

Abstract
A training set of 27 propofol (2,6-diisopropylphenol) analogues was used to construct four-dimensional (4D) quantitative structure–activity relationship (QSAR) models for three screens of biological activity: loss of righting reflex (LORR) in tadpoles, enhancement of agonist activity at the γ-aminobutyric acid type A (GABAA) receptor, and direct (agonist-independent) activation of the receptor. The three resulting 4D-QSAR models are almost identical in form, and all suggest three key ligand–receptor interaction sites. The formation of an intermolecular hydrogen bond involving the proton of the ligand–OH group is the most important binding interaction. A hydrophobic pocket binding interaction involving the six-substituent is the second most significant binding site, and a similar hydrophobic pocket binding interaction near the two-substituent is the third postulated binding site from the 4D-QSAR models. A test set of eight compounds was used to evaluate the tadpole LORR 4D-QSAR model. Those compounds highly congeneric to the training set compounds were accurately predicted. However, compounds exploring substituent sites and/or electronic structures different from the training set were less well-predicted. Overall, the results show a striking similarity between the models of the sites responsible for anesthesia and those mediating effects of the training set of propofol analogues on the GABAA receptor; it follows that the GABAA receptor is therefore the likely site of propofol’s anesthetic action.
Introduction
Propofol (2,6-diisopropylphenol) is a short-acting general anesthetic agent, effective for short surgical procedures and for the induction and maintenance of anesthesia when administered by intravenous infusion. Propofol has the pharmacokinetic advantages of rapid onset and offset of action and produces a low incidence of postoperative nausea and vomiting.1–3 Several adverse clinical effects are, however, associated with propofol infusion, including pain on injection, bradycardia, reduction in blood pressure, and a high incidence of transient apnea. Some of these side effects have been shown to be associated with the emulsion-based formulation of propofol currently marketed (Diprivan®), and this has spawned efforts to overcome the drawbacks associated with the emulsion formulation.4–6
The general anesthetic properties of propofol were first discovered in the 1970s during a screen of 97 alkylphenols in mice and rabbits.7 This study demonstrated that 2,6-dialkylphenols, particularly 2,6-di-sec-alkylphenols, were the most potent general anesthetics while other more sterically hindered analogues, such as 2,6-di-tert-butyl- and 2,6-dicyclopentylphenol, were inactive as anesthetics. Several laboratories have expanded on this initial study and analyzed a diverse range of propofol analogues, in an attempt to better understand the pharmacodynamics of propofol, and also to search for compounds with improved aqueous solubility.8–13
Although the precise molecular mechanism of action of propofol and other general anesthetics remains unclear, substantial experimental evidence indicates that γ-aminobutyric acid type A (GABAA) receptors are major targets of propofol, being implicated in both the behavioral and the pharmacological actions of this agent.14–17 GABAA receptors mediate fast inhibitory neurotransmission in the central nervous system and are composed of various combinations of subunit families (α, β, γ, δ, π, θ, and ε), which can combine in multiple ways to form a pentameric chloride ion channel complex.18–20 The actions of propofol at GABAA receptors are complex and include enhancement (“potentiation”) of GABA-induced responses, agonist-independent direct receptor activation in the absence of GABA (“direct activation”), alteration of receptor desensitization and deactivation, and channel block. 21–29 The importance of each of these actions in the induction and maintenance of general anesthesia is not completely clear although the focus has been on potentiation of GABA responses, since this occurs at clinically relevant concentrations and would result in alterations in the duration of synaptic inhibition. Propofol, like other general anesthetic drugs such as the inhaled ethers and the barbiturates, shows little or no specificity among the diverse GABAA receptor subunits.28–30
In this study, we develop a three-dimensional (3D) pharmacophore for several effects of propofol and some of its analogues: (i) activity as general anesthetics, (ii) potentiation of GABAA receptor-mediated responses, and (iii) direct activation of GABAA receptors. We apply the technique of 4D-QSAR analysis to a previously published dataset of 27 analogues, which includes complete data for LORR in Xenopus laevis tadpoles as well as actions at the human GABAA α1β2γ2 receptor.9 Propofol and other phenols have very complicated pharmacokinetics in mammals, including extensive binding to plasma proteins.1–3 As a result, much of the anesthetic data for propofol analogues in mammals is approximate and often does not include determination of plasma or brain concentrations. The LORR assay in tadpoles, which has a long history with respect to the study of general anesthetics, provides a self-consistent set of potencies for the immobilizing properties of the propofol analogues.31 The GABAA α1β2γ2s receptor represents the most common subunit combination in the mammalian central nervous system and thus serves as a likely target of the actions of propofol.18–20
4D-QSAR analysis32 is a molecular modeling method that has proved both useful and reliable in the construction of quantitative 3D pharmacophore models for a set of ligand analogues when the geometry of the corresponding receptor is not known. In particular, 4D-QSAR analysis incorporates ligand conformational flexibility, multiple alignment exploration, and exhaustive evaluation of ligand-embedded pharmacophore groupings in the QSAR model building process. In addition, a 4D-QSAR model can be used as a virtual screen to evaluate a virtual library of compounds.
In a recent study of a set of gene-regulating ecdysteroids,33 which have structural and conformational features similar to the propofol analogues, 4D-QSAR analysis was found to yield more robust and more predictive models than the popular comparative molecular field analysis (CoMFA) 3D-QSAR method.34 Thus, 4D-QSAR analysis was deemed the preferred QSAR methodology to apply to a training set of propofol analogues that are flexible, there is minimal information regarding the binding alignment of these analogues to the GABAA receptor site, and the analogues contain a reasonably large number of possible pharmacophore groupings.
4D-QSAR analysis provides a vehicle to compare quantitative 3D pharmacophore models from three different screens directly and meaningfully to one another. The capability of making these model comparisons, in turn, has permitted us to propose a pharmacological hypothesis for the anesthetic action of propofol.
Materials and Methods
1. Training Set of Propofol Analogues
Krasowski et al.9 have reported structure–activity data that has been selected to construct the training set used in the study reported in this paper. The chemical structures of the training set of 27 propofol analogues are given in Figure 1. Three measures of activities have been made for each of these analogues: (i) EC50 for potentiating submaximal (EC20) GABA responses at GABAA α1β2γ2s receptors; (ii) EC50 for directly activating GABAA α1β2γ2s receptors; and (iii) EC50 for the compounds in producing LORR in X. laevis tadpoles.
Figure 1.

Structures of the 27 propofol analogues used in the 4D-QSAR training set. Each type of activity measure is expressed as −log(EC50) in the construction of the QSAR models where EC50 is in molar units. The same compounds are inactive in all three screens, with the minor exception of phenol not showing activity for direct activation. It should be pointed out here that about half of the 27 analogues were tested as far as aqueous solubility allowed but did not show activity, even when applied for very long periods of time. The inactive compounds were monitored down to −log(EC50) = 2 (which corresponds to a ligand concentration of 10 mM). However, the lower limit in activity for each of the three measures was varied from 2 to 0 in the construction of the 4D-QSAR models. The optimum 4D-QSAR model for each activity class was obtained by setting the activity of each inactive analogue to −log(EC50) = 1.0. The −log(EC50) values for each of the analogues and each of the three activity types are given in Table 1. This manner of including class measures, as opposed to actual activity measures, in QSAR model building must be carefully monitored and kept in mind in interpreting the resultant model.
2. Receptor-Independent (RI) 4D-QSAR Analysis Applied to the Propofol Training Set
The 4D-QSAR scheme can be applied to either receptor-dependent or RI training sets. The RI 4D-QSAR formalism was applied in this study since the detailed geometry of the propofol binding site on the GABAA receptor is not known.
The current formulation of the methodology for RI 4D-QSAR analysis consists of 10 operational steps that have been described in detail in previous papers32 and are summarized here only in terms of modeling the propofol training set.
