

Abstract
Purpose
It was recently shown that phosphatidylinositol-(PI)3 kinase is upregulated in wounded rabbit corneal epithelia. Extracellular signal–regulated kinase (erk)-1 and -2 proteins and PI-3 kinase were activated in embryonic corneal epithelia after 1-hour stimulation by type I collagen. In the current investigation specific inhibitors of PI-3 kinase and mitogen-activated kinase-kinase (MEK-1 kinase) were used to determine the role of these signaling molecules in actin reorganization and collagen binding to isolated sheets of corneal epithelial tissue.
Methods
Effects of specific PI-3 kinase and MEK-1 inhibitors (LY294002, PD98059, respectively) were investigated in embryonic corneal epithelial tissues. Avian embryonic corneal epithelia were isolated as tissue sheets, organ cultured in the presence of these specific inhibitors, and stimulated with type I collagen. The tissues were evaluated for collagen-stimulated actin reorganization, erk-1 and -2 and PI-3 kinase activity, total filamentous actin accumulation, and collagen binding.
Results
The MEK-1 inhibitor PD98059 decreased erk-1 and -2 phosphorylation and blocked actin reorganization in a dose-dependent manner. The PI-3 kinase 85-kDa subunit was decreased 25% in LY294002-treated tissue, and collagen binding also decreased significantly in tissues treated with MEK-1 and PI-3 kinase inhibitors compared with control tissues. In addition, both inhibitors blocked actin cortical mat reorganization.
Conclusions
PI-3 kinase and erk-1 and -2 signaling pathways are activated and necessary for collagen binding and integrin-mediated actin reorganization in embryonic avian corneal epithelium.
Class III phosphatidylinositol (PI)-3 kinase has been implicated in regulating cell proliferation survival, metabolism, and cytoskeletal organization.1 Recently, Zhang et al.2 demonstrated that PI-3 kinase is upregulated in wounds of migrating corneal epithelial cells in the presence of epidermal growth factor (EGF), however, the mechanism by which PI-3 kinase exerts its effects are still unknown. It has been shown that focal adhesion kinase (FAK) binds PI-3 kinase3 and may be required for cell migration,4 indicating that the actin cytoskeleton becomes rearranged. In addition there is evidence in growth factor–stimulated signaling cascades that PI-3 kinase is upstream of the mitogen-activated protein (MAP) kinase pathway.5 The PI-3 kinase inhibitor LY294002 selectively blocks the adenosine triphosphate binding site in the p85 subunit of PI-3 kinase6 and inhibits FAK-promoted cell migration4 and neutrophil aggregation.7
It is well known that the MAP kinases can respond to a variety of extracellular signals, including osmotic stress, heat shock, cytokines, and mitogens.8,9 Two of the MAP kinases termed extracellular signal–regulated protein kinases (erk-1 and erk-2; also referred to as erk-1/2) translocated to the nucleus after activation to regulate the expression of various transcription factors.8 Activation of the MAP kinase pathways has been identified as a mechanism that integrins use to regulate gene expression leading to cell shape changes during cell spreading or migration.10,11 The mitogen-activated kinase-kinase (MEK-1) inhibitor PD98059 specifically blocks the dephosphorylated form of MAP kinase-kinase-1, MEK-1, but not the activated or phosphorylated form of MEK-1. In addition, PD98059 can block erk-2.12
Corneal epithelial tissues isolated without basal lamina (−BL; Figs. 1C, 1D) respond to extracellular matrix (ECM) using an actin-dependent mechanism. The basal cell surface flattens, and the disrupted actin cortical mat (ACM) reorganizes in the presence of laminin, fibronectin, or collagen within 2 hours (Fig. 1).13-17 The actin reorganization does not require new protein synthesis. The epithelia increase collagen synthesis in response to ECM stimulation but do not accumulate enough for autostimulation.13-17 In addition, we have demonstrated that the actin bundles have distinct configurations in the presence of different ECM molecules.18 We have also shown that this tissue reorganizes the actin cytoskeleton in response to bombesin or lysophosphatidic acid (LPA).18
Figure 1.
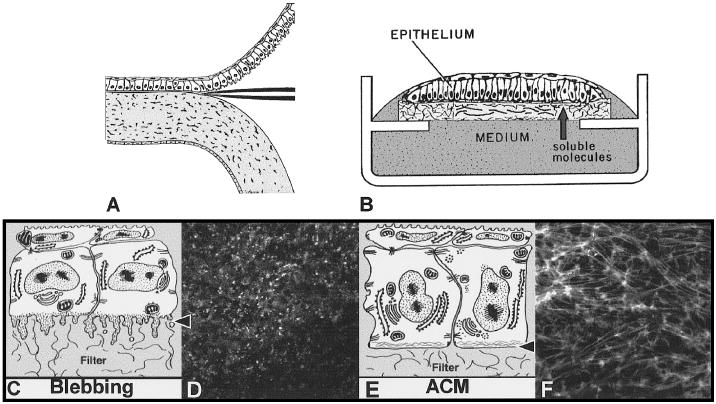
Embryonic corneal epithelium isolation (A) and culture (B). Epithelium was isolated as a sheet of tissue (A) and placed on a polycarbonate filter basal side down, and cultured at the air–medium interface (B). Epithelia isolated −BL extended basal cellular processes termed blebs (C, arrowhead). The blebs contain F-actin that appeared as punctate spots in single optical sections (D, arrowheads) taken through the basal area (C, arrowhead). If the tissue was cultured with soluble ECM molecules, the basal actin reorganized into an ACM (E, arrowhead) that appeared as bundles of F-actin in single optical sections (E, arrows) that align from cell to cell. Epithelium cultured in the presence of COL for 2 hours contained extensive actin bundles that extended from cell to cell (F) across the field. Scale bar, 10 μm. Reprinted with permission from Svoboda KKH, Orlow DL, Ashrafzadeh, A., Jirawuthiworavong, G. Zyxin and vinculin distribution at the “cell-matrix attachment complex” (CMAX) in corneal epithelial tissue are actin dependent. Anat Rec. 1999;254:336–347. Copyright © 1999, Wiley-Liss, Inc., a subsidiary of John Wiley & Sons, Inc.
