

Abstract
The endoplasmic reticulum (ER) is recognized primarily as the site of synthesis and folding of secreted and membrane-bound proteins. The ER provides stringent quality control systems to ensure that only correctly folded, functional proteins are released from the ER and that misfolded proteins are degraded. The efficient functioning of the ER is essential for most cellular activities and for survival. Stimuli that interfere with ER function can disrupt ER homeostasis, impose stress to the ER, and subsequently cause accumulation of unfolded or misfolded proteins in the ER lumen. ER transmembrane proteins detect the onset of ER stress and initiate highly specific signaling pathways collectively called the “unfolded protein response” (UPR) to restore normal ER functions. However, if ER homeostasis cannot be reestablished in response to intense or prolonged ER stress, the UPR induces ER stress-associated apoptosis to protect the organism by removing the stressed cells that produce misfolded or malfunctioning proteins. This chapter summarizes current understanding of ER stress-induced apoptosis and reliable methods to examine ER stress and apoptosis in mammalian cells. Since the liver is the major organ dealing with metabolic or pathological stress and is responsible for the detoxification of chemical compounds, the experimental protocols described here focus on identification and characterization of ER stress-induced apoptosis in mouse liver.
1. The Endoplasmic Reticulum (ER) and the Unfolded Protein Response
The endoplasmic reticulum (ER) is the site of biosynthesis for sterols, lipids, membrane-bound and secreted proteins, and glycoproteins (Gaut and Hendershot, 1993; Kaufman, 1999). In higher eukaryotes, nearly all newly synthesized proteins require folding and/or assembly in the ER prior to trafficking to specific destinations to carry out their functions. As a unique protein-folding compartment and a dynamic calcium store, the ER is very sensitive to alterations in intracellular homeostasis. A number of biochemical, physiological, and pathological stimuli, such as chemicals that disrupt protein folding, nutrient depletion and hypoxia, calcium depletion, reductive or oxidative stress, expression of secretory proteins, expression of mutant difficult-to-fold proteins, unbalanced expression of subunits of protein complexes, elevated lipids or cholesterol, DNA damage, growth arrest, and viral/bacterial infection can disrupt ER homeostasis, impose stress to the ER, and subsequently cause accumulation of unfolded or misfolded proteins in the ER lumen. The cell has evolved highly specific signaling pathways called the unfolded protein response (UPR) to alter intracellular transcriptional and translational programs to deal with the accumulation of unfolded or misfolded proteins. These pathways prevent the accumulation of unfolded protein in the ER lumen by decreasing the protein-folding load, increasing the ER protein-folding capacity, and increasing the degradation of misfolded proteins through processes of ER-associated protein degradation (ERAD) or autophagy (Kaufman, 2002; Mori, 2000; Ron and Walter, 2007). In recent years, accumulating evidence suggests that the UPR signaling pathways represent an essential component of cell differentiation and function, as well as specific adaptive responses to viruses, hormones, growth factors, nutrients, and other external stimuli.
The basic UPR pathways consist of three main signaling cascades initiated by three ER-localized prototypical stress sensors: inositol-requiring 1 (IRE1), double-stranded RNA-activated protein kinase-like ER kinase (PERK), and activating transcription factor 6 (ATF6) (Ron and Walter, 2007; Schroder and Kaufman, 2005). The stress sensors IRE1, PERK, and ATF6 all have an ER lumenal domain that senses the presence of unfolded or misfolded proteins, an ER transmembrane domain that targets the protein for localization to the ER membrane, and a cytosolic functional domain. In resting cells, all three ER stress receptors are maintained in an inactive state through their association with the ER chaperone, GRP78/BiP. When cells encounter ER stress, GRP78/BiP dissociates from the three ER stress sensors, leading to activation of the UPR.
