 (66 nM) was preincubated at 25°C for 17 min and 37 °C for 3 min with varying inhibitor concentrations in DMSO followed by the addition of [1-14C]arachidonic acid (50 µM) for 30 s at 37 °C. Reactions were terminated by solvent extraction in Et2O/CH3OH/1 M citrate, pH 4.0 (30:4:1). The phases were separated by centrifugation at 2000g for 2 min and the organic phase was spotted on a TLC plate (EMD Kieselgel 60, VWR). The plate was developed in EtOAc/CH2Cl2/glacial AcOH (75:25:1) at 4 °C. Radiolabeled products were quantified with a radioactivity scanner (Bioscan, Inc., Washington, D.C.). The percentage of total products observed at different inhibitor concentrations was divided by the percentage of products observed for protein samples preincubated for the same time with DMSO.
Cell Culture and In Vitro Intact Cell Metabolism Assay
HCT116, ATCC CCL-247™ human colorectal carcinoma cells, passage 8–18, mycoplasma negative by a pcr detection method (Sigma VenorGem,) were grown in DMEM (Invitrogen/Gibco)+10% FBS (Atlas) to 70% confluence. RAW264.7, ATCC TIB-71™ murine macrophage-like cells, passage number 8–15, mycoplasma negative by a pcr detection method were grown in DMEM+10% heat-inactivated FBS to 40% confluence (6-well plates, Sarstedt) and activated for 7 hrs in 2 ml serum-free DMEM with 200 ng/ml LPS (Calbiochem) and 10u/ml interferon gamma (Calbiochem). Human head and neck squamous cell carcinoma (HNSCC), 1483 cells (20), derived, characterized and provided by Dr. Peter Sacks, were grown at passage 8–18, mycoplasma negative by a pcr detection method, in DMEM/F12+10% FBS+ Antibiotic/Antimycotic in 6-well plates to 60% confluence. Serum-free medium (2 ml) was added, and the cells were treated with inhibitor dissolved in DMSO (0 – 5 µM, final concentration) for 30 min at 37°C followed by the addition of [1-14C]-arachidonic acid [10 µM, ~55 mCi/mmol] for 20 min at 37°C. Reactions were terminated by solvent extraction in Et2O/CH3OH/1 M citrate, pH 4.0 (30:4:1), and the organic phase was spotted on a 20×20 cm TLC plate (EMD Kieselgel 60, VWR). The plate was developed in EtOAc/CH2Cl2/glacial AcOH (75:25:1), and radiolabeled products were quantified with a radioactivity scanner (Bioscan, Inc., Washington, D.C.). The percentage of total products observed at different inhibitor concentrations was divided by the percentage of products observed for cells pre-incubated with DMSO.
Fluorescence microscopy of RAW264.7 cells or 1483 HNSCC cells
RAW264.7 cells were plated on 35 mm MatTek dishes (MatTek Corporation, Ashland, MA) such that the cells were 40% confluent and human 1483 HNSCC cells were 60% confluent on the day of the experiment. The RAW264.7 cells were activated for 6 hrs in serum-free DMEM with 200 ng/ml LPS and 10 u/ml interferon gamma. Both cell lines were incubated in 2.0 ml HBSS/Tyrode’s with 200 nM compound 1, 2, or 3 for 30 min at 37°C. To block the COX-2 active site, the cells were preincubated with 10 µM indomethacin or 5 µM celecoxib for 20 min prior to the addition of compound 1 or 2. The cells were then washed briefly three times and incubated in HBSS/Tyrode’s for 30 min at 37°C. Following the required washout period, the cells were imaged in 2.0 ml fresh HBSS/Tyrode’s on a Zeiss Axiovert 25 Microscope with the propidium iodide filter (0.5–1.0 sec exposure, gain of 2). All treatments were performed in duplicate dishes in at least three separate experiments.
Confocal microscopy of 1483 cells treated with compound 2/mitotrackerGR
1483 HNSCC were plated in MatTek dishes (MatTek Corporation, Ashland, MA) and grown to 60–70% confluence for 48 hr. DMSO or compound 2 (100 nM) was added to each dish containing 2.0 ml HBSS/Tyrode’s for 30 min at 37°C. After four quick HBSS washes, cells were incubated for 30 min in 2.0 ml HBSS/Tyrode’s and imaged with a Zeiss LSM510 confocal microscope using a 63 × 1.4 NA plan-Apochromat objective lens. To visualize cellular mitochondria, 100 nM Mitotracker GR was added for 15 min at 37°C, followed by 3 quick washes before imaging. 488 nm excitation was used to image Mitotracker GR through a 500–530 nm bandpass filter, and compound 2 was imaged using 532 nm excitation and collection through a 565–615 nm bandpass filter. The pinhole was set to 1 Airy unit and images were collected throughout the focus of the cells. To assure a full sampling of the perinuclear region, analysis was performed on the optical sections through the middle of the nucleus.
In vivo imaging of COX-2 in inflammation
Carrageenan (50 µL 1% in sterile saline) was injected in the rear left footpad of female C57BL/6 mice, followed by compound 1 or 2 (1 mg/kg, i.p.) at 24 hr post-carrageenan. Animals were imaged 3 hr later in a Xenogen IVIS 200 (DsRed filter, 1.5 cm depth, 1 sec). For comparison, animals also were dosed with compound 3, which does not inhibit COX-2. To test further the molecular target for compound 2, parallel experiments were performed using COX-2 (−/−) mice. Experiments also were performed in which compound 1 was administered to the same animals by repetitive i.p. injection on days 1,3,5, and 7 to monitor the time course of compound uptake following carrageenan induction of inflammation.