Recently, our laboratory has identified a basal-lateral membrane compartment of corneal epithelial cells: cell–matrix attachment complexes (CMAXs), which are analogous to focal adhesions in cultured fibroblasts. The actin-associated proteins α-actinin, vinculin, zyxin, FAK, paxillin, and tensin have been identified in these CMAXs18,19. The reorganization of the actin cytoskeleton in response to ECM stimulation is orchestrated through signaling cascades. The epithelia must be in contact with matrix molecules for a minimum of 15 minutes for the ACM to reform. During these 15 minutes many signaling proteins became activated.18 We also established that tyrosine phosphorylation events are required, because the c-Src and cell-permeable tyrosine kinase inhibitor herbimycin A blocks actin reorganization in a dose-dependent manner. In addition, we reported that late in the signaling cascade (60–20 minutes) two of the MAP kinase proteins (erk-1 and erk-2) become phosphorylated, and the 85-kDa subunit of PI-3 kinase appears to be upregulated in response to collagen.18 In the current research we investigated the role of these two signaling pathways in collagen-stimulated actin reorganization by using specific inhibitors to block function.
Materials and Methods
Tissue Isolation and Organ Culture
Fertile white leghorn chicken eggs (Spafas, Norwich, CT) were incubated for 8 days at 37°C. The embryos (stages 32–3420) were removed from the eggs and rinsed in Hank’s balanced saline solution (HBSS; Gibco, Grand Island, NY). The animals were handled according to National Institutes of Health guidelines and the ARVO Statement for the Use of Animals in Ophthalmic and Vision Research. Whole corneas, with some surrounding sclera, were removed from the embryos with fine forceps. Whole corneas were treated for 2 to 4 minutes at 37°C in 8.0 mg/ml trypsin and 8.0 mg/ml collagenase (type 1A; Sigma, St. Louis, MO) in Ca2+-Mg2+–free HBSS to isolate epithelial sheets −BL. After incubation in trypsin-collagenase, whole corneas were rinsed in Ham’s F-12 medium (Gibco) and HBSS. A continuous sheet of corneal epithelial cells was dissected from the underlying stroma with fine forceps (Fig. 1A) and placed basal side down on a black polycarbonate filter (3.0-mm diameter, 0.4-μm pore size; Poretics Corp., Livermore, CA).15
Corneal epithelial sheets were suspended at the air–medium interface by a triangular-shaped wire grid in an organ culture dish (Falcon, Lincoln Park, NJ). The height of the medium barely covered the apical surface of the tissue (Fig. 1B). Cultures were incubated at 37°C for 24 to 28 hours in a humidified gas mixture (5% CO2 and 95% air). The control culture medium was composed of Ham’s F-12 medium, 1% antibiotic-antimycotic (GIBCO), and 50 μM ascorbic acid (Sigma).
Organ Culture for MAP Kinase and PI-3 Kinase Inhibition Experiments
The synthetic MAP kinase inhibitor, 2-(2′-amino-3′-methoxy-phenyl)-oxanphthalen-4-one (PD98059; Calbiochem, San Diego, CA) was used at three dosages (10, 50, and 100 μM). Freshly isolated epithelia (−BL) were placed into five treatment groups (n = 20–30 epithelia per group): two groups with control medium (containing 0.01% dimethyl sulfoxide [DMSO]), and three groups treated with PD98059 (10, 50, and 100 μM). The MAP kinase inhibitor was kept as a 1-M stock solution in DMSO. All treatment groups were cultured overnight (18–24 hours) at 37°C. After the inhibitor treatment, epithelial sheets were stimulated with 100 μg/ml type I collagen (COL; Collaborative Research, Bedford, MA) either with (n = 3 experiments) or without (n = 4 experiments) inhibitor for 2 hours at 37°C and compared with stimulated (+COL) and nonstimulated control groups (NT, no treatment). Control groups contained 0.01% DMSO.
The PI-3 kinase inhibitor, 2-(4-morpholinyl)-8-phenyl-4H-1-benzopyran-4-one [also known as 2-(4-morpholinyl)-8-phenyl-chromone (LY294002; Calbiochem]6 was used in three concentrations (10, 50, and 100 nM). These experiments used the same protocol as described for the MAP kinase inhibitor, PD98059.
Organ culture experiments were repeated at least seven times, with 20 to 30 epithelia pooled in each treatment group. The epithelia were fixed and stained immediately for morphology (n = 10 epithelia per group) or placed in extraction buffer for sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) and Western blot analysis (n = 20 epithelia per group).
F-Actin Staining
Filamentous actin (F-actin) was visualized with rhodamine phalloidin (Molecular Probes, Eugene, OR). Epithelia were fixed, permeabilized, and stained with rhodamine phalloidin (1:100 dilution of stock solution) for 30 minutes at room temperature, followed by rinses in phosphate-buffered saline (PBS).18
Specimens were analyzed by upright confocal laser scanning microscope (CLSM; Leica, Deerfield, IL), equipped with an argon ion laser and a ×50 water immersion lens (P1 Fluotar; Leica; numeric aperture, 1.0) with a working distance of 100 μm. En face images were obtained of the basal cytoplasm occupied by the ACM (Fig. 1F). Additional epithelia were analyzed with a confocal microscope (model 510; Carl Zeiss, Thornwood, NY) equipped with one argon and two helium neon lasers with excitation wavelengths of 488, 543, and 633 nm. The tissue was analyzed with a ×40 water immersion lens (PlanNeo; Zeiss; numeric aperture, 0.75) with a working distance of 330 μm and a scanning area of 230.3 μm2. Confocal images were analyzed, enhanced, and stored. Images were arranged by computer (Photoshop and Pagemaker programs; Adobe, San Jose, CA).
SDS-PAGE and Western Blot Analysis
Corneal epithelia (20 per treatment group) were isolated −BL and treated as described previously in the organ culture procedure.18,19,21 The Western blots were incubated with one of the following antibodies: PI-3 kinase (250 μg/ml, Transduction Laboratories, Lexington, KY) or phosphorylated erk-1 and -2 (350 μg/ml, Promega, Madison, WI). PI-3 kinase antibodies were diluted 1:1000 in 1% milk with PBS-Tween. The anti-phospho-erk-1 and -erk-2 antibody was diluted 1:10,000 in 1% milk with PBS-Tween. The primary antibodies were detected with horseradish peroxidase (HRP) goat anti-mouse or goat anti-rabbit secondary antibodies (Transduction Laboratories) and identified with enhanced chemiluminescence (ECL, Amersham, Arlington Heights, IL) and autoradiography. The blots were washed with 1% Tween/Tris-buffered saline (TBS) and reprobed with β-tubulin as an internal loading standard.
Densitometry readings of representative Western blot analyses were analyzed by computer (NIH Image; National Institutes of Health, Bethesda, MD). Densitometry readings, expressed in optical density (OD) units, were taken of all the β-tubulin lanes and used to equalize the loading per lane. The erk-1, erk-2, and PI-3 kinase OD values were calculated and graphed from representative Western blot analyses (Figs. 2, 3).
