

Abstract
Nitric oxide synthases belong to a family of dual-flavin enzymes that transfer electrons from NAD(P)H to a variety of heme protein acceptors. During catalysis, their FMN subdomain plays a central role by acting as both an electron acceptor (receiving electrons from FAD) and an electron donor, and is thought to undergo large conformational movements and engage in two distinct protein–protein interactions in the process. This minireview summarizes what we know about the many factors regulating niric oxide synthase flavoprotein domain function, primarily from the viewpoint of how they impact electron input/output and conformational behaviors of the FMN subdomain.
Keywords: conformational equilibrium, electron flux, electron transfer, flavoprotein, global kinetic model, heme protein, heme reduction, nitric oxide, protein–protein interaction, semiquinone
Introduction
Flavoproteins are a versatile group of biological catalysts that may represent 1–3% of all genes in prokaryotic and eukaryotic genomes [1,2]. Nitric oxide synthases (NOS; EC 1.14.13.39) are members of a dual-flavin reductase family, which transfer electrons from NADPH to a variety of heme protein acceptors [3–5]. The electron transfer occurs in a linear manner from NADPH to FAD to FMN. During catalysis, the FMN subdomain plays a central role by acting as both an electron acceptor (receiving an electron from FADH2) and an electron donor (transferring an electron typically from FMNH−), and is thought to undergo large conformational movements in the process. How this process occurs and is regulated in dual-flavin enzymes like NOS is a topic of current interest.
Characteristics of NOS
NOS enzymes catalyze the NADPH- and O2-dependent conversion of L-arginine (Arg) to citrulline and nitric oxide (NO) via the intermediate N-hydroxyarginine (Scheme 1) [6–9]. There are three mammalian NOS enzymes: neuronal (nNOS), endothelial (eNOS) and inducible (iNOS). nNOS and eNOS are reversibly activated by the Ca2+-binding protein calmodulin (CaM) to enable their participation in biological signaling cascades. By contrast, iNOS binds CaM regardless of the Ca2+ concentration and can remain continuously active [7,10].
Scheme 1.

Reaction catalyzed by NOS.
NOS enzymes are homodimers (Fig. 1). Their subunits are modular and are comprised of an N-terminal ‘oxygenase domain’ (NOSoxy) that binds iron protoporphyrin IX (heme), (6R)-5,6,7,8-tetrahydro-L-biopterin (H4B) and Arg, and a C-terminal flavoprotein or reductase domain that binds NADPH, FAD and FMN. The two domains are separated by a CaM-binding motif. During catalysis, NADPH-derived electrons transfer into the FAD and FMN in each NOS subunit and then on to the ferric heme in the partner subunits of the homodimer (Fig. 1). Heme reduction, which is rate limiting for NO synthesis [11–13], enables O2 binding and substrate oxidation to occur within the NOSoxy domain [14–16]. The individual NOS domains and subdomains can be expressed separately, which has facilitated biochemical and structural studies. The protein structural elements that bind heme, Arg, H4B, CaM, NADPH, FAD and FMN have been identified based on crystallography, mutagenesis and homology studies [17–22].
Fig. 1.
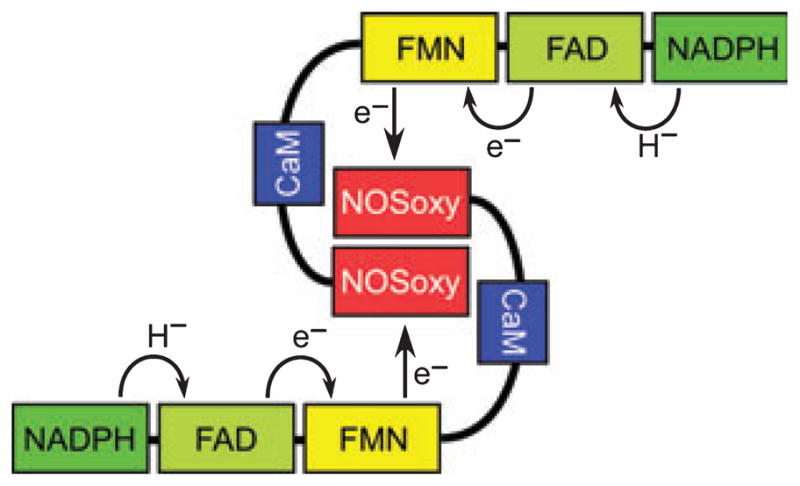
Domain arrangement and electron flow in the NOS dimer.
NOS enzymes have novel features
NOS are heme-thiolate enzymes and catalyze oxygen activation by a mechanism similar to that of the cytochrome P450 (CYP) enzymes (Fig. 2). The oxygen activation involves a two-step heme reduction with protons donated to help break the O–O bond and generate reactive heme-oxy enzyme species. However, in NOS, the second electron is provided to the heme-dioxy species by a bound H4B cofactor rather than by the flavoprotein domain [16]. The H4B radical is then reduced within the enzyme by the flavoprotein domain in order to continue catalysis [23]. NOSoxy domains also have a unique protein fold compared with CYPs, a shorter heme-binding loop and a distinct proximal heme environment with different hydrogen bonding to the cysteine heme ligand [17–19,24]. The attached flavoprotein and heme domains of NOS are also an unusual feature shared by only a handful of prokaryotic CYP proteins [4,8,25].
Fig. 2.

Simplified model of arginine hydroxylation in NOS enzymes. Ferric heme receives an electron from FMNH2/MNH− enabling oxygen binding and formation of a ferrous dioxygen species. A second electron must be delivered from H4B to eventually form a high valent iron-oxo species that hydroxylates Arg. The H4B+• radical has to be reduced before the next catalytic cycle can proceed.
In comparison, the NOS flavoprotein domain is related to a family of dual-flavin enzymes that contain FAD and FMN, and transfer NADPH-derived electrons to separate hemeprotein partners or to attached heme domains [5,14,20,22,26]. Other members from eukaryotes include cytochrome P450 reductase (CYPR) and methionine synthase reductase. Typically (except bacterial CYPBM3), these flavoproteins are isolated in their 1-electron reduced forms containing oxidized FAD and a stable FMN semiquinone radical (FMNH•). After reduction by NADPH occurs, they utilize a 3-2-1 electron-transfer cycle in which their FMN group redox cycles between its electron-accepting semiquinone form (FMNH−) and its fully reduced, electron-donating hydroquinone form (FMNH2 or FMNH−). However, the NOS flavoprotein displays a number of unique features within this enzyme family. These include NOS electron-transfer reactions being suppressed in the native state by up to three unique protein regulatory inserts: an autoinhibitory insert in the FMN domain [27–30], a C-terminal tail (CT) [31–33] and possibly a small insertion or β-finger in the connecting domain [34,35] (Fig. 3A,B). CaM binding to NOS relieves the suppression at three points in the electron-transfer sequence [36–40] (Fig. 3C). NOS electron- transfer activity can also be impacted by phosphorylation [41–46] and by extrinsic proteins like caveolin-1 [47,48], dynamin-2 [49] and heat-shock protein 90 [50]. Finally, NOS enzyme activity is controlled by self-generated NO, which binds to the NOS heme as an intrinsic feature of catalysis [12,13,51] (Fig. 4). This forces the NOS heme reduction rate (kr in Fig. 4) to remain relatively slow in order to minimize an inherent NO dioxygenase activity in NOS that destroys the NO it makes (futile cycle, Fig. 4).
Fig. 3.

(A) Domain organization in NOS and related enzymes. NOS includes regulatory elements that are absent in other closely related proteins. (B) Structure of nNOS flavoprotein domain. The FNR and FMN subdomain are shown in green and yellow, respectively. Regulatory elements (β-finger; AI, autoinhibitory insert; CT, C-terminal tail) are shown in pink. The coenzymes FMN (orange), FAD (dark blue) and NADP+ (cyan) are shown as sticks. Modeled fragments, not visible in the crystal structure, are shown in light gray. The visible parts of the hinge element between FMN and FNR subdomains are shown in dark blue. (C) CaM exerts an enhancing effect in three electron-transfer steps.
Fig. 4.

Global kinetic model for NOS catalysis. Ferric enzyme reduction (kr) is rate limiting for the biosynthetic reactions (central linear portion). kcat1 and kcat2 are the conversion rates of the FeIIO2 species to products in the Arg and NOHA reactions, respectively. The ferric heme–NO product complex (FeIIINO) can either release NO (kd) or become reduced (kr) to a ferrous heme–NO complex (FeIINO), which reacts with O2 (kox) to regenerate the ferric enzyme. Adapted from Stuehr et al. [51].
In summary, NOS enzymes display at least four features that distinguish them from other dual-flavin and heme-thiolate enzymes: (a) the FMN subdomain interacts with its partner donor and acceptor domains all within an enzyme dimer; (b) electron transfer is suppressed in the basal state and the suppression is relieved by CaM binding; (c) bound H4B provides the second electron for oxygen activation in place of the flavoprotein, and then redox cycles within NOS; and (d) heme–NO binding is an intrinsic feature of catalysis that constrains the rate of heme reduction by the flavoprotein domain. How these features shape NOS flavoprotein domain function is discussed below.
Key function of the FMN subdomain
Figure 5 depicts a three-state, two-equilibrium model that can describe FMN subdomain function in a NOS dimer. The FMN subdomain receives electrons from the NADPH/FAD subdomain (FNR) in subunit 1 (green), and then shuttles electrons to the NOSoxy domain in subunit 2 (black). This process is thought to require relatively large (70 Å) movement of the FMN subdomain [22], and to involve two reversible and temporally distinct protein binding interactions:
Fig. 5.

Model of NOS FMN subdomain function in electron transfer and heme reduction. Electron transfer in NOS can be regarded as a three-state model. Equilibrium A indicates the change between a conformation in which FNR and FMN subdomains are interacting (left) and a conformation where the FMN subdomain is deshielded and available for interaction with electron acceptors such as cytochrome c (center). Equilibrium B indicates the transition from the FMN deshielded conformation to a FMN–NOSoxy domain interacting state. See text for details.
Equilibrium A describes the FNR–FMN subdomain interaction that is required to generate FMNH− or FMNH2:
Equilibrium B describes the FMN–NOSoxy interaction that enables heme reduction:
Large movements of the FMN subdomain are constrained by two hinge elements (green, H1 & H2) that connect it to the electron-donating (FNR) and electron-accepting (NOSoxy) components within the NOS dimer. The CaM-binding site (gray box) in the H2 hinge enables CaM to influence the movements. The same face on the FMN subdomain (red) is expected to interact with each partner subdomain to receive and give electrons. Thus, at either end of a larger movement, the FMN subdomain likely engages in distinct short-range conformational sampling motions with each of its partner subdomains [52,53]. Basic tenets of this model have previously been used to describe FMN subdomain function in other dual-flavin enzymes that shuttle electrons to hemeprotein partners [54,55] and even across subunits as in the dimeric CYPR–BM3 [56,57].
