

Abstract
Phage-display selection of combinatorial libraries is a powerful technique for identifying binding ligands against desired targets. Evaluation of target binding capacity of multiple clones recovered from phage display selection to a specific target is laborious, time-consuming, and a rate-limiting step. We constructed phage-display combinatorial peptide libraries in fusion with a β-lactamase enzyme, which acts as a reporter. Linear dodecapeptide and cysteine-constrained decapeptide libraries were created at the amino-terminus of the Enterobacter cloacae P99 cephalosporinase molecule (P99 β-lactamase). The overall and positional diversity of amino acids in both libraries was similar to other phage-display systems. The libraries were selected against the extracellular domain of ErbB2 receptor (ErbB2ECD). The target-selected clones were already conjugated to an enzyme reporter, therefore, did not require subcloning or any other post-panning modifications. We used β-lactamase enzyme activity-based assays for sample normalizations and clone binding evaluation. Clones were identified that bound to purified ErbB2ECD and ErbB2-overexpressing cell-lines. The peptide sequences of the selected binding clones shared significant motifs with several rationally designed peptide mimetics and phage-display derived peptides that have been reported to bind ErbB2ECD. β-Lactamase fusion to peptides saved time and resources otherwise required by the phage-ELISA of a typical phage display screening protocol. The β-lactamase enzyme assay protocols is a one-step process that does not require secondary proteins, several steps of lengthy incubations, or washings and can be finished in a few minutes instead of hours. The clone screening protocol can be adopted for a high throughput platform. Target-specific β-lactamase-linked affinity reagents selected by this procedure can be produced in bulk, purified, and used, without any modification, for a variety of downstream applications, including targeted prodrug therapy.
Keywords: Peptide libraries, phage-display, β-lactamase reporter, enzyme-linked peptides, accelerated screening
INTRODUCTION
The generation of combinatorial libraries and their selection by display on filamentous bacteriophage coat protein has provided a powerful tool for isolating ligands against virtually any target [1–6]. Although many systems have been developed, the most common phage-display system expresses the fusion protein on the minor coat protein, pIII, or the major coat protein, pVIII [7, 8]. The size of the fusion protein varies from a short peptide to antibody fragments. Phage display of random peptides has developed into a very useful technology for the discovery of novel peptide ligands and revolutionized pharmaceutical design and means of studying molecular interactions [9–13]. Using phage-display technology, peptide ligands have been successfully isolated from a wide array of targets of biological interest, such as receptor proteins, monoclonal antibodies, and carbohydrate moieties, to name a few [12–16]. The selection of target-specific ligands through phage technology is a straightforward procedure, based on the general scheme of making a large library of diverse ligands and putting it through repeated iterations of selection (or panning) and amplification [7, 17]. The selection procedure typically yields a collection of hundreds to thousands of clones. The clones from a selected population are randomly chosen and sequenced for their random DNA inserts to determine a consensus sequence motif and/or be screened for their binding to the target. This latter task is very laborious, time-consuming, and a major rate-limiting step in a typical phage-display selection protocol [18]. Screening of clones for their binding to the target is usually done by phage-ELISA, in which an antibody against phage coat protein pVIII is used as a detection reagent. Phage-ELISA is a lengthy process that requires several steps of incubations and washings [17, 19, 20]. It is also susceptible to artifacts, owing to the presence of free pVIII protein in phage preparations and variations in the peptide expression of phage clones [21].
In order to make the whole process of clone screening simpler and faster, we constructed phage-display random-peptide libraries in fusion to β-lactamase (β-lactam hydrolase, E.C. # 3.5.2.6) enzyme as a reporter. The linear dodecapeptide (X12) and cysteine-constrained decapeptide (CX10C) libraries were created at the N-terminal end of the Enterobacter cloacae P99 cephalosporinase molecule (P99 β-lactamase), between the signal peptide and the enzyme protein. Four classes of β-lactamase (A-D) have been reported [22]; P99 β-lactamase is a class C enzyme from the Enterobacter cloacae strain P99. Peptide-β-lactamase fusions were displayed at the amino terminus of the minor coat pIII protein of filamentous (M13) phage. β-Lactamase has several characteristics that make it suitable for its use as a reporter molecule [23–25]. P99 β-lactamase has been reported to tolerate N-terminal fusions without losing its enzyme activity [26]. The periplasmic secretion of β-lactamase allows the formation of disulfide bonds for cyclic peptides and a convenient extraction of fusion protein for various applications [27]. β-Lactamase is known for high turnover of its enzyme activity with several commercially available chromogenic and fluorogenic substrates. This helped us develop a very sensitive and efficient assay for measuring concentrations and target-binding of phage-displayed or free β-lactamase-peptide fusion protein. This is for the first time the construction and selection of a random-peptide library in fusion to the amino-terminus of an enzyme reporter is being reported. The target-selected clones from these reporter-linked phage-display peptide libraries do not require any post-panning chemical or biological manipulations and can be directly screened for their binding to the target in time- and cost-efficient manner using a high throughput platform. The target-specific affinity reagents identified from these libraries can easily be produced in large quantities for their uses in a variety of downstream applications.
Present communication deals with the construction of β-lactamase-peptide fusion libraries, quality analysis of these libraries as judged by overall and positional diversity of amino acids in the random regions, selection and screening of binding ligands against the extracellular domain of the ErbB2 receptor (ErbB2ECD) as a model protein. ErbB2 (HER2/c-neu) is a member of the epidermal growth factor receptor family, which is over-expressed on the cell surface of certain cancers and correlates with poor prognosis in human breast carcinoma [28].
MATERIALS AND METHODS
1. Cell culture
MCF-7 and ErbB2-overexpressing MCF-7 (MCF-7/ErbB2) were grown in 90% minimum essential medium (Eagle) with 2 mM L-glutamine, 0.1 mM non-essential amino acids, 1 mM sodium pyruvate and 1.5 g/L sodium bicarbonate, and 10% fetal bovine serum. SK-BR-3 cells were propagated in 90% McCoy’s 5A medium (modified) with 1.5 mM L-glutamine adjusted to contain 2.2 g/L sodium carbonate, and 10% fetal bovine serum. The cells and media were obtained from the American-type culture collection (Manassas, Virginia). The MCF-7/ErbB2 cells were a gift from Dr. Lippman from Georgetown University, Washington, DC.
2. Phagemid vector
We modified our earlier reported phagemid vector [29], which expressed a wild-type β-lactamase as an N-terminal fusion to a truncated pIII protein, for the construction of the libraries used in this study. The phagemid vector was digested with restriction enzymes PstI and BamHI and the stuffer was replaced by a nucleotide sequence that would complete the pIII gene for an expression of full pIII protein containing functional N1, N2, and CT domains. The protease-cleavable linker (PAGLSEGSTIEGRGAHE) was added at the N-terminal end of pIII. The FLAG and 6xHis tags were cloned at the C-terminal end of P99 β-lactamase sequence. The expressions of different proteins were confirmed by immuno-detection following Western blotting. A schematic representation of the phagemid vector design and characteristics are presented in Figure 1.
Fig. (1). Schematic presentation of phagemid vector design and features.

The linear dodecapeptide (X12) and cysteine-constrained decapeptide (CX10C) libraries were created at the N-terminal position of Enterobacter cloacae P99 cephalosporinase (BLA) molecule, between the signal peptide and enzyme protein. The random sequences of these libraries were linked to the N-terminal end of the β-lactamase molecule with a short linker (GGGS). Restriction sites SpeI (3326–3231 bp) and AgeI (3256–3261 bp) were used for cloning. The selected site was randomized using the nnk-scheme (n= a, t, c, or g; k=g or t). The N-terminal presence of pIII signal peptide helped in periplasmic translocation of BLA. Chloramphenicol acetyltransferase (CAT) and BLA provided antibiotic resistance to transformed host bacteria in presence of chloramphenicol and cefotaxime, respectively. This vector in suppressor host TG1 E. coli with the help of helper phage (KM13)-generated phage particles expressing BLA-linker-random peptide as an N-terminal fusion protein to phage coat pIII. A protease-cleavable linker between pIII protein of phage particle and BLA was used for trypsin elution of phage following panning. The transformation of non-suppressor strain Top10 E. coli, which recognizes the amber stop (tag) between the BLA and protease-cleavable linker, with the vector allowed the expression of free BLA protein with a random peptide library at the N-terminal end, and FLAG and 6xHis tags at the C-terminal end. The magnified insert region given at the lower left of the figure is inversed in relation to the vector because the insert oligonucleotide and the gene sequences are written 5′– 3′.
