

Abstract
Growing computing capacity and algorithmic advances have facilitated the study of increasingly large biomolecular systems at longer timescales. However, with these larger, more complex systems come questions about the quality of sampling and statistical convergence. What size systems can be sampled fully? If a system is not fully sampled, can certain “fast variables” be considered well-converged? How can one determine the statistical significance of observed results? The present review describes statistical tools and the underlying physical ideas necessary to address these questions. Basic definitions and ready-to-use analyses are provided, along with explicit recommendations. Such statistical analyses are of paramount importance in establishing the reliability of simulation data in any given study.
Keywords: Error analysis, principal component, block averaging, convergence
1. Introduction
It is a well-accepted truism that the results of a simulation are only as good as the statistical quality of the sampling. To compensate for the well-known sampling limitations of conventional molecular dynamics (MD) simulations of even moderate-size biomolecules, the field is now witnessing the rapid proliferation of multi-processor computing, new algorithms, and simplified models. These changes underscore the pressing need for unambiguous measures of sampling quality. Are current MD simulations long enough to make quantitative predictions? How much better are the new algorithms than the old? Can even simplified models be fully sampled?
Overall, errors in molecular simulation arise from two factors: inaccuracy in the models, and insufficient sampling. The former is related to choices in representing the system, e.g., all-atom vs. coarse grained models, fixed charge vs. polarizable force fields, and implicit vs. explicit solvent, as well as technical details like the system size, thermodynamic ensemble, and integration algorithm used. Taken in total, these choices define the model used to represent the system of interest. The second issue, quality of sampling, is largely orthogonal to the choice of model. In some sense, assessing the quality of the sampling is a way of asking how accurately a given quantity was computed for the chosen model. While this review will focus on the issue of sampling, it is important to point out that without adequate sampling, the predictions of the forcefields remain unknown: very few conclusions, positive or negative, can be drawn from an undersampled calculation. Those predictions are embodied most directly in the equilibrium ensemble that simulations have apparently failed to produce in all but small-molecule systems 1, 2. Thus, advances in forcefield design and parameterization for large biomolecules must proceed in parallel with sampling advances and their assured quantification.
This review will attempt to acquaint the reader with the most important ideas in assessing sampling quality. We will address both the statistical uncertainty in individual observables and quantification of the global quality of the equilibrium ensemble. We will explicitly address differing approaches necessary for standard dynamics simulations, as compared to algorithms such as replica exchange, and while we use the language of molecular dynamics, virtually all of the arguments apply equally to Monte Carlo methods as well. Although this review will not specifically address path sampling, many of the ideas carry over to what amounts to equilibrium sampling of the much larger space of paths. We will recommend specific ‘best practices’, with the inevitable bias toward the authors’ work. We have tried to describe the intellectual history behind the key ideas, but the article is ultimately organized around practically important concepts.
For the convenience of less experienced readers, key terms and functions have been defined in the appendix: average, variance, correlation function, and correlation time.
1.1. Examples big and small: butane and rhodopsin
Example trajectories from small and large systems (to which we will return throughout the review) illustrate the key ideas. In fact, almost all the complexity we will see in large systems is already present in a molecule as simple as n-butane. Nevertheless, it is very valuable to look at both “very long” trajectories and some that are “not long enough.” Concerning the definition of “long,” we hope that if ‘we know it when we see it,’ then we can construct a suitable mathematical definition. Visual confirmation of good sampling is still an important check on any quantitative measure.
Butane
Let us first consider butane, as in Figure 1. Several standard molecular coordinates are plotted for a period of 1 nsec, and it is clear that several timescales less than 1 nsec are present. The very fastest motions (almost vertical in the scale of the figure) correspond to bond length and angle vibrations, while the dihedrals exhibit occasional quasi-discrete transitions. The CH3 dihedral, which reports on methyl spinning, clearly makes more frequent transitions than the main dihedral.
Figure 1.

Widely varying timescales in n-butane. Even the simple butane molecule (upper left) exhibits a wide variety of dynamical timescales, as exhibited in the three time traces. The slowest timescales are evident in the C-C-C-C dihedral, the fast motions of the C-C-C bond-angle appear to exhibit a slow undulation as well.
Perhaps the trajectory of butane’s C-C-C angle is most ambiguous, since there appears to be a slow overall undulation in addition to the rapid vibrations. The undulation appears to have a frequency quite similar to the transition rate of the main dihedral, and underscores the point that generally speaking, all degrees of freedom are coupled, as sketched in Figure 2. In the case of butane, the sampling quality of the C-C-C angle may indeed be governed by the slowest motions of the molecule, isomerization of the central torsion.
Figure 2.
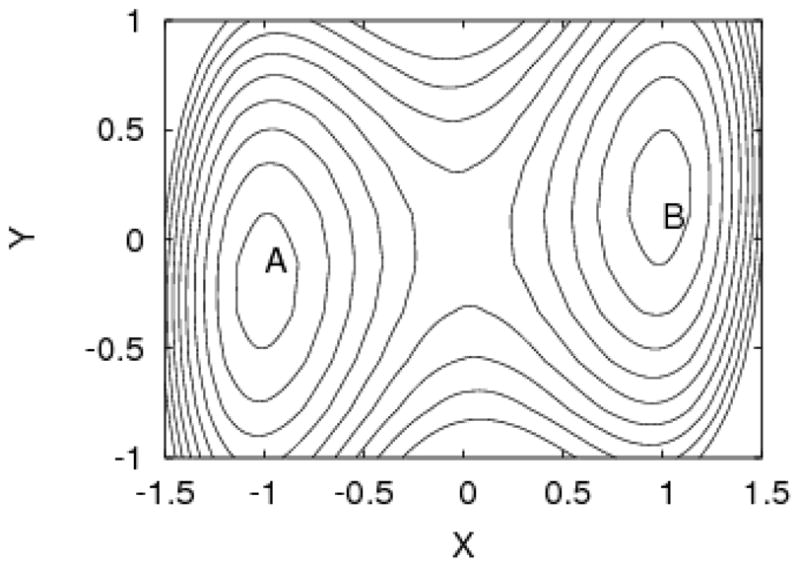
Slow and fast timescales are generally coupled. The plot shows a schematic two-state potential. The y coordinate is fast regardless of whether state A or B is occupied. However, fast oscillations of y are no guarantee of convergence because the motions in x will be much slower. In a molecule, all atoms interact – even if weakly or indirectly – and such coupling must be expected.
Rhodopsin
It is perhaps not surprising that all of the degrees of freedom are tightly coupled in a simple system like butane. It seems reasonable that this coupling may be less important in larger biomolecular systems, where there are motions on time scales ranging from femtoseconds to milliseconds; indeed, it is commonly assumed that small-scale reorganizations, such as side-chain torsions in proteins, can be computed with confidence from molecular dynamics simulations of moderate length. While this assumption is likely true in many cases, divining the cases when it holds can be extremely difficult. As a concrete example, consider the conformation of the retinal ligand in dark-state rhodopsin. The ligand is covalently bound inside the protein via Schiff base linkage to an internal lysine, and contains an aromatic hydrocarbon chain terminated by an ionone ring. This ring packs against a highly conserved tryptophan residue, and is critical to this ligand’s role as an inverse agonist.