Figure 2.
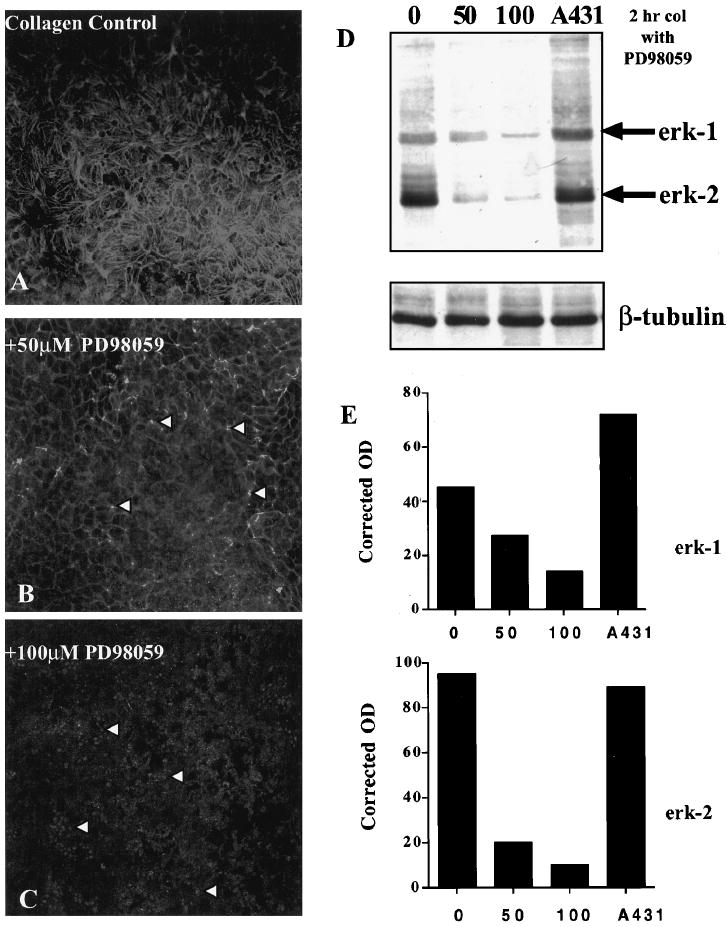
Epithelia treated with MAP kinase inhibitor, PD98059. Confocal images through the ACM optical plane (Fig. 1E, arrowhead) of whole epithelial tissue stained with FITC-phalloidin after incubation with PD98059 (50 and 100 μM; B, C) and stimulation with COL for 2 hours compared with control tissue (A). The lowest dose of PD98059 had little effect on ECM-stimulated ACM reformation; however, at the middle (B, arrowheads) and highest (C, arrowheads) doses, the blebs were more prominent. Western blot of activated erk-1 and -2 (D) showed a decrease in activity of these MAP kinase proteins from cell extracts that were treated with PD98059 in a dose-dependent manner compared with untreated epithelia (0) or control cell extracts (A431). The blot was reprobed with β-tubulin to demonstrate that the gel was loaded equally. Densitometry analyses (E) of the anti active erk-1 and -2 Western blot (D). β-Tubulin was used to obtain corrected densities (OD units) of the two proteins, erk-1 and -2. Both erk-1 and -2 decreased in a dose-dependent manner compared with uninhibited control tissue (0). Scale bar, 10 μm.
Figure 3.
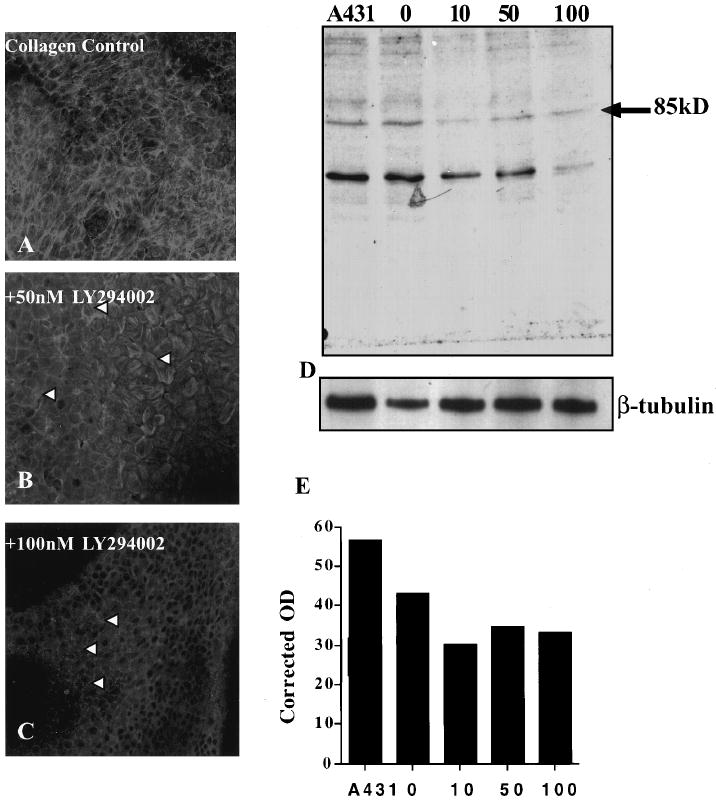
Epithelia treated with the PI-3 kinase inhibitor LY294002. Single confocal images taken through the ACM optical plane of FITC-phalloidin–stained epithelial tissue after incubation with PI-3 kinase inhibitor LY294002 (50 and 100 nM) and stimulated with COL in the presence of inhibitor for 2 hours (B, C) compared with control COL-treated epithelia (A). Tissue treated with 50 nM LY294002 (B, arrowheads), had actin filaments in the basal region of the tissue; however, they did not align from cell to cell as seen in the control tissue (A, arrowheads). In contrast, tissue treated with 100 nM LY294002 (C, arrowheads) had a complete loss of actin filament bundles in the ACM region. Western blot of the 85-kDa PI-3 kinase subunit (D) showed a decrease in the protein from cell extracts that were treated with LY294002 compared with untreated epithelia (0) or control cell extracts (A431). Densitometry analysis (E) of the PI-3 kinase Western blot (D) corrected for loading with β-tubulin. The amount of 85-kDa was reduced approximately 25% in all treated doses compared with untreated tissue (0). The protein levels did not decrease with increasing doses of LY294002. Scale bar, 10 μm.