Step 1
A 3D structure of each of the 27 propofol analogues given in Figure 1 was constructed in the neutral form using the HyperChem 5.01 software.35 Partial atomic charges were computed using the AM1 semiempirical method, also implemented in the HyperChem program. Each structure was energy-minimized using the HyperChem 5.01 quantum mechanical method without any geometric constraint. The energy-minimized structures are used as the initial structures in conformational sampling.
Step 2
The atoms of each molecule were classified into seven types of interaction pharmacophore elements (IPEs), which are defined as follows: (a) any type of atom, any (0); (b) nonpolar atom, np (1); (c) polar atom of positive charge, p+ (2); (d) polar atom of negative charge, p− (3); (e) hydrogen bond acceptor, hba (4); (f) hydrogen bond donor, hbd (5); and (g) aromatic carbon and hydrogen, ar (6). This IPE classification scheme was used to define the types of interactions involved with each pharmacophore site of the 4D-QSAR model.
Step 3
Molecular dynamic simulation, MDS, was used to sample the conformational states available to each analogue and to generate its corresponding conformational ensemble profile (CEP). The MDSs are done using the MOLSIM package36 with an extended MM2 force field.37,38 The temperature for the MDS is set at 300 K with a simulation sampling time of 40 ps with intervals of 0.001 ps for a total sampling of 40 000 conformations of each propofol analogue. The atomic coordinates of each conformation and its intramolecular energy sampled during the MDS are recorded every 0.02 ps for a total of 2000 “frames”, or steps, in the CEP of each compound.
Step 4
The current (RI) 4D-QSAR methodology uses three-ordered atom alignments to compare the molecules of a training set. Six alignments that span the entire propofol core structure were selected in this study and are defined in Table 2.
Table 2.
Set of Trial Alignments Used in Constructing the 4D-QSAR Models
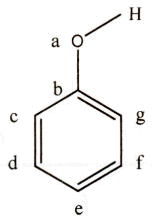
| align no. | 1st atom | 2nd atom | 3rd atom | align no. | 1st atom | 2nd atom | 3rd atom |
|---|---|---|---|---|---|---|---|
| 1 | a | b | c | 4 | b | d | f |
| 2 | b | g | f | 5a | a | b | c |
| 3a | a | b | c | 6 | c | e | g |
Because most compounds in the training set are symmetrical in structure, the variance of alignments are mainly made relative to asymmetrical compounds.
Step 5
Each conformation of an analogue from its CEP is aligned relative to a cubic lattice reference grid through the invariant coordinates of the three-ordered alignment atoms. In this study, the size of the cubic grid cell is 1 Å3, and the extent of the overall grid cell lattice was chosen to enclose each compound within the training set. The normalized occupancy of each grid cell by each IPE atom type over the CEP for a given alignment forms a unique set of QSAR descriptors referred to as grid cell occupancy descriptors, GCODs. The GCODs are computed and used as the basis set of trial 4D-QSAR descriptors in 4D-QSAR analysis.
Step 6
A 4D-QSAR analysis generates an enormous number of trial QSAR descriptors, GCODs, because of the large number of grid cells and the seven IPEs. Partial least squares (PLS) regression analysis39 is used to perform a data reduction fit between the observed dependent variable (in this study, each of the three observed biological activities) measures and the corresponding set of GCOD values.
Step 7
The M most highly weighted PLS GCOD descriptors, generated in step 6, are used to form the trial basis set for a genetic algorithm (GA) model optimization. The specific GA currently used in the 4D-QSAR software40 is a modification of the genetic function approximation (GFA).41,42 The GFA optimization is initiated using N (currently 300) randomly generated 4D-QSAR models. Mutation probability over the crossover optimization cycle is set at 10%. The smoothing factor, a GFA control variable, which specifies the number of independent variables in the QSAR models, is varied in order to determine the optimal number of descriptors in the 4D-QSAR models. The diagnostic measures used to analyze the resultant 4D-QSAR models generated by the GFA include (i) descriptor usage as a function of crossover operation, (ii) linear cross-correlation among descriptors and/or dependent variables (biological activity measures), (iii) number of significant and independent 4D-QSAR models, and (iv) indices of model significance including the correlation coefficient, r2, leave-one-out cross-validation correlation coefficient, xv-r2, and Friedman’s lack of fit (LOF).43
Non-4D-QSAR descriptors can also be included in the GFA model optimization. In this study, the logarithm of the 1-octanol/water partition coefficient, logP, was added in the form of both the linear and the square values of ClogP, which was calculated using the ClogP 4.0 program.44 The “C” in ClogP indicates that this property is calculated as opposed to measured. The ClogP values for each of the 27 propofol analogues are given as part of Table 1. It is well-known that the potency for many general anesthetics in immobilizing mammals correlates fairly well with logP, even when considering general anesthetics with diverse chemical structures. As a consequence, many traditional QSAR analyses7,12,45–47 of general anesthetics identify logP as the major independent variable that predicts anesthetic potency. The relationship between general anesthetic potency and logP, often referred to as the Meyer–Overton correlation, has often been invoked to suggest that general anesthetics act at lipid, as opposed to protein, targets. The purpose of adding logP as a trial descriptor in this study was to investigate the possible effect of the lipophilicity of the analogues to their corresponding activities.
Table 1.
−Log(EC50) of the Propofol Analogues for Potentiation of GABAA Responses, Direct Activation, and Anesthetic Action in Tadpoles
| Compd | Potentiation | Direct activation | Tadpole LORR | ClogP |
|---|---|---|---|---|
| 1, phenol | 3.0* | 1.0 | 2.8* | 1.5 |
| 2, 2,6-dimethylphenol | 3.9* | 3.7* | 4.2* | 2.5 |
| 3, 2,6-dimethylthiophenol | 1.0 | 1.0 | 1.0 | 2.9 |
| 4, 2,6-dibromophenol | 1.0 | 1.0 | 1.0 | 3.0 |
| 5, 2-isopropylphenol | 4.4* | 3.8* | 3.9* | 2.9 |
| 6, 2,6-dimethoxyphenol | 1.0 | 1.0 | 1.0 | 1.1 |
| 7, 2-isopropylthiophenol | 1.0 | 1.0 | 1.0 | 3.3 |
| 8, 2,6-diethylphenol | 4.9* | 3.9* | 4.7* | 3.4 |
| 9, 2-cyclopentylphenol | 1.0 | 1.0 | 1.0 | 3.5 |
| 10, 2-hydroxy-3-isopropylbenzoic acid | 1.0 | 1.0 | 1.0 | 3.6 |
| 11, 2-tert-butyl-6-methylphenol | 6.3* | 4.7* | 4.5* | 3.8 |
| 12, 2,6-diethylphenyl bromide | 4.7* | 3.9* | 3.8* | 5.1 |
| 13, 2,6-diethylphenyl isocyanate | 4.6* | 3.6* | 3.8* | 2.0 |
| 14, 1,3-diisopropylbenzene | 1.0 | 1.0 | 1.0 | 5.0 |
| 15, 2,6-diethylphenyl isothiocyanate | 4.8* | 3.5* | 3.8* | 5.4 |
| 16, 2,6-diisopropylphenol (propofol) | 5.7* | 5.0* | 5.7* | 4.3 |
| 17, 3,5-diisopropylcatechol | 4.4* | 3.7* | 4.3* | 3.7 |
| 18, 2,6-diisopropylphenyl isocyanate | 1.0 | 1.0 | 1.0 | 2.8 |
| 19, 2,4-di-tert-butylphenol | 1.0 | 1.0 | 1.0 | 5.1 |
| 20, 3,5-di-tert-butylphenol | 4.0* | 3.3* | 4.0* | 5.1 |
| 21, 2,6-di-tert-butylphenol | 1.0 | 1.0 | 1.0 | 5.1 |
| 22, 2,6-diisopropylphenyl isothiocyanate | 1.0 | 1.0 | 1.0 | 6.2 |
| 23, 4-iodo-2,6-diisopropylphenol | 5.0* | 4.5* | 5.6* | 5.7 |
| 24, 2,6-di-sec-butylphenol | 5.6* | 4.7* | 6.4* | 5.4 |
| 25, 2,4-di-sec-butylphenol | 4.5* | 3.6* | 5.0* | 5.4 |
| 26, 2,6-dicyclopentylphenol | 1.0 | 1.0 | 1.0 | 5.6 |
| 27, 2,6-dicyclohexylphenol | 1.0 | 1.0 | 1.0 | 6.7 |
The last column is the ClogP of each compound. Activities are expressed as −log(EC50), with EC50 in molar units. Experimentally obtained values are indicated by *. The inactive compounds have been assigned a −log(EC50) of 1.0.