3. Construction of β-lactamase N-terminal fusion library
We created linear dodecapeptide (X12) and cysteine-constrained decapeptide (CX10C) libraries at the N-terminal position of a β-lactamase molecule, between the signal peptide and enzyme protein. In the dodecapeptide library, cloning inserted 12 random amino acids at the −5 to −16 positions in reference to the β-lactamase molecule. The cysteine-constrained library has a random decapeptide, flanked with cysteine molecules at −5 and −16 positions. The phagemid vector following column purification (Qiagen, Valencia, California) was digested with SpeI and AgeI (New England Biolabs, Ipswich, Massachusetts) restriction endonucleases and small stuffers were separated from cut vector by gel electrophoresis. The vector bands were cut and the DNA was purified using a Qiagen gel extracting kit. The half-site cloning method [30] was employed for constructing the β-lactamase fusion libraries using the nnk-scheme of randomization. In this strategy, a direct-strand oligonucleotide is complemented with 2 oligonucleotides at the 5’ and 3’ ends, leaving a gap of random region (nnk) to be filled with complementary (nnm) nucleotides following bacterial transformation. Three oligonucleotides, forward and its 2 complementary half sequences, for each cloning were annealed at an equal concentrations (1:1:1) in annealing buffer (10 mM Tris-HCl, 50mM NaCl, 1mM EDTA, pH 7.5) by heating at 95°C in a heating block for 5 minutes. The required oligonucleotides were synthesized and gel-purified by Bio synthesis (Lewisville, Texas). In order to avoid clones without a randomized insert, the randomization was achieved in 2 steps. In the first step, a set of annealed 5’-phosphorylated oligonucleotides containing stop-codons (forward, ctagtcgttcctttctat tctcactctgcttaatgataatgataatgataataatgataatgataaggtggaggttcgaca; complementary, agcagagtgagaat agaaaggaacga and ccggtgtcgaacctccacc) at the place of intended randomization were cloned. In the second step, the vector containing stop-codons was purified, digested with SpeI and AgeI, and cloned with the oligonucleotides with the random region (nnk). The forward strands of 5’-phosphorylated oligonucleotides were ctagtcgttcctttctattctcactctgctnnknnknnknnknnknnknnknnk nnknnknnknnkggtggaggttcgaca and ctagtcgttcctttctattctcactctgcttgtnnknnknnknnknnknnknnknnk nnknnktgcggtggaggttcgaca for linear dodecapeptide and cysteine-constrained decapeptide libraries, respectively. The complementary strands were the same as described for the first step of cloning with oligonucleotides containing stop codons.
Electroporation-competent TG1 bacterial cells (≥1×1010 transformations/µg) were used for bacterial transformation (Stratagene, La Jolla, California). For library preparations, 30 transformations were performed using a Bio-Rad electroporator with 0.1-cm gap electroporation cuvettes at 1700 volts, according to the supplier’s instructions. The serial dilutions of 5 µl of transformed bacteria were plated on LB/Agar containing 10 µg/ml chloramphenicol and/or 0.1 µg/ml cefotaxime (Sigma Chemical Co., St Louis, Missouri) for determining the numbers of total primary and β-lactamase-active primary clones. The rest of the transformed bacteria was plated in LB/Agar bio-dishes (245×245×18 mm, BD Biosciences, Acton, Massachusetts) containing 10 µg/ml chloramphenicol and 0.1 µg/ml cefotaxime. Phage libraries were produced using β-lactamase active clones following a super-infection with KM13 helper phage [31]. KM13 phage contains a protease cleavage site between the second and third domains of their minor coat protein pIII, rendering the phage without fusion protein non-infective at the trypsin elution step of biopanning.
4. Panning of phage libraries on ErbB2ECDprotein
A mixture of equal numbers of clones from both linear dodecapeptide and cysteine-constrained decapeptide libraries was employed for panning against purified ErbB2ECD protein (Creative Biomolecules, Inc., Hopkinton, Massachusetts). The phage library was subtracted for plastic and casein binders before panning. The subtracted library was panned on ErbB2ECD immobilized in MaxiSorp™ polystyrene Immuno-plate wells (Nulge Nunc, Roskilde, Denmark). One hundred µl of ErbB2ECD solution (2.5 µg/ml PBS) was added in a 96-well microplate and incubated overnight in cold. Wells were washed 3 times with PBST and blocked with 1% casein solution (Thermo Fisher Scientific, Rockford, Illinois) for 1 hour at room temperature. Blocked wells were incubated with 100 µl subtracted phage library (~1×1012 cfu; 1:1 mixture of both X12 and CX10C libraries) for 2 hours at room temperature. Wells were washed 6 times with PBST (phosphate-buffered saline containing 0.05% Tween-20) and 2 times with PBS over a period of 30 minutes. The bound phage particles were eluted with 100 µl of trypsin solution (1 mg/ml PBS) by incubating at room temperature for 10 minutes. Eluted phage were transferred to 1 ml TG1 E. coli culture grown to an OD600 of 0.4 and incubated for 30 minutes at 37°C in a water bath without shaking. Different dilutions of a small portion of this culture were plated on LB/Agar containing chloramphenicol (10 µg/ml) and cefotaxime (0.1 µg/ml) for determining the output. The rest of the culture was centrifuged and the pellet was suspended in 100 µl LB medium and plated on LB/agar plates (150 mm) containing chloramphenicol (10 µg/ml) and cefotaxime (0.1 µg/ml). Plates were incubated overnight at 37°C and the colonies were scraped in 1 ml of 15% glycerol in LB medium. Fifty µl of this glycerol stock was added to 50 ml LB medium containing 10 µg/ml chloramphenicol and incubated with shaking at 37°C until it reached to 0.4 OD600. Ten ml of this culture was mixed with 5×1010 cfu of KM13 helper phage and incubated for 30 minutes at 37°C in a water bath without shaking. The culture was centrifuged and suspended in 50 ml LB containing chloramphenicol (10 µg/ml) and kanamycin (50 µg/ml) and incubated with shaking at 30°C overnight. The culture was centrifuged and phage particles in the supernatant were concentrated by PEG/NaCl precipitation. This phage preparation from the first round of panning was used as an input for the second round of panning. A total of 3 rounds of panning were conducted.
5. β-Lactamase sample preparation
β-Lactamase samples were prepared from bacterial hosts TG1 and TOP10 E. coli. The clones from the third round of panning, which were in TG1 host, were screened for binding to purified ErbB2ECD. The plasmids of the positive clones from this screening were used for the electro-transformation of competent TOP10 E. coli cells (Invitrogen, Carlsbad, California). TOP10 is a non-suppressor E. coli strain that recognizes amber stop at the 3’ end of 6xHis-tag sequence; therefore, it generates a free β-lactamase-FLAG-6xHis protein [32]. The transformed bacteria were plated on LB/Agar plates containing cefotaxime (0.1 µg/ml) and chloramphenicol (10 µg/ml). Randomly selected individual isolated colonies were amplified in 2xYT medium with shaking at 260 rpm for 30 hours at 30°C. The cultures were centrifuged in cold at 3000×g for 10 minutes. The bacterial pellets were frozen at −80°C with BPER-II (Invitrogen, Carlsbad, California) and bacterial protease inhibitor cocktail (Sigma Chemical Co., St. Louis, Missouri). Lysates were prepared by incubating the thawed pellets at room temperature for 90 minutes with slow shaking [29]. The lysed bacterial preparations were centrifuged at 15,000×g at 4°C for 20 minutes. Supernatants were used for β-lactamase purification, binding screening, and β-lactamase protein and enzyme activity assays. β-Lactamase was purified using nickel-sepharose columns (GE Health Care, Bio-sciences AB, Uppsala, Sweden).