The immediate response to the accumulation of unfolded or misfolded proteins is dimerization, trans-autophosphorylation, and activation of PERK protein kinase to inhibit protein biosynthesis through phosphorylation of the α subunit of eukaryotic translation initiation factor (eIF2α). When eIF2α is phosphorylated, formation of the ternary translation initiation complex eIF2/GTP/Met-tRNAiMet is prevented, leading to attenuation of mRNA translation in general to reduce the protein-folding workload on the ER. Murine cells deleted in PERK or mutated at Ser51 in eIF2α to prevent phosphorylation did not attenuate protein synthesis upon ER stress. As a consequence, these cells were not able to survive ER stress (Harding et al., 2000; Scheuner, 2001). Whereas phosphorylation of eIF2α by PERK leads to attenuation of global mRNA translation, phosphorylated eIF2α selectively stimulates translation of a specific subset of mRNAs. One mRNA that requires eIF2α phosphorylation for translation encodes ATF4, a transcription factor that activates genes encoding proteins involved in amino acid biosynthesis and transport, antioxidative stress responses, and ER stress-induced apoptosis (Harding et al., 2003).
On activation of the UPR, IRE1 is also activated through its homodimerization and trans-autophosphorylation. In mammals, IRE1 is encoded by two homologous genes: IRE1α and IRE1β. Whereas IRE1α is apparently expressed in all cells and tissues, IRE1β expression is restricted primarily to the intestinal epithelial cells (Tirasophon et al., 1998; Wang et al., 1998). Activated IRE1α can function as an endoribonuclease to initiate removal of a 26 base small intron from X-box binding protein 1 (Xbp1) mRNA. Spliced Xbp1 mRNA encodes a potent basic leucine zipper (bZIP)-containing trans-activator that induces expression of genes encoding enzymes that facilitate protein folding, secretion, or degradation (Calfon et al., 2002; Lee et al., 2003; Shen et al., 2001; Yoshida et al., 2001). Studies have identified that IRE1α-mediated stress signaling is required for many physiological processes in addition to its role in the classical UPR. IRE1α can be activated by glucose in pancreatic β cells and contributes to proinsulin synthesis and β-cell differentiation and function (Lipson et al., 2006; Scheuner and Kaufman, unpublished observation). During B-cell lymphopoesis, IRE1α is activated and required for both early and late stages of B-cell differentiation (Zhang et al., 2005). In addition, under ER stress conditions, IRE1α can serve as a scaffold to recruit protein factors, including TRAF2, BAX, and BAK, and is possibly involved in the inflammatory response and apoptosis (Hetz et al., 2006; Urano et al., 2000). ATF6 is a UPR trans-activator belonging to bZIP transcription factors of the CREB/ATF family. Under ER stress conditions, ATF6 (90 kDa) transits to the Golgi compartment where it is cleaved by site-1 protease (S1P) and site-2 protease (S2P) to generate a 50-kDa cytosolic bZIP-containing fragment that migrates to the nucleus to activate transcription of the target genes encoding ER-resident molecular chaperones, folding catalysts, and ERAD machinery (Nadanaka et al., 2004; Ye et al., 2000). There are two homologues of ATF6 in the mammalian genome, ATF6α and ATF6β. Whereas ATF6α mediates gene induction upon UPR activation, the role of ATF6β in the UPR is presently unknown (Wu et al., 2007; Yamamoto et al., 2007). Although ATF6α facilitates adaptation to ER stress and plays a role in assisting plasma cells to produce antibodies, it is dispensable for classical UPR signaling (Gunn et al., 2004; Wu et al., 2007). In addition, it has been noted that ATF6α is involved in lipid metabolism and the acute-phase response by dimerizing with other ER transmembrane bZIP transcription factors that are also regulated by proteolysis (Zeng et al., 2004; Zhang et al., 2006).
2. Endoplasmic Reticulum Stress-Induced Apoptosis
In multicellular organisms, if the translational and transcriptional adaptive responses are not sufficient to relieve the unfolded or misfolded protein load, the cell enters programmed cell death—apoptosis. It has been proposed that multiple UPR pathways contribute to ER stress-induced cell apoptosis, although the mechanisms still remain largely unknown.