Step 8
Steps 4–7 are repeated until all (six) trial alignments are included in the 4D-QSAR analysis.
Step 9
The inspection and evaluation of the entire population of 4D-QSAR models are made in this step. The objective is to identify the “best” 4D-QSAR models with respect to the set of chosen alignments. Each alignment considered will lead to a particular best 4D-QSAR model for that specific alignment. The alignment corresponding to the 4D-QSAR model with the overall highest r2 and xv-r2 measures, for all alignments tested, is selected as the best alignment. The linear cross-correlation matrix of the GCODs for the best 4D-QSAR model for the optimal alignment is then built to determine if the GCODs of this best model are correlated to one another. In this particular 4D-QSAR application, the lack of resolved inactivity measures prompted the use of additional multivariate analyses to those described above. However, it was not possible to construct better models or to glean additional insight, as compared to that realized using the “standard” 4D-QSAR statistical analyses described above.
Step 10
The final step of the 4D-QSAR formalism is to hypothesize the “active” conformation of each compound in the training set. This is achieved by first identifying all conformer states sampled for each compound, one at a time, that are within ΔE of the apparent global minimum energy conformation of the compound’s CEP. Currently, ΔE is set at 2 kcal/mol. The resultant set of energy-filtered conformations is then individually evaluated in the best 4D-QSAR. That conformation within 2 kcal/mol of the apparent global minimum that predicts the highest activity in the best 4D-QSAR model is defined as the active conformation.
The 4D-QSAR model can also be used as a virtual high throughput screen (VHTS) to predict the activities of the members of a virtual library of “similar” compounds to those of the training set.48,49 A comparison between the predicted activities from a 4D-QSAR model and the corresponding experimental values is perhaps the most stringent validation test of the 4D-QSAR model. A virtual screen of a small library of eight test compounds is carried out in this study as part of the tadpole LORR 4D-QSAR analysis.
Results
4D-QSAR models were independently constructed for each of the three types of activity screens used to evaluate the training set compounds. The details of each of the three separate 4D-QSAR models are given below.
Potentiation of GABA Responses at Human GABAA α1β2γ2s Receptors
The six alignments given in Table 2 were applied in the 4D-QSAR study. An inactivity value of −log(EC50) = 1 gave the best 4D-QSAR models, with alignment 4 yielding the overall best model. Hence, the inactivity value was set to −log(EC50) = 1 in all further studies. The corresponding best 4D-QSAR models, as assessed by the cross-validated correlation coefficient xv-r2, are given in Table 3. Figure 2A contains plots of r2 and xv-r2 as a function of the number of descriptors in a model using alignment 4. From Figure 2A, it is clear that maximum model significance is achieved with six descriptor terms, and there is no meaningful model improvement with the addition of additional descriptor terms. Thus, a six-term 4D-QSAR model, using alignment 4, yielded the best overall model, which is where n = 27, r2 = 0.836, and xv-r2 = 0.737.
Table 3.
Cross-Validated Correlation Coefficient of the Best Potentiation of GABAA Response 4D-QSAR Model for Each Alignment
| align | cross-validated correlation | coeff, xv-r2 align | cross-validated correlation coeff, xv-r2 |
|---|---|---|---|
| 1 | 0.600 | 4 | 0.737 |
| 2 | 0.571 | 5 | 0.556 |
| 3 | 0.701 | 6 | 0.690 |
Figure 2.

Plots of the number of 4D-QSAR model descriptors vs r2 and xv-r2. (a) Potentiation of GABAA responses; (b) direct activation; (c) tadpole LORR.
In eq 1, GCi(x, y, z, X) is the ith significant GCOD in the (x, y, z) coordinate having the X type IPE as defined above in step 2 of the 4D-QSAR analysis. ClogP was included in the trial descriptor pool in both linear and quadratic forms but did not survive as a significant descriptor in the 4D-QSAR model optimization process. A stereographic representation of the 3D pharmacophore embedded in the 4D-QSAR model, eq 1, is shown in Figure 3A. The reference compound in Figure 3A is 2-tert-butyl-6-methylphenol (compound 11 in Table 1) in its predicted active conformation. The activity values for potentiation, both experimentally determined and derived using eq 1, are plotted in Figure 4A. Two GCODs of the 4D-QSAR model specify pharmacophore sites that enhance activity (i.e., these have positive regression coefficients), and these correspond to (i) a hydrogen bond donor site near the OH and (ii) any atoms, with a preference for nonpolar atoms, located at certain sites involving the two- and six-substituents.
Figure 3.

Stereoviews of the 3D pharmacophores of the best 4D-QSAR models (eqs 1–3) shown relative to analogue 11, 2-tert-butyl-6-methylphenol (see Figure 1), in its respective active conformations as predicted using each of the best 4D-QSAR models. The GCODs are shown as spheres, although the actual grid cells are cubes in space. −log(EC50)-enhancing grid cells are shown as blue spheres, and grid cells that diminish −log(EC50) are shown as red spheres.
Figure 4.
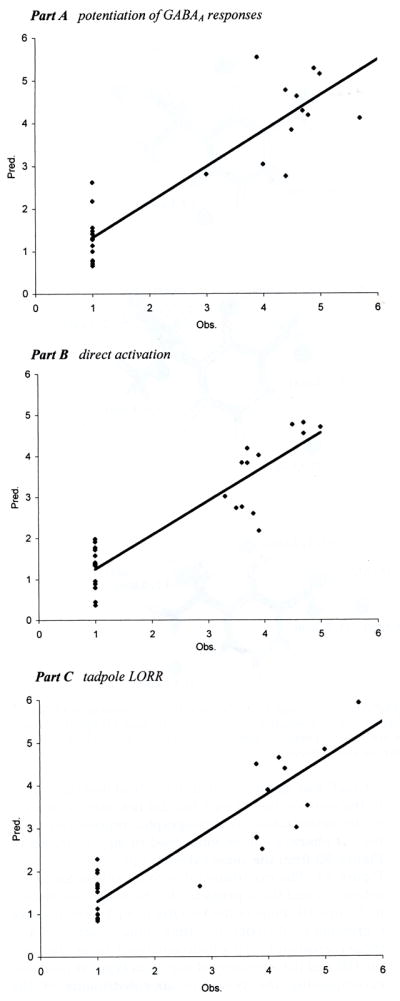
Predicted and observed activities [−log(EC50)] of each training set analogue for the three types of activities.
Four of the GCODs of eq 1 correspond to GCODs that decrease potency, since they have negative regression coefficients, and these correspond to (i) three GCODS, largely all-atom type IPEs, that apparently map the spatial size and shape restrictions on the choice of the two- and six-substituents and (ii) a polar negative IPE GCOD suggesting the region of space occupied by the GABAA receptor hydrogen bond acceptor group that is complementary to the hydrogen bond donor GCOD.