6. β-Lactamase enzyme assay
The enzyme activity of β-lactamase in different preparations was assayed by using nitrocefin (Oxoid, Basingstoke, UK) and soluble fluorocillin green (Invitrogen, Carlsbad, California) as substrates. The colorimetric assay [33] was conducted in a 96-well microplate by mixing 20 µl of enzyme preparation with 100 µl of nitrocefin solution (100 µg/ml) made in PBS containing 0.125% (w/v) n-octyl-β-D-glucopyranoside (Sigma Chemical Co., St. Louis, Missouri). The changes in the optical density were measured at 490 nm at 2-minute intervals for 10 minutes using Synergy-HT microplate reader (Biotek Instruments, Winooski, Vermont). Fluorocillin green substrate was used for a fluorometric assay of β-lactamase enzyme activity. One hundred µl fluorocillin green (10 µg/ml) solution in PBS was added to microplate wells containing β-lactamase and the fluorescence was read at 2-minute intervals for 10 minutes using ~495/525 mm filters for excitation and emission, respectively. A linear range of both assays were determined by using different amounts of β-lactamase enzyme.
7. ELISA of β-lactamase
One hundred µl of different dilutions of β-lactamase lysates were incubated overnight in cold in 96-well Ni-NTA microplates (HisSorb™; Qiagen, Valencia, California). The next day, wells were washed 4 times with PBST and blocked with Casein (Thermo Fisher Scientific, Rockford, Illinois) for 1 hour at room temperature. After washing the wells 2 times with PBS, 100 µl FLAG antibody-HRP conjugate (Sigma Chemical Co., St. Louis, Missouri), diluted 1: 20000, was added and incubated for 1 hour at room temperature. The wells were washed for 4 times with PBST and 2 times with PBS. The stable peroxidase and luminol/enhancer from SuperSignal® ELISA kit (Thermo Fisher Scientific, Rockford, Illinois) were mixed in an equal volume and added to wells for assaying the horse-radish peroxidase activity. The luminescence was read using GloRunner™ microplate luminometer (Turner BioSystems, Sunnyvale, California).
8. Screening of selected clones for binding to ErbB2ECD
The bacterial lysates were prepared from TG1 E. coli colonies following the third round of panning. β-Lactamase activities in the lysates were determined using nitrocefin as a substrate. The samples used for the binding assay were normalized for their β-lactamase activities by diluting the lysates in PBS containing bacterial protease inhibitor cocktail (Sigma Chemical Co., St. Louis, Missouri). ErbB2ECD and bovine serum albumin (BSA) were immobilized in the MaxiSorp™ polystyrene Immuno-plate wells (Nulge Nunc, Roskilde, Denmark) by incubating 100 µl protein solutions (2.5 µg/ml in PBS) overnight at 4°C. The next day, wells were washed 3 times with PBST and blocked with casein (1% w/v) solution in PBS (Thermo Fisher Scientific, Rockford, Illinois) for 1hour at room temperature. After washing the wells 2 times with PBS, 100 µl lysates of β-lactamase clones were added and incubated for 2 hours with slow shaking at room temperature. The lysates from the both TG1 E. coli that were transformed, with the vector expressing wild-type β-lactamase, and from the unselected library culture served as as negative controls. Wells were washed 4 times with PBST and once with PBS. The activity of bound β-lactamase was measured by using fluorocillin green substrate as described earlier.
9. Binding studies with human cancer cells over-expressing ErbB2
The ErbB2ECD-binding clones were studied for their binding to MCF-7, MCF7/ErbB2 and SK-BR-3 cells. Thirty thousand cells of each cell-line were plated in a 96-well cell culture plates (Corning Incorporated, Corning, New York)) and incubated overnight at 37°C in a humidified, 5% CO2 atmosphere. The next day, the cells were down-shifted to medium without fetal bovine serum and incubated at 37°C in a humidified, 5% CO2 atmosphere. After 2 hours, the medium was removed and cells were washed with PBS. The cells were fixed in 0.5% p-formaldehyde for 30 minutes at room temperature. After washing the cells fo 5 times with PBS, the wells were blocked with casein (1% w/v) solution in PBS (Thermo Fisher Scientific, Rockford, Illinois) for 1 hour at room temperature. The cells were washed again 2 times with PBS and incubated with purified free β-lactamase samples. The β-lactamase samples were prepared from the lysates of TOP10 E. coli host and were normalized for their enzyme activities by diluting the samples in PBS containing BSA (1 mg/ml) and bacterial protease inhibitor cocktail (Sigma Chemical Co., St. Louis, Missouri). The samples of wild-type β-lactamase and those prepared from unselected library clones were also included as negative controls. The cells were incubated for 2 hours with slow shaking and washed 6 times with PBS. The enzyme activities of cell-bound β-lactamase were assayed fluorometrically (495 ex/525 em) as described earlier in β-lactamase assay section.
For visualization of cell-bound β-lactamase activity, washed cells after incubation with β-lactamase clones were exposed to precipitating fluorocillin green substrate (Invitrogen, Carlsbad, California) and observed under Nikon TE2000-U inverted fluorescence microscope (Nikon Corp., Kanagawa, Japan) using 360±20 nm excitation, 535±20 nm emission, and 400 nm dichroic filters.
10. DNA sequence analysis and bioinformatics
Using a set of primers (Table 1), the DNA of the vectors after each cloning and that of the inserts of ErbB2-binding clones were sequenced. Bacterial pellet from 4-ml cultures of each selected clone was processed for plasmid purification using QIAprep® miniprep columns (Valencia, California). The cycle sequencing reactions were performed using BigDye® Terminator version 3.1 kit (Applied Biosystems, Foster City, California). The Vermont Cancer Center DNA Analysis Facility at the University of Vermont carried out automated DNA sequencing using ABI Prism® 3130xl Genetic Analyzer. The translated amino acids of N-terminal fusion peptides in ErbB2-binding β-lactamase clones were searched for significant motifs using the IBM Sequence Pattern Discovery Tool software program based on TEIRESIAS algorithm [34]. The frequency and diversity of amino acids distribution in peptide inserts from the clones of both libraries were determined by using receptor ligands contacts (RELIC) program [35].
Table 1.
Primer sequences used for DNA sequencing of the vector.
| Name | Primer sequence |
|---|---|
| M13 F (−21) | 5'-tgtaaaacgacggccagt-3' |
| M13 R | 5'-caggaaacagctatgacc-3' |
| SP6 Primer | 5'-atttaggtgacactatag-3' |
| T7 Primer | 5'-taatacgactcactataggg-3' |
| BLA-3121 | 5'-gaaacagctatgaccatgattacg-3' |
| BLA-3516 | 5'-cggtgaccagatactggc-3' |
| BLA-3931 | 5'-cgtgcaggatatggcgaa-3' |
| BLA-4106 | 5'-gagggcagcgacagtaagg-3' |
| BLA-4546 | 5'-taacgtctggaaagacgacaa-3' |
| BLA-4941 | 5'-tttatacgggcactgttactcaag-3' |
| BLA-5331 | 5'-tacagtctgacgctaaaggcaa-3' |
BLA series of primers either hybridize to P99 β-lactamase gene or to the vector regions near to this gene.
RESULTS
1. Size and quality of the libraries
The colonies of transformed bacteria that grew in the presence of both chloramphenicol and cefotaxime were considered to be primary β-lactamase-active clones. The linear dodecapeptide and cysteine-constrained decapeptide libraries had 4.9±0.32×107 and 6.1±0.44×107 primary β-lactamase-active clones, respectively. The assay of β-lactamase activity in different clones exhibited strong enzyme activities in their purified phage particles, pIII-β-lactamase fusion, and free β-lactamase protein preparations. There was ~3-fold variation in the enzyme activities among clones. However, the ratios of enzyme activity to β-lactamase protein levels in different clones were consistent (data not presented).