FITC Labeling of ECM Molecules
Fluorescein isothiocyanate (FITC, 10 mg/ml in DMSO; Sigma) was coupled to ECM protein in a 0.1-M carbonate-bicarbonate buffer (pH 9.0),18 and rat tail COL (1 mg/ml) was labeled. Excess FITC was removed by exhaustive dialysis against Ham’s F-12 medium in a sterile container. The ratio of dye to coupled protein was measured using a spectrophotometer. The biologic activity of the protein was assessed by induction of ACM shown during the morphologic assay.18 FITC-labeled ECM molecules were stored and remained stable at 4°C.
Quantitation of ECM Binding to Corneal Epithelia
Corneal epithelia were incubated with MAP kinase (100 μM) or PI-3 kinase inhibitor (100 nM) and stimulated with FITC-labeled COL for 30 or 60 minutes The tissues were rinsed to remove any nonspecifically bound COL, fixed, and stained with Texas red phalloidin. The tissues were analyzed by confocal microscope (model 510; Zeiss). Initial data were collected from three to four epithelia as a complete z series (230.3-μm2 area) through the whole tissue, recording both red and green channels simultaneously. The image files (n = 25–30 images) were projected into one image using a program that sums all the intensity data into one file. These projected images were electronically turned 90° (Fig. 4A), producing xz images of the xy data set. All pixel information was contained in these images. A densitometry computer program from the microscope manufacturer (Zeiss) was used to determine the average intensity (range = 0–255) of the pixels that intersected lines through the center of the tissue and at the basal cell surface (Fig. 4A, yellow lines). The average of data collected from epithelia (n = 4) from each treatment group was statistically analyzed using one-way analysis of variance.
Figure 4.
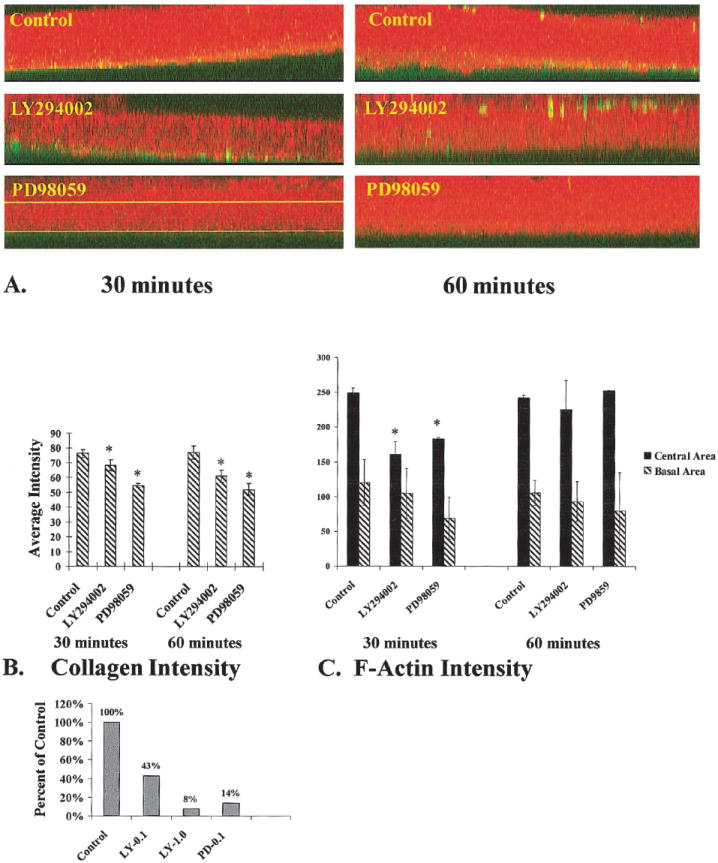
COL binding and F-actin accumulation in the presence of MAP kinase or PI-3 kinase inhibitors. Epithelia were cultured in the presence and absence of either MAP kinase (100 μM PD98059) or PI-3 kinase inhibitor (100 nM LY294002) and stimulated with FITC-labeled COL for 30 or 60 minutes. A complete z-series of confocal images were projected into one image. Then an xz image was produced from the projected three-dimensional stack (A). These final images contained all the image pixels from the complete set of z-series images. Average intensities (scale range, 0–255) of bound COL (green) and F-actin (red) were determined using a line drawn through the center of the basal cells (upper line in A, PD98059 tissue) or at the base of the epithelia (lower line in A, PD98059-treated tissue). Densitometry software was used to determine average intensities of bound COL (green channel intensity levels, n = 4 epithelia per group) cellular F-actin (red channel intensity levels, n = 4 epitheliaper group). In the basal compartment, there was a decrease in both bound COL (B, *P ≤ 0.05) and F-actin (C) in PI-3 kinase– and MAP kinase–inhibited epithelia compared with control epithelia. The actin accumulation in the center of the basal cells also demonstrated a marked difference between groups at 30 minutes (C, *P < 0.05). However, by 60 minutes, there was no difference between groups (C). Collagen-coated polystyrene beads were used to further quantitate the differences in collagen binding (D). The tissues were incubated basal side up and treated with inhibitors, and the collagen-coated beads were added to the medium for 30 minutes. The tissues were placed in a black 96-well dish (1 epithelium per well; n = 5–10 epithelia per treatment group) and analyzed with a fluorometer. The intensity of each well was analyzed with both FITC and rhodamine filter sets. The mean intensity and SDs were calculated for each group. Tissue treated with 100 nM LY294002 had 43% intensity compared with controls, whereas tissues treated with 1 μM LY294002 had less than 10% intensity. Tissues treated with 100 μM PD98059 had 14% of control intensity.
Collagen-coated polystyrene beads (2 μm; yellow-green fluorescent FluoSpheres, Molecular Probes) were incubated with corneal epithelial tissues (n = 5–10 per treatment group) organ cultured as described earlier, except that the tissues were placed basal side up on black filters. The epithelia were cultured in the presence of control medium with or without inhibitors for 2 hours, and the collagen-coated beads were added to the medium for 30 minutes in each culture dish and gently swirled. Because the medium level covered the tissue, the beads had access to the basal surface of the tissue. After 30 minutes, some tissues were removed from the organ culture dishes, rinsed, fixed, and stained with Texas red phalloidin. Some samples were rinsed and fixed without phalloidin. After all rinses, the tissues were placed into a black 96-well plate (1 epithelium per well, n = 5 to 10 epithelia per treatment group) and counted with a cytometer (Fluorocount; Packard, Meriden, CT) with filters specific for FITC and rhodamine. Background levels were established with the single-labeled samples and subtracted from the final intensity of the positive samples. This procedure eliminated any crossover between channels. The means and SDs were calculated for each treatment group, and the data were expressed as a percentage of control.