The best 4D-QSAR model, as represented by eq 1, has been statistically evaluated. A linear cross-correlation matrix of the GCODs of eq 1 was built and is given by Table 4. Any pair of GCODs having a correlation of greater than 0.50 or less than −0.50 is flagged as a highly correlated pair and indicated in bold in Table 4A. GC3 and GC4, having the correlation coefficient of 0.81, are significantly correlated to each other. Removal of either of these two significantly correlated GCODs from the descriptor pool, followed by reoptimization, yields a new set of five- and six-term 4D-QSAR models that are statistically poor (r2 < 0.60). Therefore, it appears that all six specific GCODs of the best 4D-QSAR model provide requisite composite and complementary information.
Table 4.
Linear Cross-Correlation Matrix of the GCODs Found in the Best 4D-QSAR Models
| Part A: Potentiation of GABAA Responses | ||||||
|---|---|---|---|---|---|---|
| (−1,−1,−2,hb d) | (−2,3,3, any) | (0,−1,−2,p−) | (0,−3,1,np) | (0,0,3, any) | (1,4,−4, any) | |
| (−1,−1,−2,hb d) | 1.00 | |||||
| (−2,3,3, any) | −0.15 | 1.00 | ||||
| (0,−1,−2,p−) | −0.23 | −0.07 | 1.00 | |||
| (0,−3,1,np) | −0.17 | −0.13 | 0.81 | 1.00 | ||
| (0,0,3, any) | 0.41 | −0.44 | 0.05 | 0.13 | 1.00 | |
| (1,4,−4, any) | 0.31 | −0.08 | −0.11 | −0.16 | 0.23 | 1.00 |
| Part B: Direct Activation | ||||||
| (−1,−1,−1,hbd) | (1,−2,2, any) | (0,2,−3, any) | (1,2,−4, any) | (1,3,−1, any) | (2,0,5, any) | |
| (−1, −1, −1, hbd) | 1.00 | |||||
| (1,−2,2, any) | −0.21 | 1.00 | ||||
| (0,2,−3, any) | 0.20 | −0.17 | 1.00 | |||
| (1,2,−4, any) | 0.33 | −0.18 | 0.26 | 1.00 | ||
| (1,3,−1, any) | 0.14 | −0.19 | −0.09 | 0.06 | 1.00 | |
| (2,0,5, any) | −0.16 | −0.13 | −0.10 | −0.11 | −0.35 | 1.00 |
| Part C: Tadpole LORR | ||||||
| (−1,−1,−2,hbd) | (−4,2,−2, any) | (0,−3,1, any) | (1,−4,3,np) | (1,−2,2,np) | (1,5,1, any) | |
| (−1,−1,−2,hbd) | 1.00 | |||||
| (−4,2,−2, any) | 0.13 | 1.00 | ||||
| (0,−3,1, any) | 0.04 | −0.02 | 1.00 | |||
| (1,−4,3,np) | 0.60 | 0.56 | −0.10 | 1.00 | ||
| (1,−2,2,np) | −0.27 | −0.12 | 0.09 | −0.20 | 1.00 | |
| (1,5,1, any) | −0.13 | −0.07 | −0.08 | −0.13 | −0.14 | 1.00 |
The high correlation between GC3 and GC4 involves two grid cells that are about 3 Å apart in space; yet, both contribute to activity when occupied. The correlation between these two GCODs could possibly arise from an intramolecular “allosteric” effect where occupancy at one grid cell has a coupled interaction to yield an increase/decrease in the occupancy of the other grid cell.
Direct Activation of Human GABAA α1β2γ2s Receptors
A similar 4D-QSAR study was carried out for the same training set (Table 1 and Figure 1), using the −log(EC50) for direct activation as the dependent variable. Using alignment 4 of Table 2, an inactivity value of −log(EC50) = 1 and six descriptors again yielded the optimum 4D-QSAR model. The optimum six-term 4D-QSAR model for direct receptor activation is where n = 27, r2 = 0.829, and xv-r2 = 0.765.
ClogP was again included in the trial descriptor pool in the construction of eq 2 but did not survive in GFA model optimization. A stereographic representation of the 3D pharmacophore embedded in eq 2 is shown in Figure 3B from the same external reference view as in Figure 3A. The experimental activity values for direct activation and those predicted by the model are plotted in Figure 4B. Four of the GCODs of eq 2 have positive regression coefficients (i.e., they enhance activity) and these correspond to (i) a hydrogen bond donor site near the OH and (ii) occupancy of any atom types at positions encompassing the two- and six-substituents of the analogues.
In addition, there are two GCODs with negative regression coefficients that characterize steric restrictions associated with the size and shape of the two- and six-substituents. The linear cross-correlation matrix of the GCODs of eq 2 is presented in Table 4B. None of the GCODs are significantly correlated to one another.
Tadpole LORR
A similar 4D-QSAR study was performed for the training set (Table 1 and Figure 1) using activity for inducing LORR in tadpoles as the dependent variable. Not surprisingly, in view of the significant correlation between activity in the LORR assay and activity in the two in vitro screens (Table 5), the best 4D-QSAR model for LORR was found to correspond to alignment 4, with the −log(EC50) value for inactive compounds set equal to one and with six descriptor terms in the model.
Table 5.
Linear Cross-Correlation Matrixes of the −log(EC50) Values of the Three Activity Screens
| Part A for 27 Analogues | |||
|---|---|---|---|
| potentiation | direct activation | tadpole LORR | |
| Potentiation | 1.00 | ||
| Direct activation | 0.97 | 1.00 | |
| Tadpole LORR | 0.96 | 0.96 | 1.00 |
| Part B for Active Analogues Only | |||
| potentiation | direct activation | tadpole LORR | |
| potentiation | 1.00 | ||
| Direct activation | 0.88 | 1.00 | |
| Tadpole LORR | 0.68 | 0.78 | 1.00 |
Table 5 reports the cross-correlation values among the three activity measures for (i) the entire training set and (ii) for the active analogues only. The cross-correlation values for the active analogue set are less, respectively, than those of the entire training set, but a major degree of common behavior among the three activity screens is still indicated. The cross-correlation values for the entire training set are all quite significant. Overall, Table 5 suggests that the three structure–activity profiles are markedly similar for the compounds studied.
ClogP did not survive the model optimization process, which is not surprising given the weak correlation between LORR −log(EC50) and ClogP (linear r2 = 0.31, and quadratic r2 = 0.38).
The best 4D-QSAR model for LORR is where n = 27, r2 = 0.836, and xv-r2 = 0.765. The linear cross-correlation matrix of the GCODs of eq 3 is given in Table 4C. The distance between GC1 and GC4 is about 7 Å, while GC2 and GC4 are approximately 9 Å apart. The removal of any one of these three GCODs from the trial descriptor pool results in a major loss in the quality of the resulting 4D-QSAR model (r2 < 0.50). Thus, it is concluded that the high GCOD correlation arises from a composite, and necessary, intramolecular “allosteric” effect. The stereographic representation of the embedded 3D pharmacophore of eq 3 is given in Figure 3C and is identical in format to those of eq 1/Figure 3A and eq 2/Figure 3B. The tadpole LORR −log(EC50) values, both experimentally observed and predicted using eq 3, are plotted in Figure 4C. The 4D-QSAR model given by eq 3 is very similar to both eq 1 and eq 2. There are four GCODs that have positive regression coefficients corresponding to increasing activity and define (i) a hydrogen bond donor site near the OH and (ii) three sites within the spaces of the two- and six-substituents, which define preferred occupancy sites in terms of any or nonpolar IPEs.
There are two GCODs with negative regression coefficients that define sites within the spaces of the two- and six-substituents, which should not be occupied by any and/or nonpolar IPEs.