Establishment of xenografts in nude mice
Female nude mice, NU-Fox1nu, were purchased at 6–7 weeks of age from Charles River Labs. Human 1483 HNSCC cells and HCT116 colorectal carcinoma cells were trypsinized and resuspended in cold PBS containing 30% Matrigel such that 1×106 cells in 100 µl were injected subcutaneously on the left flank. The HCT116 or 1483 xenografts required only 2–3 weeks of growth.
In vivo imaging of nude mice with xenografts
Female nude mice bearing medium-sized HCT116 or 1483 xenograft tumors on the left flank were dosed by intraperitoneal injection with 2 mg/kg compound 2 or by retro-orbital injection with 1 mg/kg compound 2. The animals were lightly anesthetized with 2% isoflurane for fluorescence imaging in the Xenogen IVIS 200 with the DSRed filter at 1.5 cm depth and 1 sec exposure (f2). For the COX-2 active site-blocking experiments, nude mice bearing 1483 xenografts were pre-dosed by i.p. injection with 2 mg/kg indomethacin at 24 hr and 1 hr prior to dosing with compound 2 (2 mg/kg, i.p.).
Pharmacokinetics of candidate compounds
Female nude mice with medium-sized 1483 HNSCC xenograft tumors on the left flank were injected i.p. with 2 mg/kg compound 2. At 0, 0.5 hr, 3 hrs, 12 hrs, and 24 hrs, the mice (n=4 for each time point, duplicate experiments) were anesthetized with isoflurane. Blood samples were taken by cardiac puncture into a heparinized syringe into a 1.5 ml heparinized tube on ice, followed by removal of the liver, kidney, contralateral leg muscle, and xenograft tumor. All organs/tissues were rinsed briefly in ice-cold PBS, blotted dry, weighed, and snap-frozen in liquid nitrogen. The blood samples were centrifuged at 4°C at 6000 rpm for 5 min, and the plasma was transferred to clean tubes and frozen at −80°C. Compound 2 was extracted by homogenizing the tissue in 100 mM Tris, pH 7.0, buffer and mixing an aliquot of the homogenate with 1.2x volume of acetonitrile. The acetonitrile was removed and the samples were dried, reconstituted and analyzed via reversed phase HPLC-UV using a Phenomenex 10 × 0.2 cm C18 or a Phenomenex 7.5 × 0.2 cm Synergi Hydro-RP column held at 40°C. The samples were quantified against a standard curve prepared by adding 2 to tissue homogenates of un-dosed animals followed by the workup described. Co-chromatography was performed with multiple columns and elution conditions as described in Supplemental Data.
In Vivo Imaging of Min Mice
C57BL/6 APC-Min mice maintained on a high fat (11%) diet for 18 weeks developed 20–30 intestinal polyps per mouse. Prior to imaging, Min mice were anesthetized (2% inhaled isoflurane) for retro-orbital injection of compound 2 at 1 mg/kg. At 2 hrs post-injection, the mice were euthanized, and the intestines were resected, washed with PBS, and fixed in 10 % formalin prior to ex vivo imaging by fluorescence dissecting microscopy (Zeiss M2Bio, Thornwood, NY) (n=5).
Results
COX-2 is a potentially ideal target for molecular imaging because its active site (and the active site of COX-1) is buried deep inside each subunit of the homodimeric protein (21–23). Access to the active site is controlled by a constriction that separates it from a large opening in the membrane-binding domain that we have termed the lobby (Supplemental Data, Figure 1). All substrates or inhibitors bind in the lobby and then diffuse through the constriction into the active site (24). The constriction is comprised of Tyr-355, Glu-524, and Arg-120 and serves as the binding site for the carboxylic acid group of substrates and certain inhibitors (25). We have reported that neutral derivatives (esters and amides) of certain carboxylic acid inhibitors (e.g., indomethacin) bind to COX-2 but not to COX-1 (26). A three-dimensional structure of COX-2 complexed to such a conjugate has not been solved but structures of related complexes suggest the indomethacin unit binds in the active site with the tethered amide breeching the constriction and projecting into the lobby (22, 27). These structural and functional analyses provide the design principles for construction of COX-2-targeted imaging agents.
Figure 1. Labeling of COX-2-expressing cells by compound 2.

The experimental protocols are described in Materials and Methods. a, 1483 HNSCC cells treated with 200 nM compound 2 for 30 min. b, 1483 HNSCC cells pretreated with 5 µM celecoxib for 20 min prior to compound 2 treatment. c, Confocal microscopy of 1483 HNSCC cells treated with both mitotrackerGR in blue (mitochondria) and compound 2 in red (perinuclear).
Synthesis of Candidate Compounds and Cellular Imaging
Three carboxylic acid cores – i.e., indomethacin, a celecoxib carboxylic acid derivative, and an indolyl carboxamide analog of indomethacin – were tethered through a series of alkylenediamines, piperazines, polyethylene glycol, or phenylenediamines to a diverse range of fluorophores. The fluorophores attached included dansyl, dabsyl, coumarin, fluorescein, rhodamine, alexa-fluor, nile blue, cy5, cy7, near IR and IR dyes as well as lanthanide chelators. Nearly 200 compounds were synthesized and each conjugate was tested for its ability to selectively inhibit COX-2 in assays using purified proteins in vitro. Promising molecules were tested for their ability to inhibit COX-2 in lipopolysaccharide-treated RAW264.7 macrophages. Preliminary experiments indicated that indomethacin conjugates bound most tightly and selectively to COX-2, therefore most of the compounds synthesized were derived from this core.