Results
Embryonic Chick Corneal Epithelia General Morphologic Characteristics
At day 8 (stage 34) of development, the embryonic chick corneal epithelium is composed of only two cell layers unlike the mature stratified corneal epithelium. The apical layer comprises flat hexagonal periderm cells, whereas the basal cells are cuboidal. The basal cells have a well-defined basal lamina at the basal surface.22 Confocal microscopy studies have demonstrated that F-actin is located in peridermal microvilli, at basal-lateral cell–cell junctions within interdigitating process and in the ACM near the basal surface (Fig. 1F).16 Previous studies have established that ACM reformation occurs within 2 hours of ECM stimulation through an actin-dependent mechanism.13,16 The epithelia must be in contact with matrix molecules for a minimum of 15 minutes for the ACM to reform in 2 hours. During the initial 15 minutes, many signaling proteins become tyrosine phosphorylated, including p190 Rho-GAP, FAK, paxillin, tensin, and 42- and 44-kDa MAP kinase proteins.18
MAP Kinase and ECM-Stimulated Actin Reorganization
MAP kinases can respond to a variety of extracellular signals, including osmotic stress, heat shock, cytokines, and mitogens.8,9 The MAP kinase pathway has been identified as a downstream pathway for integrins to regulate gene expression leading to actin cytoskeletal-mediated cell shape changes including cell spreading or migration.10,11
To determine whether the erk-1/2–MAP kinase pathway was involved in collagen-stimulated actin reformation, we cultured epithelia in the presence of the MEK-1 inhibitor PD98059 for 24 hours before a 2-hour stimulation period with COL (with or without inhibitor). The inhibitor-treated epithelia were compared with parallel collagen-stimulated cultures (Figs. 2A, 2B, 2C).
Single confocal sections at the ACM optical plane (Fig. 1E, arrowhead) demonstrated a dose-dependent decrease in actin filament bundles (Figs 2B, 2C, arrowheads) compared with control samples (Fig. 2A). Overall actin distribution and cell shape morphology of whole tissue did not change in the presence of PD98059. Epithelia exposed to the lowest dose (10 μM) had normal COL-stimulated actin reformation (data not shown). However, epithelia treated with higher doses did not have organized actin, and the basal surface was covered by blebs detected as punctate aggregates of actin in confocal micrographs (Figs. 2B, 2C, arrowheads).
Activated erk-1/2 detected with an antibody that specifically binds phosphorylated erk-1/2 proteins increased in the presence of COL compared with the controls. The changes in actin reorganization in the presence of PD98059 were accompanied by decreases in activated erk-1 and -2 (Fig. 2D) in a dose-dependent manner. Cell lysates from A431 cells were used as positive controls on all Western blot analyses (Figs. 2, 3). Erk-2 activity was similar in the two control cell lysates. Densitometry analysis of epithelia treated with MEK inhibitor demonstrated a 5- to 10-fold decrease in active erk-2, whereas active erk-1 was decreased only 2- to 3-fold. β-Tubulin staining (Figs. 2, 3) was used to determine that the lanes were loaded equally. If the inhibitor was washed out before COL stimulation, levels of erk-1/2 activity decreased, and the ACM did not reform, indicating that the inhibitor had a long-term downstream effect on this whole tissue.
PI-3 Kinase and ACM Reorganization
Class III PI-3 kinases have been implicated in regulating cell proliferation survival, metabolism, and cytoskeletal organization.1 To determine whether the PI-3 kinase cascade was involved in ECM-stimulated actin reorganization, we cultured epithelia in the presence of LY294002 for 24 hours before COL stimulation (2 hours, with or without the inhibitor). The morphology of epithelia treated with LY294002 (Figs. 3B, 3C) was compared with parallel experiments of control epithelia isolated −BL and stimulated with COL (Fig. 3A).
The PI-3 kinase inhibitor altered ACM reformation in a dose-dependent manner (Figs. 3A, 3B, 3C, arrowheads). Of note, there were no changes in overall actin distribution or cell shape of the peridermal cells or apical region of the basal cells at all dosages. At the optical plane of the ACM, there was no change in actin reorganization at the lowest dose (10 nM) compared with control tissues (data not shown). Actin formed bundles after exposure to the medium dose (50 nM, Fig. 3B, arrowheads); however, the bundles do not align from cell to cell as seen in the control samples (Fig. 3A) and ACM formation appeared to be disrupted. At the highest concentration tested (100 nM), there was a complete breakdown in actin bundle formation (Fig. 3C). The actin cytoskeleton did not reorganize at all and had disorganized blebbing morphology (Fig. 3C, arrowheads) indicated by the punctate distribution of the F-actin.
This disruption in actin reorganization was accompanied by a decrease in the detectability of the 85-kDa PI-3 kinase subunit on Western blot analysis (Fig. 3D). Densitometry analysis of the Western blot demonstrated a 25% decrease in the 85-kDa PI-3 kinase subunit in inhibitor-treated epithelia compared with control epithelia (Fig. 3E). Of interest, increasing the dose did not further decrease the presence of the 85-kDa PI-3 kinase subunit. In addition, an approximately 60-kDa protein was consistently labeled with the p85 antibody. This may be a degradation product, because it also decreased in tissue treated with the PI-3 kinase inhibitor (Fig. 3D). In washout experiments in which the inhibitor was absent during the final 2-hour COL stimulation, the presence of this subunit did not decrease substantially (data not shown), even though the actin did not reorganize (Figs. 3A, 3C). This indicates that the kinase activity was decreased even when the 85-kDa subunit was present.
Collagen Binding and F-Actin Accumulation
We have established a morphologic assay to measure the amount of ECM binding to the basal surface of corneal epithelial cells.18 In these experiments, the ECM protein was labeled with FITC using a standard protocol. The FITC label did not interfere with integrin signaling, because the epithelial tissues reorganized the ACM in 2 hours, similar to tissues treated with unlabeled ECM proteins.18
Control epithelia had the highest intensity levels for collagen binding (Fig. 4B), when measured by image analysis (NIH Image). There was a decrease in COL binding at the basal cell surface (Fig. 4B, *P ≤ 0.05) in tissues treated with either inhibitor; however, the MAP kinase inhibitor had a greater effect on COL binding. These results were confirmed in binding studies that used 3H-labeled COL and collagen-coated beads quantitated with a microplate fluorescence reader (Fig. 4D). In this study, the 100-nM dose of LY294002 decreased collagen-coated bead binding more than 50%, whereas a 10-fold higher dose (1 μM) decreased collagen-coated bead binding to less than 10% of that in control tissue (Fig. 4D). The tissues treated with 100 μM PD98059 also had reduced collagen-coated bead binding (14% of control). This independent assay confirmed the confocal analysis demonstrating that inhibiting these two pathways decreased collagen binding to corneal epithelia.
