

Potassium channel interacting proteins (KChIP) are Ca2+-binding proteins that originally were identified as auxiliary subunits for KV4 channels. KV4 channels encode the voltage gated A-current (IA) in neuronal tissue and the fast, transient outward current (Ito,f) in cardiac tissue. Recently, we have reported that KChIP2 functionally modulates the cardiac CaV1.2-governed L-type Ca2+ current (ICa,L) through a direct interaction between KChIP2 and the amino-terminus of CaV1.2. Here, we show that KChIP2 and CaV1.2 co-immunoprecipitate enhancing the biochemical support for our previous finding. Using gene-chip and real-time PCR techniques, we find that KChIP2−/− mice have an increased transcriptional activity of the calcium channel β2 subunit, CACNB2, whereas the expression of CaV1.2 is preserved. Although Ito,f is absent and ICa,L is decreased in myocytes from KChIP2−/− mice, the action potential morphology is not altered. Furthermore, we show that the ventricular effective refractory period (VERP) is comparable in wild-type (53 ± 5 ms) and KChIP2−/− mice (48 ± 3 ms; p > 0.05). In summary, our findings document a novel function of KChIP2 and expand our insights into the in vivo modulation of cardiac ion currents.
The K+ channel interacting proteins (KChIPs) are small (216–270 amino acids) cytosolic, calcium-binding proteins that were initially identified as subunits for the voltage-gated A-type K+ current in neuronal tissue and the transient outward K+ current in cardiac tissue.1 The pore-forming protein complex governing these K+ currents is KV4. KChIPs, dipeptidyl aminopeptidase-like proteins2 (DPP), KVβ1,3 and potentially several other protein structures assemble in macromolecular structures with KV4 to generate and regulate the native current.3
Four KCHIP genes have been identified in humans and mice.1,4,5 The proteins are characterized by a conserved Ca2+-binding region, which places the KChIPs in the family of small Ca2+-binding proteins, including Ca2+-binding protein 1,6,7 calmodulin8,9 and neuronal Ca2+ sensor 1.10 The conserved Ca2+ binding domain of the KChIPs contains 4 EF hands, of which only the last 3 bind Ca2+.1 A highly variable amino-terminal sequence preceding the Ca2+ binding domain distinguishes the KChIPs from other Ca2+-binding proteins and confers variability among the KChIPs themselves.6 The 4 KCHIP genes can, by use of alternative splicing or alternative use of transcription initiation sites, encode several protein isoforms.5 KChIP2 is the only KChIP expressed in the heart1 where 4 and 5 transcript isoforms have been identified in mouse and man, respectively.5
Recently, we have shown that KChIP2, in addition to facilitating KV4-mediated current in vivo,11,12 also modulates the cardiac L-type Ca2+ current (ICa,L).13 The hypothesis was triggered by the finding that KChIP2 immunoprecipitates with CaV1.2 α1C from brain tissue (Fig. 1). We found that ICa,L density is significantly reduced in myocytes disaggregated from KChIP2−/− mice compared to myocytes from wild-type (WT) mice. The decreased current density in the absence of KChIP2 did not result from reduced protein expression or trafficking of the pore-forming CaV1.2 α1C subunit. Rather, we showed that the channel modulation results from a direct interaction between KChIP2 and the intracellular amino-terminus of the α1C subunit. We were unable to show a direct association between CaV1.2 and KChIP2 in cardiac tissue, despite the demonstrated functional effects.13 Several reasons could underlie this, including antibodies with higher affinity for neuronal proteins and higher concentration of ion-channel proteins in brain. However, KChIP2 does copurify with the amino-terminus of CaV1.2 when co-expressed in a bacterial system.13 Moreover, co-expression of KChIP2 increased the current density governed by recombinant CaV1.2 in a transfected, mammalian cell line, further substantiating our finding that KChIP2 directly augments ICa,L.13 We proposed a model, in which KChIP2 indirectly increases the open probability of CaV1.2 by alleviating the inhibitory effects of the CaV1.2 amino-terminus, as previously characterized.14 Deleting the amino-terminus of CaV1.2 causes a large increase in the whole-cell current amplitude,13,15 and studies have shown that the β2 subunit of ICa,L in part increases CaV1.2 open probability by interacting with the amino-terminus.14 Additional studies showed that KChIP2 could not fully replace the β2 subunit, likely due to the pivotal role of β2 in CaV1.2 trafficking.16,17 Nevertheless, β2 and possibly KChIP2 both increase the open probability of CaV1.2 via modulation of the aminoterminal inhibitory segment of CaV1.2. Single-channel recordings of CaV1.2 are required to determine the exact mechanism of KChIP2-induced augmentation of whole-cell Ca2+ current.