The ring’s orientation, relative to the hydrocarbon chain, is largely described by a single torsion, and one might expect that this kind of local quantity would be relatively easy to sample in a molecular dynamics simulation. The quality of sampling for this torsion would also seem easy to assess, because as for most torsions, there are three stable states. However, Figure 3 shows that this is not the case, because of coupling between fast and slow modes. The upper frame of Figure 3 shows a time series of this torsion from a molecular dynamics simulation of dark state rhodopsin 3; the three expected torsional states (g+, g−, and t) are all populated, and there are a number of transitions, so most practitioners would have no hesitation in concluding that a) the trajectory is reasonably well sampled, and b) that all three states are frequently populated, with g− the most likely and trans the least. The middle panel, however, shows the same trajectory extended to 150 ns; it too seems to suggest a clear conclusion, in this case that the transitions in the first 50 ns are part of a slow equilibration, but that once the protein has relaxed the retinal is stable in the g− state. The bottom panel, containing the results of extending the trajectory to 1600 ns, suggests yet another distinct conclusion, that g− and t are the predominant states, and rapidly exchange with each other, on the nanosecond scale. The black line on this graph, showing the time series smoothed using a 10 ns window, seems to suggest that the there is an additional relaxation occurring, with the population of the trans state increasing.
Figure 3.
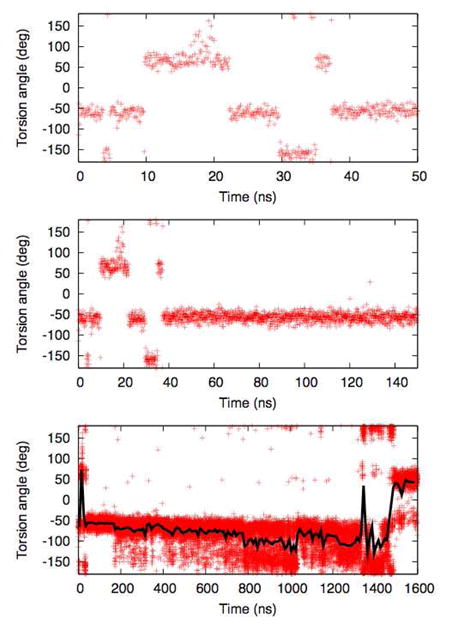
Time series for the torsion connecting the ionone ring to the chain of rhodopsin’s retinal ligand. All three panels show the same trajectory, cut at 50 ns, 150 ns, and 1600 ns, respectively. The black line in the bottom panel shows the data smoothed over a 10 ns window.
These results highlight the difficulties involved in assessing the convergence of single observables. No amount of visual examination of the upper and middle panels would have revealed the insufficiency of the sampling (although it is interesting to note that the ‘effective sample size’ described below is not too large). Rather, it is only after the fact, in light of the full 1600 ns trajectory, that the sampling flaws in the shorter trajectories become obvious. This highlights the importance of considering time scales broadly when designing and interpreting simulations. This retinal torsion is a local degree of freedom, and as such should relax relatively quickly, but the populations of its states are coupled to the conformation of the protein as a whole. As a result, converging the sampling for the retinal requires reasonable sampling of the protein’s internal degrees of freedom, and is thus a far more difficult task than it would first appear.
1.2. Absolute vs. relative convergence
Is it possible to describe a simulation as absolutely converged? From a statistical point of view, we believe the answer is clearly “no”, except in those cases where the correct answer is already known by other means. Whether a simulation employs ordinary MD or a much more sophisticated algorithm, so long as the algorithm correctly yields canonical sampling according to the Boltzmann factor, one can expect the statistical quality will increase with the duration of the simulation. In general, the statistical uncertainty of most conceivable molecular simulation algorithms will decay inversely with the square-root of simulation length. The square-root law should apply once a stochastic simulation process is in the true sampling regime – i.e., once it is long enough to produce multiple properly distributed statistically independent configurations.
The fundamental perspective of this review is that simulation results are not absolute, but rather are intrinsically accompanied by statistical uncertainty 4–8. Although this view is not novel, it is at odds with informal statements that a simulation is “converged.” Beyond quantification of uncertainty for specific observables, we also advocate quantification of overall sampling quality in terms of the “effective sample size” 8 of an equilibrium ensemble 9, 10.
As a conceptual rule-of-thumb, any estimate for the average of an observable which is found to be based on fewer than ~20 statistically independent configurations (or trajectory segments) should be considered unreliable. There are two related reasons for this. First, any estimate of the uncertainty in the average based on a small number of observations will be unreliable because this uncertainty is based on the variance, which converges more slowly than the observable (i.e., average) itself. Secondly, any time the estimated number of statistically independent observations (i.e., effective sample size) is ~20 or less, both the overall sampling quality and the sample-size estimate itself must be considered suspect. This is again because sample-size estimation is based on statistical fluctuations that are, by definition, poorly sampled with so few independent observations.
Details and ‘best practices’ regarding these concepts will be given below.
1.3. Known unknowns
Lack of ergodicity – unvisited regions of configuration space
No method known to the authors can report on a simulation’s failure to visit an important region of configuration space unless these regions are already known in advance. Thus, we instead focus on assessing sampling quality in the regions of space that have been visited. One can hope that the generation of many effectively independent samples in the known regions of configuration space with a correct algorithm is good “insurance” against having missed parts of the space – but certainly it is no guarantee. Larger systems are likely to have more thermodynamically relevant substates, and may thus require more independent samples even in the absence of significant energetic barriers.
Small states rarely visited in dynamical simulation
This issue is also related to ergodicity, and is best understood through an example. Consider a potential like that sketched in Figure 4, with two states of 98% and 2% population at the temperature of interest. A “perfect” simulation capable of generating fully independent configurations according to the associated Boltzmann factor would simply yield 2 of every 100 configurations in the small state, on average. However, a finite dynamical simulation behaves differently. As the barrier between the states gets larger, the frequency of visiting the small state will decrease exponentially. Thus, estimating an average like x will be very difficult – since the small state might contribute appreciably. Further, quantifying the uncertainty could be extremely difficult if there are only a small number of visits to the small state – because the variance will be poorly estimated.
Figure 4.

Cartoon of a landscape for which dynamical simulation is intrinsically difficult to analyze. As the barrier between the states gets higher, the small state requires exponentially more dynamical sampling, even though the population may be inconsequential. It would seem that, in principle, a cutoff should be chosen to eliminate “unimportant” states from analysis. In any complex molecular system, there will always be extremely minor but almost inaccessible basins.
1.4. Non-traditional simulation methods
The preceding discussion applied implicitly to what we classify as dynamical simulations – namely, those simulations in which all correlations in the final trajectory arise because each configuration is somehow generated from the previous one. This time-correlated picture applies to a broad class of algorithms: MD, Langevin and Brownian dynamics, as well as traditional Monte Carlo (MC, also known as Markov-chain Monte Carlo). Even though MC may not lead to true physical dynamics, all the correlations are sequential.
However, in other types of molecular simulation, any sampled configuration may be correlated with configurations not sequential in the ultimate “trajectory” produced. That is, the final result of some simulation algorithms is really a list of configurations, with unknown correlations, and not a true trajectory in the sense of a time series.