Studying conformational equilibrium A
Equilibrium A is critical because it helps define electron entry and exit from the FMN subdomain. Obtaining the KeqA and associated kon and koff kinetic parameters for the FNR–FMN subdomain complex is a worthwhile and important goal. To date, conformational studies on the NOS flavoprotein domain have involved ensemble measures with the bound FMN poised in its oxidized, semiquinone and hydroquinone states. These studies measured fluorescence intensity of the oxidized flavins, the interaction of bound FMNH• with a soluble paramagnetic agent by EPR spectroscopy, and rates and extent of reaction of bound FMNH2 (FMNH−) with cytochrome c in single turnover or pre-steady-state conditions by stopped-flow spectroscopy [58–62]. In general, these methods can report on any dual-flavin enzyme that is poised in the 0-, 1- and 4-electron reduced states which, practically speaking, are the reduced states most attainable for experimentation. Some strengths and limitations of the measures have been discussed recently [63]. The flavin fluorescence and EPR methods provide semiquantitative information regarding equilibrium A that is useful for comparative studies, whereas the stopped-flow/cytochrome c method can provide quantitative estimates of KeqA and in some cases measures of koff for the FMN subdomain (Fig. 5), as recently reported for eNOS and nNOS (described below) [58]. Experimentally, it is challenging to study equilibrium A because dual-flavin enzymes are difficult to poise in all the intermediate states that are likely to be populated during catalysis. For example, this includes the 2- and 3-electron reduced state, with accompanying variations in NADP(H) binding site occupancy. Recently, Salerno and colleagues discussed a kinetic modeling approach that might help to address these issues [64].
Electron flux and equilibrium A
In general, electron flux through a protein depends on the rates of electron input and output, with either process being rate limiting. In the case of the NOS flavoprotein (or for dual-flavin enzymes in general), the question becomes, how is the electron flux affected by the rate of FMNH2 formation and by the rate of FMNH2 (or FMNH−) reaction with the electron acceptor? Electron flux through NOS enzymes can be measured by the steady-state activities of cytochrome c reduction, NO synthesis and/or accompanying rates of NADPH or O2 consumption. Among these, cytochrome c reductase activity is the most straightforward way to measure electron flux through the flavoprotein domain. This is because cytochrome c is reduced very slowly by the FNR subdomain [62], and instead is reduced by the FMN subdomain only when it contains FMNH2 or FMNH−, in a quasi-irreversible single-electron transfer reaction that is rapid, not rate limiting, and that can occur only when the FMN subdomain is in an open or deshielded conformation away from its partner subdomains [58,59,63]. By contrast, electron flux measures that rely on a ‘downstream’ event like NOS heme reduction (or subsequent NO synthesis activity) are more complicated to interpret, because heme reduction is relatively slow, CaM dependent and subject to thermodynamic constraints [65], and NO synthesis activity is a culmination of many steps that are prone to influences beyond conformational equilibrium A [51].
The features that make cytochrome c reductase activity an excellent measure of electron flux also make it a useful predictor (but never proof) of changes in equilibrium A in dual-flavin enzymes. Figure 6 contains curves showing how electron flux through the FMN subdomain of a dual-flavin enzyme, as measured by cytochrome c reductase activity, might change with the value of KeqA, according to a simple kinetic model (Fig. 6A). One can compare the model with the equilibrium A that is depicted in Fig. 5, with k1 = koff and k2 = kon. The calculated kobs values shown in Fig. 6B assume that there are fast rates of electron input (k3) and output (k4 = 1000 s−1) relative to the rates of conformational change for the FMN subdomain (k1 + k2 = 10 s−1), and also that any change in the FMN redox state (FMNH2 versus FMNH•) does not change the k1 or k2 values. Each curve in Fig. 6 was calculated using a different electron input rate (k3, the rate of FMNH2 formation). Calculations of the concentrations of each species with time were carried out using GEPASI v. 3.30 [66]. The model predicts that there is always a Keq position for maximum electron flux through the enzyme. On either side of this optimum, the electron flux drops off because either the formation rate (k2) or dissociation rate (k1) of the FNR–FMN subdomain complex becomes slower. At relatively fast rates of FMNH2 formation, electron flux through the flavoprotein is primarily a function of the rates of conformational change (k1, k2) that determine KeqA. However, when the rate of conformational change begins to approach the rate of FMNH2 formation (either from speeding up k1 and k2 or by slowing FMNH2 formation), then the rate of electron input (k3) becomes an important factor for governing the electron flux, and consequently electron flux would be more sensitive to changes in the rate of FMNH2 formation. Thus, one could envision three ways that electron flux through the FMN subdomain might be controlled in a dual-flavin enzyme: changing the ratio or speed of k1 and k2, changing the rate of FMNH2 formation or by a combination of these effects. In addition, further tuning could be achieved if the changes in the FMN redox state that occur during catalysis (FMNH2 versus FMNH•) do cause the k1 or k2 values to change.
Fig. 6.

Model and simulations of cytochrome c reduction by NOS enzymes. (A) Scheme of cytochrome c reduction. The model uses four kinetic rates: dissociation (k1) and association (k2) of the FMN and FNR subdomains; FMNH• reduction rate (k3) and cytochrome c reduction rate (k4). For simplicity, k1 and k2 are assumed to be independent of the flavin reduction state, k4 is assumed to be much faster than the conformational equilibrium so the backwards rates are negligible, oxidized cytochrome c concentration is constant and in 100-fold excess. (B) Apparent rates of steady-state cytochrome c reduction for different FMNH• reduction (k3) values. kobs values were determined by fitting the apparent change in the concentration of reduced cytochrome c versus time to a straight line. The percentage of deshielding is (k1/(k1 + k2)) × 100. See text for details.
Factors that may modulate equilibrium A and/or the FMNH2 formation rate
Although the model in Fig. 6 is conceptually useful, the situation is more complicated in dual-flavin enzymes because of a number of factors, including the k1 and k2 of KeqA possibly being influenced by changes in the FAD and FMN reduction state or by changes in NADP(H)-binding site occupancy during catalysis. Another factor to consider is the thermodynamic driving force to generate FMNH2. The midpoint potential of the FMNH2/FMNH couple in NOS enzymes (and in most other dual-flavin enzymes) is similar to the FADH2/FADH couple and is somewhat more negative than the FADH/FAD couple [67]. These data indicate that a relatively poor driving force exists for FMNH2 buildup, which then occurs to different incomplete extents as the flavoprotein cycles through its 3- and 2-electron reduced states during catalysis. This, in turn, can impact the rate of electron exit and flux through the flavoprotein. Two studies have investigated how changes in the flavin midpoint potentials may alter FMNH2 formation and the resultant electron flux through the FMN subdomain of NOS [68,69].
Factors that may alter equilibrium A conformational rates k1 and k2 and/or alter the rate of electron input into the NOS flavoprotein are listed in Table 1. These include proteins, small molecules, NOS regulatory inserts and point mutations. For many of the factors, our only indication currently that they might alter KeqA is from their changing the steady-state cytochrome c reductase activity. Thus, more work needs to be done to obtain measures of KeqA and the associated k1 and k2 values for dual-flavin enzymes, particularly when they are poised in all catalytically relevant intermediate redox states (1-, 2-, or 3-electron reduced), perhaps ultimately using single molecule spectroscopic approaches.
Table 1.
Factors that may alter conformational equilibrium A, B and/or the rate of electron input in nitric oxide synthase (NOS) enzymes.a AI, autoinhibitory insert; B2R, bradykinin receptor B2; CaM, calmodulin; CT, C-terminal tail; HSP-90, heat-shock protein 90; iNOS, inducible nitric oxide synthase; ND, not determined; NA, not applicable; ?, different modifications (mutation, deletion) gave different results.
|
KeqA |
KeqB |
||||||
|---|---|---|---|---|---|---|---|
| Cyt c reduction |
Flavin reduction rate |
Heme reduction |
NO synthesis |
||||
| Factor | −CaM | +CaM | −CaM | +CaM | +CaM | +CaM | Ref |
| CaM | NA | ↑b | NA | ↑ | ↑ | ↑ | [37–39,59] |
| NADPH | ↓b | =b | ↑ | ↑ | = | ↑ | [58,59,61,90,99] |
| CTc | ↓ | ↓d | ↓ | ↓e,i =n | ↑n =e,i | ↓i ↑e,ne | [31–33,102,105] |
| AI(e/n)c | ↓ | ↓e↑n | ND | ND | ND | ↓e ↑n | [28,102,105,115] |
| β-finger (e/n/iNOS) | ? | ? | ND | ND | ND | ? | [34,35] |
| R1229E nNOS | ↑b | =b | ↑ | ↑ | ND | ↓ | [77] |
| F1395S nNOS | ↑b | =b | ↑ | = | ↓ | ↓ | [61,72,116] |
| R1400E nNOS | ↑b | ↑b | ↑ | ↓ | ↑ | ↓ | [60] |
| S1412D nNOS | ↑ | ↑ | ↑ | = | ↑ | ↓ | [88] |
| S1179D eNOS | ↑ | ↑ | ND | ND | ND | ↑ | [117] |
| Caveolin-1 | ↓ | ↓ | ND | ND | ND | ↓ | [47,48] |
| HSP-90 | =f | ↑f | ND | ND | ND | ↑ | [50,118–122] |
| Dynamin-2 | ND | ↑ | ND | ND | ND | ↑ | [49,123] |
| B2R | ND | = | ND | ND | ND | ↓ | [124,125] |
Unless otherwise stated, cytochrome c reduction and NO synthesis changes correspond to steady-state measurements, flavin reduction and heme reduction rates are derived from stopped-flow experiments. e, i or n refer to studies on eNOS, iNOS or nNOS, respectively. For an extensive list of proteins that interact with NOS the reader is referred to other reviews [10,126,127]. Regarding NOS phosphorylation, only the phosphorylation mimics S1179D eNOS and S1412D nNOS are shown; for more detailed information, see Hayashi et al. [44] and Mount et al. [128].
Pre-steady-state cytochrome c reduction measurements.
The effect of the element is inferred from deletion mutants, therefore the effects reported in the table are the opposite of the observed effects.