The representation of different amino acids in the random region of both libraries was determined and the data in comparison to their expected values are presented in Figure 2. The expected values were calculated based on the number of codon available for a given amino acid under the nnk-scheme of cloning. The observed frequencies of amino acid residues should be similar to expected values if the phage-host system treats each peptide equally, without bias. The results showed that the observed frequencies of most of the amino acids in both libraries were close to their calculated frequencies. However, some overrepresentation (1.5- to 2-fold) of glycine, phenylalanine and valine, and underrepresentation (1/2 to 1/3) of asparagine, glutamine, and lysine were observed. These results show overall fair representation of all the amino acids in the random regions of both libraries.
Fig. (2). Frequency of occurrence of different amino acids in the random regions of peptide libraries.

Observed frequencies of amino acid occurrence in both the libraries were compared with their expected frequencies based on the availability of codons for a given amino acid. Observed frequencies were calculated in the following manner: the number of times a given amino acid appeared in the analyzed random inserts (n) ÷ the total count of amino acids (n × 10 or 12). Expected frequencies were calculated based on 32 codons available in the nnk-scheme of cloning. Amino acids are represented as single-letter codes.
Figure 3 presents the diversity of different amino acids at each position (1 through 10 or 12) of random peptides in both libraries. Diversity is a statistical measure of the proportion of the 20 possible amino acids that are observed at a given position. If a given position in all analyzed sequences shows 1 amino acid, the diversity score is 0.05 (1/20); if it is populated in equal proportions by all 20 amino acids, the score is 1.0 (20/20). The data presented here clearly show good amino acid diversity at different positions in the random region of both libraries. A notable low diversity was seen at position 1 of both libraries, indicating preference for certain amino acids at this position. Furthermore, the amino acid diversity was a little lower at positions 5, 6 and 7 in cysteine-constrained library as compared to the respective positional diversity in linear dodecamer library.
Fig. (3). Positional amino acid diversity in peptide libraries.

The diversity of different amino acids at each position (1 through 10 or 12) in the randomized regions of both linear dodecapeptide (X12) and cysteine-constrained decapeptide (CX10C) libraries as presented is a statistical measure of the proportion of the 20 possible amino acids that are observed at a given position. If a position is populated equally by all the 20 amino acids, the score is 1.0 (20/20). The data represent positional diversity in the occurrence of amino acids ± SD. Amino acids are represented as single-letter codes.
2. Identification of ErbB2ECD-binding clones
The screening of the clones for their binding to immobilized ErbB2ECD was based on a very simple and fast assay of bound β-lactamase enzyme activity using bacterial lysates. The binding study of 50 clones from the third round of panning identified 12 clones that bound specifically to the ErbB2ECD (Figure 4). These clones exhibited significantly higher binding to ErbB2ECD in comparison to BSA. The ErbB2ECD-positive clones did not bind to casein or casein blocked plastic wells (data not presented). The lysates of bacterial culture of unselected library and of bacteria transformed with vector expressing wild-type β-lactamase, which served as negative controls, did not show significant binding to ErbB2ECD.
Fig. (4). Screening of clones for their binding to extracellular domain of ErbB2 (ErbB2-ECD).

Randomly selected 50 clones following the third round of panning were screened for their binding to purified ErbB2-ECD and bovine serum albumin (BSA) proteins. BSA was used as a negative control protein. The screening was based on assaying the protein-bound β-lactamase activities. The clones expressing wild-type β-lactamase (Wt-BLA; without fusion peptide) and the unselected library were used as negative clone controls. The bars represent β-lactamase activity as the change in relative fluorescence unit (RFU)/min ± SE. *p≤0.05, significance of difference in the clone binding to ErbB2-ECD and BSA were determined using Student’s T-test.
3. Amino acid sequences of ErbB2-binding clones and their significant motifs
The amino acid sequences of ErbB2ECD-binding clones as deduced from their insert DNA sequencing are presented in Table 2. All of the 12 selected clones were unique. There was a selection of an equal number of ErbB2ECD-binding clones from linear dodecapeptide and cysteine-constrained decapeptide libraries. It shows no preference of selection between these two 2 libraries for the given target and the panning technique used in this study.
Table 2.
Amino acid sequences of ErbB2ECD-binding clones
| Clone number |
Amino acid sequence |
|---|---|
| 111 | CSPGGVVSVCIC |
| 113 | MCGVCLSAQRWT |
| 119 | CFSGCGWVVKWC |
| 120 | CGERFPLVDPCC |
| 128 | CSLRGLDLRCFC |
| 129 | VSCPWLKYSGAL |
| 132 | CWLPVVVGSRCC |
| 134 | VWGLPSCTLAHG |
| 139 | QSIFVDTMFRGS |
| 145 | DDLSYLMVPGLL |
| 149 | CVLGGLSVVFEC |
| 150 | SGLWWLGVDILG |
Amino acids are represented by single-letter code.
The ErbB2ECD-binding sequences from this study were aligned with the ErbB2ECD-binding peptide sequences published from other studies (Table 3). Some of the sequences from this study (clones 111, 113, 120, 129, 134, 139 and 149) appear to share strong motifs with the previously reported ErbB2ECD-binding peptides.
Table 3.
Alignments of ErbB2ECD-binding peptides from present study with some other peptides, which have been reported to bind ErbB2ECD, for determination of consensus sequence motifs.
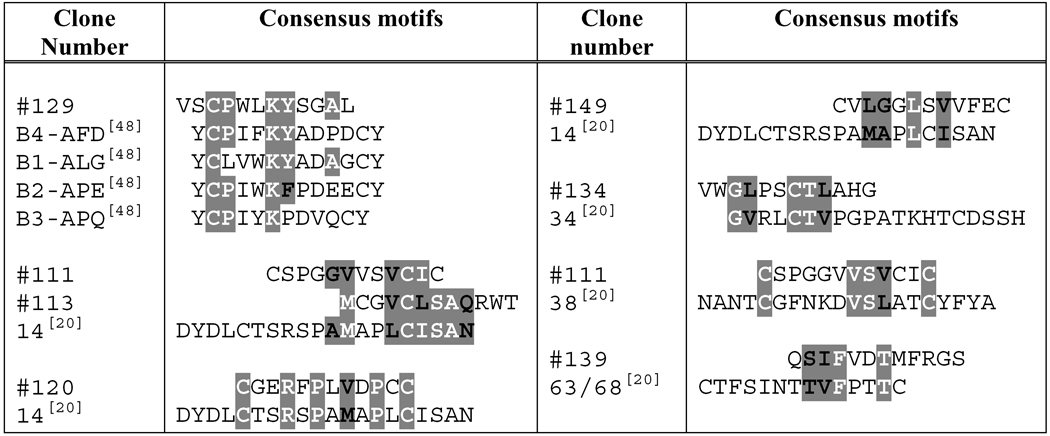 |
Amino acids sharing consensus motifs are highlighted. Amino acids which are exactly the same are given in white bold letters and those represent conserved substitutions are in black bold letters. Amino acids are represented as single letter codes.
Clones selected from this study.
Reference numbers related to the reported peptides are presented in parentheses.
Table 4 presents alignments of certain ErbB2ECD-binding insert-peptide sequences with EGF-like domain of ErbB receptor ligands and with certain conserved regions of ErbB receptors. Although no natural ligand for ErbB2 receptor has been reported, it is interesting to find the alignment of two ErbB2ECD-binding clones, 129 (VSCPWLKYSGAL) and 132 (CWPVVVGSRCC), with a conserved region of EGF-like domain of other ErbBs receptor ligands; namely, EGF, TNF-α, β-cellulin, neuregulin-1 and neuregulin-4. Some of the selected ErbB2-binding sequences also aligned with conserved regions of other ErbB receptors which are known to form dimers with ErbB2. The insert-peptides of clones 132 (CWPVVVGSRCC), 145 (DDLSYLMVPGLL) and 149 (CVLGGLSVVFEC) closely aligned with a conserved sequence (54/56 – 83/85) of L1 domain of ErbB1, ErbB3 and ErbB4 receptors. The insert-peptides of both clones 111 (CSPGGVVSVCIC) and 113 (MCGVCLSAQRWT) showed consensus motifs at 2 places within a short conserved region of 32 amino acids in the S2 domain of these ErbB receptors.