One increasingly popular method which leads to non-dynamical trajectories is replica exchange Monte Carlo or molecular dynamics 11–13, which employs parallel simulations at a ladder of temperatures. The “trajectory” at any given temperature includes repeated visits from a number of (physically continuous) trajectories wandering in temperature space. Because the continuous trajectories are correlated in the usual sequential way, their intermittent – i.e., non-sequential – visits to the various specific temperatures produce non-sequential correlations when one of those temperatures is considered as a separate ensemble or “trajectory.”14 Less prominent examples of non-dynamical simulations occur in a broad class of polymer-growth algorithms (e.g., References 15–17).
Because of the rather perverse correlations that occur in non-dynamical methods, there are special challenges in analyzing statistical uncertainties and sampling quality. This issue has not been well explored in the literature; see however 10, 18, 19. We therefore present some tentative thoughts on non-dynamical methods, based primarily on the notion that independent simulations appear to provide the most definitive means for analyzing non-dynamical simulations. In the case of replica exchange, understanding the difference between “mixing” and sampling will prove critical to any analysis.
2. Error estimation in single observables
One of the main goals of biomolecular simulation is the estimation of ensemble averages, which should always be qualified by estimates of statistical uncertainty. We will review the two main approaches to estimating uncertainty in averages, but a general note of caution should be repeated. Because all variables can be correlated in a complex system, the so-called “fast” variables may not be as fast as they appear based on standard error estimation techniques: see Figure 2. As in the examples of the rhodopsin dihedral, above, even a single coordinate undergoing several transitions may not be well sampled. Also, investigators should be wary of judging overall sampling quality based on a small number of observables unless they are specifically designed to measure ensemble quality, as discussed below.
The present discussion will consider an arbitrary observable f, which is a function of the configuration x of the system being simulated. The function f(x) could represent a complex measure of an entire macromolecule, such as the radius of gyration, or it could be as simple as a single dihedral or distance.
Our focus will be on time correlations and block averaging. The correlation-time analysis has been in use for some decades 7, including to analyze the first protein MD simulation 20, and it embodies the essence of all the single-observable analyses known to the authors. The block averaging approach 5, 21 is explicitly described below because of its relative simplicity and directness in estimating error using simple variance calculations. Block averaging “short cuts” the need to calculate a correlation time explicitly, although timescales can be inferred from the results. Similarly, the “ergodic measure” of Thirumalai and coworkers 22–24, not described here, uses variances and correlation times implicitly.
Both the correlation time analysis and the block averaging scheme described below assume that a dynamical trajectory is being analyzed. Again, by “dynamical” we only mean that correlations are “transmitted” via sequential configurations – which is not true in a method like replica exchange.
2.1 Correlation time analysis
The correlation-time analysis of a single observable has a very intuitive underpinning. Consider first that dynamical simulations (e.g., molecular and Langevin dynamics), as well as ‘quasi-dynamical’ simulations (e.g., typical Monte Carlo 25), create trajectories that are correlated solely based on the sequence of the configurations. As described in the Appendix, the correlation time τf measures the length of simulation time – whether for physical dynamics or Monte Carlo – required for the trajectory to lose “memory” of earlier values of f. Therefore, the correlation time τf for the specific observable f provides a basis for estimating the number of statistically independent values of f present in a simulation of length tsim, namely . By itself, would suggest good sampling for the particular observable f.
The correlation time is computed from the correlation function (see Appendix), and it is useful to consider an example. Figure 5 shows the time correlation functions computed for individual state lifetimes as measured by a 100 ns simulation of butane. Specifically, for each snapshot from the trajectory, the central torsion was classified as trans, g+, or g−. A time series was then written for each state, with a value of 1 if the system was in that state and 0 otherwise. The autocorrelation functions for each of those time series are shown in Figure 5. All three correlation functions drop smoothly to zero within 200 ps, suggesting that a 100 ns simulation should contain a very large number of independent samples. However, the populations for the 3 states over the course of the trajectory are 0.78, 0.10, and 0.13 for the trans, g+, and g− states, respectively. The g+ and g− states are physically identical, and thus should have the same populations in the limit of perfect sampling. Thus even a very long simulation of a very simple system is incapable of estimating populations with high precision.
Figure 5.
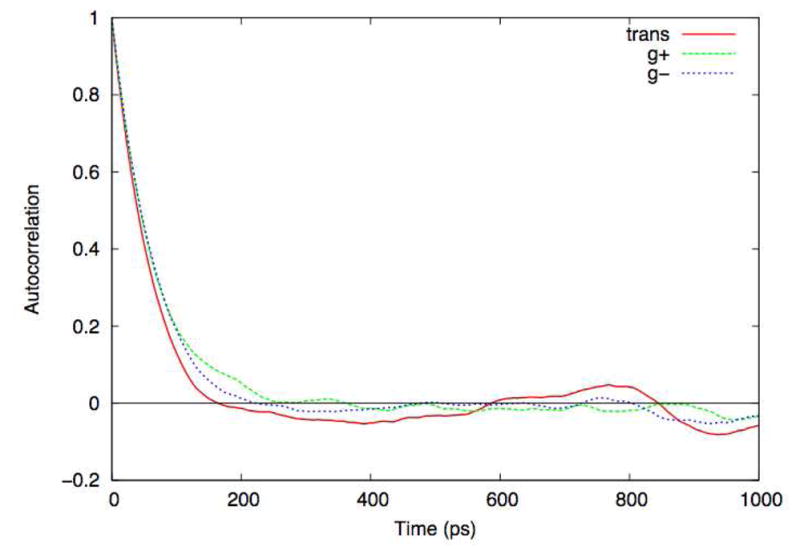
State autocorrelations computed from 100 ns butane simulations. The central torsion was labeled as either trans, g+, or g−, and the autocorrelation function for presence in each state was computed.
To obtain an estimate of the statistical uncertainty in an average 〈f〉, the correlation time τf must be used in conjunction with the variance (square of the standard deviation; see Appendix) of the observable. By itself, the standard deviation only gives the basic scale or range of fluctuations in f, which might be much larger than the uncertainty of the average 〈f〉. In other words, it is possible to know very precisely the average of a quantity that fluctuates a lot: as an extreme example, imagine measuring the average height of buildings in Manhattan. In a dynamical trajectory, the correlation time τf provides the link between the range of fluctuations and the precision (uncertainty) in an average, which is quantified by the standard error of the mean, SE,
| (1) |
In this notation, is the number of independent samples contained in the trajectory, tsim and is the length of the trajectory. The standard error can be used to approximate confidence intervals, with a rule of thumb being that ±2SE represents roughly a 95% confidence interval 26. The actual interval depends on the underlying distribution and the sampling quality as embodied in .; see reference 25 for a more careful discussion.
It has been observed that the simple relation between correlation time and sampling quality embodied in the estimate is actually too conservative in typical cases 27. That is, even though the simulation may require a time τf to “forget” its past (with respect to the observable f), additional information beyond a single estimate for f is obtained in the period of a single correlation time – i.e., from partially correlated configurations. However, the improvement in sampling quality is modest – the effective sample size may be double the estimate based simply on τf. Such subtleties are accounted for automatically in the block-averaging analysis described below.
Understanding the correlation time analysis, as well as the habitual calculation of correlation functions and times, is extremely useful. Yet the analysis has weaknesses for quantifying uncertainty that suggest relying on other approaches for generating publication-quality error bars. First, like any single-observable analysis, the estimation of correlation times may fail to account for slow timescales in observables not considered: recall the rhodopsin example. Second, the calculation of correlation times becomes less reliable in precisely those situations of greatest interest – when a second, slower timescale enters the intrinsically noisier tail of the correlation function. The third weakness was already described: a lack of full accounting for all statistical information in the trajectory. These latter considerations suggest that a block-averaging procedure, described next, is a preferable analysis of a single observable.