Indomethacin conjugates to dansyl, dabsyl, coumarin, fluorescein, and rhodamine-derived fluorophores exhibited promising COX-2 inhibition and selectivity both in vitro and in intact cells. The carboxy-X-rhodamine (6-ROX- and 5-ROX)-based conjugates, 1 and 2, displayed the best balance of cellular activity and optical properties (λex = 581 nm, λemit = 603 nm) and were used for all subsequent experiments (Table 1). A detailed kinetic analysis indicated that 1 and 2 require lengthy preincubations with COX-2 to achieve maximal inhibition but once bound they dissociate very slowly (Supplemental Data, Figure 2). Thus, they are slow, tight-binding inhibitors with very low rates of association and dissociation. Compounds 1 and 2 were less potent that celecoxib or rofecoxib as inhibitors of COX-2 (Table 1). A negative control molecule (3) was synthesized that contained 6-ROX bound to indomethacin through a shorter ethylenediamine tether, which eliminated COX-2 inhibition (Table 1).
Table 1. Biochemical properties of carboxy-X-rhodamine derivatives.
Assays were conducted as described in Materials and Methods. IC50’s for inhibition of ovine COX-1 or human COX-2. Compounds also were tested in intact RAW264.7 macrophages (Cell IC50). ND – no inhibition detected up to 5 µM. NT – not tested
| Compd No. |
Structure | Purified Enzyme IC50(µM) |
Cell IC50 (µM) |
|
|---|---|---|---|---|
| COX-1 | COX-1 | |||
| 1 |
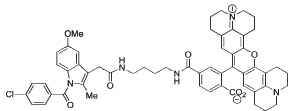
|
> 66 | 0.83 | 0.36 |
| 2 | 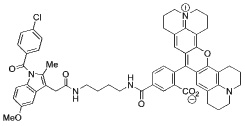 |
> 66 | 0.7 | 0.31 |
| 3 | 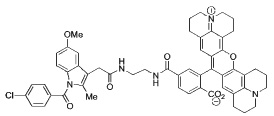 |
> 66 | > 4 | ND |
| 4 | 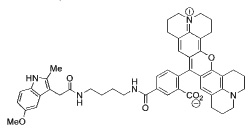 |
> 66 | > 4 | ND |
| 5 | 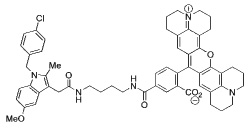 |
> 66 | > 4 | ND |
| 6 | 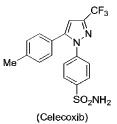 |
0.92 | 0.21 | NT |
| 7 | 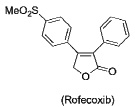 |
> 66 | 0.14 | NT |
| 8 | 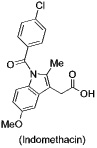 |
0.05 | 0.75 | 0.01 |
Figure 2. In vivo labeling of COX-2-expression in inflammation by compound 1, 2, or 3.

a, C57BL/6 mouse with carrageenan-induced inflammation in the left foot pad. The left mouse was dosed with the negative control molecule 3 (1 mg/kg, i.p.) and the right mouse was dosed with compound 1 (1 mg/kg, i.p.). Both mice were imaged at 3 hr post-injection. b, Fold increase of fluorescence in inflamed vs. contralateral paw of wild-type and COX-2(−/−) mice at 3 hr post-injection of compound 2 (1 mg/kg, i.p.) (n = 6). c, Carrageenan was injected in the rear left footpads of female C57BL/6 mice, followed by dosing compound 1 (1 mg/kg i.p.) 24 hr later. Animals were reinjected with compound 1 at 3, 5, and 7 days post-carrageenan (n = 9). Mice were imaged at 3 hr after compound injection. The plot shows the fold-increase of fluorescence in swollen vs. contralateral foot (n=6).
The human head and neck cancer cell line, 1483, which expresses high levels of COX-2 (28), exhibited strong labeling with compounds 1 or 2 (Figure 1a). Preincubation of the cells with the COX-2-selective inhibitor, celecoxib, prevented labeling of 1483 cells by either compound (Figure 1b). In all of these in vitro experiments, the labeling appeared to be intracellular, so confocal microscopy was performed to verify the localization. Incubation of compound 2 with 1483 cells led to perinuclear labeling of membraneous structures that appeared to be endoplasmic reticulum or Golgi (Figure 1c). The perinuclear labeling correlated well to multiple previous reports of the intracellular localization of COX-2 (29–32). Incubation of the same cells with Mitotracker™ showed that the mitochondria did not co-localize with compound 2 (Figure 1c).