All but one report indicate decreased cytochrome c reduction + CaM in ΔCT nNOS [33].
All but one report indicates increased NO synthesis in ΔCT eNOS [105].
Only eNOS data [121], not determined for iNOS or nNOS.
Relationship between CT, bound NADPH and equilibrium A in NOS
Among the factors listed in Table 1, only the roles of CaM, the CT and bound NADPH have been studied in detail. An interesting and possibly novel connection appears to link regulation of KeqA by the CT and bound NADPH. Basically, the CT of nNOS and eNOS contain a conserved Arg residue whose side chain makes a salt bridge interaction with the 2′-phosphate of bound NADP(H) [22]. Mutagenesis studies suggest that this interaction helps transduce the effect of bound NADP(H) on KeqA (it causes KeqA to decrease), presumably by strengthening the CT to act as a clasp for the FMN subdomain [60]. NADP(H) binding may have a similar influence on KeqA in the related enzyme CYPR [55,70], although it has no CT regulatory element. This suggests that multiple modes of regulation are in play, even for the relatively fundamental circumstance of NADP(H) binding. Some other modes have been explored in the FNR, CYPR and NOS enzymes [61,71–76].
Is there a correlation between NOS reductase activity and KeqA?
That a relationship exists between the KeqA and the cytochrome c reductase activity of the CaM-free reductase domain of neuronal NOS (nNOSr) was first considered based on measures taken with the 4-electron reduced nNOS flavoprotein in three different states (NADPH-free/CaM-free, NADPH-bound/CaM-free and CaM-bound) [59]. Subsequent measures made with CT point mutants of nNOS (R1400E, R1400S or F1395S) [60,61], and nNOS mutants possessing graded CT truncations [33], allowed the relationship to be examined over a wider range of KeqA than was previously possible. Figure 7 shows that a good correlation appears to exist (R = 0.96) between the cytochrome c reductase activities of the various CaM-free nNOS flavoproteins and their degree of FMN deshielding, which is directly related to the KeqA for each flavoprotein (greater FMN deshielding = higher KeqA). Curiously, several of the CaM-free mutant enzymes depicted in Fig. 7 appear to be in a super-deshielded state compared with the CaM-bound wild-type nNOSr. This may be at odds with more recent data [63,77], indicating that the FMN deshielding level in CaMbound nNOSr is near its maximal value, because it is similar in magnitude to the isolated FMN subdomain, which should exhibit the maximal possible FMN deshielding. This discrepancy may reflect the inherent difficulty in precisely measuring FMN shielding and the value for KeqA in nNOS, because of its small dynamic range (FMN shielding values only range between 50 and near 100%) [63,77]. At this point, the data suggest that KeqA and the associated kon and koff conformational rates are primary factors in regulating the cytochrome c reductase activity of NOS enzymes, particularly in the CaM-free state.
Fig. 7.

Correlation between nNOS cytochrome c reductase activity and FMN deshielding. The figure plots relative cytochrome c reductase activities of various CaM-free nNOS flavoproteins and CaM-bound wild-type versus their degree of FMN deshielding. All values are relative to NADPH-bound wild-type enzyme, which was given activity and shielding values of unity. Line is a least squares best fit. Adapted from Tiso et al. [33].
Do the conformational motions describing equilibrium A limit electron flux through NOS enzymes?
Daff and colleagues [59] first proposed that the conformational opening of the FNR–FMN subdomain complex (koff in Fig. 5 and k1 in Fig. 6) might limit electron flux through the NOS flavoprotein, and they presented the first data to support such a mechanism. There have been several subsequent investigations, culminating in a recent report by Ilagan et al. [58] that provides the first ensemble rate measures (Table 2) for the conformational steps in the nNOS and eNOS flavoproteins (dissociation and association of the 4-electron reduced FNR–FMN subunit complex, kon and koff in Fig. 5). Remarkably, the results suggest that koff is the sole kinetic parameter that limits steady-state electron flux to cytochrome c for both the CaM-free eNOS and nNOS flavoproteins (Table 2). So the answer to the question posed above is yes, in the CaM-free nNOS and eNOS, the rate of FMNH2 formation appears to be relatively fast and not rate limiting, and instead a specific conformational step (koff, dissociation of the reduced FMN subdomain) is rate limiting for cytochrome c reductase activity. How these conformational movements are regulated in NOS, and whether similar conformational motions may limit electron flux through other dual-flavin enzymes, are exciting questions that could be approached through similar experimental means.
Table 2.
Parameters describing conformational equilibrium A for the 4-electron reduced nNOS and eNOS flavoproteins. a CaM, calmodulin; ND, not determined.
Does the rate of electron input (rate of FMNH2 formation) limit electron flux through NOS enzymes?
As noted above, for CaM-free eNOS and nNOS, the answer to this question appears to be no [58]. But in the CaM-bound enzymes, or in other dual-flavin enzymes, it remains an open question. Electron input into NOS has been studied by monitoring flavin reduction kinetics [33,60–62,78]. Hydride transfer from NADPH to FAD is relatively fast and does not limit the rate of FMNH2 formation or electron flux through NOS, except in mutants that retard this hydride transfer [62]. FMN reduction is often difficult to discern because of its similar spectral properties to the bound FAD. In addition, the observed rate of FMN reduction in a dual-flavin enzyme may depend to a variable extent on the KeqA parameter kon, which is the formation rate of the FNR–FMN subdomain complex (Fig. 5). The kinetics of interflavin electron transfer (FAD and FMN) in dual-flavin enzymes has been studied using a T-jump method [79] and by observing rates of flavin semiquinone formation (FADH• and/or FMNH•) during pre-equilibrium reduction reactions with NADPH [61,80–85]. In such studies, kobs ranged from 20 to 100 s−1 at 10°C for nNOS, but appeared to be slower in eNOS. Several factors appear to influence the rate of flavin reduction in NOS enzymes (Table 1). CaM increased the rates of NOS flavin reduction in most studies. The mechanism appears to involve specific domains of CaM [86,87]. A faster interflavin electron transfer may conceivably help explain how CaM increases electron flux through NOS enzymes (cytochrome c reductase activity; the effect of changing k3 in Fig. 6). Indeed, some correlation exists between the rates of flavin reduction and the cytochrome c reductase activity of nNOS bound to a series of CaM analogs [88,89]. However, it is difficult to interpret these data because a means to exclusively alter the rate of FMNH2 formation without causing coincident changes in conformational equilibrium A and in the kon and koff parameters is still unavailable. Indeed, CaM shifts equilibrium A in NOS enzymes to the more open conformation, and therefore likely increases the koff parameter of equilibrium A [58] and may possibly increase the kon parameter as well. Unfortunately, the shift in KeqA caused by CaM prevented an accurate measure of the koff parameter in CaM-bound eNOS and nNOS [58], and thus prevented assessment of the relative importance of conformational change rates versus rates of FMNH2 formation in limiting electron flux through the CaM-bound NOS enzymes. In general, as the kon and koff of equilibrium A increase, it becomes more probable that the rate of electron input (specifically, FMNH2 formation) or some other step like NADP+ release, as in F1395S nNOS [61] and the analogous CYPR mutants [73], will limit electron flux through NOS or other dual-flavin enzymes. Further work should continue to clarify this issue.
Creating an intrinsic set point for equilibrium A
Common structural features in dual-flavin enzymes may determine their set points for KeqA. Among these are complementary charge pairing interactions that are present to various extents in the FNR–FMN subdomain interface, including the interface in NOS (Fig. 8). Point mutations that neutralize charge pairing or introduce charge-repelling interactions may increase the KeqA set point to various degrees, at least as judged by the increase in cytochrome c reductase activity that they cause [90]. Remarkably, CaM-free eNOS and nNOS have significantly different set points for KeqA [58] (Table 2), but CaM binding shifted the KeqA of eNOS to a value closer to that of nNOS (Table 2). Their different basal set points for equilibrium A explain why eNOS has much slower electron flux through its FMN subdomain (as measured by cytochrome c reductase activity) [58]. The structural basis for their different set points is unclear at this point, but may certainly involve apparent differences in their CT and autoinhibitory insert elements, or elsewhere in the enzyme.
Fig. 8.

Complementary charges in the FMN–FNR subdomain interface. The electrostatic potential surfaces of the FMN (left) and FNR (right) subdomains show complementary negative charges in the FMN surface that interact with a positively charged surface patch in the FNR module. Adapted from Panda et al. [90].
Changing the set point for KeqA may influence electron flux through the NOS flavoprotein in interesting ways (Fig. 6). For example, the basal set points of eNOS and nNOS, although different from one another, appear to both lie to the left of their optimum, and support a suboptimal electron flux. CaM binding shifts their KeqA set points to a value that supports increased electron flux. According to this model, introducing a mutation that shifts the intrinsic set point, say, by weakening the FNR–FMN subunit interaction, would be expected to boost electron flux through either of the CaM-free NOS enzymes. However, this is only true to a point, because the mutation could conceivably cause the KeqA to shift so far that upon CaM binding, the mutant KeqA would lie beyond the optimum, and therefore would actually support a slower electron flux in the CaM-bound versus CaM-free state. Real-life examples may already exist, in particular the FNR–FMN subdomain interface mutant R1229E nNOS [77] and the nNOS CT truncation mutant tr1397 [32,33]. In these cases, the rate of FMNH2 formation may be limited by a conformational change, namely, the kon for FNR–FMN subdomain complex formation may be so slow that it becomes rate limiting for FMNH2 formation during the steady state (also see k2 in Fig. 6A). A means to measure the reduction state of the bound FMN (FMNH2 versus FMNH•) during steady-state catalysis in dual-flavin enzymes would be generally useful, as was done in other flavoproteins modified to contain reporter flavin analogs [91]. In any case, the set point for KeqA is a fundamental parameter whose varied settings [58] could both up- and downregulate electron flux through the dual-flavin enzymes.
Conformational equilibrium B
We know comparatively little about the FMN–NOSoxy interaction and the associated equilibrium described by KeqB (Fig. 5). A crystal structure of this domain–domain interaction is not available. Nevertheless, a conserved electropositive surface on the NOSoxy domain is proposed to provide a potential docking site for the FMN subdomain [18], and this idea is supported by limited mutagenesis studies [92]. Combining the known structures of nNOS flavoprotein, the NOSoxy dimer and CaM when it is bound to the eNOS binding peptide, Garcin et al. [22] constructed a model for full-length nNOS that indicates that an allowable large motion of the FMN module could bring the FMN cofactor within an acceptable electron-transfer distance from the heme in the partner NOSoxy domain. Although this model suggests feasibility, whether it is an accurate depiction of the FMN–NOSoxy interaction is still unclear. However, recent crystal structures of CYPR mutants now support the feasibility of the long-range movement that is required for the FMN subdomain to support heme reduction in NOS enzymes [85].