2.2 Block averaging
When executed properly, the block-averaging analysis automatically corrects two of the weaknesses in correlation-time estimates of the error based on Eq. (1). In particular, any slow timescales present in the time series for the particular observable are accounted for (although only in regard to the observable studied). Second, because block averaging uses the full trajectory, it naturally includes all the information present. The block averaging analysis was first reported by Flyvbjerg and Petersen 5, who credited the previously un-published idea to others.
The approach can be described simply (although it is not easily understood from the original reference). A trajectory with N = M · n snapshots is divided into M segments (“blocks”), with an initial very short block length, such as n =1 (See Fig 6). The average of the observable is calculated for each block yielding M values for 〈f〉i, with i =1, …, M. The block length n is gradually increased and the set of block averages is recalculated for each length. Further, for each value of n, the standard deviation among the block averages, σn, is used to calculate a running estimate of the overall standard error, namely,
Figure 6.
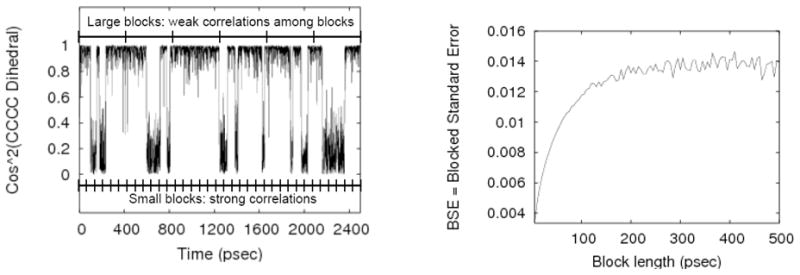
The block averaging procedure considers a full range of block sizes. Small blocks will tend to be highly correlated with neighboring blocks, whereas blocks longer than important correlation times will only be weakly correlated. The block averaging analysis implicitly detects such correlations in the BSE plot at right. The true statistical uncertainty (standard error) is obtained when BSE reaches a plateau in the right plot, reflecting essentially independent blocks. Note that the correlation time of ~100–200 psec essentially can be “read” from the BSE plot. The BSE plot was calculated from 50 nsec of data.
| (2) |
This is the standard error in estimates of the mean based on blocks (trajectory segments) of length n. Clearly, for small n (and large M = N/n) when consecutive blocks are highly correlated, BSE greatly under-estimates the statistical error, since Eq. (2) only yields the true standard error when all M blocks are statistically independent. On the other hand, once the blocks are essentially independent of one another, (i.e., when the block length is substantially greater than the correlation time, n ≫ τ f/Δt), BSE will cease to vary with n and become a reliable estimator of the true SE. . Fig. 6 illustrates this behavior for a trigonometric function of butane’s main (C-C-C-C) dihedral.
The function BSE(f, n) therefore increases monotonically with n and asymptotes to the true standard error associated with 〈f〉, as seen in Fig. 6. Thus, a plot of BSE(f, n) includes a “signal” as to whether or not the error estimate has converged, which is not subject to the extremes of numerical uncertainty associated with the tail of a correlation function. Furthermore, the block-averaging analysis directly includes all trajectory information (all frames).
The only weakness of the block-averaging approach, which is minor in our opinion, is that it does not directly render the correlation time. Having the correlation time in hand provides important physical intuition. Nevertheless, we note that the correlation time can be estimated cleanly using the block averaging results. Specifically, using the trajectory f (t), one can directly calculate the variance σf and then solve for using Eq. (1). The correlation time is then given approximately by , which will somewhat under-estimate the correlation time (as noted implicitly by Harris and Berg 27) perhaps by a factor of ~2.
It is not uncommon for researchers to use the name “block averaging” to describe a second, far simpler procedure. In this case, a single time series is split into M blocks, and the variance between the averages for those blocks is presented as the uncertainty. However, unlike the true block averaging protocol described above, this procedure is not statistically meaningful, because the single block size is chosen arbitrarily; it is only by systematically varying the block size that one can reliably draw conclusions about the uncertainty.
2.3 Summary – Single Observables in dynamical simulations
Several points are worth emphasizing: (i) Single observables should not be used to assess overall sampling quality. (ii) The central ideas in single-observable analysis are that the correlation time separates statistically independent values of the observable, and that one would like to have many statistically independent “measurements” of the observable – i.e., . (iii) The block-averaging analysis is simple to implement and provides direct estimation of statistical uncertainty. We recommend that the correlation time and effective sample size also be estimated to ensure . (iv) In a correlation time analysis, one wants to ensure the total simulation time is a large multiple of the correlation time – i.e., tsim/τf ≫1.
2.4. Analyzing single observables in non-dynamical simulations
As discussed earlier, the essential fact about data from non-dynamical simulations (e.g., replica exchange and polymer growth methods) is that a configuration occurring at one point in the “trajectory” may be highly correlated with another configuration anywhere else in the final list of configurations. Similarly, a configuration could be fully independent of the immediately preceding or subsequent configurations. To put it most simply, the list of configurations produced by such methods is not a time series, and so analyses based on the explicit or implicit notion of a correlation time (time correlations are implicit in block averaging) cannot be used.
From this point of view, the only truly valid analysis of statistical errors can be obtained by considering independent simulations. Ideally, such simulations would be started from different initial conditions to reveal “trapping” (failure to explore important configurational regions) more readily. Running multiple simulations appears burdensome, but it is better than excusing “advanced” algorithms from appropriate scrutiny. Of course, rather than multiplying the investment in computer time, the available computational resources can be divided into 10 or 20 parts. All these parts, after all, are combined in the final estimates of observable averages. Running independent trajectories is an example of an “embarrassingly parallel” procedure, which is often the most efficient use of a standard computer cluster. Moreover, if a simulation method is not exploring configuration space well in a tenth of the total run time, then it probably is not performing good sampling anyway.
How can statistical error be estimated for a single observable from independent simulations? There seems little choice but to calculate the standard error in the mean values estimated from each simulation using Eq. (1), where the variance is computed among the averages from the independent simulations and is set to the number of simulations. In essence, each simulation is treated as a single measurement, and presumed to be totally independent of the other trajectories. Importantly, one can perform a “reality check” on such a calculation because the variance of the observable can also be calculated from all data from all simulations – rather than from the simulation means. The squared ratio of this absolute variance to the variance of the means yields a separate (albeit crude) estimate of the number of independent samples. This latter estimate should be of the same order as, or greater than, the number of independent simulations, indicating that each “independent” simulation indeed contained at least one statistically independent sample of the observable.
It is interesting to observe that, in replica exchange simulations, the physically continuous trajectories (which wander in temperature) can be analyzed based on time-correlation principles 10, 14. Although each samples a non-traditional ensemble, it is statistically well defined and can be used as a proxy for the regular ensemble. A more careful analysis could consider, separately, those segments of each continuous trajectory at the temperature of interest. The standard error among these estimates could be compared to the true variance, as above, to estimate sampling quality. A detailed discussion of these issues in the context of weighted histogram analysis of replica exchange simulation is given by Chodera et al.14.