A concurrent decrease in F-actin in the basal area was also observed in tissues treated with either inhibitor (Fig. 4C, *P ≤ 0.05). The MAP kinase inhibitor PD98059 had the least F-actin accumulation in the basal cell area measured (Fig. 4C). The same epithelial samples (n = 3 per treatment group) were used to determine the mean F-actin accumulation in the central region of the basal cells, by using densitometry software (Fig. 4D). The control epithelia (Fig. 4D) were saturated (mean intensity levels, 250–255 OD units) by 30 minutes, whereas the inhibitor-treated tissues were significantly less, having mean intensity values of 160 and 170 at 30 minutes (Fig. 4D). However, by 60 minutes there was no difference between groups, indicating that the F-actin polymerization was delayed but not inhibited. Of interest, these doses of inhibitors blocked ACM reformation or actin bundling in the basal membrane area (Figs. 3C, 4C).
In summary, we have shown that the PI-3 kinase and MAP kinase signaling pathways are critical for actin polymerization and reorganization in the embryonic corneal epithelium. Furthermore, disrupting these signal pathways decreased the amount of collagen binding to the basal cell surface.
Discussion
These experiments establish that PI-3 kinase and MEK-1, or one of the downstream kinases (erk-1 and or erk-2) are necessary for not only ECM-stimulated actin bundle formation but also collagen binding. Actin bundles are formed when actin filaments cross-link into arrays with the long axis of filaments in parallel to each other. This actin bundle formation takes place in corneal epithelial sheets as they migrate over wound beds.23 During epithelial migration to cover wounds, PI-3 kinase expression was elevated2 and vinculin protein increased.24 Treatment of the corneal wounds with EGF increased PI-3 kinase expression in the rabbit corneal wound model.2 This embryonic whole-epithelium model differs from the wound-healing models, in that just the epithelial sheet is exposed to a single stimulating molecule. We have shown that this epithelial tissue reorganizes the actin cytoarchitecture in response to specific ECM, lipid, and neuropeptide molecules.18
This simple epithelial model has specialized cell–cell and cell–matrix domains that are responsible for stability, differentiation, and communication from the microenvironment. Using this whole corneal epithelial model, we previously determined that the reorganization of the basal actin cytoskeleton in response to specific ECM molecules is a tyrosine phosphorylation-dependent mechanism involving signaling molecules p190 Rho-GAP, FAK, paxillin, tensin, MAP kinase (erk 1/2), and PI-3 kinase.18 In the current investigation, we further characterized the MAP kinase (erk 1/2) and PI-3 kinase pathways by using specific inhibitors for these proteins. This is the first study to show that blocking these downstream signaling pathways not only decreased actin bundle formation but also decreased collagen binding.
It has been shown in a prior study that FITC-labeled ECM molecules bound to whole floating epithelial sheets and reorganized the basal cell surface after a 3-hour incubation. Furthermore, the bound ECM could be competitively removed with only similar molecules, indicating a specific receptor subtype was used for each ECM molecule class.14 We extended these experiments and determined that 15 minutes was the minimum ECM binding time that was necessary for the cells to reorganize the basal cell surface in 2 hours.18 As stated previously, the same assay was used to determine that blocking MAP kinase (erk-1/2) and PI-3 kinase pathways also decreased collagen binding.
We had previously demonstrated that PI-3 kinase protein levels increased in response to collagen, whereas MAP kinase protein levels did not increase.18 In contrast, the activated forms of erk-1/2 increased slightly in response to collagen stimulation.18 FAK interacts with the phospholipid-signaling pathway through PI-3 kinase.3 PI-3 kinase phosphorylates PIP (PI[4]phosphate) or PIP2 (PI[4,5]bisphosphate) at the D3 position to generate respectively, PI(3,4)P2 or PI(3,4,5)P3.25 These phospholipid byproducts have been implicated in downstream signaling of cytoskeletal reorganization through interactions with profilin, gelsolin (Fig. 5), and Rac.26 In addition to its phosphorylation properties, PI-3 kinase can also act as a ser/thr protein kinase.26 In NIH 3T3 mouse fibroblasts, FAK was found to be associated with PI-3 kinase, and tyrosine phosphorylated the p85 subunit of PI-3 kinase in vitro (Fig. 5). The binding affinity between FAK and PI-3 kinase increased when FAK was autophosphorylated. During cell adhesion, there was increased tyrosine phosphorylation of the p85 subunit of PI-3 kinase.27
Figure 5.
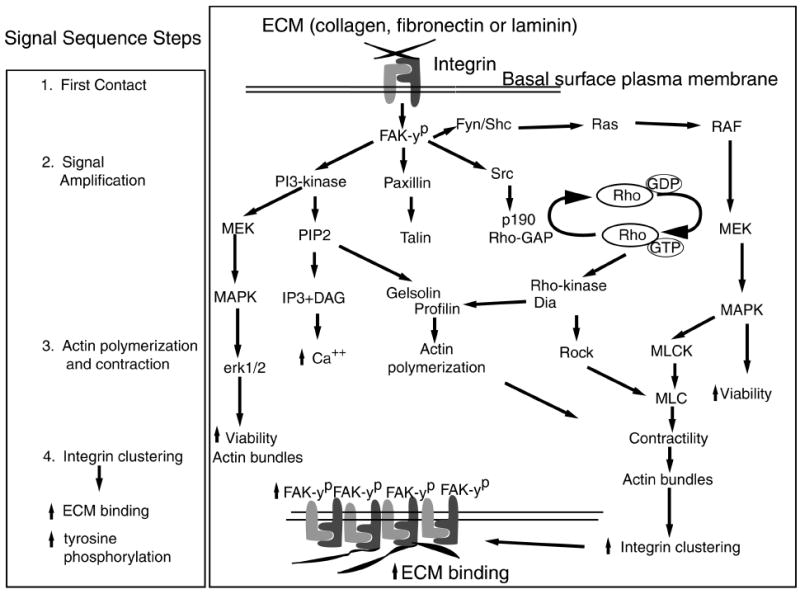
The proposed signaling hypothesis. The stages of epithelial response to ECM were divided into four parts: 1) first contact, 2) signal amplification, 3) actin polymerization and contraction, and 4) integrin clustering leading to increased ECM binding and further signal amplification. Viewing the diagram from top to bottom, the first contact events occur at the basal cell surface but are drawn at the top of the diagram for illustration of the hypothesis. The first contact events include ECM binding and tyrosine phosphorylation of FAK. Signal amplification is represented by FAK phosphorylating the surrounding kinases and proteins (PI-3 kinase, paxillin, Fyn/ Shc, Src). These proteins amplify the signal further by stimulating their downstream targets leading to increased actin polymerization and contraction. The actin rearrangements aid increased integrin clustering and ECM binding producing a stronger intracellular signal through tyrosine phosphorylation.