Measuring the FMN–NOSoxy interaction and KeqB
NO synthesis activity is too complex to be a reliable indicator of the FMN–NOSoxy interaction. Measuring heme reduction is better but is still indirect and may have inherent limitations. Measuring the rate and extent of back electron transfer from the ferrous NOS heme to FMNsq following flash photolysis of CO can indicate precisely the rate of electron transfer, but cannot reveal the extent of the FMN–NOSoxy interaction [93–95]. Recently, Ilagan et al. [63] investigated KeqB by studying single-turnover electron-transfer reactions between a fully-reduced FMN–NOSoxy construct of nNOS and excess cytochrome c. Their evidence shows that KeqB is poised at values far below unity in nNOS, such that the dissociated conformation predominates and the KeqB value is little changed in the presence or absence of bound CaM. Thus, broad differences appear to exist in the set points of KeqA and KeqB in NOS enzymes, and in how the two set points are regulated. The FMN–NOSoxy complex formation described by KeqB appears to be infrequent and/or transient in practically all circumstances, such that the FMN subdomain may interact far less with NOSoxy than it does with the FNR subdomain in a NOS homodimer. These concepts are consistent with the poor ability of isolated nNOS flavoprotein and nNOSoxy domains to interact with one another and catalyze heme reduction or NO synthesis when they are mixed together [96], and is consistent with NOS enzymes having slow rates of heme reduction compared with other flavo-heme proteins [51]. Moreover, this likely distinguishes NOS from related flavoproteins that do not have attached heme acceptor domains and thus make higher affinity interactions between their FMN subdomains and their detached electron acceptor partners (e.g. the interaction of CYPR with heme oxygenase 1) [97,98]. Additional measurements of KeqB and the associated conformational rates in NOS enzymes will certainly improve our understanding of this essential FMN subdomain interaction.
Relationship of KeqA to equilibrium B and to NOS heme reduction
At the limit, KeqA can impact KeqB, heme reduction and NO synthesis because the reduced FMN subdomain must become dissociated from the FNR sub-domain in order to interact with NOSoxy and to reduce the heme (Fig. 5). However, the lowest possible rates for the FMN subdomain dissociation step (koff) in the CaM-bound eNOS and nNOS are ~1 and 20 s−1, respectively [58] (Table 2), and these rates are still 4–10 times faster than the observed rates of heme reduction in the CaM-bound eNOS or nNOS at the same temperature and conditions (0.1 and 5 s−1, respectively) [99,100]. This indicates that the electron transfer from the reduced FMN subdomain to the NOS heme is considerably less efficient than is its electron transfer to cytochrome c, which has turnover numbers of 1 and 20 s−1 for CaM-bound eNOS and nNOS, respectively, under the same conditions [58]. Indeed, greatly increasing the KeqA in nNOS via CT truncations enables only a small NO synthesis by the CaM-free enzyme [33]. This, and a variety of other evidence [33,51,68,90,99,101–103] suggest that shifting KeqA toward the FMN-deshielded state is not enough on its own to support heme reduction and NO synthesis in nNOS. Instead, additional and distinct effects on the FMN–NOSoxy interaction must be required, and the effects of CaM binding cannot be totally ascribed to the flavoprotein domain as suggested by others. Interestingly, these additional CaM effects need not cause a significant change in KeqB [63], but could rather have more subtle effects on structural elements that restrict motions of the FMN subdomain or present physical barriers that prevent the FMN subdomain from docking in a subset of conformations that allow electron transfer to the NOSoxy heme.
Factors that may regulate equilibrium B
Table 1 lists factors that may influence KeqB in NOS enzymes, mostly as indicated by their effects on NO synthesis activity or on the heme reduction rate. A few are discussed below.
Calmodulin
CaM has been assumed to promote the FMN–NOSoxy interaction, as judged by its ability to trigger NOS heme reduction and NO synthesis. Early hypotheses that the autoinhibitory insert and CT elements were critical in the process are not supported by deletion studies showing that NOS mutants missing either one or both of these control elements for the most part require CaM for NO synthesis, and then achieve an NO synthesis activity that is ≥ 50% of wild-type [28,30,31,102,104,105]. Studies with CaM variants [60,86–89,106–110] indicate that several structural features of CaM may be important. However, the recent results of Ilagan et al. [63] suggest that CaM binding may not alter KeqB to a great extent, implying it may primarily function through additional mechanisms.
Connecting hinge domains
The composition of the two hinges that connect the FMN subdomain in NOS enzymes (H1 and H2 in Fig. 5) defines the allowable movements of the FMN subdomain and thus controls the FMN–NOSoxy interaction (equilibrium B). This in turn may greatly impact the extent and rate of heme reduction in NOS enzymes. Precedent includes flavocytochrome b2, where altering its hinge length caused a 10-fold change in the heme reduction rate [111–114]. The FMN–FNR subdomain hinge (H1 in Fig. 5) is one of the least conserved motifs and is shorter in eNOS than in nNOS. Swapping the H1 hinge of nNOS into eNOS increased its heme reduction rate and increased its NO synthesis activity fourfold [99]. This confirms that the NOS H1 is a structural element that helps define the FMN–NOSoxy interaction, but whether it impacts KeqB is still unclear. Analogous studies have been carried out on the H1 hinge of CYPR [55,85].
Challenge of H4B reduction
During NO synthesis, the NOS FMN subdomain must provide an electron to reduce the ferric heme and the H4B radical at two distinct points during the catalytic cycle (Fig. 2). A recent study found that reduction of the H4B radical in nNOS requires CaM binding and occurs at a rate similar to ferric heme reduction [23]. These results, along with distance constraints suggesting that direct electron transfer from the FMN subdomain to the H4B radical would be too slow, led the authors to propose a through-heme model for H4B radical reduction by the FMN subdomain in NOS (Fig. 9). This mechanism essentially has the heme porphyrin ring acting as a wire to deliver an electron from the FMN subdomain to the H4B radical. It eliminates the problem of electron transfer over a long distance, and also eliminates the need to invoke a separate docking site for the FMN subdomain on NOSoxy or the need for the flavoprotein to sense when an electron is required by the heme versus the H4B radical at discreet steps in the reaction cycle (Fig. 2). Because reduction of the H4B radical presents a novel function for the FMN subdomain, it will be important to further test the validity, kinetics and thermodynamics of the through-heme pathway in NOS enzymes.
Fig. 9.
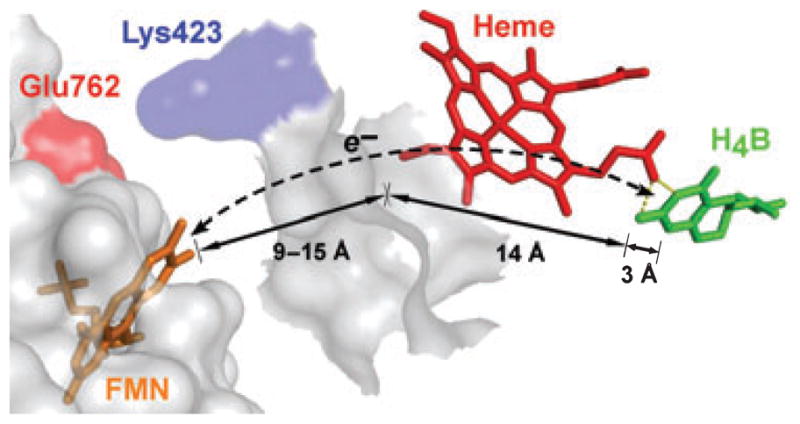
Through-heme model for H4B radical reduction in NOS. H4B is 17 Å away from the putative FMN-docking surface. Placing the FMN domain in conformations where Lys423 and Glu762 are in close contact enables feasible distances (9–15 Å) for FMN to heme electron transfer but too long (26–32 Å) for direct FMN to H4B electron transfer. It is proposed that electron transfer proceeds through heme (dashed line) involving two short-distance (< 15 and 3 Å) electron transfer steps. Adapted from Wei et al. [23].
Conclusions
Although the NOS flavoprotein domain has fundamental structural, thermodynamic and mechanistic features in common with the dual-flavin family of reductases, there are unique aspects related to NO synthesis that constrain and shape its function. Both common and unique features govern electron flux through the NOS flavoprotein domain. Many of these appear to act by influencing a conformational equilibrium (KeqA) that defines the interaction between the FMN subdomain and the FNR subdomain, although some may also influence the rate of electron import into the FMN subdomain and the resulting formation of FMNH2. The extent to which KeqA or the rate of FMNH2 formation influences electron flux through the NOS flavoprotein can vary depending on the circumstances. However, the KeqA, and specifically the dissociation rate of the reduced FMN subdomain, appears to be the primary factor that determines electron flux through the CaM-free nNOS and eNOS flavoproteins. A second conformational equilibrium (KeqB) defines the interaction of the reduced FMN subdomain with the NOSoxy domain that is required for heme reduction and NO synthesis. This equilibrium appears to have a different set point and regulation compared to KeqA, but has not been as thoroughly studied. An intrinsic heme–NO binding event occurs in NOS enzymes during catalysis and is likely to restrict the electron transfer function (heme reduction) of the NOS FMN subdomain relative to its function in related dual-flavin enzyme systems.
Acknowledgments
We thank past and present members of the Stuehr lab for their efforts and valuable discussions, and National Institutes of Health grants GM51491, CA53914 and HL76491 for financial support.