2.1. The PERK/eIF2α pathway
The most well studied of ER stress-induced apoptotic pathways is mediated by a transcription factor called CHOP/GADD153, which is activated primarily through the PERK/eIF2α UPR pathway. CHOP (C/EBP homologous protein) is a bZIP-containing transcription factor that was identified as a member of the CCAAT/enhancer binding protein (C/EBP) family (Ron and Habener, 1992). CHOP is also known as growth arrest and DNA damage-inducible gene 153 (GADD153), although it is induced by ER stress more than by growth arrest or DNA damage. Upon ER stress, activated PERK phosphorylates eIF2α, which subsequently attenuates general mRNA translation through reducing the efficiency of AUG codon recognition. As a consequence, eIF2α phosphorylation can produce quantitative, as well as qualitative (through alternate initiator AUG codon recognition), changes in proteins (Kaufman, 2004). However, a few specific mRNAs that harbor regulatory sequences in their 5′-untranslated regions, for example, the internal ribosomal entry site in the cationic amino acid transporter 1 (Cat1) mRNA, require eIF2α phosphorylation for efficient translation (Harding et al., 2000; Hinnebusch, 2000; Scheuner, 2001; Yaman et al., 2003). Another mRNA that requires eIF2α phosphorylation encodes ATF4, a cAMP response element-binding transcription factor (C/EBP). Under prolonged or severe ER stress, ATF4 induces the transcription factor CHOP, a well-recognized proapoptotic factor (Harding et al., 2003) (Fig. 20.1). In addition, ATF6 can also contribute to induction of the Chop gene (Wu et al., 2007). CHOP-deficient cells are protected from ER stress-induced apoptosis (Zinszner et al., 1998). Overexpression of CHOP induces cell cycle arrest or apoptosis by regulating the expression of multiple genes encoding proapoptotic factors, including death receptor 5 (DR5), Tribbles homolog 3 (TRB3), carbonic anhydrase VI (CAVI) and BCL2 family proteins (Matsumoto et al., 1996; McCullough et al., 2001; Ohoka et al., 2005; Sok et al., 1999; Yamaguchi and Wang, 2004). Dimerization of CHOP and cAMP-responsive element binding protein (CREB) suppresses the expression of Bcl2 (McCullough et al., 2001), which opposes induction of Bcl2 by Akt/CREB (Pugazhenthi et al., 2000). A decrease in the cellular level of BCL2 may increase the susceptibility of mitochondria to the proapoptotic effects of BH3-only proteins. Evidence suggests that CHOP forms heterodimers with C/EBPα under ER stress conditions to induce transcription of Bim, a proapoptotic BH3-only member of the BCL2 family, and is essential for ER stress-induced apoptosis in both cultured cells and within the whole animal (Puthalakath et al., 2007). In addition, CHOP directly activates GADD34, which subsequently dephosphorylates eIF2α and leads to apoptosis by promoting protein synthesis in stressed cells (Marciniak et al., 2004). CHOP also contributes to apoptosis through activating ERO1α, an ER oxidase that promotes hyperoxidization of the ER (Marciniak et al., 2004).
Figure 20.1.

ER stress-induced apoptotic pathways. Under ER stress, PERK is activated by homodimerization and autophosphorylation. Activated PERK phosphorylates eIF2α, which subsequently induces proapoptotic factors ATF4 and CHOP. CHOP induces expression of numerous genes encoding proapoptotic factors, including DR5, TRB3, CAVI, and BCL2 family proteins that promote mitochondria-mediated apoptosis. CHOP also induces expression of GADD34 and ERO1α, leading to apoptosis by promoting protein synthesis and oxidation in the stressed ER. On activation of the UPR, IRE1α recruits TRAF2 and leads to activation of the JNK-mediated apoptotic pathway. ER stress induces BAX and BAK localization and oligomerization at the ER, which promotes calcium efflux into the cytoplasm. Calcium uptake into the mitochondrial matrix depolarizes the inner membrane and leads to transition of the outer membrane permeability pore. This causes cytochrome c release and Apaf-1-dependent activation of the apoptosome, leading to apoptosis. In addition, it has been proposed that ER stress induces dissociation of TRAF2 from procaspase-12 residing on the ER membrane, allowing caspase-12 activation to mediate apoptosis. Mito, mitochondria; Cyt-C, cytochrome c; pCP12, procaspase-12; CP12, caspase-12; CP9, caspase-9; CP-3, caspase-3.