3. Overall Sampling Quality in Simulations
In contrast to measures of convergence that reflect a single local observable, e.g. a torsion angle, some methods focus on the global sampling quality. For a simulation of a macromolecule, the distinction would be between asking “how well do I know this particular quantity?” and “how well have I explored the conformational space of the molecule?” The latter question is critical, in that if the conformational space is well sampled, most physical quantities should be known well.
This review will describe two classes of analyses of overall sampling quality: (i) qualitative and visual techniques, which are mainly useful in convincing oneself a simulation is not sufficiently sampled; and (ii) quantitative analyses of sampling, which estimate the “effective sample size.”
3.1 Qualitative and Visual Analyses of Overall Sampling Effectiveness
There are a number of techniques that, although they cannot quantitatively assess convergence or statistical uncertainty, can give tremendous qualitative insight. While they cannot tell the user that the simulation has run long enough, they can quickly suggest that the simulation has not run long enough. Thus, while they should not replace more rigorous methods like block averaging and sample-size estimation, they are quite useful.
Scalar RMSD Analyses
One of the simplest methods is the comparison of the initial structure of the macromolecule to that throughout the trajectory via a distance measure such as the root mean square deviation (RMSD). This method is most informative for a system like a folded protein under native conditions, where the molecule is expected to spend the vast majority of the time in conformations quite similar to the crystal structure. If one computes the RMSD time series against the crystal structure, one expects to see a rapid rise due to thermal fluctuations, followed by a long plateau or fluctuations about a mean at longer time scales. If the RMSD time series does not reach a steady state, the simulation is either a) still equilibrating, or b) drifting away from the starting structure. In any event, until the system assumes a steady-state value – one that may fluctuate significantly, but has no significant trend – the system is clearly not converged. Indeed, one can argue that under that circumstance equilibrium sampling has not yet even begun. However, beyond this simple assessment, RMSD is of limited utility, mostly because it contains little information about what states are being sampled; a given RMSD value maps a 3N-dimensional hypersphere of conformation space (for N atoms) to a single scalar, and for all but the smallest RMSD values this hypersphere contains a broad range of structures. Moreover, the limiting value for the RMSD cannot be known in advance. We know the value should be non-zero and not large, but the expected plateau value is specific to the system studied, and will vary not only between macromolecules, but also with changes to simulation conditions such as temperature and solvent.
An improvement is to use a windowed RMSD function as a measure of the rate of conformation change. Specifically, for a given window length (e.g., 10 consecutive trajectory snapshots), the average of the all of the pairwise RMSDs (or alternatively, the average deviation from the average over that interval) is computed as a function of time. This yields a measure of conformational diversity over time, and can more readily reveal conformational transitions.
All-to-all RMSD Analysis
A more powerful technique is to compute the RMSDs for all pairs of snapshots from the trajectory and plot them on a single graph28. Figure 7 shows the results of such a plot, made using the alpha-carbon RMSD computed from a 1.6 μs all-atom simulation of dark state rhodopsin in an explicit lipid membrane3. The plot reveals a hierarchical block structure along the diagonal; this suggests that the protein typically samples within a substate for a few hundred nanoseconds, and then rapidly transitions to a new conformational well. However, with the exception of two brief excursions occurring around 280 ns and 1150 ns into the trajectory, the system never appears to leave and then return to a given substate. This suggests that this simulation, although very long by current standards, probably has not fully converged.
Figure 7. All-to-all RMSD for rhodopsin alpha-carbons.
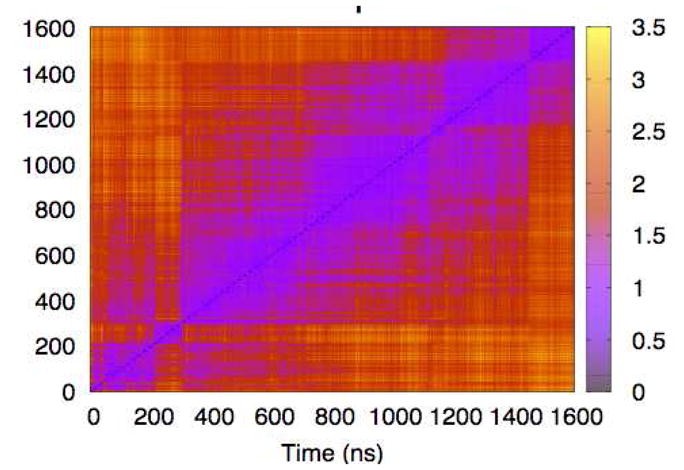
Using the color scale at right, this matrix visually indicates time intervals over which configurations are similar – i.e., blue-violet in color.
Cluster counting
A more general approach, also based on pairwise distances, would be to use cluster analysis. Although a general discussion of the many clustering algorithms presently in use is beyond the scope of this manuscript, for our purposes we define clustering to be any algorithm that divides an ensemble into sets of self-similar structures. One application of clustering to the assessment of convergence came from Daura et al, who measured the rate of discovery of new clusters over the course of a trajectory; when this rate became very small, the simulation was presumed to be reasonably well converged29. However, a closer look reveals this criterion to be necessary but not sufficient to guarantee good sampling. While it is true that a simulation that is still exploring new states is unlikely to have achieved good statistics (at least for a reasonable definition of “states”), simply having visited most of the thermodynamically relevant states is no guarantee that a simulation will produce accurate estimates of observables.
“Structural histogram” of clusters
As discussed by Lyman and Zuckerman9, not only must clusters be visited, but it is important that the populations of those regions be accurately reproduced, since the latter provide the weights used to compute thermodynamic averages. In a procedure building on this idea, one begins by performing a cluster analysis on the entire trajectory to generate a vocabulary of clusters or bins. The cluster/bin populations can be arrayed as a one-dimensional “structural histogram” reflecting the full configuration-space distribution. Structural histograms from parts of the trajectory are compared to one computed for the full trajectory, and plotting on a log-scale gives the variation in kT units, indicating the degree of convergence.
Principal components analysis
Principal component analysis (PCA) is another tool that has been used extensively to analyze molecular simulations. The technique, which attempts to extract the large scale characteristic motions from a structural ensemble, was first applied to biomolecular simulations by Garcia30, although an analogous technique was used by Levy et al.31. The first step is the construction of the 3N × 3N (for an N-atom system) fluctuation correlation matrix
where xi represents the a specific degree of freedom (e.g. the z-coordinate of the 23rd atom) and the overbar indicates the average structure. This task is commonly simplified by using a subset of the atoms from the molecule of interest (e.g. the α-carbons from the protein backbone). The matrix is then diagonalized to produce the eigenvalues and eigenvectors; the eigenvectors represent characteristic motions for the system, while each eigenvalue is the mean square fluctuation along its corresponding vector. The system fluctuations can then be projected onto the eigenvectors, giving a new time series in this alternative basis set. The diagonalization and time series projection can be performed efficiently using singular value decomposition, first applied to principal component analysis of biomolecular fluctuations by Romo et al. 32.
In biomolecular systems characterized by fluctuations around a single structure (e.g. equilibrium dynamics of a folded protein), a small number of modes frequently account for the vast majority of the motion. As a result, the system’s motions can be readily visualized, albeit abstractly, by plotting the time series for the projections of the first two or three modes. For example, projecting the rhodopsin trajectory described above3 onto its two largest principle modes yields Figure 8. As with the all-to-all RMSD plots (see Figure 7), this method readily reveals existence of a number of substates, although temporal information is obscured. A well-sampled simulation would exhibit a large number of transitions among substates, and the absence of significant transitions can readily be visualized by plotting principal components against time. It is important to note that this method does not depend on the physical significance or statistical convergence of the eigenvectors themselves, which is reassuring because previous work has shown that these vectors can be extremely slow to converge.1, 33 Rather, for these purposes the modes serve as a convenient coordinate system for viewing the motions.