Figure 1.
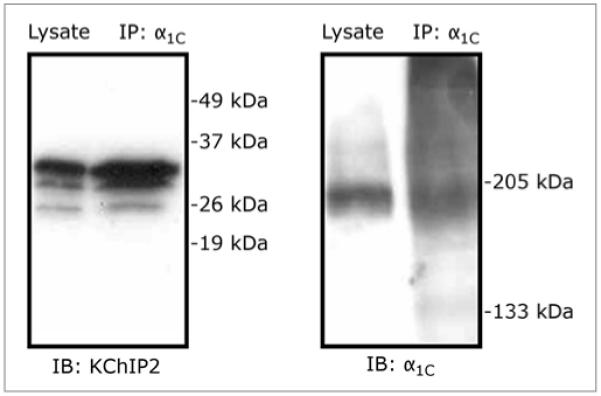
Co-immunoprecipitation showing a biochemical association between CaV1.2 α1C and KChIP2. Tissue samples from adult rat brain were lysed and immunoprecipitated (IP) with anti-α1C, according to methods described earlier.22 The lysate lanes represent total tissue lysates. The membranes are immunoblotted (IB) using anti-KChIP2 (left) and anti-α1C (right).
Microarray analysis with Mouse Genome 430A 2.0 array chips (Affymetrix) of cDNA from total RNA isolated from hearts of 4 KChIP2−/− and 4 C57BL6/J mice revealed that transcription of several genes was comparatively altered in the KChIP2−/− mice. We used quantitative, real-time PCR to verify the microarray results for the tran-scriptional activity of CaV1.2 and a number of associated subunits in KChIP2−/− mice (Fig. 2). As anticipated, KChIP2 mRNA contents in KChIP2−/− were below the detection levels of both assays, confirming the successful deletion of all KChIP2 isoforms (Fig. 2 and Table 1). Despite the increased protein levels of CaV1.2 that we observed in KChIP2−/− ventricles,13 we found no increases in mRNA levels of CaV1.2 in either assay. This would be compatible with an increased protein stability or decreased protein degradation in the absence of KChIP2; however further experimental work is required to test this hypothesis and elucidate the mechanism. Transcriptional activity of the β2 subunit was increased in KChIP2−/− ventricles, compared to WT ventricles (p < 0.05 in both assays; Fig. 2).
Figure 2.

Analysis of the molecular remodelling in KChIP2−/− ventricles reveals a significantly increased transcriptional activity of the β2 subunit. The transcription levels of ICa,L subunits and associated proteins in WT (n = 6) and KChIP2−/− (n = 6) ventricles were examined using quantitative real-time PCR, as previously described.11 Values are normalized to cyclophilin A level; error bars represents SEM; *p < 0.05. All samples are tested in triplicate. Primers and abbreviations are provided in Table 1. These analyses revealed that β2 expression is increased significantly, whereas KChIP2 mRNA could not be detected in KChIP2−/− ventricular tissue.
Table 1.
Primers used for quantitative real-time PCR
| Gene | Forward primer | Reverse primer |
|---|---|---|
| CACNA1C (CaV1.2) |
TGACTACCTGACTAGGGATTGGTCTA | TGCTCTAGGTTCCCTTCTGTTTTG |
| CACNB2 (β2) | TTGCAAGAACACTGCAATTGG | GGGCCAGTTTATCAGCTGCTA |
| CACNA2D2 (α2δ2) |
TCCAAGACCAGCGCCTTAAG | ACGTTCCACTAACTGCTGTGTAATG |
| KChIP2 | AGCGTGGAGGATGAGTTTGAAC | TTCCCCGAAGAATCACTGACA |
| CACNG6 (γ6) | AGCGAGAGGGCAAGATCAAG | TTGGTGGTTCGCTGGAAGAT |
| Calm | AGCCTTCTCCCTCTTCGACAA | TAATCATGTCCTGCAGCTCCG |
| Camk2d | CAGACTTCGGCTTAGCCATAGAA | GGTGTGCCAGCAAAACCAA |
| Camk2g | TTGAAGACATTGTGGCCAGAGA | GCCCAGATGTCCACAGGTTT |
| Cyclophilin A | TGGCGGCAGGTCCATCTA | TCCACAATGTTCATGCCTTCTT |
Abbreviations: Calm, calmodulin; Camk2d and -g, Ca2+/calmodulin-dependent protein kinase II, subtype d and g, respectively.