2.2. The IRE1α pathway
The IRE1α/XBP1-mediated UPR pathway plays a prosurvival role through the induction of ER chaperones, ER folding catalysts, and ERAD machinery. However, in response to ER stress, IRE1α can recruit the adaptor protein tumor necrosis factor receptor-associated factor 2 (TRAF2) (Urano et al., 2000). This interaction recruits c-Jun NH2-terminal inhibitory kinase (JIK), which can interact with both IRE1α and TRAF2 (Yoneda et al., 2001). The IRE1α – TRAF2 complex formed during ER stress can recruit the apoptosis signal-regulating kinase, which can relay various stress signals to the downstream mitogen-activated protein kinases JNK and p38 (Nishitoh et al., 1998, 2002). Overexpression of JIK promotes interaction between IRE1α and TRAF2 and JNK activation in response to tunicamycin, whereas overexpression of an inactive JIK mutant inhibits JNK activation (Yoneda et al., 2001). JNK activation is known to influence the cell-death machinery through phosphorylation of BCL2 family proteins, including BCL2 and BIM, which subsequently promote cell death programs (Davis, 2000). These observations led to the hypothesis that ER stress-induced, IRE1α- and TRAF2-dependent activation of JNK is one of apoptotic pathways in cells under ER stress, although there are no compelling data to confirm a requirement for the IRE1α-mediated apoptotic pathway in a physiological context (Fig. 20.1).
2.3. Caspases and BCL2 family proteins
Caspases also participate in ER stress-induced apoptosis (Fig. 20.1). In mice, procaspase-12 is localized on the cytosolic side of the ER membrane and is cleaved and activated by ER stress (Nakagawa and Yuan, 2000; Nakagawa et al., 2000). TRAF2 has been shown to interact with procaspase-12 under ER stress conditions or by overexpression of IRE1α. By this way, TRAF2 promotes the clustering of procaspase-12 at the ER membrane (Yoneda et al., 2001). It has been proposed that, during ER stress, caspase-12 activation requires the dissociation of procaspase-12 from TRAF2, which may subsequently be recruited to IRE1α (Yoneda et al., 2001). Calpains, a family of Ca2+-dependent cysteine proteases, may play a role in caspase-12 activation (Nakagawa and Yuan, 2000; Rao et al., 2001). Calpain-deficient murine embryonic fibroblasts (MEFs) have reduced ER stress-induced caspase-12 activation and are resistant to ER stress-associated apoptosis (Tan et al., 2006). In addition to calpain, caspase-7 translocates from the cytosol to the cytoplasmic side of the ER membrane under ER stress and interacts with cleaved caspase-12, leading to its activation (Rao et al., 2001). Activated caspase-12 activates caspase-9, which, in turn, forms the apoptosome with released cytochrome c and Apaf-1 to activate cell death executor caspase-3, leading to apoptosis. Although initial studies suggest that caspase-12-deficient cells are partially resistant to ER stress-induced apoptosis (Nakagawa et al., 2000), more recent studies showed that caspase-12−/− mice are not protected from cell death induced by ER stress (Saleh et al., 2006). In addition, the requirement for caspase-12 in apoptosis in human cells is open to question, as the human caspase-12 gene contains several inactivating mutations (Fischer et al., 2002). It is possible that caspase-4 mediates ER stress-induced apoptosis in human cells (Hitomi et al., 2004; Kim et al., 2006). Finally, ER stress-induced apoptosis may be mediated by calpain, but not by caspases, based on the observation that calpain inhibitors, but not a pan-caspase inhibitor, blocked tunicamycin and thapsigargin-induced apoptosis (Sanges and Marigo, 2006).