Table 4.
Alignments of selected ErbB2ECD-binding clones with EGF-like domain of ErbB-receptor ligands and with certain conserved regions of extracellular domain of other ErbB-receptors.
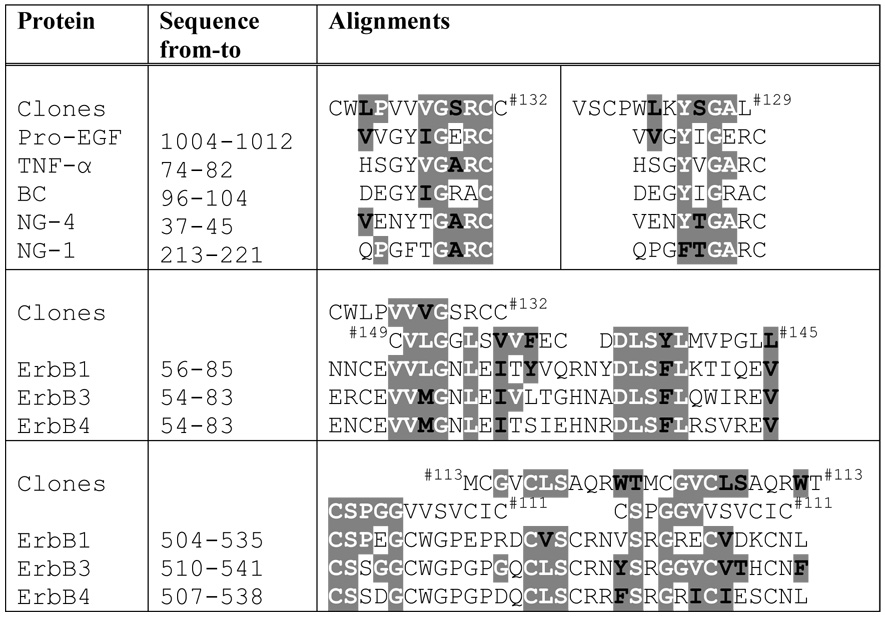 |
Amino acids sharing consensus motifs are highlighted. Amino acids which are exactly the same are given in white bold letters and those represent conserved substitutions are in black bold letters. Amino acids are represented by single-letter codes. BC, β-Cellulin; NG-1, Neuregulin-1; NG-4, Neuregulin-4.
ErbB2ECD-selected clone from this study.
4. Binding of ErbB2ECD-positive clones to human cancer cells over-expressing ErbB2
The clones that were found to bind ErbB2ECD were purified as free β-lactamase proteins, independent of pIII fusion, and studied for their binding to human cancer cells MCF-7/ErbB2 and SK-BR-3, known for their higher expression of ErbB2. The MCF-7 cell-line was used as a control, as these cells show minimal ErbB2 expression. The binding data presented in Figure 5 are derived from fluorometric readings from the assay of cell-bound β-lactamase activity using soluble fluorocillin green as a substrate. Nine of the 12 ErbB2-positive clones showed significantly higher binding to MCF-7/ErbB2 and SK-BR-3 cells, as compared to MCF-7 cells. Clones 128 and 149 did not show any preferential binding to either of the 2 ErbB2 over-expressing cells, and clone 120 showed slightly higher binding to only SK-BR-3 cells in comparison to MCF-7 cells. The negative controls of the clones expressing wild-type β-lactamase and unselected random library did not show any significant binding to ErbB2 over-expressing cells.
Fig. (5). Binding of selected ErbB2ECDpositive clones to ErbB2 over-expressing cells.

The clones that were found to bind purified ErbB2ECD were studied for their binding to ErbB2 over-expressing MCF-7/ErbB2 and SK-BR-3 cell-lines. The MCF-7 cell-line that is known to exhibit minimal ErbB2 expression was used for negative control cells. The screening was based on assaying the cell-bound β-lactamase activities. The clones expressing wild-type β-lactamase (Wt-BLA; without fusion peptide) and unselected library were used as negative clone controls. The bars represent β-lactamase activity as change in relative fluorescence unit (RFU)/min ± SE. *p≤0.05, significance of difference in the binding of clones to MCF-7 in comparison to MCF-7/ErbB2 or SK-BR-3 cells were determined using Student’s T-test. ErbB2ECD, extracellular domain of ErbB2.
The bindings of ErbB2ECD-positive clones to these cell-lines were also studied microscopically using a substrate that produces precipitated fluorescence product. The binding results of this microscopic study as represented by clones 113 and 150 are presented in Figure 6. The results clearly show higher bindings of these clones to MCF-7/ErbB2 and SK-BR-3 cells in comparison to MCF-7 cells. The negative controls of clones expressing wild-type β-lactamase or unselected library did not show any appreciable binding.
Fig. (6). Fluorescence imaging of ErbB2 over-expressing cells for their binding to ErbB2ECDpositive clones.

The figure shows binding of representative clones 113 and 150 to MCF-7/ErbB2 and SK-BR-3 cells, which are known to over-express the ErbB2 receptor. High intensities of fluorescence are seen in both MCF-7/ErbB2 and SK-BR-3 cells in comparison to minimally expressive MCF-7 cells. Negative controls of wild-type β-lactamase (Wt-BLA) and unselected library clones did not show higher fluorescence in MCF-7/ErbB2 and SK-BR-3 cells. The precipitating green fluorescence is produced by the β-lactamase activity associated with clones.
DISCUSSION
Using phage-display technology, we generated libraries of random peptides in fusion to P99 β-lactamase enzyme as a reporter molecule. For the construction of these libraries, we employed pIII signal peptide of Sec-pathway for P99 β-lactamase translocation. Our Western blot analysis of phage proteins showed significantly higher phage display of β-lactamase-pIII fusion protein (~1.43 vs 1.17 out of 5 pIII/phage) by pIII signal peptide as compared to DsbA signal peptide of SRP pathway [36], which has been reported to improve phage presentation of certain proteins up to 700-fold [37]. The size of both the linear dodecapeptide and cysteine-constrained decapeptide libraries, as represented by the number of primary clones, was close to the size found in the phage-display libraries we used in our previous studies [6, 20]. It was remarkable to find high numbers of random peptide clones exhibiting the active β-lactamase fusion enzyme. Analyses of free β-lactamase fusion protein, independent of phage, from randomly selected clones showed no changes in the enzyme activity due to peptide fusion. DNA sequencing of random inserts of 160 clones from both libraries did not show any clone without a peptide insert and all of them were in frame. The success may be attributed to the strategy of using 2-step cloning in which stop-codon containing nucleotides were cloned in the first step, and to the selection of a β-lactamase-active library in the presence of cefotaxime.
The overall representations of different amino acids in random regions of both libraries were very good, as only a few amino acids exhibited a little over- or under-representation. The frequencies of occurrence of different amino acids in phage-display random libraries depend not only on the availability of the number of codons for a given amino acid but also on the other factors, including phage-host biology. Rodi et al. [38] analyzed commercially available phage-display linear dodecapeptide (PhD-12™) and cysteine-constrained heptapeptide (PhD-CX7C™) libraries (New England Biolabs, Ipswich, Masachusetts) and reported overall depression in the occurrence of cysteine, arginine, and glycine and an overabundance of proline, threonine, and histidine residues. The analyses of amino acid diversity at each position in the random regions of our linear dodecapeptide and cysteine-constrained decapeptide libraries revealed a good diversity at all the positions except at the first position, indicating preferences to certain residues. We found fewer glutamine, arginine, proline, and isoleucine and an abundance of glycine, valine, and phenylalanine residues at this position. An under-representation of arginine and proline at the first position in random peptides was observed in PhD-12™ and PhD-CX7C™ libraries as well [38]. A strong bias against positively charged residues at the amino-terminus of random peptide pIII libraries has been reported for membrane insertion [39, 40]. Proline at the first position is known to act as an inhibitor of the signal peptidase enzyme, which cleaves the signal peptide from the mature protein subsequent to membrane insertion [39, 41–43]. The analyses of clone insert sequences indicated that the complexities of random peptide libraries in fusion to β-lactamase were consistent with standards of other types of phage-display library systems and they were suitable for selection studies [38].