In addition to a reduced ICa,L, the KChIP2−/− mice had no residual KV4.2-mediated Ito,f;11,12 however we were unable to identify any differences in action potential morphology in multicelluar preparations.11 This is in accordance with ECG analysis performed by Kuo et al.12 and by us,13 showing a J-wave elevation in KChIP2−/− mice, but no difference in QT intervals between WT and KChIP2−/− mice. Furthermore, the ventricular effective refractory period (VERP), established at a basic pacing rate of 100 ms is 53 ± 5 ms in anesthetized WT mice (n = 5) compared to 48 ± 3 ms in KChIP2−/− mice (n = 7; p > 0.05). Hence, although the amplitudes of several depolarizing and repolarizing currents are altered, the overall repolarization reserve seems preserved.11,18
It has been reported that KChIP2−/− mice have an increased incidence of ventricular polymorphic tachyarrhythmias in response to programmed electrical stimulation.12 Interestingly, KV4.2−/− mice have no arrhythmogenic phenotype,19,20 suggesting that the augmented susceptibility to arrhythmia in KChIP2−/− mice does not result exclusively from the absence of a functional Ito,f. Given the prominent J-wave elevation, the KChIP2−/− mice may serve as a model of the early repolarization syndrome, a familial disease characterized by ventricular ectopic activity and sudden death related to an accentuation of the J wave.21
Acknowledgements
We gratefully acknowledge Dr. Geoffrey S. Pitt (Duke University Medical Center, Durham, North Carolina, USA) and Dr. Michael R. Rosen (Columbia University, New York, New York, USA) for critical comments and discussions in the preparation of this manuscript. The present experimental work was performed during Dr. Thomsen’s tenure as Research Fellow of the Heart Rhythm Society. This work was supported by US Public Health Service/National Heart, Lung and Blood Institute grants HL-67101 and HL-28958 (to Michael R. Rosen) and HL-088089 and HL-071165 (to Geoffrey S. Pitt).
References
- 1.An WF, Bowlby MR, Betty M, Cao J, Ling HP, Mendoza G, et al. Modulation of A-type potassium channels by a family of calcium sensors. Nature. 2000;403:553–6. doi: 10.1038/35000592. [DOI] [PubMed] [Google Scholar]
- 2.Nadal MS, Ozaita A, Amarillo Y, Vega-Saenz de Miera E, Ma Y, Mo W, et al. The CD26-related dipeptidyl aminopeptidase-like protein DPPX is a critical component of neuronal A-type K+ channels. Neuron. 2003;37:449–61. doi: 10.1016/s0896-6273(02)01185-6. [DOI] [PubMed] [Google Scholar]
- 3.Nerbonne JM, Kass RS. Molecular physiology of cardiac repolarization. Physiol Rev. 2005;85:1205–53. doi: 10.1152/physrev.00002.2005. [DOI] [PubMed] [Google Scholar]
- 4.Morohashi Y, Hatano N, Ohya S, Takikawa R, Watabiki T, Takasugi N, et al. Molecular cloning and characterization of CALP/KChIP4, a novel EF-hand protein interacting with presenilin 2 and voltage-gated potassium channel subunit KV4. J Biol Chem. 2002;277:14965–75. doi: 10.1074/jbc.M200897200. [DOI] [PubMed] [Google Scholar]
- 5.Pruunsild P, Timmusk T. Structure, alternative splicing and expression of the human and mouse KCNIP gene family. Genomics. 2005;86:581–93. doi: 10.1016/j.ygeno.2005.07.001. [DOI] [PubMed] [Google Scholar]
- 6.Burgoyne RD, Weiss JL. The neuronal calcium sensor family of Ca2+-binding proteins. Biochem J. 2001;353:1–12. [PMC free article] [PubMed] [Google Scholar]
- 7.Haeseleer F, Imanishi Y, Sokal I, Filipek S, Palczewski K. Calcium-binding proteins: intracellular sensors from the calmodulin superfamily. Biochem Biophys Res Commun. 2002;290:615–23. doi: 10.1006/bbrc.2001.6228. [DOI] [PubMed] [Google Scholar]
- 8.Zuhlke RD, Pitt GS, Deisseroth K, Tsien RW, Reuter H. Calmodulin supports both inactivation and facilitation of L-type calcium channels. Nature. 1999;399:159–62. doi: 10.1038/20200. [DOI] [PubMed] [Google Scholar]