Abbreviations
- CaM
calmodulin
- CT
C-terminal tail
- CYP
cytochrome P450
- CYPR
cytochrome P450 reductase
- eNOS
endothelial nitric oxide synthase
- FADH•
one-electron reduced (semiquinone) FAD
- FADH2
two-electron reduced (hydroquinone) FAD
- FMNH•
one-electron reduced (semiquinone) FMN
- FMNH2/FMNH−
two-electron reduced (hydroquinone) FMN
- FNR
ferredoxin NADP+ reductase-like subdomain
- H4B
(6R)-5,6,7,8-tetrahydro-L-biopterin
- iNOS
inducible nitric oxide synthase
- nNOS
neuronal nitric oxide synthase
- nNOSr
reductase domain of neuronal NOS
- NO
nitric oxide
- NOS
nitric oxide synthase
- NOSoxy
oxygenase domain of NOS
References
- 1.De Colibus L, Mattevi A. New frontiers in structural flavoenzymology. Curr Opin Struct Biol. 2006;16:722–728. doi: 10.1016/j.sbi.2006.10.003. [DOI] [PubMed] [Google Scholar]
- 2.Joosten V, van Berkel WJ. Flavoenzymes. Curr Opin Chem Biol. 2007;11:195–202. doi: 10.1016/j.cbpa.2007.01.010. [DOI] [PubMed] [Google Scholar]
- 3.Gutierrez A, Grunau A, Paine M, Munro AW, Wolf CR, Roberts GC, Scrutton NS. Electron transfer in human cytochrome P450 reductase. Biochem Soc Trans. 2003;31:497–501. doi: 10.1042/bst0310497. [DOI] [PubMed] [Google Scholar]
- 4.Girvan HM, Waltham TN, Neeli R, Collins HF, McLean KJ, Scrutton NS, Leys D, Munro AW. Flavocytochrome P450 BM3 and the origin of CYP102 fusion species. Biochem Soc Trans. 2006;34:1173–1177. doi: 10.1042/BST0341173. [DOI] [PubMed] [Google Scholar]
- 5.Murataliev MB, Feyereisen R, Walker FA. Electron transfer by diflavin reductases. Biochim Biophys Acta. 2004;1698:1–26. doi: 10.1016/j.bbapap.2003.10.003. [DOI] [PubMed] [Google Scholar]
- 6.Alderton WK, Cooper CE, Knowles RG. Nitric oxide synthases: structure, function and inhibition. Biochem J. 2001;357:593–615. doi: 10.1042/0264-6021:3570593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Stuehr DJ. Mammalian nitric oxide synthases. Biochim Biophys Acta. 1999;1411:217–230. doi: 10.1016/s0005-2728(99)00016-x. [DOI] [PubMed] [Google Scholar]
- 8.Gorren ACF, Mayer B. Nitric-oxide synthase: a cytochrome P450 family foster child. Biochim Biophys Acta Gen Subj. 2007;1770:432–445. doi: 10.1016/j.bbagen.2006.08.019. [DOI] [PubMed] [Google Scholar]
- 9.Li H, Poulos TL. Structure–function studies on nitric oxide synthases. J Inorg Biochem. 2005;99:293–305. doi: 10.1016/j.jinorgbio.2004.10.016. [DOI] [PubMed] [Google Scholar]
- 10.Roman LJ, Martasek P, Masters BS. Intrinsic and extrinsic modulation of nitric oxide synthase activity. Chem Rev. 2002;102:1179–1190. doi: 10.1021/cr000661e. [DOI] [PubMed] [Google Scholar]
- 11.Miller RT, Martasek P, Omura T, Masters BSS. Rapid kinetic studies of electron transfer in the three isoforms of nitric oxide synthase. Biochem Biophys Res Commun. 1999;265:184–188. doi: 10.1006/bbrc.1999.1643. [DOI] [PubMed] [Google Scholar]
- 12.Santolini J, Adak S, Curran CM, Stuehr DJ. A kinetic simulation model that describes catalysis and regulation in nitric-oxide synthase. J Biol Chem. 2001;276:1233–1243. doi: 10.1074/jbc.M006858200. [DOI] [PubMed] [Google Scholar]
- 13.Santolini J, Meade AL, Stuehr DJ. Differences in three kinetic parameters underpin the unique catalytic profiles of nitric-oxide synthases I, II, and III. J Biol Chem. 2001;276:48887–48898. doi: 10.1074/jbc.M108666200. [DOI] [PubMed] [Google Scholar]
- 14.Masters BS, McMillan K, Sheta EA, Nishimura JS, Roman LJ, Martasek P. Neuronal nitric oxide synthase, a modular enzyme formed by convergent evolution: structure studies of a cysteine thiolate-liganded heme protein that hydroxylates L-arginine to produce NO. as a cellular signal. FASEB J. 1996;10:552–558. doi: 10.1096/fasebj.10.5.8621055. [DOI] [PubMed] [Google Scholar]
- 15.Marletta MA. Nitric oxide synthase: aspects concerning structure and catalysis. Cell. 1994;78:927–930. doi: 10.1016/0092-8674(94)90268-2. [DOI] [PubMed] [Google Scholar]
- 16.Wei CC, Crane BR, Stuehr DJ. Tetrahydrobiopterin radical enzymology. Chem Rev. 2003;103:2365–2383. doi: 10.1021/cr0204350. [DOI] [PubMed] [Google Scholar]
- 17.Raman CS, Li H, Martasek P, Kral V, Masters BS, Poulos TL. Crystal structure of constitutive endothelial nitric oxide synthase: a paradigm for pterin function involving a novel metal center. Cell. 1998;95:939–950. doi: 10.1016/s0092-8674(00)81718-3. [DOI] [PubMed] [Google Scholar]
- 18.Crane BR, Arvai AS, Ghosh DK, Wu C, Getzoff ED, Stuehr DJ, Tainer JA. Structure of nitric oxide synthase oxygenase dimer with pterin and substrate. Science. 1998;279:2121–2126. doi: 10.1126/science.279.5359.2121. [DOI] [PubMed] [Google Scholar]
- 19.Fischmann TO, Hruza A, Niu XD, Fossetta JD, Lunn CA, Dolphin E, Prongay AJ, Reichert P, Lundell DJ, Narula SK, et al. Structural characterization of nitric oxide synthase isoforms reveals striking activesite conservation. Nat Struct Biol. 1999;6:233–242. doi: 10.1038/6675. [DOI] [PubMed] [Google Scholar]
- 20.Zhang J, Martasek P, Paschke R, Shea T, Masters BSS, Kim JJ. Crystal structure of the FAD/NADPH-binding domain of rat neuronal nitric-oxide synthase. Comparisons with NADPH-cytochrome P450 oxidoreductase. J Biol Chem. 2001;276:37506–37513. doi: 10.1074/jbc.M105503200. [DOI] [PubMed] [Google Scholar]
- 21.Aoyagi M, Arvai AS, Tainer JA, Getzoff ED. Structural basis for endothelial nitric oxide synthase binding to calmodulin. EMBO J. 2003;22:766–775. doi: 10.1093/emboj/cdg078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Garcin ED, Bruns CM, Lloyd SJ, Hosfield DJ, Tiso M, Gachhui R, Stuehr DJ, Tainer JA, Getzoff ED. Structural basis for isozyme-specific regulation of electron transfer in nitric-oxide synthase. J Biol Chem. 2004;279:37918–37927. doi: 10.1074/jbc.M406204200. [DOI] [PubMed] [Google Scholar]
- 23.Wei CC, Wang ZQ, Tejero J, Yang YP, Hemann C, Hille R, Stuehr DJ. Catalytic reduction of a tetrahydrobiopterin radical within nitric-oxide synthase. J Biol Chem. 2008;283:11734–11742. doi: 10.1074/jbc.M709250200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Crane BR, Arvai AS, Gachhui R, Wu C, Ghosh DK, Getzoff ED, Stuehr DJ, Tainer JA. The structure of nitric oxide synthase oxygenase domain and inhibitor complexes. Science. 1997;278:425–431. doi: 10.1126/science.278.5337.425. [DOI] [PubMed] [Google Scholar]
- 25.Munro AW, Girvan HM, McLean KJ. Cytochrome P450 – redox partner fusion enzymes. Biochim Biophys Acta. 2007;1770:345–359. doi: 10.1016/j.bbagen.2006.08.018. [DOI] [PubMed] [Google Scholar]
- 26.Porter TD. An unusual yet strongly conserved flavoprotein reductase in bacteria and mammals. Trends Biochem Sci. 1991;16:154–158. doi: 10.1016/0968-0004(91)90059-5. [DOI] [PubMed] [Google Scholar]
- 27.Salerno JC, Harris DE, Irizarry K, Patel B, Morales AJ, Smith SM, Martasek P, Roman LJ, Masters BS, Jones CL, et al. An autoinhibitory control element defines calcium-regulated isoforms of nitric oxide synthase. J Biol Chem. 1997;272:29769–29777. doi: 10.1074/jbc.272.47.29769. [DOI] [PubMed] [Google Scholar]
- 28.Daff S, Sagami I, Shimizu T. The 42-amino acid insert in the FMN domain of neuronal nitric-oxide synthase exerts control over Ca(2+)/calmodulin-dependent electron transfer. J Biol Chem. 1999;274:30589–30595. doi: 10.1074/jbc.274.43.30589. [DOI] [PubMed] [Google Scholar]
- 29.Lane P, Gross SS. The autoinhibitory control element and calmodulin conspire to provide physiological modulation of endothelial and neuronal nitric oxide synthase activity. Acta Physiol Scand. 2000;168:53–63. doi: 10.1046/j.1365-201x.2000.00654.x. [DOI] [PubMed] [Google Scholar]
- 30.Montgomery HJ, Romanov V, Guillemette JG. Removal of a putative inhibitory element reduces the calcium-dependent calmodulin activation of neuronal nitric-oxide synthase. J Biol Chem. 2000;275:5052–5058. doi: 10.1074/jbc.275.7.5052. [DOI] [PubMed] [Google Scholar]
- 31.Roman LJ, Miller RT, de La Garza MA, Kim JJ, Masters BSS. The C-terminus of mouse macrophage inducible nitric-oxide synthase attenuates electron flow through the flavin domain. J Biol Chem. 2000;275:21914–21919. doi: 10.1074/jbc.M002449200. [DOI] [PubMed] [Google Scholar]
- 32.Roman LJ, Martasek P, Miller RT, Harris DE, de La Garza MA, Shea TM, Kim JJ, Masters BS. The C termini of constitutive nitric-oxide synthases control electron flow through the flavin and heme domains and affect modulation by calmodulin. J Biol Chem. 2000;275:29225–29232. doi: 10.1074/jbc.M004766200. [DOI] [PubMed] [Google Scholar]
- 33.Tiso M, Tejero J, Panda K, Aulak KS, Stuehr DJ. Versatile regulation of neuronal nitric oxide synthase by specific regions of its C-terminal tail. Biochemistry. 2007;46:14418–14428. doi: 10.1021/bi701646k. [DOI] [PubMed] [Google Scholar]