The selection of these libraries on the purified ErbB2ECD protein was done by standard protocol of repeated panning and amplification. The presence of a protease-cleavable linker between β-lactamase and the phage coat pIII protein helped in the elution of target-bound phage by trypsin treatment. Trypsin, at the same time, inactivated the phage pIII molecules that originated from the KM13 helper phage, as they have this protease-cleavable linker between N1-N2 and CT domains of pIII protein [31]. N1 and N2 domains are essential for the bacterial infection through pIII. Thus, the treatment with trypsin renders nonviable any phage not carrying a fusion to the coat protein pIII. However, phage which consists of both helper phage encoded proteins and fusion coat proteins still remained infective upon trypsin elution. This strategy allowed selective amplification of phage displaying pIII-fusion protein. The panning of these libraries on purified ErbB2ECD identified several binding clones that bind specifically to the target. The bacterial lysates of selected clones were used for the screening of clone binding to the ErbB2ECD protein, based on assaying residual β-lactamase activity. This protocol offered several advantages over standard phage-binding protocol. The process of lysate preparation is quick and simple in comparison to the protocol of phage production from individual clones. A typical screening protocol with phage clones requires phage quantification for sample normalization and for target-binding assessment, which is usually done with the help of phage-ELISA [44]. Phage-ELISA is a lengthy time-consuming process that requires antibodies and several rounds of incubations and washings [14]. In the present investigation, we used β-lactamase enzyme activities of the samples for normalization and binding screening. It was a savings on the time and resources that would have been needed for the phage-ELISA protocol. Unlike Phage-ELISA, our enzyme-based screening does not require a secondary protein, several steps of lengthy incubations and washings, and can be finished more rapidly. Our studies on the ELISA of β-lactamase protein showed a very good correlation between protein levels and enzyme activities of different clones, indicating no modification in enzyme activities owing to peptide fusions. The enzyme-based screening of clones for their target binding has been reported earlier also, using alkaline phosphatase enzyme as a reporter [45–47]. However, in these reports the fusion of target-selected clones to a reporter was a whole separate post-panning process that required several steps related to the amplification, vector modifications, subcloning and bacterial transformation. In our protocol, the target-selected clones from a panning do not need these additional steps as they are already in fusion with the reporter enzyme and are ready for the screening.
The binding study identified 12 unique clones that bound to ErbB2ECD specifically. The alignment of amino acid sequences of these binding peptides with other previously reported ErbB2ECD-binding peptides revealed several significant consensus sequence motifs. Of particular interest is clone 129 (VSCPWLKYSGAL), which shared a significant motif (CPXXKY/F) with the peptide mimetics that were rationally designed based on a dimerizing site of ErbB receptors [48]. These peptide mimetics have been reported to specifically bind ErbB ectodomains, block inter-receptor interactions and inhibit signaling. The extracelluler domain of ErbB2 has been reported to homodimerize and preferentially heterodimerize with the ectodomains of other ErbB family of receptors [49], indicating the presence of certain amino acid motifs on ErbBs that may bind to ErbB2ECD. Clones 132 (CWLPVVVGSRCC), 145 (DDLSYLMVPGLL) and 149 (CVLGGLSVVFEC) shared 2 significant motifs within 31 conserved amino acids of L1 domain of ErbB1 (EGFR/HER1), ErbB3 (HER3) and ErbB4 (HER4) receptors. Clones 111 (CSPGGVVSVCIC) and 113 (MCGVCLSAQRWT) each shared 3 significant motifs within 32 conserved amino acids of S2 domain of these ErbB receptors. These results indicate possible involvement of ErbB2 dimerization sites in our clone binding; however, further investigations involving deletion and site-directed mutagenesis are needed before the relevance of these motifs in relation to target binding can be affirmed. Since ErbB2 does not have a known biological ligand to date, we compared the ErbB2-binding sequences with known ligands to other ErbB family receptors [28, 49]. The alignments analyses revealed that clones 129 (VSCPWLKYSGAL) and 132 (CWLPVVVGSRCC) shared significant motifs with EGF, β-cellulin, TNF-α, neuregulin-1, and −4. It is interesting to note that both clones shared the motifs with EGF-like regions of these biological ligands.
In a standard phage-display protocol, once a phage clone is identified as a binder, its DNA insert is sequenced, and the deduced amino acid sequences are usually synthesized and chemically modified (e.g., biotinylated, fluorescently tagged, enzyme-linked) for downstream investigations. Another approach is bioconjugation, in which the DNA insert encoding a target-selected polypeptide is subcloned into another vector in such a way that the polypeptide-reporter fusion protein is produced. Green fluorescent proteins [50–52], alkaline phosphatase [53–55] and β-galactosidase [56] are some of the examples of fusion partners used in such studies. In the present study, clones that showed positive binding were expressed in a suppresser host, purified as free peptide-β-lactamase fusion proteins, and used directly without any additional modification for binding studies on ErbB2-overexpressing human cancer cells. The presence of an amber stop codon (tag) between the hexahistidine and protease-cleavable linker allowed the expression of free peptide-β-lactamase protein [32]. The presence of 6xHis and FLAG tags in the free peptide-fusion protein has been very useful, particularly in the absence of a commercially available antibody against P99 β-lactamase, for purification, physical estimation, and tracking of peptide-fusion clones. The binding assessment of selected clones to human cancer cells was done in a 96-well format by assaying the β-lactamase enzyme activity of residual cell-bound peptide-fusion clones. Several ErbB2ECD-selected clones exhibited higher binding to ErbB2-overexpressing MCF-7 (MCF-7/ErbB2) and SK-BR-3 cells in comparison to negative control MCF-7 cells, which are known for their minimal ErbB2 expression [14]. The soluble fluorescent product of Fluorocillin™ Green substrate was measured for assaying the enzyme activity of cell-bound peptide-β-lactamase clones. A modified version of this substrate that produces precipitating green fluorescent product was used for microscopic study of clone binding in these cell-lines. The results of cell-imaging confirmed the higher binding of selected clones to ErbB2-overexpressing cells in comparison to minimally-expressive cells, thus exhibiting the feasibility of microscopic studies on β-lactamase activity-based clone-binding.
The whole process of clone screening for their binding to purified target protein or to cells, as described in present study, is very simple and fast and can be easily adopted to analyze hundreds or thousands of clones in a high throughput manner. β-Lactamase is a high turnover enzyme with several available chromogenic and fluorogenic substrates [57–60] that can be suitably used for conducting a very fast, sensitive, and efficient screening of target-binding of phage-display or free β-lactamase-peptide fusion protein. In the present study we used nitrocefin, a chromogenic substrate, for sample normalizations and Fluorocillin™ Green, a fluorogenic substrate, for clone binding studies. Both substrates are known for their very high sensitivity towards β-lactamase; however, the linear range was bigger in the case of Fluorocillin™ Green, as is true with any fluorometric analysis. The peptide-β-lactamase ligands identified by our procedure can also be directly used with esterified derivatives of fluorescence substrates such as CCF2 and CCF4, which have been shown to localize individual cells containing fewer than 100 β-lactamase molecules [61]. These substrates may be useful in real-time monitoring of target-bound β-lactamase clone internalization in living cells. Target-specific β-lactamase-linked affinity reagents selected by our procedure can be used for additional applications, such as ELBA competition assay, immunohistochemistry, flow cytometry, and Western blotting. Peptide-β-lactamase fusion ligands can be easily produced in bulk and since it is a small single domain enzyme, it is very stable and easy to purify [62].