The mouse macrophage-like cell line, RAW 264.7, does not express COX-2 and exhibited very weak labeling with 1 or 2 (e.g., Supplemental Data, Figure 3a) whereas lipopolysaccharide-pretreated cells labeled more strongly (Supplemental Data, Figure 3b). The labeling of the COX-2 expressing RAW cells by 1 or 2 was prevented by pretreatment of the cells with indomethacin (Supplemental Data, Figure 3c) or celecoxib, which block the COX-2 active site. Importantly, no labeling was observed when either control or lipopolysaccharide-pretreated RAW cells were incubated with compound 3, which does not inhibit COX-2 (Supplemental Data, Figure 3d). The extent of compound 2 uptake increased at 4 hr with the appearance of COX-2 protein. A further increase in uptake was not observed at 7 hr although there was higher COX-2 protein as detected by Western blotting (Supplemental Data, Figure 4). Comparison of the amount of 2 taken up at 7 hr to the amount of COX-2 estimated by Western blotting in the lipopolysaccharide-treated cells suggested a stoichiometry of binding of 0.90 (Supplemental Data).
Figure 3. In vivo labeling of COX-2-expressing xenografts by compound 2.

a, Nude mice with HCT116 xenograft (COX-2 negative), or b, 1483 xenograft (COX-2 positive) were dosed (retro-orbital) with 1 mg/kg compound 2 and imaged at 3.5 hrs post-injection. c, Nude mice with 1483 xenografts were pre-dosed with DMSO prior to injection of compound 2 (2 mg/kg, intraperitoneal), or d, pre-dosed with indomethacin (2 mg/kg, intraperitoneal) 24 hrs and 1 hr prior to compound 2 and imaged at 3 hr post-injection (Xenogen IVIS, DsRed filter, 1 s, f2, 1.5 cm depth). The emission observed around the peritoneal cavity in Figures 3c and d is due to residual 2 at the site of injection.
Figure 4. Analysis of fluorescent material in xenografts and several mouse tissues.

a, representative HPLC-UV chromatogram (detection = 581 nm) of 1483 tumor extract (4 hr post-administration) revealed a single major fluorescent compound that coeluted with compound 2 (15.3 min). The peak eluting at 13.3 min integrates for less than 5% of the peak at 15.3 min. Non-specific fluorescent peaks were eluted at or near the void volume of the column. Inset – the Q1 mass spectrum of the extracted fluorescent material was identical to that of an authentic standard of 2. b, Time-course and distribution of compound 2 in various mouse tissues in vivo.
Imaging Carrageenan-Induced Inflammation
Compounds 1 and 2 appeared promising based on these in vitro imaging experiments, so their potential for in vivo imaging was evaluated using carrageenan-induced inflammation in the mouse footpad, human tumor xenografts in nude mice, and spontaneous tumors arising in mouse models. The mouse footpad model is well-documented for the role of COX-2-derived prostaglandins as a major driving force for the acute edema that results 24 hr after carrageenan injection into the paw (33). One of the significant advantages of this animal model of inflammation is the ability to image the inflamed footpad in comparison to the non-inflamed contralateral footpad, which does not express COX-2. We injected female C57BL/6 mice with 50 µl 1% carrageenan in the rear left footpad, followed by compound 1 or 2 (1 mg/kg, i.p.) at 24 hrs post-carrageenan. Animals were imaged 3 hrs later in a Xenogen IVIS 200 (DsRed filter, 1.5 cm depth, 1 sec). Both compounds 1 and 2 targeted the swollen footpad with an average 4.5-fold increase in fluorescence over that of the contralateral, uninjected footpad (Figure 2). For comparison, animals also were dosed with compound 3, which does not inhibit COX-2. In Figure 2a, the left mouse was dosed with compound 3 and the right mouse with compound 1. Compound 3 yielded minimal fluorescence in the inflamed paw compared to the contralateral paw whereas compound 1 yielded a strong signal in the inflamed paw. To test further the molecular target of compounds 1 or 2, parallel experiments were performed using mice bearing targeted deletions in COX-2. Figure 2b depicts the fold difference in the compound 2-derived fluorescence signal in the 24-hr carrageenan-injected footpad over the control footpad for wild type vs. COX-2 (−/−) mice. The COX-2 null mice consistently showed approximately a 40% increase in signal in the swollen footpad apparently due to non-specific binding. This contrasts with a 400–600% increase in the swollen footpad in wild-type mice. Finally, experiments were conducted to evaluate the uptake of compound 1 during resolution of inflammation. Following carrageenan injection, 1 was administered i.p. 1, 3, 5, and 7 days later and the animals were imaged. Uptake of 1 was maximal at 24 hr but declined thereafter (Figure 2c). Attempts to estimate active COX-2 protein by quantification of prostaglandins in paw extracts were unsuccessful because of poor recovery.
Imaging COX-2-Expressing Tumors
The results in the footpad inflammation model demonstrate that COX-2-targeted fluorescent conjugates are taken up in inflamed paws of COX-2 expressing mice but not in COX-2 null animals. We next evaluated the ability of these compounds to target COX-2 in human tumor xenografts. Female nude mice were injected in the left flank with HCT-116 or 1483 cells and the xenografts were allowed to grow to approximately 750–1000 mm3. Animals were dosed by retroorbital injection with compound 2 (1 mg/kg) then lightly anesthetized with 2% isoflurane in preparation for imaging. No fluorescence was observed during the first 60 min post-injection, but signal was reproducibly detected in the COX-2 expressing 1483 tumors starting at 3–5 hr and persisted as long as 26 hr post-injection. At 3.5 hr post-injection, the HCT116 tumor, which does not express COX-2 (34), showed minimal fluorescence (Figure 3a) whereas the 1483 tumor exhibited bright fluorescence (Figure 3b). In another control experiment, nude mice bearing 1483 xenografts were treated with the fluorophore alone, 5-ROX (2 mg/kg, intraperitoneal), which is neither an inhibitor of COX-2 nor COX-1. No signal from 5-ROX alone accumulated in the tumors at any time point. This result demonstrated that the fluorophore moiety was not responsible for the tumor uptake of compound 2 supporting the conclusion that the difference in labeling of 1483 and HCT116 xenografts is due their differential in COX-2 expression.