- 34.Knudsen GM, Nishida CR, Mooney SD, Ortiz de Montellano PR. Nitric-oxide synthase (NOS) reductase domain models suggest a new control element in endothelial NOS that attenuates calmodulindependent activity. J Biol Chem. 2003;278:31814–31824. doi: 10.1074/jbc.M303267200. [DOI] [PubMed] [Google Scholar]
- 35.Jones RJ, Smith SM, Gao YT, DeMay BS, Mann KJ, Salerno KM, Salerno JC. The function of the small insertion in the hinge subdomain in the control of constitutive mammalian nitric-oxide synthases. J Biol Chem. 2004;279:36876–36883. doi: 10.1074/jbc.M402808200. [DOI] [PubMed] [Google Scholar]
- 36.Abu-Soud HM, Yoho LL, Stuehr DJ. Calmodulin controls neuronal nitric-oxide synthase by a dual mechanism. Activation of intra- and interdomain electron transfer. J Biol Chem. 1994;269:32047–32050. [PubMed] [Google Scholar]
- 37.Kobayashi K, Tagawa S, Daff S, Sagami I, Shimizu T. Rapid calmodulin-dependent interdomain electron transfer in neuronal nitric-oxide synthase measured by pulse radiolysis. J Biol Chem. 2001;276:39864–39871. doi: 10.1074/jbc.M102537200. [DOI] [PubMed] [Google Scholar]
- 38.Sagami I, Daff S, Shimizu T. Intra-subunit and inter-subunit electron transfer in neuronal nitric-oxide synthase: effect of calmodulin on heterodimer catalysis. J Biol Chem. 2001;276:30036–30042. doi: 10.1074/jbc.M104123200. [DOI] [PubMed] [Google Scholar]
- 39.Panda K, Ghosh S, Stuehr DJ. Calmodulin activates intersubunit electron transfer in the neuronal nitric-oxide synthase dimer. J Biol Chem. 2001;276:23349–23356. doi: 10.1074/jbc.M100687200. [DOI] [PubMed] [Google Scholar]
- 40.Beaumont E, Lambry JC, Blanchard-Desce M, Martasek P, Panda SP, van Faassen EE, Brochon JC, Deprez E, Slama-Schwok A. NO formation by neuronal NO-synthase can be controlled by ultrafast electron injection from a nanotrigger. ChemBioChem. 2009;10:690–701. doi: 10.1002/cbic.200800721. [DOI] [PubMed] [Google Scholar]
- 41.Chen ZP, Mitchelhill KI, Michell BJ, Stapleton D, Rodriguez-Crespo I, Witters LA, Power DA, Ortiz de Montellano PR, Kemp BE. AMP-activated protein kinase phosphorylation of endothelial NO synthase. FEBS Lett. 1999;443:285–289. doi: 10.1016/s0014-5793(98)01705-0. [DOI] [PubMed] [Google Scholar]
- 42.Dimmeler S, Fleming I, Fisslthaler B, Hermann C, Busse R, Zeiher AM. Activation of nitric oxide synthase in endothelial cells by Akt-dependent phosphorylation. Nature. 1999;399:601–605. doi: 10.1038/21224. [DOI] [PubMed] [Google Scholar]
- 43.Fulton D, Gratton JP, McCabe TJ, Fontana J, Fujio Y, Walsh K, Franke TF, Papapetropoulos A, Sessa WC. Regulation of endothelium-derived nitric oxide production by the protein kinase Akt. Nature. 1999;399:597–601. doi: 10.1038/21218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hayashi Y, Nishio M, Naito Y, Yokokura H, Nimura Y, Hidaka H, Watanabe Y. Regulation of neuronal nitric-oxide synthase by calmodulin kinases. J Biol Chem. 1999;274:20597–20602. doi: 10.1074/jbc.274.29.20597. [DOI] [PubMed] [Google Scholar]
- 45.Butt E, Bernhardt M, Smolenski A, Kotsonis P, Frohlich LG, Sickmann A, Meyer HE, Lohmann SM, Schmidt HH. Endothelial nitric-oxide synthase (type III) is activated and becomes calcium independent upon phosphorylation by cyclic nucleotide-dependent protein kinases. J Biol Chem. 2000;275:5179–5187. doi: 10.1074/jbc.275.7.5179. [DOI] [PubMed] [Google Scholar]
- 46.Lane P, Gross SS. Disabling a C-terminal autoinhibitory control element in endothelial nitric-oxide synthase by phosphorylation provides a molecular explanation for activation of vascular NO synthesis by diverse physiological stimuli. J Biol Chem. 2002;277:19087–19094. doi: 10.1074/jbc.M200258200. [DOI] [PubMed] [Google Scholar]
- 47.Ghosh S, Gachhui R, Crooks C, Wu C, Lisanti MP, Stuehr DJ. Interaction between caveolin-1 and the reductase domain of endothelial nitric-oxide synthase. Consequences for catalysis. J Biol Chem. 1998;273:22267–22271. doi: 10.1074/jbc.273.35.22267. [DOI] [PubMed] [Google Scholar]
- 48.Sato Y, Sagami I, Shimizu T. Identification of caveolin-1-interacting sites in neuronal nitric-oxide synthase. Molecular mechanism for inhibition of NO formation. J Biol Chem. 2004;279:8827–8836. doi: 10.1074/jbc.M310327200. [DOI] [PubMed] [Google Scholar]
- 49.Cao S, Yao J, McCabe TJ, Yao Q, Katusic ZS, Sessa WC, Shah V. Direct interaction between endothelial nitric-oxide synthase and dynamin-2. Implications for nitric-oxide synthase function. J Biol Chem. 2001;276:14249–14256. doi: 10.1074/jbc.M006258200. [DOI] [PubMed] [Google Scholar]
- 50.Garcia-Cardena G, Fan R, Shah V, Sorrentino R, Cirino G, Papapetropoulos A, Sessa WC. Dynamic activation of endothelial nitric oxide synthase by Hsp90. Nature. 1998;392:821–824. doi: 10.1038/33934. [DOI] [PubMed] [Google Scholar]
- 51.Stuehr DJ, Santolini J, Wang ZQ, Wei CC, Adak S. Update on mechanism and catalytic regulation in the NO synthases. J Biol Chem. 2004;279:36167–36170. doi: 10.1074/jbc.R400017200. [DOI] [PubMed] [Google Scholar]
- 52.Leys D, Basran J, Talfournier F, Sutcliffe MJ, Scrutton NS. Extensive conformational sampling in a ternary electron transfer complex. Nat Struct Biol. 2003;10:219–225. doi: 10.1038/nsb894. [DOI] [PubMed] [Google Scholar]
- 53.Toogood HS, Leys D, Scrutton NS. Dynamics driving function: new insights from electron transferring flavoproteins and partner complexes. FEBS J. 2007;274:5481–5504. doi: 10.1111/j.1742-4658.2007.06107.x. [DOI] [PubMed] [Google Scholar]
- 54.Wang M, Roberts DL, Paschke R, Shea TM, Masters BS, Kim JJ. Three-dimensional structure of NADPH-cytochrome P450 reductase: prototype for FMN- and FAD-containing enzymes. Proc Natl Acad Sci USA. 1997;94:8411–8416. doi: 10.1073/pnas.94.16.8411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Grunau A, Geraki K, Grossmann JG, Gutierrez A. Conformational dynamics and the energetics of protein–ligand interactions: role of interdomain loop in human cytochrome P450 reductase. Biochemistry. 2007;46:8244–8255. doi: 10.1021/bi700596s. [DOI] [PubMed] [Google Scholar]
- 56.Neeli R, Girvan HM, Lawrence A, Warren MJ, Leys D, Scrutton NS, Munro AW. The dimeric form of flavocytochrome P450 BM3 is catalytically functional as a fatty acid hydroxylase. FEBS Lett. 2005;579:5582–5588. doi: 10.1016/j.febslet.2005.09.023. [DOI] [PubMed] [Google Scholar]
- 57.Kitazume T, Haines DC, Estabrook RW, Chen B, Peterson JA. Obligatory intermolecular electron-transfer from FAD to FMN in dimeric P450BM-3. Biochemistry. 2007;46:11892–11901. doi: 10.1021/bi701031r. [DOI] [PubMed] [Google Scholar]
- 58.Ilagan RP, Tiso M, Konas DW, Hemann C, Durra D, Hille R, Stuehr DJ. Differences in a conformational equilibrium distinguish catalysis by the endothelial and neuronal nitric-oxide synthase flavoproteins. J Biol Chem. 2008;283:19603–19615. doi: 10.1074/jbc.M802914200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Craig DH, Chapman SK, Daff S. Calmodulin activates electron transfer through neuronal nitric-oxide synthase reductase domain by releasing an NADPH-dependent conformational lock. J Biol Chem. 2002;277:33987–33994. doi: 10.1074/jbc.M203118200. [DOI] [PubMed] [Google Scholar]
- 60.Tiso M, Konas DW, Panda K, Garcin ED, Sharma M, Getzoff ED, Stuehr DJ. C-terminal tail residue Arg1400 enables NADPH to regulate electron transfer in neuronal nitric-oxide synthase. J Biol Chem. 2005;280:39208–39219. doi: 10.1074/jbc.M507775200. [DOI] [PubMed] [Google Scholar]
- 61.Konas DW, Zhu K, Sharma M, Aulak KS, Brudvig GW, Stuehr DJ. The FAD-shielding residue Phe1395 regulates neuronal nitric-oxide synthase catalysis by controlling NADP+ affinity and a conformational equilibrium within the flavoprotein domain. J Biol Chem. 2004;279:35412–35425. doi: 10.1074/jbc.M400872200. [DOI] [PubMed] [Google Scholar]
- 62.Konas DW, Takaya N, Sharma M, Stuehr DJ. Role of Asp(1393) in catalysis, flavin reduction, NADP(H) binding, FAD thermodynamics, and regulation of the nNOS flavoprotein. Biochemistry. 2006;45:12596–12609. doi: 10.1021/bi061011t. [DOI] [PubMed] [Google Scholar]
- 63.Ilagan RP, Tejero J, Aulak KS, Ray SS, Hemann C, Wang ZQ, Gangoda M, Zweier JL, Stuehr DJ. Regulation of FMN subdomain interactions and function in neuronal nitric oxide synthase. Biochemistry. 2009;48:3864–3876. doi: 10.1021/bi8021087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ghosh DK, Vogelien DL, Assan-Menash IB, Couser LM, Dinsmore NE, Rogers AG, Weinberg JB, Salerno JC. Function of the reductase unit of nitric oxide synthase: catalytic model. Nitric Oxide. 2008;19:S50. [Google Scholar]
- 65.Presta A, Weber-Main AM, Stankovich MT, Stuehr D. Comparative effects of substrates and pterin cofactor on the heme midpoint potential in inducible and neuronal nitric oxide synthases. J Am Chem Soc. 1998;120:9460–9465. [Google Scholar]