A peptide-β-lactamase fusion selected against cancer-specific targets can also be used for prodrug therapy of cancer in conjunction with suitable prodrug substrates. Cephalosporin prodrugs of mechanistically diverse anticancer agents, such as doxorubicin [63], taxol [64], platinum complexes [65], phenylenediamine mustard [66], and vinblastine [67] have been successfully used with β-lactamase in experimental studies. The hydrolytic cleavage of β-lactam ring by β-lactamase enzyme causes a secondary reaction that triggers the release of anticancer drug at the C-3’ position of the cephalosporin nucleus. P99 β-lactamase has been reported to show versatility and very high activity towards a broad spectrum of cephalosporins and their prodrug conjugates, some of which have been used in preclinical studies on antibody-directed prodrug therapy (ADEPT) of cancer [63, 66–69]. The development of a cancer target-specific peptide in fusion with catalytically active P99 β-lactamase molecule should find use as an alternative to antibody-β-lactamase conjugates in enzyme prodrug therapy. The development of bioluminogenic substrate [70] of β-lactamase also allows the use of phage-display selected peptide-β-lactamase fusion ligands for target-specific in vivo imaging applications.
In conclusion, we present a very simple, cost-effective, and efficient method for an accelerated screening of clones selected from phage-display combinatorial peptide libraries that were constructed in fusion to β-lactamase as a reporter. The clone screening protocol can be adopted for a high throughput platform using liquid handling robots. The selected enzyme-linked affinity reagents from these libraries can be produced in bulk, purified, and used for a variety of downstream applications without any modification.
Acknowledgements
This work was supported by the National Institutes of Health [RO1CA112091] and the SD Ireland Cancer Research Fund.
REFERENCES
- 1.Bradbury A. Trends Biotechnol. 1999;17:137. doi: 10.1016/s0167-7799(98)01289-x. [DOI] [PubMed] [Google Scholar]
- 2.de Bruin R, Spelt K, Mol J, Koes R, Quattrocchio F. Nat. Biotechnol. 1999;17:397. doi: 10.1038/7959. [DOI] [PubMed] [Google Scholar]
- 3.Smothers JF, Henikoff S, Carter P. Science. 2002;298:621. doi: 10.1126/science.298.5593.621. [DOI] [PubMed] [Google Scholar]
- 4.Hoogenboom HR, Chames P. Immunol Today. 2000;21:371. doi: 10.1016/s0167-5699(00)01667-4. [DOI] [PubMed] [Google Scholar]
- 5.Petrenko V. Expert opinion on drug delivery. 2008;5:825. doi: 10.1517/17425247.5.8.825. [DOI] [PubMed] [Google Scholar]
- 6.Shukla GS, Krag DN. Oncol. Rep. 2005;13:757. [PubMed] [Google Scholar]
- 7.Smith GP, Petrenko VA. Chem Rev. 1997;97:391. doi: 10.1021/cr960065d. [DOI] [PubMed] [Google Scholar]
- 8.Paschke M. Appl. Microbiol Biotechnol. 2006;70:2. doi: 10.1007/s00253-005-0270-9. [DOI] [PubMed] [Google Scholar]
- 9.Shukla GS, Krag DN. Expert opinion on biological therapy. 2006;6:39. doi: 10.1517/14712598.6.1.39. [DOI] [PubMed] [Google Scholar]
- 10.Aina OH, Liu R, Sutcliffe JL, Marik J, Pan CX, Lam KS. Molecular pharmaceutics. 2007;4:631. doi: 10.1021/mp700073y. [DOI] [PubMed] [Google Scholar]
- 11.Sidhu SS, Koide S. Curr. Opin. Struct. Biol. 2007;17:481. doi: 10.1016/j.sbi.2007.08.007. [DOI] [PubMed] [Google Scholar]
- 12.Newton J, Deutscher SL. Handbook of experimental pharmacology. 2008:145. doi: 10.1007/978-3-540-77496-9_7. [DOI] [PubMed] [Google Scholar]
- 13.Landon LA, Zou J, Deutscher SL. Current drug discovery technologies. 2004;1:113. doi: 10.2174/1570163043335108. [DOI] [PubMed] [Google Scholar]
- 14.Pero SC, Shukla GS, Armstrong AL, Peterson D, Fuller SP, Godin K, Kingsley-Richards SL, Weaver DL, Bond J, Krag DN. Int. J. Cancer. 2004;111:951. doi: 10.1002/ijc.20306. [DOI] [PubMed] [Google Scholar]
- 15.Taki T, Ishikawa D, Ogino K, Tanaka M, Oku N, Asai T, Popa I, Portoukalian J. Biochim. Biophys. Acta. 2008;1780:497. doi: 10.1016/j.bbagen.2008.01.005. [DOI] [PubMed] [Google Scholar]
- 16.Rowley MJ, O'Connor K, Wijeyewickrema L. Biotechnol. Annu. Rev. 2004;10:151. doi: 10.1016/S1387-2656(04)10006-9. [DOI] [PubMed] [Google Scholar]
- 17.Tonikian R, Zhang Y, Boone C, Sidhu SS. Nature protocols. 2007;2:1368. doi: 10.1038/nprot.2007.151. [DOI] [PubMed] [Google Scholar]
- 18.Buckler DR, Park A, Viswanathan M, Hoet RM, Ladner RC. Drug discovery today. 2008;13:318. doi: 10.1016/j.drudis.2007.10.012. [DOI] [PubMed] [Google Scholar]
- 19.Kay BK, Kasanov J, Yamabhai M. Methods. 2001;24:240. doi: 10.1006/meth.2001.1185. [DOI] [PubMed] [Google Scholar]
- 20.Shukla GS, Krag DN. J. Drug Target. 2005;13:7. doi: 10.1080/10611860400020464. [DOI] [PubMed] [Google Scholar]
- 21.Kretzschmar T, Geiser M. Gene. 1995;155:61. doi: 10.1016/0378-1119(94)00897-2. [DOI] [PubMed] [Google Scholar]
- 22.Jacoby GA, Munoz-Price LS. N. Engl. J. Med. 2005;352:380. doi: 10.1056/NEJMra041359. [DOI] [PubMed] [Google Scholar]
- 23.Hribar G, Smilovic V, Zupan AL, Gaberc-Porekar V. Biotechniques. 2008;44:477. doi: 10.2144/000112730. [DOI] [PubMed] [Google Scholar]
- 24.Campbell RE. Trends Biotechnol. 2004;22:208. doi: 10.1016/j.tibtech.2004.03.012. [DOI] [PubMed] [Google Scholar]
- 25.Qureshi SA. Biotechniques. 2007;42:91. doi: 10.2144/000112292. [DOI] [PubMed] [Google Scholar]
- 26.Siemers NO, Kerr DE, Yarnold S, Stebbins MR, Vrudhula VM, Hellstrom I, Hellstrom KE, Senter PD. Bioconjug. Chem. 1997;8:510. doi: 10.1021/bc9700751. [DOI] [PubMed] [Google Scholar]
- 27.Harding FA, Liu AD, Stickler M, Razo OJ, Chin R, Faravashi N, Viola W, Graycar T, Yeung VP, Aehle W, Meijer D, Wong S, Rashid MH, Valdes AM, Schellenberger V. Mol. Cancer Ther. 2005;4:1791. doi: 10.1158/1535-7163.MCT-05-0189. [DOI] [PubMed] [Google Scholar]
- 28.Gschwind A, Fischer OM, Ullrich A. Nature reviews. 2004;4:361. doi: 10.1038/nrc1360. [DOI] [PubMed] [Google Scholar]
- 29.Shukla GS, Murray CJ, Estabrook M, Shen GP, Schellenberger V, Krag DN. Int. J. Cancer. 2007;120:2233. doi: 10.1002/ijc.22138. [DOI] [PubMed] [Google Scholar]