Nude mice with 1483 xenografts were pretreated with either DMSO or indomethacin in DMSO (2 mg/kg, intraperitoneal) prior to compound 2 dosing (2 mg/kg, intraperitoneal). At 3 hr post-injection, the DMSO-pretreated mice showed strong fluorescence in their tumors (Figure 3c) compared to weak signals in the tumors of the indomethacin-pretreated mice (Figure 3d). In the mouse xenograft model, indomethacin was able to block 92 ± 6% (n = 8) of the COX-2- expressing tumor uptake of compound 2.
We next investigated whether the COX-2 inhibitory activity of our imaging probes correlated with their in vivo efficacy in targeting COX-2-expressing tumors. Nude mice bearing 1483 xenografts were dosed (2 mg/kg,intraperitoneal) with compound 4 (no COX inhibition at 3 µM), compound 5 (30% COX-2 inhibition at 3 µM), and compound 2 (90% COX-2 inhibition at 3 µM) (Table 1). At 3.5 hr post-injection, fluorescence from the tumor region was directly proportional to the compound potency as a COX-2 inhibitor (Supplemental Data, Figure 5).
Figure 5. In vivo labeling of COX-2-expression in intestinal polyps by compound 2.

C57BL/6J-Min/+ mice bearing small intestinal polyps were euthanized at 2 hr after retro-orbital injection of compound 2 (1 mg/kg), and small intestines were washed, opened, and examined by dissecting fluorescence microscopy. a, Section of small intestine with no polyp, 90 ms exposure. b, Single polyp, 90 ms exposure. c, Polyp cluster, 90 ms exposure.
Experiments were conducted to determine the identity of the fluorescent material(s) detected in vivo and to monitor the time course of its distribution and tissue uptake following injection of compound 2 into nude mice bearing 1483 human tumor xenografts. Extracts of plasma, liver, kidney, tumor, and adjacent muscle were quantitatively analyzed by HPLC at different times after intraperitoneal administration of the compound. A single fluorescent compound was detected in all the extracts, which coeluted with a standard of 2 in multiple HPLC systems. This compound displayed an identical mass spectrum to the unmetabolized parent molecule, 2 (Figure 4a). The time courses of uptake and distribution of compound 2 in plasma and various tissues are displayed in Figure 4b. Compound 2 was rapidly distributed following ip administration and reached nearly maximal levels in plasma, liver, and kidney 30 min after injection. Compound 2 levels declined substantially over the course of 12–24 hr to a small fraction of its initial levels in all three of these compartments. In contrast, the time course for uptake of compound 2 into the 1483 tumors lagged substantially and required approximately 3 hr to reach near maximal levels. The levels of 2 remained relatively high in the tumor so by 24 hours the tumor levels were as high as the levels in liver or kidney. This indicates both slow uptake and release of 2 into and out of the tumor.
APCmin mice bear the same mutation (Apc−) that is causative for familial adenomatous polyposis in human beings and these mice primarily develop small intestinal tumors that express COX-2 (35, 36). Crossing APCmin mice with COX-2 (−/−) mice reduces intestinal tumor development by 85% and treatment of APCmin mice with COX-2 inhibitors also reduces tumorigenesis (37, 38). APCmin mice (18–20 weeks old, fed a high-fat diet) were injected retroorbitally with compound 2 (1 mg/kg) and after 2 hr, animals were sacrificed and their intestines removed. The tissue was washed thoroughly with PBS, opened longitudinally, and imaged. Figure 5a shows the low background fluorescence of a section of small intestine without polyps. A single polyp (Figure 5b) and a 5-polyp cluster (Figure 5c) displayed high fluorescence, with greatly increased detection compared to bright field visualization..The signal enrichment of compound 2 in the polyps was estimated to be > 50:1. COX-2 expression in the polyps appears to be required for this selective uptake although other factors beside the level of COX-2 protein may contribute to the relative enrichment over surrounding normal tissue.
Discussion
These studies demonstrate the feasibility of specific in vivo targeting of COX-2 in inflammatory lesions and tumors using organic fluorophores tethered to indomethacin through an amide linkage. Compounds 1 and 2 display a very high degree of selectivity of uptake by inflammatory tissue and tumors in live animals relative to surrounding normal tissue or muscle as determined by either imaging or mass spectrometry. This selectivity appears greater than that reported in previous literature reports of fluorescent tumor imaging in which the ratio of tumor fluourescence was compared to muscle fluorescence (39). Uptake of our compounds requires the expression of COX-2 at the target site and declines as the level of COX-2 decreases. Although uptake into inflamed or tumor tissue appears to be slower than expected from simple distribution in the body, the kinetics of compound release appear to be extremely slow, thus leading to a detectable buildup of the label. Similar results are observed by both imaging compounds 1 or 2 (Figures 2 and 3) or by direct quantitative analysis of compound 2 (Figure 4 and Supplemental Data Figure S4).