- 66.Mendes P. GEPASI: a software package for modelling the dynamics, steady states and control of biochemical and other systems. Comput Appl Biosci. 1993;9:563–571. doi: 10.1093/bioinformatics/9.5.563. [DOI] [PubMed] [Google Scholar]
- 67.Noble MA, Munro AW, Rivers SL, Robledo L, Daff SN, Yellowlees LJ, Shimizu T, Sagami I, Guillemette JG, Chapman SK. Potentiometric analysis of the flavin cofactors of neuronal nitric oxide synthase. Biochemistry. 1999;38:16413–16418. doi: 10.1021/bi992150w. [DOI] [PubMed] [Google Scholar]
- 68.Dunford AJ, Rigby SEJ, Hay S, Munro AW, Scrutton NS. Conformational and thermodynamic control of electron transfer in neuronal nitric oxide synthase. Biochemistry. 2007;46:5018–5029. doi: 10.1021/bi7001339. [DOI] [PubMed] [Google Scholar]
- 69.Garnaud PE, Koetsier M, Ost TW, Daff S. Redox properties of the isolated flavin mononucleotideand flavin adenine dinucleotide-binding domains of neuronal nitric oxide synthase. Biochemistry. 2004;43:11035–11044. doi: 10.1021/bi049312v. [DOI] [PubMed] [Google Scholar]
- 70.Grunau A, Paine MJ, Ladbury JE, Gutierrez A. Global effects of the energetics of coenzyme binding: NADPH controls the protein interaction properties of human cytochrome P450 reductase. Biochemistry. 2006;45:1421–1434. doi: 10.1021/bi052115r. [DOI] [PubMed] [Google Scholar]
- 71.Deng Z, Aliverti A, Zanetti G, Arakaki AK, Ottado J, Orellano EG, Calcaterra NB, Ceccarelli EA, Carrillo N, Karplus PA. A productive NADP+ binding mode of ferredoxin–NADP+ reductase revealed by protein engineering and crystallographic studies. Nat Struct Biol. 1999;6:847–853. doi: 10.1038/12307. [DOI] [PubMed] [Google Scholar]
- 72.Dunford AJ, Marshall KR, Munro AW, Scrutton NS. Thermodynamic and kinetic analysis of the isolated FAD domain of rat neuronal nitric oxide synthase altered in the region of the FAD shielding residue Phe1395. Eur J Biochem. 2004;271:2548–2560. doi: 10.1111/j.1432-1033.2004.04185.x. [DOI] [PubMed] [Google Scholar]
- 73.Gutierrez A, Doehr O, Paine M, Wolf CR, Scrutton NS, Roberts GC. Trp-676 facilitates nicotin-amide coenzyme exchange in the reductive half-reaction of human cytochrome P450 reductase: properties of the soluble W676H and W676A mutant reductases. Biochemistry. 2000;39:15990–15999. doi: 10.1021/bi002135n. [DOI] [PubMed] [Google Scholar]
- 74.Nogues I, Tejero J, Hurley JK, Paladini D, Frago S, Tollin G, Mayhew SG, Gomez-Moreno C, Ceccarelli EA, Carrillo N, et al. Role of the C-terminal tyrosine of ferredoxin–nicotinamide adenine dinucleotide phosphate reductase in the electron transfer processes with its protein partners ferredoxin and flavodoxin. Biochemistry. 2004;43:6127–6137. doi: 10.1021/bi049858h. [DOI] [PubMed] [Google Scholar]
- 75.Piubelli L, Aliverti A, Arakaki AK, Carrillo N, Ceccarelli EA, Karplus PA, Zanetti G. Competition between C-terminal tyrosine and nicotinamide modulates pyridine nucleotide affinity and specificity in plant ferredoxin–NADP(+) reductase. J Biol Chem. 2000;275:10472–10476. doi: 10.1074/jbc.275.14.10472. [DOI] [PubMed] [Google Scholar]
- 76.Tejero J, Perez-Dorado I, Maya C, Martinez-Julvez M, Sanz-Aparicio J, Gomez-Moreno C, Hermoso JA, Medina M. C-terminal tyrosine of ferredoxin –NADP+ reductase in hydride transfer processes with NAD(P)+/H. Biochemistry. 2005;44:13477–13490. doi: 10.1021/bi051278c. [DOI] [PubMed] [Google Scholar]
- 77.Welland A, Garnaud PE, Kitamura M, Miles CS, Daff S. Importance of the domain–domain interface to the catalytic action of the NO synthase reductase domain. Biochemistry. 2008;47:9771–9780. doi: 10.1021/bi800787m. [DOI] [PubMed] [Google Scholar]
- 78.Knight K, Scrutton NS. Stopped-flow kinetic studies of electron transfer in the reductase domain of neuronal nitric oxide synthase: re-evaluation of the kinetic mechanism reveals new enzyme intermediates and variation with cytochrome P450 reductase. Biochem J. 2002;367:19–30. doi: 10.1042/BJ20020667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Gutierrez A, Munro AW, Grunau A, Wolf CR, Scrutton NS, Roberts GC. Interflavin electron transfer in human cytochrome P450 reductase is enhanced by coenzyme binding. Relaxation kinetic studies with coenzyme analogues. Eur J Biochem. 2003;270:2612–2621. doi: 10.1046/j.1432-1033.2003.03633.x. [DOI] [PubMed] [Google Scholar]
- 80.Matsuda H, Iyanagi T. Calmodulin activates intramolecular electron transfer between the two flavins of neuronal nitric oxide synthase flavin domain. Biochim Biophys Acta. 1999;1473:345–355. doi: 10.1016/s0304-4165(99)00193-2. [DOI] [PubMed] [Google Scholar]
- 81.Guan ZW, Kamatani D, Kimura S, Iyanagi T. Mechanistic studies on the intramolecular one-electron transfer between the two flavins in the human neuronal nitric-oxide synthase and inducible nitric-oxide synthase flavin domains. J Biol Chem. 2003;278:30859–30868. doi: 10.1074/jbc.M301929200. [DOI] [PubMed] [Google Scholar]
- 82.Guan ZW, Iyanagi T. Electron transfer is activated by calmodulin in the flavin domain of human neuronal nitric oxide synthase. Arch Biochem Biophys. 2003;412:65–76. doi: 10.1016/s0003-9861(03)00009-2. [DOI] [PubMed] [Google Scholar]
- 83.Yamamoto K, Kimura S, Shiro Y, Iyanagi T. Interflavin one-electron transfer in the inducible nitric oxide synthase reductase domain and NADPH-cytochrome P450 reductase. Arch Biochem Biophys. 2005;440:65–78. doi: 10.1016/j.abb.2005.05.027. [DOI] [PubMed] [Google Scholar]
- 84.Nishino Y, Yamamoto K, Kimura S, Kikuchi A, Shiro Y, Iyanagi T. Mechanistic studies on the intramolecular one-electron transfer between the two flavins in the human endothelial NOS reductase domain. Arch Biochem Biophys. 2007;465:254–265. doi: 10.1016/j.abb.2007.05.021. [DOI] [PubMed] [Google Scholar]
- 85.Hamdane D, Xia C, Im SC, Zhang H, Kim JJ, Waskell L. Structure and function of an NADPH-cytochrome P450 oxidoreductase in an open conformation capable of reducing cytochrome P450. J Biol Chem. 2009;284:11374–11384. doi: 10.1074/jbc.M807868200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Newman E, Spratt DE, Mosher J, Cheyne B, Montgomery HJ, Wilson DL, Weinberg JB, Smith SM, Salerno JC, Ghosh DK, et al. Differential activation of nitric-oxide synthase isozymes by calmodulin–troponin C chimeras. J Biol Chem. 2004;279:33547–33557. doi: 10.1074/jbc.M403892200. [DOI] [PubMed] [Google Scholar]
- 87.Spratt DE, Newman E, Mosher J, Ghosh DK, Salerno JC, Guillemette JG. Binding and activation of nitric oxide synthase isozymes by calmodulin EF hand pairs. FEBS J. 2006;273:1759–1771. doi: 10.1111/j.1742-4658.2006.05193.x. [DOI] [PubMed] [Google Scholar]
- 88.Adak S, Santolini J, Tikunova S, Wang Q, Johnson JD, Stuehr DJ. Neuronal nitric-oxide synthase mutant (Ser-1412 → Asp) demonstrates surprising connections between heme reduction, NO complex formation, and catalysis. J Biol Chem. 2001;276:1244–1252. doi: 10.1074/jbc.M006857200. [DOI] [PubMed] [Google Scholar]
- 89.Gachhui R, Abu-Soud HM, Ghosh DK, Presta A, Blazing MA, Mayer B, George SE, Stuehr DJ. Neuronal nitric-oxide synthase interaction with calmodulin –troponin C chimeras. J Biol Chem. 1998;273:5451–5454. doi: 10.1074/jbc.273.10.5451. [DOI] [PubMed] [Google Scholar]
- 90.Panda K, Haque MM, Garcin-Hosfield ED, Durra D, Getzoff ED, Stuehr DJ. Surface charge interactions of the FMN module govern catalysis by nitric-oxide synthase. J Biol Chem. 2006;281:36819–36827. doi: 10.1074/jbc.M606129200. [DOI] [PubMed] [Google Scholar]
- 91.Nisimoto Y, Motalebi S, Han CH, Lambeth JD. The p67(phox) activation domain regulates electron flow from NADPH to flavin in flavocytochrome b(558) J Biol Chem. 1999;274:22999–23005. doi: 10.1074/jbc.274.33.22999. [DOI] [PubMed] [Google Scholar]
- 92.Shimanuki T, Sato H, Daff S, Sagami I, Shimizu T. Crucial role of Lys(423) in the electron transfer of neuronal nitric-oxide synthase. J Biol Chem. 1999;274:26956–26961. doi: 10.1074/jbc.274.38.26956. [DOI] [PubMed] [Google Scholar]
- 93.Feng C, Thomas C, Holliday MA, Tollin G, Salerno JC, Ghosh DK, Enemark JH. Direct measurement by laser flash photolysis of intramolecular electron transfer in a two-domain construct of murine inducible nitric oxide synthase. J Am Chem Soc. 2006;128:3808–3811. doi: 10.1021/ja0578606. [DOI] [PubMed] [Google Scholar]
- 94.Feng C, Tollin G, Holliday MA, Thomas C, Salerno JC, Enemark JH, Ghosh DK. Intraprotein electron transfer in a two-domain construct of neuronal nitric oxide synthase: the output state in nitric oxide formation. Biochemistry. 2006;45:6354–6362. doi: 10.1021/bi060223n. [DOI] [PubMed] [Google Scholar]