Evidence suggests that ER stress-induced apoptosis is closely linked to mitochondrial mechanisms involving the BCL2 family proteins, including BCL2, BAX, and BAK, that are associated with both mitochondria and ER membranes (Krajewski et al., 1993; Zong et al., 2003). Two pathways have been proposed by which BAX and BAK may promote apoptosis in response to ER stress. During ER stress, BAX and BAK undergo conformational changes and oligomerization in the ER membrane (Zong et al., 2003). This leads to a disruption of ER Ca2+ stores, causing an increase in the Ca2+ concentration in the cytosol (Scorrano et al., 2003). Increased cytosolic Ca2+ flux can activate m-calpain, which cleaves and activates procaspase-12 as mentioned earlier. However, the significance of this pathway in vivo remains to be explored. The second pathway involves Ca2+ uptake into the mitochondrial matrix, leading to depolarization of mitochondrial inner membrane and cytochrome c release and formation of the apoptosome to activate caspase-3, DNA fragmentation, and cell death (Crompton, 1999) (Fig. 20.1).
3. Methods Used to Identify and Characterize ER Stress-Induced Apoptosis In Vivo
Most studies on ER-associated apoptosis were performed using pharmacological toxins, such as tunicamycin or thapsigargin, that cause severe ER stress. Whether those pathways indeed contribute to ER stress-induced cell death in more physiological settings, such as the mouse liver under metabolic stress, remains largely unclear. Of the pathways implicated in ER stress-induced apoptosis, only the significance of the PERK/eIF2α pathway has been confirmed in several physiological models, including pancreas β-cell death in diabetes and macrophage cell death in atherosclerotic lesions (Feng et al., 2003; Harding et al., 2001; Scheuner, 2001; Zhou et al., 2005). It has been accepted that the ER stress-inducible proapoptotic transcription factor CHOP plays a primary role in ER stress-induced apoptosis. Reliable methods have been established to characterize ER stress-induced apoptosis in vitro and in vivo. This section discusses the established experimental methods for the identification and characterization of PERK/eIF2α-mediated CHOP-dependent apoptosis in vivo. Because the liver is the major organ exposed to metabolic stress and plays a major role in drug detoxification, the approaches discussed here focus on identification and characterization of ER stress signaling and stress-induced apoptosis in the mouse liver.
3.1. Experimental protocols used to analyze UPR pathways associated with apoptosis in mouse liver
To induce ER stress in the liver, wild-type mice with C57BL/6J strain background (3 months old) are given a single intraperitoneal injection of 1 μg/g body weight of tunicamycin in 150 mM dextrose. At 24 or 36 h after injection, the mice are euthanized, and the livers are dissected for the following experiments: (1) approximately 50 mg fresh liver tissue is homogenized for the purification of liver total RNA using Trizol reagent (Invitrogen); (2) approximately 100 mg fresh liver tissue is homogenized for preparation of liver protein samples for Western blot assay or immunoprecipatation (IP)-Western blot assay; (3) a small piece of fresh liver tissue (about 50 mg) is washed briefly with phosphate-buffered saline (PBS) and then fixed in 4% PBS-buffered formalin. Fixed liver samples are paraffin embedded, and 4-μm sections are prepared and mounted on glass slides for immunohistochemical staining. The remaining liver tissue is snap frozen in liquid nitrogen and stored at 80 °C for future use.