Figure 8.
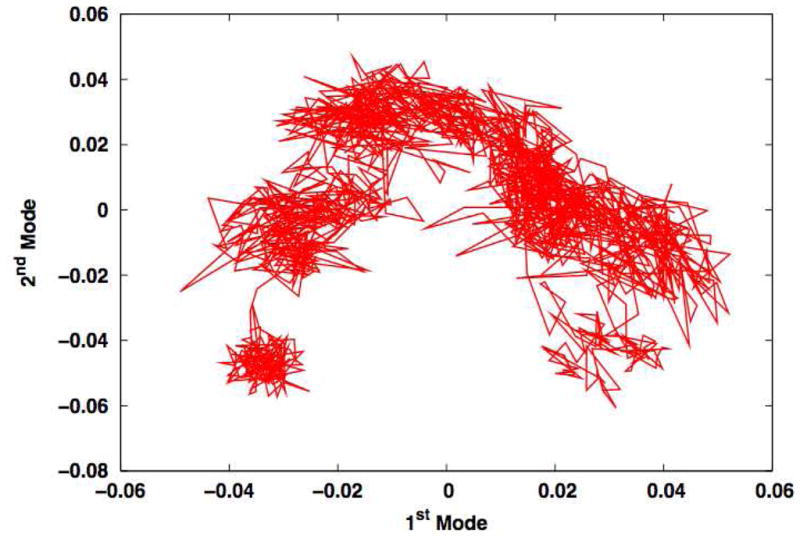
Projection of rhodopsin fluctuations onto the first two modes derived from principal component analysis. As with Figure 7, this method directly visualizes substates in the trajectory.
PCA can also be used to quantify the degree of similarity in the fluctuations of two trajectories (or two portions of a single trajectory). The most rigorous measure is the covariance overlap suggested by Hess 1, 34, 35
which compares the eigenvalues λ and eigenvectors v computed from two datasets A and B. The overlap ranges from 0, in the case where the fluctuations are totally dissimilar, to 1, where the fluctuation spaces are identical. Physically, the overlap is in essence the sum of all the squared dot products of all pairs of eigenvectors from the two simulations, weighted by the magnitudes of their displacements (the eigenvalues) and normalized to go from 0 to 1. Hess used this quantity as an internal measure of convergence, comparing the modes computed from subsets of a single trajectory to that computed from the whole35. More recently, Grossfield et al. computed the principal components from 26 independent 100 ns simulations of rhodopsin, and used the covariance overlap to quantify the similarity of their fluctuations, concluding that 100 ns is not sufficient to converge the fluctuations of even individual loops1. Although these simulations are not truly independent (they used the same starting structure for the protein, albeit with different coordinates for the lipids and water), the results again reinforce the point that the best way to assess convergence is through multiple repetitions of the same system.
3.2 Quantifying Overall Sampling Quality: The Effective Sample Size
To begin to think about the quantification of overall sampling quality – i.e., the quality of the equilibrium ensemble – it is useful to consider “ideal sampling” as a reference point. In the ideal case, we can imagine having a perfect computer program which outputs single configurations drawn completely at random and distributed according the appropriate Boltzmann factor for the system of interest. Each configuration is fully independent of all others generated by this ideal machinery, and is termed “i.i.d.” – independent and identically distributed.
Thus, given an ensemble generated by a particular (non-ideal) simulation, possibly consisting of a great many “snapshots,” the key conceptual question is: To how many i.i.d. configurations is the ensemble equivalent in statistical quality? The answer is the effective sample size 1, 9, 10 which will quantify the statistical uncertainty in every slow observable of interest – and many “fast” observables also, due to coupling, as described earlier.
The key practical question is: How can the sample size be quantified? Initial approaches to answering this question were provided by Grossfield and coworkers 1 and by Lyman and Zuckerman 10. Grossfield et al. employed a bootstrap analysis to a set of 26 independent trajectories for rhodopsin, extending the previous “structural histogram” cluster analysis 10 into a procedure for estimating sample size. They compared the variance in a cluster’s population from the independent simulations to that computed using a bootstrap analysis (bootstrapping is a technique where a number of artificial data sets are generated by choosing points randomly from an existing data set 36). Because each data point in the artificial data sets is truly independent, comparison of the bootstrap and observed variances yielded estimates of the number of independent data points (i.e., effective sample size) per trajectory. The results were astonishingly small, with estimates ranging from 2–10 independent points, depending on the portion of the protein examined. Some of the numerical uncertainties in the approach may be improved by considering physical states rather than somewhat arbitrary clusters; see below.
Lyman and Zuckerman suggested a related method for estimating sample size 10. Firstly, they pointed out that binomial and related statistics provided an analytical means for estimating sample size from cluster-population variances, instead of the bootstrap approach. Secondly, they proposed an alternative analysis specific to dynamical trajectories, but which also relied on comparing observed and ideal variances. In particular, by generating observed variances from “frames” in a dynamical trajectory separated by a fixed amount of time, it can be determined whether those time-separated frames are statistically independent. The separation time is gradually increased until ideal statistics are obtained, indicating independence. The authors denoted the minimum time for independence the “structural decorrelation time” to emphasize that the full configuration-space ensemble was analyzed based on the initial clustering/binning.
Looking to the future: Can state populations provide a “universal indicator”?
The ultimate goal for sample size assessment (and thus estimation of statistical error) is a “universal” analysis, which could be applied blindly to dynamical or non-dynamical simulations and reveal the effective size. Current work in the Zuckerman group (unpublished) suggests a strong candidate for a universal indicator of sample size is the variance observed from independent simulations in the populations of physical states. Physical states are to be distinguished from the more arbitrary clusters discussed above, in that a state is characterized by relatively fast timescales internally, but slow timescales for transitions between states. (Note that proximity by RMSD or similar distances does not indicate either of these properties.) There are two reasons to focus on populations of physical states: (i) the state populations arguably are the fundamental description of the equilibrium ensemble, especially considering that (ii) as explained below, relative state populations cannot be accurate unless detailed sampling within states is correct. Of course, determining physical states is non-trivial but apparently surmountable 37.
We claim that if you know state populations, you have sampled well – at least in an equilibrium sense. Put another way, we believe it is impossible to devise an algorithm – dynamical or non-dynamical – that could correctly sample state populations without sampling correctly within states. The reason is that the ratio of populations of any pair of states depends on the ensembles internal to the states. This ratio is governed/defined by the ratio of partition functions for the states, i and j, which encompass the non-overlapping configuration-space volumes Vi and V j, namely,
| (3) |
This ratio cannot be estimated without sampling within both states – or effectively doing so 38, 39. Note that this argument does not assume that sampling is performed dynamically.
If indeed the basic goal of equilibrium sampling is to estimate state populations, then these populations can act as the fundamental observables amenable to the types of analyses already described. In practical terms, following 10, a binomial description of any given state permits the effective sample size to be estimated from the populations of the state recorded in independent simulations – or from effectively independent segments of a sufficiently long trajectory. This approached will be described shortly in a publication.