Virtual Screen
Figure 5 lists the test set of eight compounds for virtual screening using eq 3. The tadpole LORR activities of these eight propofol analogues were measured after eq 3 was constructed. The −log(EC50) values predicted using eq 3, and measured experimentally in the tadpole LORR screen, are given in Table 6. Perhaps not surprisingly, compounds 1, 3, 6, and 7 of the test set, which are similar to the training set compounds in chemical structure, are predicted well. However, the predictions for compounds 2, 5, 6, and 8 are poor, and each of these compounds differs significantly from the chemical structures sampled by the training set. Compounds 2 and 5 have 4-propyl and 5-methyl groups, respectively. These two structural features are only present in analogues 19, 25 and 17, 20, respectively (see Figure 1), in the training set. This observation may indicate ligand substituents at positions other than the two- and six-positions are not only tolerated but can enhance tadpole LORR potency. For compound 8 of the test set, two alignments were considered in which either the 1-OH or the 4-NH2 was considered as providing the hydrogen bond donor group specified by eq 3. It appears that the two bulky benzyl groups at two- and six-positions prevent the 1-OH from forming the hydrogen bond with the receptor. However, a hydrogen bond might be formed instead using the 4-NH2 substituent by “flipping over” the ligand into a binding mode in which the 4-NH2 replaces the “normal” 1-OH group.
Figure 5.
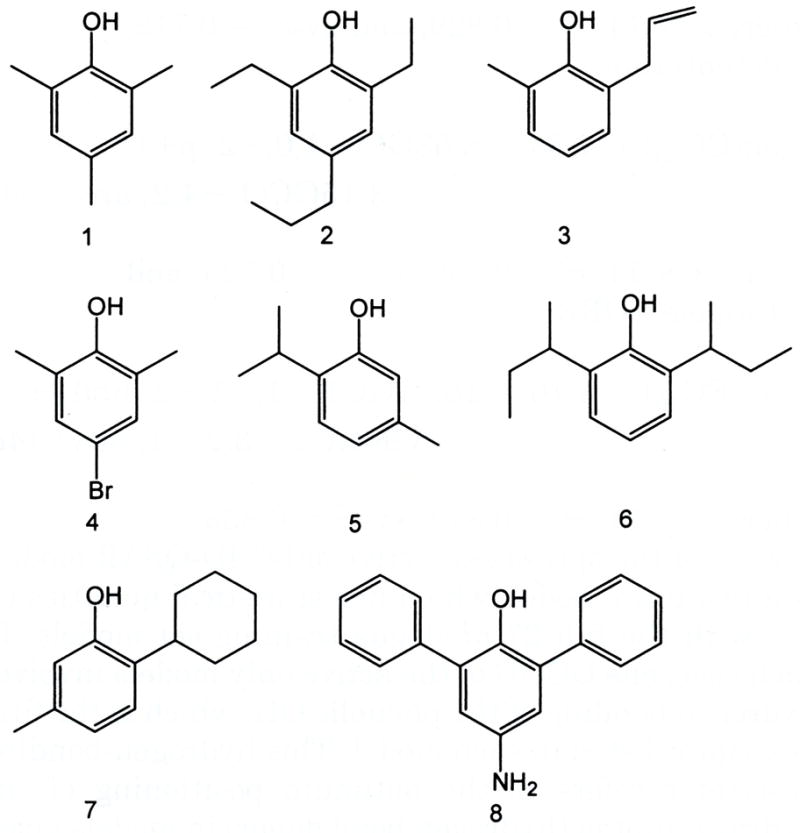
Eight propofol analogues of the test set for the tadpole LORR 4D-QSAR model.
Table 6.
Common Grid Cells and/or Grid Cells within 1 Å of One Another in Space for the Three 4D-QSAR Models of Each Activity Screen
| Common Grid Cells in Space | ||
|---|---|---|
| Potentiation and tadpole LORR | Direct activation and tadpole LORR | |
| Grid cells | (−1,−1,−2,hbd), (0,−3,1, any) | (1,−2,2, any) |
| Grid Cells within 1 Å in Space | ||
| Potentiation and direct activation | Direct activation and tadpole LORR | |
| Grid cells | (−1,−1,−2,hbd), (−1,−1,−1,hbd) | (−1,−1,−2,hbd), (−1,−1,−1,hbd) |
| (0,−3,1, any), (1,−2,2, any) | ||
4D-QSAR Analysis of Active Analogues Only
The rather inexact manner of assigning activity values to the inactive analogues was a cause for concern over the course of the study, and after the −log(EC50) predictions of the best 4D-QSAR models for each of the three types of activity screens were analyzed, the possibility of a general prediction problem was perceived. All three 4D-QSAR models underestimate the −log(EC50) of active compounds and overestimate the potency of inactive compounds. In some ways, this behavior is not too surprising since there are 13 inactive compounds in the training set all of which are assigned a −log(EC50) = 1. The regression fitting is seemingly not sufficiently flexible to accommodate all of the inactive analogues, while simultaneously describing the most active analogues using a six-term regression model.
To model the features of the active compounds in isolation, 4D-QSAR models were constructed for the 14 active compounds (−log(EC50) > 2.0) for each of the three screens (see Figure 1 and Table 1). Alignment 4 was applied to each of the three active analogue training sets. The best 4D-QSAR models are as follows:
Direct Activiation where n = 14, r2 = 0.829, and xv-r2 = 0.712;
Potentiation where n = 14, r2 = 0.890, xv-r2 = 0.725; and
Tadpole LORR where n = 14, r2 = 0.872, xv-r2 = 0.835.
Each of the optimized “active only” 4D-QSAR models is a two-term model, which has statistical qualities on par with the full 27 analogue training set models. In each case, one GCOD of the active only models involves hydrogen bonding of the phenolic OH, which is the first descriptor listed in each model. This hydrogen-bonding descriptor refers to the optimum positioning of the hydroxyl proton (hydrogen bond donor) in models given by eqs 4a and 4c and where to leave space about the OH, presumably for the hydrogen bond acceptor group of the receptor, in eq 4b. The other GCOD descriptor of each model maps out a favorable position for either the two- or six-position substituent. Overall, each of the 4D-QSAR models of the active analogue set, expressed by eqs 4a–c, are, respectively, subset models of the 4D-QSAR models for the complete training set and each particular activity endpoint. A stereographic representation of eq 4c is given in Figure 6A using the same external viewing frame of reference as in the other stereographic views of the other 4D-QSAR models reported in this paper. The reference molecule in Figure 6A is again 2-tert-butyl-6-methylphenol.
Figure 6.
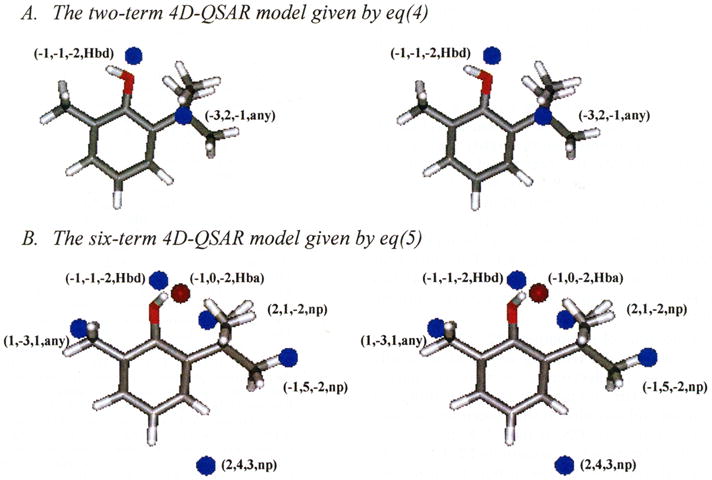
4D-QSAR models for the active tadpole LORR analogues of the training set. The GCODs are shown in the same manner as in Figure 3, and the reference analogue is number 11 of Table 1 in its active conformation as predicted by the corresponding 4D-QSAR model.