3.1.1. Phosphorylation of IRE1α and PERK
IRE1α and PERK are primary UPR transducers, and phosphorylation of IRE1α and PERK is a hallmark of ER stress and activation of the UPR. Typically, this phosphorylation can be analyzed by an upward mobility shift upon sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) and Western immunoblot analysis. Phosphorylated PERK and IRE1α can be monitored by the phospho-PERK-specific antibody (Cell Signaling) and the phospho-IRE1α specific antibody (raised in our laboratory) to probe Western blots. However, because of their low expression levels, detection of endogenous levels of IRE1α or PERK in tissues is difficult. Unfortunately, the anti-IRE1α or anti-PERK antibodies tested were not able to detect endogenous IRE1α or PERK protein in mouse liver tissue by direct immunoblot analysis. Therefore, to increase the sensitivity of detecting IRE1α or PERK by Western blot analysis, sequential immunoprecipitation and immunoblot analysis of detergent tissue lysates can be performed. We have raised murine anti-human IRE1α antibodies that can detect endogenous levels of IRE1α in murine liver tissue by IP-Western blot analysis (Kaufman et al., 2002; Tirasophon et al., 2000). In addition, a polyclonal rabbit anti-PERK PITK-289 antibody (kindly provided by Dr. Yuguang Shi, Lilly Research Laboratories, Eli Lilly and Co., Indianapolis, IN) (Shi et al., 1999) or a commercial antibody (Cell Signaling) can be used to detect endogenous PERK activation though IP-Western blot assay.
Sample preparation
(1) Approximately 100 mg of fresh liver tissue from normal mice or mice treated with tunicamycin (Tm) is transferred into an ice-cold potter homogenizer. (2) Ice cold NP-40 lysis buffer (700 μl) (1% NP-40, 50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 0.05% SDS, 0.5 mM Na vanadate, 100 mM NaF, 50 mM β-glycerophosphate, and 1 mM phenyl-methylsulfonyl fluoride (PMSF) supplemented with protease inhibitors (EDTA-free Complete Mini, Roche)] is added into the homogenizer containing the liver tissue. (3) The liver tissue is homogenized on ice and sonicated, and the liver tissue lysate is incubated on ice for 40 min. (4) The liver lysate is centrifuged at 4 °C at 10,000 rpm for 20 min. The supernatant is transferred into a clean, ice-cold 1.5-ml Eppendorf tube for IP-Western blot analysis for IRE1α or PERK. (6) The protein concentration of a diluted aliquot is determined by the Bradford assay.
Detection of IRE1α protein
(1) Murine anti-human IRE1α antibody (1 μl) raised in our laboratory (Tirasophon et al., 2000) is incubated with 20 μl protein A beads in NP-40 lysis buffer for 1 h on an end-over-end rotator at room temperature. (2) The antibody-bound beads are washed by adding 500 μl lysis buffer in 1.5-ml Eppendorf tubes, rotating at 4 °C for 10 min, and then sedimenting in a microcentrifuge for 30 s at 8200g. The supernatant is aspirated and the tube with the bead pellet is placed on ice. The sample is washed an additional two times in a similar manner. (3) The liver tissue samples prepared as described earlier (about 700 μl) are added into the tubes with washed antibody-bound beads and are then incubated for 3 h at room temperature or overnight at 4 °C on a rotator. (4) After incubation, the beads are washed with three times with 1 ml lysis buffer and one time with 1 ml PBS. (5) The solution from the tube containing the beads is removed and then 20 μl of 2 × SDS-PAGE sample buffer (100 mM Tris, pH 6.8, 20% glycerol, 4% SDS, 0.2% bromophenol blue, 200 mM dithiothreitol) is added to the tube followed by heating at 95° for 5 min. (6) Denatured proteins are separated by SDS-PAGE on 10% Tris-glycine polyacrylamide gels and transferred to a 0.45-μm PVDF membrane (RPN1416F, GE Healthcare). (7) The blots are incubated with the same murine anti-human IRE1α antibody at a 1:1000 dilution in 0.1% Tween 20-PBS buffer (8 g NaCl, 0.2 g KCl 1.44 g Na2HPO4, and 0.24 g KH2PO4 diluted to 1000 ml distilled H2O, pH 7.4) containing 5% skim milk or overnight at 4 °C followed by incubation with a 1:3000 dilution of horse-radish peroxidase (HRP)-conjugated anti-mouse antibody (NA9310V, GE Healthcare) for 2 h at room temperature. (8) Membrane-bound antibodies are detected by an enhanced chemiluminescence (ECL) detection reagent (RPN2106, GE Healthcare). The murine IRE1α protein is detected as one major band migrating with a molecular weight of ~120 kDa. After Tm treatment, the size of the IRE1α protein is increased slightly because of autophosphorylation.