To achieve this success, nearly two hundred compounds were evaluated as candidate COX-2-targeted imaging agents. While a significant percentage demonstrated COX-2 inhibitory activity against purified protein, only a fraction of these compounds inhibited COX-2 activity in intact cells, and of those, most did not possess fluorescence properties suitable for in vivo imaging. Among the compounds that emerged from our development pathway, only compounds 1 and 2 exhibited sufficient metabolic stability to survive long enough to distribute to inflammatory lesions or xenograft tumors. The low overall success rate (~1%) likely underscores why COX-2-targeted imaging agents have proven difficult to develop.
The specificity for COX-2 binding of these compounds was illustrated by multiple observations: (a) only cultured cells that express COX-2 took up COX-2 beacons and uptake was inhibited by the COX inhibitors indomethacin and celecoxib. The intracellular localization of the probes matches that of COX-2 protein and the stoichiometry of uptake was approximately 0.9 molecule of beacon per subunit; (b) uptake into inflamed over non-inflamed tissue was blocked by indomethacin pretreatment of the animals and was not observed in COX-2 (−/−) animals. No uptake was observed with a close structural analog of 1 that does not inhibit COX-2; (c) uptake into COX-2 expressing tumors was blocked by indomethacin pretreatment of the animals and a correlation was found between the amount of light emission from the tumor and the COX-2 inhibitory potency of the beacon. The non-targeted fluorophores 5-ROX and 6-ROX did not accumulate in COX-2-expressing xenografts; and (d) greater than 95% of the fluorescent material present in the tumors is the unmetabolized parent compound. Thus, in vitro and in vivo studies provide strong support for the conclusion that binding to COX-2 is the major determinant of uptake into inflamed, premalignant, or malignant tissue. Although the stoichiometry of compound 2 binding to COX-2 protein was estimated to be 0.9 in activated RAW cells, such high stoichiometry cannot be assumed in all situations. The extent of uptake in cells, inflamed tissue, or tumors will depend upon a number of factors such as the permeability of COX-2- expressing cells to the probe, kinetics of binding and release from the COX-2 active site, vascularization of the tissue, and possible expulsion of the probes by transporters. Further studies will be needed to explore in greater detail the quantitative aspects of the use of these compounds in vivo.
Compounds 1 and 2 represent the first feasible reagents for clinical detection of tissues containing high levels of COX-2 in settings amenable to fluorescent excitation and analysis by surface measurement or endoscopy (e.g., skin, esophagus, intestine, bladder). Although such COX-2 beacons will not be useful for applications in internal organs that are not accessible for optical imaging, their development provides rigorous proof-of-concept for the feasibility of molecular targeting of COX-2 in inflammatory lesions, premalignant lesions, and tumors.
Supplementary Material
Acknowledgements
We are grateful to to S.K.Dey for COX-2 null animals and to Melissa Turman for assistance with molecular graphics.
Footnotes
This work was supported by research and center grants from the National Institutes of Health (CA86283, CA89450, CA105296, CA68485, CA60867, CA126588, CA111469, and GM72048), the Medical Free-Electron Laser Program of the US Department of Defense, XL TechGroup, and New York Crohn’s Foundation.
References
- 1.Weissleder R, Ntziachristos V. Shedding light onto live molecular targets. NatMed. 2003;9:123–128. doi: 10.1038/nm0103-123. [DOI] [PubMed] [Google Scholar]
- 2.Tanabe K, Zhang Z, Ito T, Hatta H, Nishimoto S. Current molecular design of intelligent drugs and imaging probes targeting tumor-specific microenvironments. OrgBiomolChem. 2007;5:3745–3757. doi: 10.1039/b711244k. [DOI] [PubMed] [Google Scholar]
- 3.Crofford LJ. COX-1 and COX-2 tissue expression: implications and predictions. The JRheumatol. 1997;49:15–19. [PubMed] [Google Scholar]
- 4.Dannenberg AJ, Lippman SM, Mann JR, Subbaramaiah K, DuBois RN. Cyclooxygenase-2 and epidermal growth factor receptor: pharmacologic targets for chemoprevention. JClinOncol. 2005;23:254–266. doi: 10.1200/JCO.2005.09.112. [DOI] [PubMed] [Google Scholar]
- 5.Marnett LJ. The COXIB Experience: A Look in the Rear-View Mirror. AnnuRevPharmacolToxicol. 2009;49:265–290. doi: 10.1146/annurev.pharmtox.011008.145638. [DOI] [PubMed] [Google Scholar]