Equation 4c was intentionally expanded to a six descriptor term 4D-QSAR model in order to see which additional GCODs might appear in the model. where n = 14, r2 = 0.998, and xv-r2 = 0.995.
The resultant overfit 4D-QSAR model expressed by eq 5 is quite similar to both eqs 3 and 4c, which can be inferred by comparing the descriptor terms in the 4D-QSAR model equations to one another, and Figure 6B to Figures 3C (eq 3) and 6A (eq 4). The significant figures of fit are given in brackets as part of eq 5. Figure 6B is a stereographic plot of the embedded pharmacophore in eq 5 in the same format as given for eq 4 in Figure 6A. GC3 and GC5 of eq 5 have positive regression coefficients, which support the inference of a nonpolar binding site at the six-position of propofol. GC4 of eq 5 has a positive regression coefficient and corresponds to a binding site near the two-position of propofol. GC1 and GC2 of eq 5, which most likely define the hydrogen-bonding profile of the –OH to the receptor site, again dominate in eq 5.
Discussion
There are two major findings from this study. First, relative lipophilicity, as measured by ClogP, is not found to be an activity determinant/correlate in any of the three biological screens used to evaluate the training set of propofol analogues. Second, distinct 3D pharmacophores, which are indicative of specific ligand–receptor binding, are found for all three measures of activities made on the members of the training set. Moreover, these pharmacophores, and the corresponding 4D-QSAR models from which they are derived, are all very similar to one another. This second finding, in turn, suggests that (i) the propofol binding site on the GABAA receptor is likely to be the site mediating its anesthetic effects, (ii) it is also likely that the GABAA receptor represents the major site of action of propofol in causing loss of righting reflex in tadpoles, and (iii) there is a common mechanism of action governing the behavior of the propofol analogues of the training set for all three activity screens used in this work.
The 4D-QSAR models and corresponding 3D pharmacophores for the three activity screens have several specific features worth noting. First, there is a high similarity of the locations of the GCODs of each of the best 4D-QSAR models for the three activity screens. This similarity in the GCODs between the models can be discerned from Table 7, in which common GCODs within 1 Å of one another for the three models, eqs 1–3, are reported.
Table 7.
Predicted and Observed Activities [−log(EC50)] for the Tadpole LORR Test Seta
| compd | obs(−logEC50) | pred(−logEC50) |
|---|---|---|
| 1 | 4.0 | 4.1 |
| 2 | inactive | 5.8 |
| 3 | 4.5 | 4.8 |
| 4 | 4.5 | 4.3 |
| 5 | 4.3 | 1.6 |
| 6 | inactive | 3.9 |
| 7 | 4.5 (lethal) | 4.1 |
| 8 | 4.5 (slow) | −2.0/11.8 |
Activities are expressed as −log(EC50), with EC50 in molar units. Accurate predictions are shown in bold. Two different alignments were tried for compound 8. The first one was to put OH at the first position, and the second one was to turn the ring and put NH2 at the first position.
The most important feature of the binding of propofol analogues to the GABAA receptor involves the proton of the –OH group forming a hydrogen bond to an acceptor group on the GABAA receptor. Replacement of the proton of the 1-OH group decreases significantly the ligand–receptor affinity.9,12 This ligand–receptor hydrogen bond is highly directional in space relative to the ligand as evidenced by the GCODs with p− and hba IPEs near the hbd GCOD having negative regression coefficients in eqs 1 and 5. The substituents at the two- and six-positions can alter the propensity of the –OH group to adopt the preferred intermolecular hydrogen-bonding geometry.
The identification of a specific intermolecular hydrogen bond as a critical determinant of propofol analogue potency represents a significant advance over previous QSAR analyses of anesthetic potency, which almost universally find molecular lipophilicity to be the major determinant of activity.7,12,45–47 This finding perhaps relates to the advantages of 4D-QSAR analysis over the (2D) QSAR analyses applied in previous studies. Although few studies have investigated the importance of hydrogen bonding and anesthetic potency, one prior study supports a major role for hydrogen bonding in anesthetic–receptor interactions. Abraham et. al. found the ability to accept a hydrogen bond to be a negative determinant of the anesthetic potencies of a large series of linear and cyclic alkanes, alcohols, and ethers (although not including propofol).47
Each of the 4D-QSAR models given by eqs 1–3, as well as eq 5, indicates a binding region at the two-substituent position. Overall, this binding site is characterized by one or two potency-enhancing GCODs, most likely involving nonpolar atoms, and GCODs defining steric restrictions associated with a “wall” of the receptor site. The numbers and locations of the GCODs vary among the 4D-QSAR models. This type of variability among the GCODs from model to model may be due to limited information within the SAR of the training set. However, this diffuse representation of a likely nonpolar binding site across the 4D-QSAR models is also consistent with mapping a hydrophobic pocket ligand–receptor interaction. Hydrophobic pockets are often irregular and/or flexible in shape, as are the substituents fitting into them. Thus, there are multiple similar ways to reasonably describe this interaction.
The final binding site near the six-substituent site seems to be identical in form to that of the two-substituent binding site, namely, a hydrophobic pocket binding interaction. This binding site, overall, seems somewhat more important than the two-substituent site (at least for the training set studied) as can be judged from the regression coefficients of eqs 1–3. Also, this binding site and that of the OH make up eq 4 that is a 4D-QSAR model for the active analogues only. The two-substituent binding site is not present in eq 4. A comparison of the 3D pharmacophores of eqs 1–3 shown in Figure 3 suggests that the hydrophobic pocket for the two-substituent is smaller than the one for the six-substituent.
Each of the 4D-QSAR models have GCODs that are not associated with the one-, two-, or six-position binding sites. These “spurious” GCODs are located near the three-, four-, and/or five-ring positions. It is not possible to discern if these GCODs are providing significant pharmacophore information since the number of analogues in the training set that sample these regions of space are small. These descriptors will be given special attention as the current 4D-QSAR models are evolved and as additional analogues are made and tested.
Finally, there are no outliers in eqs 1–4. However, as mentioned above, eqs 1–3 correspond to 4D-QSAR models that tend to underestimate highly potent analogues and overestimate the activity of analogues of marginal potency. Using eq 3, only four of the eight predictions of the test set given in Figure 5 and Table 6 are accurate. The four correct predictions are for analogues that are highly consistent in chemical structure to members of the training set. The four poor predictions each correspond to an analogue that has structural features not found, or very poorly sampled, in the training set.
The active nonphenol compounds 12, 13, and 15 of Table 1 stand out in the training set as structurally distinct. A variety of electronic, as well as spatial, properties of all of the compounds in the training set were determined and included in the descriptor set used to build and optimize the QSAR model. None of these non-4D-QSAR descriptors for compounds 12, 13, and 15 appear unusual relative to the other training set compounds, but none of these descriptors survived in the model optimization process. The activity of the active nonphenol compounds would seem to arise from favorable electrostatic interactions (as opposed to hydrogen bonding) of their one-position groups with the receptor.
The next step in this research will be to evolve the current 4D-QSAR models of the propofol analogues to include contributions from the three-, four-, and five-substituent sites, as well as to try and build in descriptors that model changes in electronic structure and resultant GABAA receptor binding interactions. The extended models may include classification rules and/or consensus models in order to realize reliable predictions over a range of propofol chemistry.