Detection of PERK protein
(1) The rabbit anti-PERK PITK-289 antibody (1 μl) (Shi et al., 1998) or the rabbit anti-PERK antibody (1 μl) from Cell Signaling is incubated with 20 μl protein A beads in NP-40 lysis buffer for 1 h on a end-over-end rotator at room temperature. (2) Follow the steps for PERK immunoprecipitation and transfer to PVDF membranes as described earlier for IRE1α. (3) The blots are incubated with the same PERK antibody at a 1:1000 dilution for 3 h at room temperature or overnight at 4 °C, followed by incubation of HRP-conjugated anti-rabbit antibody (NA9340V, GE Healthcare) for 2 h at room temperature. (4) Membrane-bound antibodies are detected by the ECL detection reagent (RPN 2106, GE Healthcare). The murine PERK protein is detected migrating with a molecular mass of ~ 170 kDa. After Tm treatment, the size of PERK protein is increased slightly because of autophosphorylation.
3.1.2. Xbp1 mRNA splicing
On activation of the UPR, the primary UPR transducer IRE1α is activated and functions as an endoribonuclease to remove a 26 base small intron from the human or murine X-box binding protein 1 (Xbp1) mRNA to encode a potent UPR trans-activator (Fig. 20.2A). Because of the difficulty in detecting endogenous IRE1α protein, quantitative analysis of spliced and total Xbp1 mRNA is the most convenient and reliable method to measure activation of the IRE1α-mediated UPR pathway.
Figure 20.2.

Quantitative analysis of Xbp1mRNA splicing. (A) Schematic representation of unspliced and spliced forms of murine Xbp1 mRNAs and the protein coding regions. (B) Semiquantitative RT-PCR analysis of unspliced and spliced Xbp1 mRNAs in murine liver. Wild-type C57Bl/6J mice at 3 month of age were injected intraperitoneally with 2 μg/gram body weight of tunicamycin in150 mM dextrose. At 24 h after injection, total liver RNA samples were prepared for RT-PCR analysis. Wild-type (+/+) and Ire1α knockout (−/−) MEFs treated with Tm (5 μg/ml) for 8 h were included as controls. (C) Quantitative real-time RT-PCR analysis of unspliced and spliced Xbp1 mRNAs in murine livers. The liver total RNA samples were same as described in B. Fold changes in mRNA levels were determined after normalization to internal control β-actin mRNA levels.
Accurate and sensitive methods have been developed for the quantification of spliced and total Xbp1 mRNA in mammalian cells or tissue using conventional reverse transcription (RT) polymerase chain reaction (PCR) or real-time RT-PCR: (1) Total RNA is isolated from liver tissue with the Trizol reagent (Invitrogen). (2) Synthesis of cDNA from murine total RNA is performed using a multiscribe reverse transcriptase kit (Bio-Rad). The reaction mixture (20 μl) contains 500 ng total RNA, 4 μl 5× iScript reaction mix, and 1 μl reverse transcriptase. (3) The reverse transcription reaction is incubated at 25 °C for 10 min, followed by incubation at 48 °C for 30 min, and then reverse transcriptase is inactivated at 85 °C for 5 min. (4) The reaction mix is diluted 10-fold by the addition of 180 μl nuclease-free water. The diluted cDNA mix from the reverse transcription reaction is subjected to semiquantitative PCR or quantitative real-time PCR to determine levels of spliced and total Xbp1 mRNAs.