One algorithm for blindly approximating physical states has already been proposed 37, although the method requires the number of states to be input. In work to be reported soon, Zhang and Zuckerman developed a simple procedure for approximating physical states that does not require input of the number of states. In several systems, moreover, it was found that sample-size estimation is relatively insensitive to the precise state definitions (providing they are reasonably physical, in terms of the timescale discussion above). The authors are therefore optimistic that a “benchmark” blind, automated method for sample-size characterization will be available before long.
3.3 Analyzing non-standard simulations – e.g., replica exchange
The essential intuition regarding non-standard/non-dynamical simulations such as replica exchange has been given in our discussion of single observables: in brief, a given configuration in a “trajectory” may be highly correlated with much “later” configurations, yet not correlated with intervening configurations. Therefore, a reliable analysis must be based multiple independent simulations – which is perhaps less burdensome than it first seems, as discussed above.
We believe such simulations should be analyzed using state-population variances. This approach, after all, is insensitive to the origins of the analyzed “trajectories” and any internal time correlations or lack thereof. No method that relies explicitly or implicitly on time correlations would be appropriate.
Replica exchange simulations, because of their growing popularity, merit special attention. While their efficacy has been questioned recently19, 40, our purpose here is solely to describe appropriate analyses. To this end, a clear distinction must be drawn between “mixing” (accepted exchanges) and true sampling. While mixing is necessary for replica exchange to be more efficient than standard dynamics (otherwise each temperature is independent), mixing in no way suggests good sampling has been performed. This can be clearly appreciated from a simple “thought experiment” of a two-temperature replica exchange simulation of the double square well potential of Figure 9. Assume the two replicas have been initiated from different states. Because the states are exactly equal in energy, every exchange will be accepted. Yet if the barrier between the states is high enough, no transitions will occur in either of the physically continuous trajectories. In such a scenario, replica exchange will artifactually predict 50% occupancy of each state. A block averaging or time-correlation analysis of a single temperature will not diagnose the problem. As suggested in the single-observable discussion, some information on under-sampling may be gleaned from examining the physically continuous trajectories. The most reliable information, however, will be obtained by comparing multiple independent simulations; Sec. III.D explains why this is cost efficient.
Figure 9.
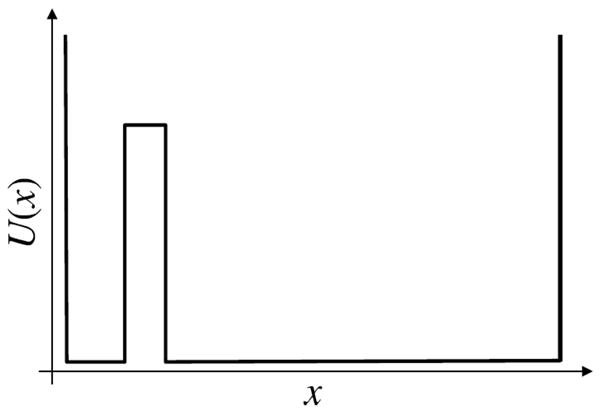
A cartoon of two states differing only in entropy. Generally, in any simulation, energetic effects are much easier to handle than entropic. The text describes the challenge of analyzing errors in replica-exchange simulations when only entropy distinguishes two energetically equal states.
4. Recommendations
General. When possible, perform multiple simulations, making the starting conformations as independent as possible. This is recommended regardless of the sampling technique used.
Single observables. Block averaging is a simple, relatively robust procedure for estimating statistical uncertainty. Visual and correlation analyses should also be performed.
Overall sampling quality – heuristic analysis. If the system of interest can be thought of as fluctuating about one primary structure (e.g., a native protein), use qualitative tools, such as projections onto a small number of PCA modes or all-to-all RMSD plots to simplify visualization of trajectory quality. Such heuristic analyses can readily identify undersampling as a small number of transitions.
Overall sampling quality – quantitative analysis. For dynamical trajectories, the “structural decorrelation time” analysis 10 can estimate the slowest timescale affecting significant configuration-space populations and hence yield the effective sample size. For non-dynamical simulations, a variance analysis based on multiple on runs is called for. Analyzing the variance in populations of approximate physical states appears to be promising as a benchmark metric.
General. No amount of analysis can rescue an insufficiently sampled simulation. A smaller system or simplified model that has been sampled well may be more valuable than large detailed model with poor statistics.
Acknowledgments
D.M.Z. would like to acknowledge in-depth conversations with Edward Lyman and Xin Zhang, as well as assistance in preparing the figures. Insightful discussions were also held with Divesh Bhatt, Ying Ding, Artem Mamonov, and Bin Zhang. Support for D.M.Z. was provided by the NIH (Grants GM076569 and GM070987) and the NSF (Grant MCB-0643456). A.G. would like to thank Tod Romo for helpful conversations and assistance in figure preparation.
Appendix
For reference, we provide brief definitions and discussions of basic statistical quantities: the mean, variance, auto-correlation function, and auto-correlation time.
Mean
The mean is simply the average of a distribution, which accounts for the relative probabilities of different values. If a simulation produces a correct distribution of values of the observable f, then relative probabilities are accounted for in the set of N values sampled. Thus the mean 〈f〉 is estimated via
| (4) |
where fi is the ith value recorded in the simulation.
Variance
The variance of a quantity f, which is variously denoted by , var(f), or σ2 (f), measures the intrinsic range of fluctuations in a system. Given N properly distributed samples of f, the variance is defined as the average squared deviation from the mean:
| (5) |
The factor of N −1 in the denominator reflects that the mean is computed from the samples, rather than supplied externally, and one degree of freedom is effectively removed.
The square root of the variance, the standard deviation, σ f, thus quantifies the width or spread in the distribution; it has the same units as f itself, unlike the variance. Except in specialized analyses (such a block averaging) the variance does not quantify error. As an example, the heights of college students can have a broad range – i.e., large variance – while the average height can be known with an error much smaller than the standard deviation.
Auto-correlation function
The auto-correlation function quantifies, on a unit scale, the degree to which a quantity is correlated with values of the same quantity at later times. The function can be meaningfully calculated for any dynamical simulation, in the sense defined earlier, and therefore including Monte Carlo. We must consider a set of time-ordered values of the observable of interest, so that fj = f(t = jΔt), with j =1, 2, …, N and Δt the time step between frames. (For MC simulations, one can simply set Δt ≡ 1). The average amount of auto-correlation between “snapshots” separated by a time t′ is quantified by
| (6) |
where the sum must prevent the argument of the second f from extending beyond N. Note that for t′ = 0, the numerator is equal to the variance, and the correlation is maximal at the value cf (0) = 1. As t′ increases significantly, for any given j, the later values of f are as likely to be above the mean as below it – independent of fj since the later values have no “memory” of the earlier value. Thus, the correlation function begins at one and decays to zero for long enough times. It is possible for cf to become negative at intermediate times – which suggests a kind of oscillation of the values of f.
Correlation time
The (auto-)correlation time τf quantifies the amount of time necessary for simulated (or even experimental) values of f to lose their “memory” of earlier values. In terms of the auto-correlation function, we can say roughly that the correlation time is smallest t′ value for which cf (t′) ≪ 1 for all subsequent times (within noise). More quantitatively, the correlation time can be defined via
| (7) |
where the numerical integration must be handled carefully due to the noise in the long-time tail of the correlation function. More approximately, the correlation time can be fit to a presumed functional form, such as an exponential or a sum of exponentials, although it is not necessarily easy to pre-determine the appropriate form 41.