It may be possible to modify the molecular structure of propofol in order to yield drugs that can discriminate between specific GABAA receptor subunit combinations. Developing such analogues may optimize anesthetic activity and reduce side effects or result in drugs with selective sedative, anxiolytic, or anticonvulsant properties.50 In addition, although no specific antagonists to the actions of propofol or other general anesthetics have yet been identified, experimental evidence supports the possibility of 51,52 developing selective antagonists to anesthetic modulatory sites on GABAA receptors. A propofol antagonist would represent an invaluable research probe and could be used to test the involvement of the GABAA receptor in the in vivo action of propofol.
Acknowledgments
Funding for this study was provided by National Institutes of Health Grant P01-GM 62195 to N.L.H., by the C.V. Starr Foundation (New York, NY) and the Rice Foundation (Skokie, IL) to N.L.H., and by National Institute of Mental Health training fellowship MH11504 to M.D.K. X.H. and A.J.H. also gratefully acknowledge support from the Laboratory of Molecular Modeling & Design at UIC and from The Chem21 Group, Inc.
References
- 1.Bryson HM, Fulton BR, Faulds D. Propofol. An update of its use in anaesthesia and conscious sedation. Drugs. 1995;50:513–559. doi: 10.2165/00003495-199550030-00008. [DOI] [PubMed] [Google Scholar]
- 2.Langley MS, Heel RC. Propofol. Drugs. 1988;35:334–372. doi: 10.2165/00003495-198835040-00002. [DOI] [PubMed] [Google Scholar]
- 3.Rees DC, Hill DR. Drugs in anesthetic practice. Ann Rep Med Chem. 1996;31:41–50. [Google Scholar]
- 4.Prankerd RJ, Stella VJ. Use of oil-in-water emulsions as a vehicle for parenteral drug administration. J Parenter Sci Technol. 1990;44:139–149. [PubMed] [Google Scholar]
- 5.Doenicke AW, Roizen MF, Rau J, Kellermann W, Babl J. Reducing pain during propofol injection: the role of the solvent. Anesth Analg. 1996;82:472–474. doi: 10.1097/00000539-199603000-00007. [DOI] [PubMed] [Google Scholar]
- 6.Doenicke AW, Roizen MF, Rau J, O’Connor M, Kugler J, Klotz U, Babl J. Pharmacokinetics and pharmacodynamics of propofol in a new solvent. Anesth Analg. 1997;85:1399–1403. doi: 10.1097/00000539-199712000-00040. [DOI] [PubMed] [Google Scholar]
- 7.James R, Glen JB. Synthesis, biological evaluation, and preliminary structure–activity considerations of a series of alkylphenols as intravenous anesthetic agents. J Med Chem. 1980;23:1350–1357. doi: 10.1021/jm00186a013. [DOI] [PubMed] [Google Scholar]
- 8.Cooke A, Anderson A, Buchanan K, Byford A, Gemmell D, Hamilton N, McPhail P, Miller S, Sunaram H, Vijn P. Water-soluble propofol analogues with intravenous anaesthetic activity. Bioorg Med Chem Lett. 2001;11:927–930. doi: 10.1016/s0960-894x(01)00088-9. [DOI] [PubMed] [Google Scholar]
- 9.Krasowski MD, Jenkins A, Flood P, Kung AY, Hopfinger AJ, Harrison NL. The general anesthetic potencies of a series of propofol analogues correlate with potency for potentiation of GABA current at the GABAA receptor but not with lipid solubility. J Pharmacol Exp Ther. 2001;297:338–351. [PubMed] [Google Scholar]
- 10.Lingamaneni R, Krasowski MD, Jenkins A, Truong T, Giunta AL, Blackbeer J, MacIver MB, Harrison NL, Hemmings HC. Anesthetic properties of 4-iodopropofol: implications for mechanisms of anesthesia. Anesthesiology. 2001;94:1050–1057. doi: 10.1097/00000542-200106000-00020. [DOI] [PubMed] [Google Scholar]
- 11.Sanna E, Motzo C, Usala M, Serra M, Dazzi L, Maciocco E, Trapani G, Latrofa A, Liso G, Biggio G. Characterization of the electrophysiological and pharmacological effects of 4-iodo-2,6-diisopropylphenol, a propofol analogue devoid of sedative-anaesthetic properties. Br J Pharmacol. 1999;126:1444–1454. doi: 10.1038/sj.bjp.0702449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Trapani G, Latrofa A, Franco M, Altomare C, Sanna E, Usala M, Biggio G, Liso G. Propofol analogues. Synthesis, relationships between structure and affinity at GABAA receptor in rat brain, and differential electrophysiological profile at recombinant human GABAA receptors. J Med Chem. 1998;41:1846–1854. doi: 10.1021/jm970681h. [DOI] [PubMed] [Google Scholar]
- 13.Anderson A, Belelli D, Bennett DJ, Buchanan KI, Casula A, Cooke A, Feilden H, Gemmell DK, Hamilton NM, Hutchinson EJ, Lambert JJ, Maidment MS, Mcquire R, McPhail P, Miller S, Muntoni A, Peters JA, Sansbury FH, Stevenson D, Sundaram H. α-Amino acid phenolic ester derivatives:novel water-soluble general anesthetic agents which allosterically modulate GABAA receptors. J Med Chem. 2001;44:2582–2591. doi: 10.1021/jm010903i. [DOI] [PubMed] [Google Scholar]
- 14.Franks NP, Lieb WR. Molecular and cellular mechanisms of general anaesthesia. Nature. 1994;367:607–614. doi: 10.1038/367607a0. [DOI] [PubMed] [Google Scholar]
- 15.Krasowski MD, Harrison NL. General anaesthetic actions on ligand-gated ion channels. Cell Mol Life Sci. 1999;55:1278–1303. doi: 10.1007/s000180050371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Trapani G, Altomare C, Sanna E, Biggio G, Liso G. Propofol in anesthesia. Mechanism of action, structure–activity relationships, and drug delivery. Curr Med Chem. 2000;7:249–271. doi: 10.2174/0929867003375335. [DOI] [PubMed] [Google Scholar]
- 17.Tanelian DL, Kosek P, Mody I, MacIver MB. The role of the GABAA receptor/chloride channel complex in anesthesia. Anesthesiology. 1993;78:757–776. doi: 10.1097/00000542-199304000-00020. [DOI] [PubMed] [Google Scholar]
- 18.Whiting PJ, Bonnert TP, McKernan RM, Farrar S, Le Bourdelles B, Heavens RP, Smith DW, Hewson L, Rigby MR, Sirinathsinghji DJ, Thompson SA, Wafford KA. Molecular and functional diversity of the expanding GABAA receptor gene family. Ann N Y Acad Sci. 1999;868:645–653. doi: 10.1111/j.1749-6632.1999.tb11341.x. [DOI] [PubMed] [Google Scholar]
- 19.Barnard EA, Skolnick P, Olsen RW, Mohler H, Sieghart W, Biggio G, Braestrup C, Bateson AN, Langer SZ. International union of pharmacology. XV. Subtypes of γ-aminobutyric acidA receptors: classification on the basis of subunit structure and receptor function. Pharmacol Rev. 1998;50:291–313. [PubMed] [Google Scholar]
- 20.McKernan RM, Whiting PJ. Which GABAA-receptor subtypes really occur in the brain? Trends Neurosci. 1996;19:139–143. doi: 10.1016/s0166-2236(96)80023-3. [DOI] [PubMed] [Google Scholar]
- 21.Concas A, Santoro G, Mascia MP, Serra M, Sanna E, Biggio G. The general anesthetic propofol enhances the function of γ-aminobutyric acid-coupled chloride channel in the rat cerebral cortex. J Neurochem. 1990;55:2135–2138. doi: 10.1111/j.1471-4159.1990.tb05807.x. [DOI] [PubMed] [Google Scholar]