- 6.Kandil HM, Tanner G, Smalley W, Halter S, Radhika A, Dubois RN. Cyclooxygenase-2 expression in Barrett's esophagus. DigDisSci. 2001;46:785–789. doi: 10.1023/a:1010700400960. [DOI] [PubMed] [Google Scholar]
- 7.Eberhart CE, Coffey RJ, Radhika A, Giardiello FM, Ferrenbach S, DuBois RN. Up-regulation of cyclooxygenase 2 gene expression in human colorectal adenomas and adenocarcinomas. Gastroenterology. 1994;107:1183–1188. doi: 10.1016/0016-5085(94)90246-1. [DOI] [PubMed] [Google Scholar]
- 8.Kargman SL, O'Neill GP, Vickers PJ, Evans JF, Mancini JA, Jothy S. Expression of prostaglandin G/H synthase-1 and -2 protein in human colon cancer. Cancer Res. 1995;55:2556–2559. [PubMed] [Google Scholar]
- 9.Edelman MJ, Watson D, Wang X, et al. Eicosanoid modulation in advanced lung cancer: cyclooxygenase-2 expression is a positive predictive factor for celecoxib + chemotherapy-- Cancer and Leukemia Group B Trial 30203. JClinOncol. 2008;26:848–855. doi: 10.1200/JCO.2007.13.8081. [DOI] [PubMed] [Google Scholar]
- 10.Toyokuni T, Kumar JS, Walsh JC, et al. Synthesis of 4-(5-[18F]fluoromethyl-3-phenylisoxazol-4-yl)benzenesulfonamide, a new [18F]fluorinated analogue of valdecoxib, as a potential radiotracer for imaging cyclooxygenase-2 with positron emission tomography. BioorgMedChemLett. 2005;15:4699–4702. doi: 10.1016/j.bmcl.2005.07.065. [DOI] [PubMed] [Google Scholar]
- 11.Tanaka M, Fujisaki Y, Kawamura K, et al. Radiosynthesis and evaluation of 11C-labeled diaryl-substituted imidazole and indole derivatives for mapping cyclooxygenase-2. BiolPharmBull. 2006;29:2087–2094. doi: 10.1248/bpb.29.2087. [DOI] [PubMed] [Google Scholar]
- 12.Majo VJ, Prabhakaran J, Simpson NR, Van Heertum RL, Mann JJ, Kumar JS. A general method for the synthesis of aryl [11C]methylsulfones: potential PET probes for imaging cyclooxygenase-2 expression. BioorgMedChemLett. 2005;15:4268–4271. doi: 10.1016/j.bmcl.2005.06.080. [DOI] [PubMed] [Google Scholar]
- 13.Prabhakaran J, Underwood MD, Parsey RV, et al. Synthesis and in vivo evaluation of [18F]-4-[5-(4-methylphenyl)-3-(trifluoromethyl)-1H-pyrazol-1-yl]benzenesulfonamide as a PET imaging probe for COX-2 expression. BioorgMedChem. 2007;15:1802–1807. doi: 10.1016/j.bmc.2006.11.033. [DOI] [PubMed] [Google Scholar]
- 14.Wuest F, Kniess T, Bergmann R, Pietzsch J. Synthesis and evaluation in vitro and in vivo of a 11C-labeled cyclooxygenase-2 (COX-2) inhibitor. BioorgMedChem. 2008;16:7662–7670. doi: 10.1016/j.bmc.2008.07.016. [DOI] [PubMed] [Google Scholar]
- 15.Schuller HM, Kabalka G, Smith G, Mereddy A, Akula M, Cekanova M. Detection of overexpressed COX-2 in precancerous lesions of hamster pancreas and lungs by molecular imaging: implications for early diagnosis and prevention. ChemMedChem. 2006;1:603–610. doi: 10.1002/cmdc.200500032. [DOI] [PubMed] [Google Scholar]
- 16.de Vries EF, van Waarde A, Buursma AR, Vaalburg W. Synthesis and in vivo evaluation of 18F-desbromo-DuP-697 as a PET tracer for cyclooxygenase-2 expression. JNuclMed. 2003;44:1700–1706. [PubMed] [Google Scholar]
- 17.de Vries EF, Doorduin J, Dierckx RA, van Waarde A. Evaluation of [11C]rofecoxib as PET tracer for cyclooxygenase 2 overexpression in rat models of inflammation. NuclMedBiol. 2008;35:35–42. doi: 10.1016/j.nucmedbio.2007.07.015. [DOI] [PubMed] [Google Scholar]
- 18.de Vries EF. Imaging of cyclooxygenase-2 (COX-2) expression: potential use in diagnosis and drug evaluation. CurrPharmDes. 2006;12:3847–3856. doi: 10.2174/138161206778559650. [DOI] [PubMed] [Google Scholar]
- 19.Timofeevski SL, Prusakiewicz JJ, Rouzer CA, Marnett LJ. Isoform-selective interaction of cyclooxygenase-2 with indomethacin amides studied by real-time fluorescence, inhibition kinetics, and site-directed mutagenesis. Biochemistry. 2002;41:9654–9662. doi: 10.1021/bi0203637. [DOI] [PubMed] [Google Scholar]
- 20.Sacks PG, Parnes SM, Gallick GE, et al. Establishment and characterization of two new squamous cell carcinoma cell lines derived from tumors of the head and neck. Cancer Res. 1988;48:2858–2866. [PubMed] [Google Scholar]
- 21.Picot D, Loll PJ, Garavito RM. The X-ray crystal structure of the membrane protein prostaglandin H2 synthase-1. Nature. 1994;367:243–249. doi: 10.1038/367243a0. [DOI] [PubMed] [Google Scholar]
- 22.Luong C, Miller A, Barnett J, Chow J, Ramesha C, Browner MF. Flexibility of the NSAID binding site in the structure of human cyclooxygenase-2. Nature StructBiol. 1996;3:927–933. doi: 10.1038/nsb1196-927. [DOI] [PubMed] [Google Scholar]