Contributor Information
Alan Grossfield, Email: alan_grossfield@urmc.rochester.edu.
Daniel M. Zuckerman, Email: ddmmzz@pitt.edu.
Bibliography
- 1.Grossfield A, Feller SE, Pitman MC. Proteins-Structure Function and Bioinformatics. 2007;67:31–40. doi: 10.1002/prot.21308. [DOI] [PubMed] [Google Scholar]
- 2.Shirts MR, Pande VS. J Chem Phys. 2005:122. doi: 10.1063/1.1873592. [DOI] [PubMed] [Google Scholar]
- 3.Grossfield A, Pitman MC, Feller SE, Soubias O, Gawrisch K. J Mol Biol. 2008;381:478–486. doi: 10.1016/j.jmb.2008.05.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Binder K, Heermann DW. Editon edn. Berlin: 1988. [Google Scholar]
- 5.Flyvbjerg H, Petersen HG. The Journal of Chemical Physics. 1989;91:461–466. [Google Scholar]
- 6.Ferrenberg AM, Landau DP, Binder K. J Stat Phys. 1991;63:867–882. [Google Scholar]
- 7.Binder K, Heermann DW. Monte Carlo Simulation in Statistical Physics: An Introduction. 3. Springer; Berlin: 1997. [Google Scholar]
- 8.Janke W. Quantum simulations of complex many-body systems: From theory to algorithms. In: Grotendorst J, Marx D, Muramatsu A, editors. John von Neumann Institute for Computing. Vol. 10. Julich: Editon edn; 2002. pp. 423–445. [Google Scholar]
- 9.Lyman E, Zuckerman DM. Biophys J. 2006;91:164–172. doi: 10.1529/biophysj.106.082941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lyman E, Zuckerman DM. J Phys Chem B. 2007;111:12876–12882. doi: 10.1021/jp073061t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Swendsen RH, Wang JS. Physical Review Letters. 1986;57:2607. doi: 10.1103/PhysRevLett.57.2607. [DOI] [PubMed] [Google Scholar]
- 12.Geyer CJ. Proceedings of the 23rd symposium on the interface; 1991. [Google Scholar]
- 13.Sugita Y, Okamoto Y. Chemical Physics Letters. 1999;314:141–151. [Google Scholar]
- 14.Chodera JD, Swope WC, Pitera JW, Seok C, Dill KA. J Chem Theory Comput. 2007;3:26–41. doi: 10.1021/ct0502864. [DOI] [PubMed] [Google Scholar]
- 15.Wall FT, Erpenbeck JJ. The Journal of Chemical Physics. 1959;30:634–637. [Google Scholar]
- 16.Grassberger P. Physical Review E (Statistical Physics, Plasmas, Fluids, and Related Interdisciplinary Topics) 1997;56:3682–3693. doi: 10.1103/physreve.53.3682. [DOI] [PubMed] [Google Scholar]
- 17.Liu JS. Monte Carlo Strategies in Scientific Computing. Springer; New York: 2002. [Google Scholar]
- 18.Denschlag R, Lingenheil M, Tavan P. Chemical Physics Letters. 2008;458:244–248. [Google Scholar]
- 19.Nymeyer H. J Chem Theory Comput. 2008;4:626–636. doi: 10.1021/ct7003337. [DOI] [PubMed] [Google Scholar]
- 20.McCammon JA, Gelin BR, Karplus M. Nature. 1977;267:585–590. doi: 10.1038/267585a0. [DOI] [PubMed] [Google Scholar]
- 21.Kent DR, Muller RP, Anderson AG, Goddard WA, Feldmann MT. Journal of Computational Chemistry. 2007;28:2309–2316. doi: 10.1002/jcc.20746. [DOI] [PubMed] [Google Scholar]
- 22.Mountain RD, Thirumalai D. J Phys Chem. 1989;93:6975–6979. [Google Scholar]
- 23.Thirumalai D, Mountain RD. Physical Review A. 1990;42:4574. doi: 10.1103/physreva.42.4574. [DOI] [PubMed] [Google Scholar]
- 24.Mountain RD, Thirumalai D. Physica A. 1994;210:453–460. [Google Scholar]
- 25.Berg BA. Markov chain Monte Carlo simulations and their statistical analysis. World Scientific; New Jersey: 2004. [Google Scholar]
- 26.Spiegel MR, Schiller J, Srinivasan RA. Schaum’s outline of probability and statistics. 2. McGraw Hill; New York: 2000. [Google Scholar]
- 27.Berg BA, Harris RC. Computer Physics Communications. 2008;179:443–448. [Google Scholar]
- 28.Garcia AE. Phys Rev Lett. Vol. 68. Editon edn; 1992. pp. 2696–2699. [DOI] [PubMed] [Google Scholar]
- 29.Smith LJ, Daura X, Gunsteren WFv. Proteins. 2002;48:487–496. doi: 10.1002/prot.10144. [DOI] [PubMed] [Google Scholar]
- 30.Garcia AE. Phys Rev Lett. 1992;68:2696–2699. doi: 10.1103/PhysRevLett.68.2696. [DOI] [PubMed] [Google Scholar]
- 31.Levy RM, Srinivasan AR, Olson WK, Mccammon JA. Biopolymers. 1984;23:1099–1112. doi: 10.1002/bip.360230610. [DOI] [PubMed] [Google Scholar]
- 32.Romo TD, Clarage JB, Sorensen DC, Phillips GN. Proteins. 1995;22:311–321. doi: 10.1002/prot.340220403. [DOI] [PubMed] [Google Scholar]
- 33.Balsera MA, Wriggers W, Oono Y, Schulten K. J Phys Chem-Us. 1996;100:2567–2572. [Google Scholar]
- 34.Faraldo-Gomez JD, Forrest LR, Baaden M, Bond PJ, Domene C, Patargias G, Cuthbertson J, Sansom MSP. Proteins-Structure Function and Bioinformatics. 2004;57:783–791. doi: 10.1002/prot.20257. [DOI] [PubMed] [Google Scholar]
- 35.Hess B. Physical Review E. 2002;65:031910. doi: 10.1103/PhysRevE.65.031910. [DOI] [PubMed] [Google Scholar]
- 36.Efron B, Tibshirani RJ. An introduction to the bootstrap. Chapman and Hall. CRC; Boca Raton, FL: 1998. [Google Scholar]
- 37.Chodera JD, Singhal N, Pande VS, Dill KA, Swope WC. The Journal of Chemical Physics. 2007;126:155101–155117. doi: 10.1063/1.2714538. [DOI] [PubMed] [Google Scholar]
- 38.Voter AF. J Chem Phys. 1985;82:1890–1899. [Google Scholar]
- 39.Ytreberg FM, Zuckerman DM. J Phys Chem B. 2005;109:9096–9103. doi: 10.1021/jp0510692. [DOI] [PubMed] [Google Scholar]
- 40.Zuckerman DM, Lyman E. J Chem Theory Comput. 2006;2:1200–1202. doi: 10.1021/ct600297q. [DOI] [PubMed] [Google Scholar]
- 41.Boon JP, Yip S. Molecular Hydrodynamics. Dover; New York: 1992. [Google Scholar]