- 22.Hales TG, Lambert JJ. The actions of propofol on inhibitory amino acid receptors of bovine adrenomedullary chromaffin cells and rodent central neurones. Br J Pharmacol. 1991;104:619–628. doi: 10.1111/j.1476-5381.1991.tb12479.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bai D, Zhu G, Pennefather P, Jackson MF, MacDonald JF, Orser BA. Distinct functional and pharmacological properties of tonic and quantal inhibitory postsynaptic currents mediated by γ-aminobutyric acidA receptors in hippocampal neurons. Mol Pharmacol. 2001;59:814–824. doi: 10.1124/mol.59.4.814. [DOI] [PubMed] [Google Scholar]
- 24.Orser BA, Wang LY, Pennefather PS, MacDonald JF. Propofol modulates activation and desensitization of GABAA receptors in cultured murine hippocampal neurons. J Neurosci. 1994;14:7747–7760. doi: 10.1523/JNEUROSCI.14-12-07747.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bai D, Pennefather PS, MacDonald JF, Orser BA. The general anesthetic propofol slows deactivation and desensitization of GABAA receptors. J Neurosci. 1999;19:10635–10646. doi: 10.1523/JNEUROSCI.19-24-10635.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hara M, Kai Y, Ikemoto Y. Propofol activates GABAA receptor-chloride ionophore complex in dissociated hippocampal pyramidal neurons of the rat. Anesthesiology. 1993;79:781–788. doi: 10.1097/00000542-199310000-00021. [DOI] [PubMed] [Google Scholar]
- 27.Hara M, Kai Y, Ikemoto Y. Enhancement by propofol of the γ-aminobutyric acidA response in dissociated hippocampal pyramidal neurons of the rat. Anesthesiology. 1994;81:988–994. doi: 10.1097/00000542-199410000-00026. [DOI] [PubMed] [Google Scholar]
- 28.Jones MV, Harrison NL, Pritchett DB, Hales TG. Modulation of the GABAA receptor by propofol is independent of the γsubunit. J Pharmacol Exp Ther. 1995;274:962–968. [PubMed] [Google Scholar]
- 29.Sanna E, Mascia MP, Klein RL, Whiting PJ, Biggio G, Harris RA. Actions of the general anesthetic propofol on recombinant human GABAA receptors: influence of receptor subunits. J Pharmacol Exp Ther. 1995;274:353–360. [PubMed] [Google Scholar]
- 30.Davies PA, Kirkness EF, Hales TG. Modulation by general anaesthetics of rat GABAA receptors comprised of α1 β3 and β3 subunits expressed in human embryonic kidney 293 cells. Br J Pharmacol. 1997;120:899–909. doi: 10.1038/sj.bjp.0700987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Downes H, Courogen PM. Contrasting effects of anesthetics in tadpole bioassays. J Pharmacol Exp Ther. 1996;278:284–296. [PubMed] [Google Scholar]
- 32.Hopfinger AJ, Wang S, Tokarski JS, Jin B, Albuquerque M, Madhav PJ, Duraiswami C. Construction of 3D-QSAR models using the 4D-QSAR analysis formalism. J Am Chem Soc. 1997;119:10509–10524. [Google Scholar]
- 33.Ravi M, Hopfinger AJ, Hormann RE, Dinan L. 4D-QSAR analysis of a set of ecdysteroids and a comparison to CoMFA modeling. J Chem Inf Comput Sci. 2001;41:1587–1604. doi: 10.1021/ci010076u. [DOI] [PubMed] [Google Scholar]
- 34.Cramer RD, Patterson DE, Bunce JD. Comparative molecular field analysis (CoMFA). 1. Effect of shape on binding of steroids to carrier proteins. J Am Chem Soc. 1988;110:5959–5965. doi: 10.1021/ja00226a005. [DOI] [PubMed] [Google Scholar]
- 35.HyperChem Program Release 501 for Windows. Hypercube, Inc; 1996. [Google Scholar]
- 36.Doherty DC. MOLSIM User’s Guide. The Chem21 Group, Inc; Lake Forest, IL: 1997. [Google Scholar]
- 37.Allinger NL. Conformational analysis. 130. MM2. A hydrocarbon force field utilizing V1 and V2 torsional terms. J Am Chem Soc. 1977;99:8127–8134. [Google Scholar]
- 38.Hopfinger AJ, Pearlstein RA. Molecular mechanics force-field parametrization precedures. J Comput Chem. 1984;5:486–492. [Google Scholar]
- 39.Glen WG, Dunn WJ, III, Scott DR. Principal components analysis and partial least squares. Tetrahedron Comput Methods. 1989;2:349–354. [Google Scholar]
- 40.4D-QSAR User’s Manual, version 2.0. The Chem21 Group, Inc; Lake Forest, IL: 1997. [Google Scholar]
- 41.Rogers D. G/SPLINES. A hybrid of Friedman’s multivariate adaptive regression splines (MARS) algorithm with Holland’s genetic algorithm. Proceedings of the Fourth International Conference on Genetic Algorithm; San Diego. 1991. pp. 38–46. [Google Scholar]
- 42.Rogers D, Hopfinger AJ. Application of genetic function approximation to quantitative structure–activity relationships and quantitative structure–property relationships. J Chem Inf Comput Sci. 1994;34:854–866. [Google Scholar]
- 43.Friedman J. Technical Report No 102. Laboratory for Computational Statistics, Department of Statistics, Stanford University; Stanford, CA: 1988. Multivariate adaptive regression splines. revised 1990. [Google Scholar]
- 44.Medicinal Chemistry Software Project. Medchem Software Manual, Release 5.50. Pomona College; Claremont, CA: 1996. [Google Scholar]
- 45.Hansch C, Dunn WJ. Linear relationships between lipophilic character and biological activity of drugs. J Pharm Sci. 1972;61:1–19. doi: 10.1002/jps.2600610102. [DOI] [PubMed] [Google Scholar]
- 46.Janoff AS, Pringle MJ, Miller KW. Correlation of general anesthetic potency with solubility in membranes. Biochim Biophys Acta. 1981;649:125–128. doi: 10.1016/0005-2736(81)90017-1. [DOI] [PubMed] [Google Scholar]
- 47.Abraham MH, Lieb WR, Franks NP. Role of hydrogen bonding in general anesthesia. J Pharm Sci. 1991;80:719–724. doi: 10.1002/jps.2600800802. [DOI] [PubMed] [Google Scholar]
- 48.Walters WP, Stahl MT, Murcko MA. Virtual screening an overview. Drug Discovery Today. 1998;3:160–194. [Google Scholar]
- 49.Hopfinger AJ, Reaka A, Venkatarangan P, Duca JS, Wang S. Construction of a virtual high throughput screen by 4D-QSAR analysis: application to a combinatorial library of glucose inhibitors of glycogen phosphorylase b. J Chem Inf Comput Sci. 1999;39:1151–1160. [Google Scholar]
- 50.Rudolph U, Crestani F, Mohler H. GABAA receptor subtypes: dissecting their pharmacological functions. Trends Pharmacol Sci. 2001;22:188–194. doi: 10.1016/s0165-6147(00)01646-1. [DOI] [PubMed] [Google Scholar]
- 51.Beckstead MJ, Phelan R, Mihic SJ. Antagonism of inhalant and volatile anesthetic enhancement of glycine receptor function. J Biol Chem. 2001;276:24959–24964. doi: 10.1074/jbc.M011627200. [DOI] [PubMed] [Google Scholar]
- 52.Krasowski MD, Nishikawa K, Nikolaeva N, Lin A, Harrison NL. Methionine 286 in transmembrane domain 3 of the GABAA receptor β subunit controls a binding cavity for propofol and other alkyphenol general anesthetics. Neuropharmacology. 2001;41:952–964. doi: 10.1016/s0028-3908(01)00141-1. [DOI] [PMC free article] [PubMed] [Google Scholar]