- 9.Peterson BZ, DeMaria CD, Adelman JP, Yue DT. Calmodulin is the Ca2+ sensor for Ca2+-Dependent inactivation of L-type calcium channels. Neuron. 1999;22:549–58. doi: 10.1016/s0896-6273(00)80709-6. [DOI] [PubMed] [Google Scholar]
- 10.Zhou H, Yu K, McCoy KL, Lee A. Molecular mechanism for divergent regulation of CaV1.2 Ca2+ channels by calmodulin and Ca2+-binding protein-1. J Biol Chem. 2005;280:29612–9. doi: 10.1074/jbc.M504167200. [DOI] [PubMed] [Google Scholar]
- 11.Thomsen MB, Sosunov EA, Anyukhovsky EP, Özgen N, Boyden PA, Rosen MR. Deleting the accessory subunit KChIP2 results in loss of Ito,f and increased IK,slow that maintains normal action potential configuration. Heart Rhythm. 2009;6:370–7. doi: 10.1016/j.hrthm.2008.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kuo HC, Cheng CF, Clark RB, Lin JJ, Lin JL, Hoshijima M, et al. A defect in the KV channel-interacting protein 2 (KChIP2) gene leads to a complete loss of Ito and confers susceptibility to ventricular tachycardia. Cell. 2001;107:801–13. doi: 10.1016/s0092-8674(01)00588-8. [DOI] [PubMed] [Google Scholar]
- 13.Thomsen MB, Wang C, Ozgen N, Wang HG, Rosen MR, Pitt GS. Accessory subunit KChIP2 modulates the cardiac L-type calcium current. Circ Res. 2009;104:1382–9. doi: 10.1161/CIRCRESAHA.109.196972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kanevsky N, Dascal N. Regulation of maximal open probability is a separable function of CaVβ subunit in L-type Ca2+ channel, dependent on NH2 terminus of alpha1C (CaV1.2α) J Gen Physiol. 2006;128:15–36. doi: 10.1085/jgp.200609485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wei X, Neely A, Olcese R, Lang W, Stefani E, Birnbaumer L. Increase in Ca2+ channel expression by deletions at the amino terminus of the cardiac α1C subunit. Receptors Channels. 1996;4:205–15. [PubMed] [Google Scholar]
- 16.Chien AJ, Zhao X, Shirokov RE, Puri TS, Chang CF, Sun D, et al. Roles of a membrane-localized β subunit in the formation and targeting of functional L-type Ca2+ channels. J Biol Chem. 1995;270:30036–44. doi: 10.1074/jbc.270.50.30036. [DOI] [PubMed] [Google Scholar]
- 17.Colecraft HM, Alseikhan B, Takahashi SX, Chaudhuri D, Mittman S, Yegnasubramanian V, et al. Novel functional properties of Ca2+ channel β subunits revealed by their expression in adult rat heart cells. J Physiol. 2002;541:435–52. doi: 10.1113/jphysiol.2002.018515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Roden DM. Taking the “idio” out of “idiosyncratic”: predicting torsades de pointes. Pacing Clin Electrophysiol. 1998;21:1029–34. doi: 10.1111/j.1540-8159.1998.tb00148.x. [DOI] [PubMed] [Google Scholar]
- 19.Nerbonne JM. Studying cardiac arrhythmias in the mouse—a reasonable model for probing mechanisms? Trends Cardiovasc Med. 2004;14:83–93. doi: 10.1016/j.tcm.2003.12.006. [DOI] [PubMed] [Google Scholar]
- 20.Guo W, Jung WE, Marionneau C, Aimond F, Xu H, Yamada KA, et al. Targeted deletion of KV4.2 eliminates Ito,f and results in electrical and molecular remodeling, with no evidence of ventricular hypertrophy or myocardial dysfunction. Circ Res. 2005;97:1342–50. doi: 10.1161/01.RES.0000196559.63223.aa. [DOI] [PubMed] [Google Scholar]
- 21.Haissaguerre M, Derval N, Sacher F, Jesel L, Deisenhofer I, de Roy L, et al. Sudden cardiac arrest associated with early repolarization. N Engl J Med. 2008;358:2016–23. doi: 10.1056/NEJMoa071968. [DOI] [PubMed] [Google Scholar]
- 22.Wang HG, George MS, Kim J, Wang C, Pitt GS. Ca2+/calmodulin regulates trafficking of CaV1.2 Ca2+ channels in cultured hippocampal neurons. J Neurosci. 2007;27:9086–93. doi: 10.1523/JNEUROSCI.1720-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]