- 30.Cwirla SE, Peters EA, Barrett RW, Dower WJ. Proc. Natl. Acad. Sci. U. S. A. 1990;87:6378. doi: 10.1073/pnas.87.16.6378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kristensen P, Winter G. Fold. Des. 1998;3:321. doi: 10.1016/S1359-0278(98)00044-3. [DOI] [PubMed] [Google Scholar]
- 32.Hayashi N, Kipriyanov S, Fuchs P, Welschof M, Dorsam H, Little M. Gene. 1995;160:129. doi: 10.1016/0378-1119(95)00218-u. [DOI] [PubMed] [Google Scholar]
- 33.Duez C, Frere JM, Ghuysen JM, Van Beeumen J, Delcambe L, Dierickx L. Biochim. Biophys. Acta. 1982;700:24. doi: 10.1016/0167-4838(82)90287-4. [DOI] [PubMed] [Google Scholar]
- 34.Rigoutsos I, Floratos A. Bioinformatics. 1998;14:55. doi: 10.1093/bioinformatics/14.1.55. [DOI] [PubMed] [Google Scholar]
- 35.Mandava S, Makowski L, Devarapalli S, Uzubell J, Rodi DJ. Proteomics. 2004;4:1439. doi: 10.1002/pmic.200300680. [DOI] [PubMed] [Google Scholar]
- 36.Shukla GS, Krag DN. J. Mol. Recognit. 2009;22:425–436. doi: 10.1002/jmr.957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Steiner D, Forrer P, Stumpp MT, Pluckthun A. Nat. Biotechnol. 2006;24:823. doi: 10.1038/nbt1218. [DOI] [PubMed] [Google Scholar]
- 38.Rodi DJ, Soares AS, Makowski L. J. Mol. Biol. 2002;322:1039. doi: 10.1016/s0022-2836(02)00844-6. [DOI] [PubMed] [Google Scholar]
- 39.Yamane K, Mizushima SJ. Biol. Chem. 1988;263:19690. [PubMed] [Google Scholar]
- 40.Peters EA, Schatz PJ, Johnson SS, Dower WJ. J. Bacteriol. 1994;176:4296. doi: 10.1128/jb.176.14.4296-4305.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pluckthun A, Knowles JR. J. Biol. Chem. 1987;262:3951. [PubMed] [Google Scholar]
- 42.Barkocy-Gallagher GA, Cannon JG, Bassford PJ., Jr J. Biol. Chem. 1994;269:13609. [PubMed] [Google Scholar]
- 43.Nilsson I, von Heijne G. FEBS Lett. 1992;299:243. doi: 10.1016/0014-5793(92)80124-y. [DOI] [PubMed] [Google Scholar]
- 44.Shukla GS, Krag DN. J Immunoassay Immunochem. 2005;26:89. doi: 10.1081/ias-200051990. [DOI] [PubMed] [Google Scholar]
- 45.Wright RM, Dudas D, Gavin B, Dottavio D, Hexham JM, Lake P. J. Immunol. Methods. 2001;253:223. doi: 10.1016/s0022-1759(01)00390-8. [DOI] [PubMed] [Google Scholar]
- 46.Han Z, Karatan E, Scholle MD, McCafferty J, Kay BK. Comb. Chem. High Throughput Screen. 2004;7:55. doi: 10.2174/138620704772884823. [DOI] [PubMed] [Google Scholar]
- 47.Gonzalez-Techera A, Umpierrez-Failache M, Cardozo S, Obal G, Pritsch O, Last JA, Gee SJ, Hammock BD, Gonzalez-Sapienza G. Bioconjug. Chem. 2008;19:993. doi: 10.1021/bc700279y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Berezov A, Chen J, Liu Q, Zhang HT, Greene MI, Murali R. J. Biol .Chem. 2002;277:28330. doi: 10.1074/jbc.M202880200. [DOI] [PubMed] [Google Scholar]
- 49.Roskoski R., Jr Biochem. Biophys. Res. Commun. 2004;319:1. doi: 10.1016/j.bbrc.2004.04.150. [DOI] [PubMed] [Google Scholar]
- 50.Griep RA, van Twisk C, van der Wolf JM, Schots A. J. Immunol .Methods. 1999;230:121. doi: 10.1016/s0022-1759(99)00131-3. [DOI] [PubMed] [Google Scholar]
- 51.Morino K, Katsumi H, Akahori Y, Iba Y, Shinohara M, Ukai Y, Kohara Y, Kurosawa Y. J. Immunol. Methods. 2001;257:175. doi: 10.1016/s0022-1759(01)00462-8. [DOI] [PubMed] [Google Scholar]
- 52.Alting-Mees MA, Risseeuw EP, Liu E, Desautels M, Crosby WA, Hemmingsen SM. Methods Mol. Biol. 2006;313:97. doi: 10.1385/1-59259-958-3:097. [DOI] [PubMed] [Google Scholar]
- 53.Chuman Y, Uren A, Cahill J, Regan C, Wolf V, Kay BK, Rubin JS. Peptides. 2004;25:1831. doi: 10.1016/j.peptides.2004.07.010. [DOI] [PubMed] [Google Scholar]
- 54.Kuhne SA, Hawes WS, La Ragione RM, Woodward MJ, Whitelam GC, Gough KC. J. Clin. Microbiol. 2004;42:2966. doi: 10.1128/JCM.42.7.2966-2976.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Velappan N, Martinez JS, Valero R, Chasteen L, Ponce L, Bondu-Hawkins V, Kelly C, Pavlik P, Hjelle B, Bradbury AR. J. Immunol. Methods. 2007;321:60. doi: 10.1016/j.jim.2007.01.011. [DOI] [PubMed] [Google Scholar]
- 56.El-Mousawi M, Tchistiakova L, Yurchenko L, Pietrzynski G, Moreno M, Stanimirovic D, Ahmad D, Alakhov V. J. Biol. Chem. 2003;278:46681. doi: 10.1074/jbc.M308681200. [DOI] [PubMed] [Google Scholar]
- 57.O'Callaghan CH, Morris A, Kirby SM, Shingler AH. Antimicrob. Agents Chemother. 1972;1:283. doi: 10.1128/aac.1.4.283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Jones RN, Wilson HW, Novick WJ, Jr, Barry AL, Thornsberry C. J. Clin. Microbiol. 1982;15:954. doi: 10.1128/jcm.15.5.954-958.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gao W, Xing B, Tsien RY, Rao J. JAm. Chem. Soc. 2003;125:11146. doi: 10.1021/ja036126o. [DOI] [PubMed] [Google Scholar]
- 60.Xing B, Khanamiryan A, Rao J. J. Am. Chem. Soc. 2005;127:4158. doi: 10.1021/ja042829+. [DOI] [PubMed] [Google Scholar]
- 61.Zlokarnik G, Negulescu PA, Knapp TE, Mere L, Burres N, Feng L, Whitney M, Roemer K, Tsien RY. Science. 1998;279:84. doi: 10.1126/science.279.5347.84. [DOI] [PubMed] [Google Scholar]
- 62.Cartwright SJ, Waley SG. Biochem. J. 1984;221:505. doi: 10.1042/bj2210505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Svensson HP, Vrudhula VM, Emswiler JE, MacMaster JF, Cosand WL, Senter PD, Wallace PM. Cancer Res. 1995;55:2357. [PubMed] [Google Scholar]
- 64.Vrudhula VM, Kerr DE, Siemers NO, Dubowchik GM, Senter PD. Bioorg. Med. Chem. Lett. 2003;13:539. doi: 10.1016/s0960-894x(02)00935-6. [DOI] [PubMed] [Google Scholar]
- 65.Hanessian S, Wang J. Can. J. Chem. 1993;71:896. [Google Scholar]
- 66.Kerr DE, Schreiber GJ, Vrudhula VM, Svensson HP, Hellstrom I, Hellstrom KE, Senter PD. Cancer Res. 1995;55:3558. [PubMed] [Google Scholar]
- 67.Meyer DL, Jungheim LN, Law KL, Mikolajczyk SD, Shepherd TA, Mackensen DG, Briggs SL, Starling JJ. Cancer Res. 1993;53:3956. [PubMed] [Google Scholar]
- 68.Vrudhula VM, Svensson HP, Kennedy KA, Senter PD, Wallace PM. Bioconjug. Chem. 1993;4:334. doi: 10.1021/bc00023a005. [DOI] [PubMed] [Google Scholar]
- 69.Vrudhula VM, Svensson HP, Senter PD. J. Med .Chem. 1995;38:1380. doi: 10.1021/jm00008a016. [DOI] [PubMed] [Google Scholar]
- 70.Yao H, So MK, Rao J. Angew. Chem. Int. Ed. Engl. 2007;46:7031. doi: 10.1002/anie.200701931. [DOI] [PubMed] [Google Scholar]