We hypothesized that the MAP kinase and PI-3 kinase signaling pathways are required for ECM-stimulated ACM reorganization in intact embryonic avian corneal epithelia (Fig. 5). The current experiments confirm this hypothesis: Blocking either one of these kinases inhibited ACM reorganization in this tissue. In a related study, Zhang et al.2 demonstrated that expression of PI-3 kinase mRNA and protein increased in adult wounded rabbit corneas. The increased expression was transient and peaked at 36 hours, when the epithelial cells are actively migrating to cover the wound bed. In a similar rat corneal wound model, vinculin was increased dramatically from 6 to 18 hours after wounding24 and was found at the leading edge of the migrating epithelial cells. More recently, E-cadherin was localized to these leading-edge cells, and the actin filaments appeared to align from cell to cell, forming a ring around the wound.23
Our data also support the hypothesis that epithelial cells respond to ECM through a sequence of events. We hypothesize that this sequence of cellular response includes at least four steps (Fig. 5). The sequence can be divided into: 1) first contact, 2) signal amplification, 3) actin polymerization and contraction, and 4) integrin clustering that leads to increased ECM binding and increased tyrosine phosphorylation (Fig. 5). First contact with ECM from the basal cell surface (note that this portion of the schematic drawing is upside down to accommodate the hypothesis) causes integrin-mediated FAK phosphorylation that in turn phosphorylates the surrounding proteins (paxillin, Fyn/shc, and Src)5 and leads to signal amplification. FAK also binds PI-3 kinase3 and is upstream of the MAP kinase pathway.5 Src phosphorylates p190RhoGAP, inactivating its GAP function that may allow RhoGTP to stay active longer, promoting further signal amplification.28,29 Activated RhoGTP binds to downstream kinases such as Rho-associated coiled coil-containing protein kinase (p160ROCK) and p140 diaphanous (p140Dia) to increase the third step, actin polymerization and contraction (Fig. 5).30,31 We hypothesize that actin reorganization assists integrin clustering, allowing more ECM binding that increases FAK phosphorylation and other signal transduction events (Fig. 5).
This sequence of signaling events is supported by data collected from the embryonic corneal epithelial model showing that ECM proteins bind to the basal surface in clusters gradually. The ECM clusters become larger and brighter with time but are not uniform (Fig. 4). Tyrosine phosphorylation of the signaling proteins also increases gradually. For example, FAK phosphorylation may not be detectable until 15 minutes after stimulation.18
Additional evidence for the hypothesis is that decreasing Rho protein levels in corneal epithelia prevented bleb retraction and actin bundle reorganization but did not completely abolish tyrosine phosphorylation of integrin-mediated signaling proteins. However, decreasing Rho diminished tyrosine phosphorylation, which indicates that Rho was necessary for amplification of the signal initiated by ECM–integrin interactions. Of note, decreasing Raf protein levels did not block ACM reorganization or tyrosine phosphorylation.32 The present research demonstrated that inhibiting MEK 1 blocked actin reorganization, and therefore the MAP kinase pathway could be stimulated with multiple upstream alternative pathways (for example, the PI-3 kinase and/or the Ras-Raf pathway, Fig. 5)
In conclusion, the data recorded in this study support the current signaling hypothesis (Fig. 5) that ECM binding to integrins initiates a tyrosine phosphorylation-dependent pathway. When MAP kinase or PI-3 kinase was inhibited, actin reorganization was blocked. We also established an in vitro whole-tissue culture system that was responsive to ECM stimulation and could be readily used to determine the role of signaling molecules in actin cytoskeletal reorganization and ECM binding. Future studies will concentrate on delineating other specific proteins and their interactions in the signaling cascade with the overall objective of achieving an understanding of the role of matrix, integrins, and/or other receptor molecules in corneal epithelial cell biology.
Acknowledgments
The authors thank Petra Moessner and Andrew Campbell for technical assistance, Laura Main and Brad Whitman (Carl Zeiss, Inc.) for assistance in quantitation of the confocal data, and Paul C. Dechow for assistance with statistics.
Supported by National Institutes of Health Grants EY08886 (KKHS) and AG05706 (WRR).