- 95.Feng C, Roman LJ, Hazzard JT, Ghosh DK, Tollin G, Masters BS. Deletion of the autoregulatory insert modulates intraprotein electron transfer in rat neuronal nitric oxide synthase. FEBS Lett. 2008;582:2768–2772. doi: 10.1016/j.febslet.2008.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Rozhkova EA, Fujimoto N, Sagami I, Daff SN, Shimizu T. Interactions between the isolated oxygenase and reductase domains of neuronal nitric-oxide synthase: assessing the role of calmodulin. J Biol Chem. 2002;277:16888–16894. doi: 10.1074/jbc.M200642200. [DOI] [PubMed] [Google Scholar]
- 97.Higashimoto Y, Sakamoto H, Hayashi S, Sugishima M, Fukuyama K, Palmer G, Noguchi M. Involvement of NADPH in the interaction between heme oxygenase-1 and cytochrome P450 reductase. J Biol Chem. 2005;280:729–737. doi: 10.1074/jbc.M406203200. [DOI] [PubMed] [Google Scholar]
- 98.Wang J, de Montellano PR. The binding sites on human heme oxygenase-1 for cytochrome P450 reductase and biliverdin reductase. J Biol Chem. 2003;278:20069–20076. doi: 10.1074/jbc.M300989200. [DOI] [PubMed] [Google Scholar]
- 99.Haque MM, Panda K, Tejero J, Aulak KS, Fadlalla MA, Mustovich AT, Stuehr DJ. A connecting hinge represses the activity of endothelial nitric oxide synthase. Proc Natl Acad Sci USA. 2007;104:9254–9259. doi: 10.1073/pnas.0700332104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Stuehr DJ, Wei CC, Santolini J, Wang Z, Aoyagi M, Getzoff ED. Radical reactions of nitric oxide synthases. Biochem Soc Symp. 2004;71:39–49. doi: 10.1042/bss0710039. [DOI] [PubMed] [Google Scholar]
- 101.Feng CJ, Tollin G, Hazzard JT, Nahm NJ, Guillemette JG, Salerno JC, Ghosh DK. Direct measurement by laser flash photolysis of intraprotein electron transfer in a rat neuronal nitric oxide synthase. J Am Chem Soc. 2007;129:5621–5629. doi: 10.1021/ja068685b. [DOI] [PubMed] [Google Scholar]
- 102.Roman LJ, Masters BS. Electron transfer by neuronal nitric oxide synthase is regulated by concerted interaction of calmodulin and two intrinsic regulatory elements. J Biol Chem. 2006;281:23111–23118. doi: 10.1074/jbc.M603671200. [DOI] [PubMed] [Google Scholar]
- 103.Daff S. Calmodulin-dependent regulation of mammalian nitric oxide synthase. Biochem Soc Trans. 2003;31:502–505. doi: 10.1042/bst0310502. [DOI] [PubMed] [Google Scholar]
- 104.Nishida CR, de Montellano PR. Control of electron transfer in nitric-oxide synthases. Swapping of autoinhibitory elements among nitric-oxide synthase isoforms. J Biol Chem. 2001;276:20116–20124. doi: 10.1074/jbc.M101548200. [DOI] [PubMed] [Google Scholar]
- 105.Chen PF, Wu KK. Structural elements contribute to the calcium/calmodulin dependence on enzyme activation in human endothelial nitric-oxide synthase. J Biol Chem. 2003;278:52392–52400. doi: 10.1074/jbc.M305469200. [DOI] [PubMed] [Google Scholar]
- 106.Gribovskaja I, Brownlow KC, Dennis SJ, Rosko AJ, Marletta MA, Stevens-Truss R. Calcium-binding sites of calmodulin and electron transfer by inducible nitric oxide synthase. Biochemistry. 2005;44:7593–7601. doi: 10.1021/bi0474517. [DOI] [PubMed] [Google Scholar]
- 107.Stevens-Truss R, Beckingham K, Marletta MA. Calcium binding sites of calmodulin and electron transfer by neuronal nitric oxide synthase. Biochemistry. 1997;36:12337–12345. doi: 10.1021/bi970973k. [DOI] [PubMed] [Google Scholar]
- 108.Stevens-Truss R, Marletta MA. Interaction of calmodulin with the inducible murine macrophage nitric oxide synthase. Biochemistry. 1995;34:15638–15645. doi: 10.1021/bi00048a006. [DOI] [PubMed] [Google Scholar]
- 109.Lee SJ, Beckingham K, Stull JT. Mutations at lysine 525 of inducible nitric-oxide synthase affect its Ca2+-independent activity. J Biol Chem. 2000;275:36067–36072. doi: 10.1074/jbc.M003935200. [DOI] [PubMed] [Google Scholar]
- 110.Lee SJ, Stull JT. Calmodulin-dependent regulation of inducible and neuronal nitric-oxide synthase. J Biol Chem. 1998;273:27430–27437. doi: 10.1074/jbc.273.42.27430. [DOI] [PubMed] [Google Scholar]
- 111.Sharp RE, Chapman SK, Reid GA. Modulation of flavocytochrome b2 intraprotein electron transfer via an interdomain hinge region. Biochem J. 1996;316:507–513. doi: 10.1042/bj3160507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Sharp RE, Chapman SK, Reid GA. Deletions in the interdomain hinge region of flavocytochrome b2: effects on intraprotein electron transfer. Biochemistry. 1996;35:891–899. doi: 10.1021/bi950457z. [DOI] [PubMed] [Google Scholar]
- 113.Sharp RE, White P, Chapman SK, Reid GA. Role of the interdomain hinge of flavocytochrome b2 in intra- and inter-protein electron transfer. Biochemistry. 1994;33:5115–5120. doi: 10.1021/bi00183a015. [DOI] [PubMed] [Google Scholar]
- 114.White P, Manson FD, Brunt CE, Chapman SK, Reid GA. The importance of the interdomain hinge in intramolecular electron transfer in flavocytochrome b2. Biochem J. 1993;291:89–94. doi: 10.1042/bj2910089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Chen PF, Wu KK. Characterization of the roles of the 594-645 region in human endothelial nitric-oxide synthase in regulating calmodulin binding and electron transfer. J Biol Chem. 2000;275:13155–13163. doi: 10.1074/jbc.275.17.13155. [DOI] [PubMed] [Google Scholar]
- 116.Adak S, Sharma M, Meade AL, Stuehr DJ. A conserved flavin-shielding residue regulates NO synthase electron transfer and nicotinamide coenzyme specificity. Proc Natl Acad Sci USA. 2002;99:13516–13521. doi: 10.1073/pnas.192283399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.McCabe TJ, Fulton D, Roman LJ, Sessa WC. Enhanced electron flux and reduced calmodulin dissociation may explain ‘calcium-independent’ eNOS activation by phosphorylation. J Biol Chem. 2000;275:6123–6128. doi: 10.1074/jbc.275.9.6123. [DOI] [PubMed] [Google Scholar]
- 118.Bender AT, Silverstein AM, Demady DR, Kanelakis KC, Noguchi S, Pratt WB, Osawa Y. Neuronal nitric-oxide synthase is regulated by the hsp90-based chaperone system in vivo. J Biol Chem. 1999;274:1472–1478. doi: 10.1074/jbc.274.3.1472. [DOI] [PubMed] [Google Scholar]
- 119.Yoshida M, Xia Y. Heat shock protein 90 as an endogenous protein enhancer of inducible nitric-oxide synthase. J Biol Chem. 2003;278:36953–36958. doi: 10.1074/jbc.M305214200. [DOI] [PubMed] [Google Scholar]
- 120.Song Y, Zweier JL, Xia Y. Heat-shock protein 90 augments neuronal nitric oxide synthase activity by enhancing Ca2+/calmodulin binding. Biochem J. 2001;355:357–360. doi: 10.1042/0264-6021:3550357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Takahashi S, Mendelsohn ME. Calmodulin-dependent and -independent activation of endothelial nitric-oxide synthase by heat shock protein 90. J Biol Chem. 2003;278:9339–9344. doi: 10.1074/jbc.M212651200. [DOI] [PubMed] [Google Scholar]
- 122.Gratton JP, Fontana J, O’Connor DS, Garcia-Cardena G, McCabe TJ, Sessa WC. Reconstitution of an endothelial nitric-oxide synthase (eNOS), hsp90, and caveolin-1 complex in vitro. Evidence that hsp90 facilitates calmodulin stimulated displacement of eNOS from caveolin-1. J Biol Chem. 2000;275:22268–22272. doi: 10.1074/jbc.M001644200. [DOI] [PubMed] [Google Scholar]
- 123.Cao S, Yao J, Shah V. The proline-rich domain of dynamin-2 is responsible for dynamin-dependent in vitro potentiation of endothelial nitric-oxide synthase activity via selective effects on reductase domain function. J Biol Chem. 2003;278:5894–5901. doi: 10.1074/jbc.M212546200. [DOI] [PubMed] [Google Scholar]
- 124.Golser R, Gorren AC, Leber A, Andrew P, Habisch HJ, Werner ER, Schmidt K, Venema RC, Mayer B. Interaction of endothelial and neuronal nitric-oxide synthases with the bradykinin B2 receptor. Binding of an inhibitory peptide to the oxygenase domain blocks uncoupled NADPH oxidation. J Biol Chem. 2000;275:5291–5296. doi: 10.1074/jbc.275.8.5291. [DOI] [PubMed] [Google Scholar]
- 125.Ju H, Venema VJ, Marrero MB, Venema RC. Inhibitory interactions of the bradykinin B2 receptor with endothelial nitric-oxide synthase. J Biol Chem. 1998;273:24025–24029. doi: 10.1074/jbc.273.37.24025. [DOI] [PubMed] [Google Scholar]
- 126.Kone BC, Kuncewicz T, Zhang W, Yu ZY. Protein interactions with nitric oxide synthases: controlling the right time, the right place, and the right amount of nitric oxide. Am J Physiol Renal Physiol. 2003;285:F178–F190. doi: 10.1152/ajprenal.00048.2003. [DOI] [PubMed] [Google Scholar]
- 127.Dudzinski DM, Michel T. Life history of eNOS: partners and pathways. Cardiovasc Res. 2007;75:247–260. doi: 10.1016/j.cardiores.2007.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Mount PF, Kemp BE, Power DA. Regulation of endothelial and myocardial NO synthesis by multisite eNOS phosphorylation. J Mol Cell Cardiol. 2007;42:271–279. doi: 10.1016/j.yjmcc.2006.05.023. [DOI] [PubMed] [Google Scholar]