- 23.Kurumbail RG, Stevens AM, Gierse JK, et al. Structural basis for selective inhibition of cyclooxygenase-2 by anti-inflammatory agents. Nature. 1996;384:644–648. doi: 10.1038/384644a0. [DOI] [PubMed] [Google Scholar]
- 24.Kurumbail RG, Kiefer JR, Marnett LJ. Cyclooxygenase enzymes: catalysis and inhibition. CurrOpinStructBiol. 2001;11:752–760. doi: 10.1016/s0959-440x(01)00277-9. [DOI] [PubMed] [Google Scholar]
- 25.Bhattacharyya DK, Lecomte M, Rieke CJ, Garavito RM, Smith WL. Involvement of arginine 120, glutamate 524, and tyrosine 355 in the binding of arachidonate and 2- phenylpropionic acid inhibitors to the cyclooxygenase active site of ovine prostaglandin endoperoxide H synthase-1. JBiolChem. 1996;271:2179–2184. doi: 10.1074/jbc.271.4.2179. [DOI] [PubMed] [Google Scholar]
- 26.Kalgutkar AS, Crews BC, Rowlinson SW, et al. Biochemically based design of cyclooxygenase-2 (COX-2) inhibitors: facile conversion of nonsteroidal antiiflammatory drugs to potent and highly selective COX-2 inhibitors. ProcNatlAcadSciUSA. 2000;97:925–930. doi: 10.1073/pnas.97.2.925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Harman CA, Turman MV, Kozak KR, Marnett LJ, Smith WL, Garavito RM. Structural basis of enantioselective inhibition of cyclooxygenase-1 by S-alpha-substituted indomethacin ethanolamides. JBiolChem. 2007;282:28096–28105. doi: 10.1074/jbc.M701335200. [DOI] [PubMed] [Google Scholar]
- 28.Zweifel BS, Davis TW, Ornberg RL, Masferrer JL. Direct evidence for a role of cyclooxygenase 2-derived prostaglandin E2 in human head and neck xenograft tumors. Cancer Res. 2002;62:6706–6711. [PubMed] [Google Scholar]
- 29.Regier MK, DeWitt DL, Schindler MS, Smith WL. Subcellular localization of prostaglandin endoperoxide synthase-2 in murine 3T3 cells. ArchBiochemBiophys. 1993;301:439–444. doi: 10.1006/abbi.1993.1168. [DOI] [PubMed] [Google Scholar]
- 30.Coffey RJ, Hawkey CJ, Damstrup L, et al. Epidermal growth factor receptor activation induces nuclear targeting of cyclooxygenase-2, basolaterial release of prostaglandins, and mitogenesis in polarizing colon cancer cells. ProcNatlAcadSciUSA. 1997;94:657–662. doi: 10.1073/pnas.94.2.657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Spencer AG, Woods JW, Arakawa T, Singer II, Smith WL. Subcellular localization of prostaglandin endoperoxide H synthases-1 and -2 by immunoelectron microscopy. JBiolChem. 1998;273:9886–9893. doi: 10.1074/jbc.273.16.9886. [DOI] [PubMed] [Google Scholar]
- 32.Mbonye UR, Wada M, Rieke CJ, Tang HY, Dewitt DL, Smith WL. The 19-amino acid cassette of cyclooxygenase-2 mediates entry of the protein into the endoplasmic reticulum-associated degradation system. JBiolChem. 2006;281:35770–35778. doi: 10.1074/jbc.M608281200. [DOI] [PubMed] [Google Scholar]
- 33.di Meglio P, Ianaro A, Ghosh S. Amelioration of acute inflammation by systemic administration of a cell-permeable peptide inhibitor of NF-kappaB activation. Arthritis and rheumatism. 2005;52:951–958. doi: 10.1002/art.20960. [DOI] [PubMed] [Google Scholar]
- 34.Sheng HM, Shao JY, Kirkland SC, et al. Inhibition of human colon cancer cell growth by selective inhibition of cyclooxygenase-2. JClinInvest. 1997;99:2254–2259. doi: 10.1172/JCI119400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Moser AR, Pitot HC, Dove WF. A dominant mutation that predisposes to multiple intestinal neoplasia in the mouse. Science. 1990;247:322–324. doi: 10.1126/science.2296722. [DOI] [PubMed] [Google Scholar]
- 36.Williams CS, Luongo C, Radhika A, et al. Elevated cyclooxygenase-2 levels in Min mouse adenomas. Gastroenterology. 1996;111:1134–1140. doi: 10.1016/s0016-5085(96)70083-5. [DOI] [PubMed] [Google Scholar]
- 37.Oshima M, Dinchuk JE, Kargman S, et al. Suppression of intestinal polyposis in ApcΔ716 knockout mice by inhibition of cyclooxygenase 2 (COX-2) Cell. 1996;87:803–809. doi: 10.1016/s0092-8674(00)81988-1. [DOI] [PubMed] [Google Scholar]
- 38.Jacoby RF, Seibert K, Cole CE, Kelloff G, Lubet RA. The cyclooxygenase-2 inhibitor celecoxib is a potent preventive and therapeutic agent in the min mouse model of adenomatous polyposis. Cancer Res. 2000;60:5040–5044. [PubMed] [Google Scholar]
- 39.Bugaj JE, Achilefu S, Dorshow RB, Rajagopalan R. Novel fluorescent contrast agents for optical imaging of in vivo tumors based on a receptor-targeted dye-peptide conjugate platform. JBiomedOpt. 2001;6:122–133. doi: 10.1117/1.1352748. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.