References
- 1.Leevers SJ, Vanhaesebroeck B, Waterfield MD. Signalling through phosphoinositide 3-kinases:the lipids take centre stage. Curr Opin Cell Biol. 1999;11:219–225. doi: 10.1016/s0955-0674(99)80029-5. [DOI] [PubMed] [Google Scholar]
- 2.Zhang Y, Liou GI, Gulatti AK, Akhtar RA. Expression of phosphatidylinositol 3-kinase during EGF-stimulated wound repair in rabbit corneal epithelium. Invest Ophthalmol Vis Sci. 1999;40:2819–2826. [PubMed] [Google Scholar]
- 3.Chen H, Guan J. Association of focal adhesion kinase with its potential substrate phosphatidylinositol 3-kinase. Proc Natl Acad Sci USA. 1994;91:10148–10152. doi: 10.1073/pnas.91.21.10148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Reiske HR, Kao SC LAC, et al. Requirement of phosphatidylinositol 3-kinase in focal adhesion kinase-promoted cell migration. J Biol Chem. 1999;274:12361–12366. doi: 10.1074/jbc.274.18.12361. [DOI] [PubMed] [Google Scholar]
- 5.Giancotti FG, Ruoslahti E. Integrin signaling. Science. 1999;285:1028–1032. doi: 10.1126/science.285.5430.1028. [DOI] [PubMed] [Google Scholar]
- 6.Vlahos CJ, Matter WF, Hui KY, Brown RF. A specific inhibitor of phosphatidylinositol 3-kinase, 2-(4-morpholinyl)-8-phenyl-4H-1-benzopyran-4-one (LY294002) J Biol Chem. 1994;269:5241–5248. [PubMed] [Google Scholar]
- 7.Vlahos CJ, Matter WF, Brown RF, et al. Investigation of neutrophil signal transduction using a specific inhibitor of phosphatidylinositol 3-kinase. J Immunol. 1995;154:2413–2422. [PubMed] [Google Scholar]
- 8.Garrington TP, Johnson GL. Organization and regulation of mitogen-activated protein kinase signaling pathways. Curr Opin Cell Biol. 1999;11:211–218. doi: 10.1016/s0955-0674(99)80028-3. [DOI] [PubMed] [Google Scholar]
- 9.Kortenjann M, Shaw PE. The growing family of MAP kinases:regulation and specificity. Crit Rev Oncog. 1995;6:99–115. [PubMed] [Google Scholar]
- 10.Schwartz MA, Baron V. Interactions between mitogenic stimuli, or, a thousand and one connections. Curr Opin Cell Biol. 1999;11:197–202. doi: 10.1016/s0955-0674(99)80026-x. [DOI] [PubMed] [Google Scholar]
- 11.Robinson MJ, Cobb MH. Mitogen-activated protein kinase pathways. Curr Opin Cell Biol. 1997;9:180–186. doi: 10.1016/s0955-0674(97)80061-0. [DOI] [PubMed] [Google Scholar]
- 12.Alessi DR, Cuenda A, Cohen P, Dudley DT, Saltiel AR. PD 098059 is a specific inhibitor of the activation of mitogen-activated protein kinase kinase in vitro and in vivo. J Biol Chem. 1995;270:27489–27494. doi: 10.1074/jbc.270.46.27489. [DOI] [PubMed] [Google Scholar]
- 13.Sugrue SP, Hay ED. Response of basal epithelial cell surface and cytoskeleton to solubilized extracellular matrix molecules. J Cell Biol. 1981;91:45–54. doi: 10.1083/jcb.91.1.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sugrue SP, Hay ED. The identification of extracellular matrix (ECM) binding sites on the basal surface of embryonic corneal epithelium and the effect of ECM binding on epithelial collagen production. J Cell Biol. 1986;102:1907–1916. doi: 10.1083/jcb.102.5.1907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Svoboda KK, Hay ED. Embryonic corneal epithelial interaction with exogenous laminin and basal lamina is F-actin dependent. Dev Biol. 1987;123:455–469. doi: 10.1016/0012-1606(87)90403-9. [DOI] [PubMed] [Google Scholar]
- 16.Svoboda KK. Embryonic corneal epithelial actin alters distribution in response to laminin. Invest Ophthalmol Vis Sci. 1992;33:324–333. [PMC free article] [PubMed] [Google Scholar]
- 17.Khoory W, Wu E, Svoboda KK. Intracellular relationship between actin and alpha-actinin in a whole corneal epithelial tissue. J Cell Sci. 1993;106:703–717. doi: 10.1242/jcs.106.3.703. [DOI] [PubMed] [Google Scholar]
- 18.Svoboda KKH, Orlow DL, Chu CL, Reenstra WR. ECM stimulated actin bundle formation in embryonic corneal epithelia is tyrosine phosphorylation dependent. Anat Rec. 1999;254:348–359. doi: 10.1002/(sici)1097-0185(19990301)254:3<348::aid-ar5>3.0.co;2-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Svoboda KKH, Orlow DL, Ashrafzadeh A, Jirawuthiworavong G. Zyxin and vinculin distribution at the “cell-extracellular matrix attachment complex” (CMAX) in corneal epithelial tissue are actin dependent. Anat Rec. 1999;254:336–347. doi: 10.1002/(sici)1097-0185(19990301)254:3<336::aid-ar4>3.0.co;2-d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hamburger V, Hamilton HL. A series of normal development in the chick embryo. J Morphol. 1951;88:49–92. [PubMed] [Google Scholar]
- 21.Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 22.Hay ED, Revel JP. Fine structure of the developing avian cornea. Monogr Dev Biol. 1969:16–46. [PubMed] [Google Scholar]
- 23.Danjo Y, Gipson IK. Actin “purse string” filaments are anchored by E-cadherin-mediated adherens junctions at the leading edge of the epithelial wound, providing coordinated cell movement. J Cell Sci. 1998;111:3323–3332. doi: 10.1242/jcs.111.22.3323. [DOI] [PubMed] [Google Scholar]
- 24.Zieske JD, Bukusoglu G, Gipson I. Enhancement of vinculin synthesis by migrating stratified squamous epithelium. J Cell Biol. 1989;109:571–576. doi: 10.1083/jcb.109.2.571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Clark EA, Brugge JS. Integrins and signal transduction pathways:the road taken. Science. 1995;268:233–239. doi: 10.1126/science.7716514. [DOI] [PubMed] [Google Scholar]
- 26.Divecha N, Irvine RF. Phospholipid signaling. Cell. 1995;80:269–278. doi: 10.1016/0092-8674(95)90409-3. [DOI] [PubMed] [Google Scholar]
- 27.Chen Q, Kinch MS, Lin TH, Burridge K, Juliano RL. Integrin-mediated cell adhesion activates mitogen-activated protein kinases. J Biol Chem. 1994;269:26602–26605. [PubMed] [Google Scholar]
- 28.Hu KQ, Settleman J. Tandem SH2 binding sites mediate the Ras-GAP-RhoGAP interaction:a conformational mechanism for SH3 domain regulation. EMBO J. 1997;16:473–483. doi: 10.1093/emboj/16.3.473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chang JH, Gill S, Settleman J, Parsons SJ. c-Src regulates the simultaneous rearrangement of actin cytoskeleton, p190RhoGAP, and p120RasGAP following epidermal growth factor stimulation. J Cell Biol. 1995;130:355–368. doi: 10.1083/jcb.130.2.355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Watanabe N, Kato T, Fujita A, Ishizaki T, Narumiya S. Cooperation between mDia1 and ROCK in Rho-induced actin reorganization. Nat Cell Biol. 1999;1:136–143. doi: 10.1038/11056. [DOI] [PubMed] [Google Scholar]
- 31.Watanabe N, Madaule P, Reid T, et al. p140mDia, a mammalian homologue of Drosophila diaphanous, is a target protein for Rho small GTPase and is a ligand for profilin. EMBO J. 1997;16:3044–3056. doi: 10.1093/emboj/16.11.3044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Reenstra WR, Campbell AO, Orlow DL, Svoboda KKH. Fibronectin stimulated actin reorganization in corneal epithelial sheets is Rho but not Raf dependent [ARVO Abstract] Invest Ophthalmol Vis Sci. 1998;39(4):S1038. Abstract nr 4793. [Google Scholar]