For conventional semiquantitative RT-PCR, 10 μl of diluted cDNA template(25 ng)ismixedwith200 μM of each dNTP,300 nM forward and reverse primers, 5 μl PCR reaction buffer (10 × concentration) with 15 mM MgCl2, 2.6 units Taq enzyme, and nuclease-free water to a total volume of 50 μl. The PCR cycle starts with a 2-min incubation at 95 °C, then 25 cycles for 30 s at 94°C, 30 s at 55 °C, and 45 s at 72 °C; this is followed by a final incubation at 72 °C for 7 min. The forward primer for PCR amplification of spliced and total mouse Xbp1 mRNA is 5′-ACACGCTTGGGAATGGACAC-3′, and the reverse primer is 5′-CCATGGGAAGATGTTCTGGG -3′. PCR products are separated by electrophoresis on a 3% agarose gel and visualized by ethidium bromide staining. The size of amplified unspliced Xbp1 mRNA is 170 bp, and the size of amplified spliced Xbp1 mRNA is 144 bp (Fig. 20.2B).
For quantitative real-time RT-PCR, a pair of real-time PCR primers is designed for the quantification of mouse Xbp1 mRNA splicing. The forward primer sequence is 5′-GAGTCCGCAGCAGGTG-3′. This primer was designed to span the 26 base intron, thus can only anneal to the spliced Xbp1 transcript. The reverse primer sequence is 5′-GTGTCAGAGTC-CATGGGA-3′, which is 70 bases downstream of the forward primer. This pair of real-time PCR primers can specifically amplify the spliced form of murine Xbp1 mRNA. Pairs of real-time PCR primers are also designed for quantification of the total Xbp1 mRNA level. The forward primer sequence is 5′-AAGAACACGCTTGGGAATGG-3′, and the reverse primer sequence is 5′-ACTCCCCTTGGCCTCCAC-3′. This pair of real-time PCR primers can amplify both unspliced and spliced forms of Xbp1 mRNA. In addition, a pair of primers is used to quantitate β-actin mRNA by real-time RT-PCR as an internal control: the forward primer is 5′-GATCTGGCACCACACCTTCT-3′ and the reverse primer is 5′-GGGGTGTTGAAGGTCTCAAA-3′.
SYBR green PCR master mix (Bio-Rad) is used to set up the quantitative real-time PCR reaction. The reaction (20 μl) contains 500 nM forward and reverse primers for Xbp1 or β-actin transcripts, 12.5 ng cDNA templates made from murine liver total RNA, and 10 μl SYBR Green Supermix (50 mM KCl, 20 mM Tris-HCl, pH 8.4, 0.2 mM of each dNTP, 25U/ml iTaq DNA polymerase, 3 mM MgCl2, SYBR green 1, 10 nM fluorescein and stabilizers). The thermal cycle parameters are as follow: step 1, 95 °C for 10 min; step 2, 95 °C for 15 s; and step 3, 59 °C for 1 min. Step 2 is repeated for 40 cycles. The final step is incubation at 4 °C to terminate the reaction. Data are analyzed with the iCycler iQ real-time PCR detection system (Bio-Rad) according to the manufacturer’s instructions. Figure 20.2C shows quantification of the spliced Xbp1 mRNA in murine liver with or without Tm treatment. The spliced form of Xbp1 mRNA was increased significantly in murine liver after Tm treatment compared to that of control liver.
3.1.3. GRP78/BiP induction
It has been well established that induction of GRP78/BiP is a marker of ER stress and a central regulator of the activation of the UPR transducers (IRE1α, PERK, and ATF6). The following are methods to measure GRP78/BiP transcript and protein in murine liver.
Semiquantitative conventional RT-PCR analysis of GRP78/BiP transcripts
(1) Murine liver cDNA mix is prepared from total liver RNA as described in Section 3.1.2. (2) A 10-μl aliquot of diluted cDNA (25 ng) template is mixed with 200 μM of each dNTP, 300 nM forward and reverse primers, 5 μl PCR reaction buffer (10 × concentration, with 15 mM MgCl2), 2.6 U Taq enzyme, and nuclease-free water to a final volume of 50 μl. (3) The PCR cycle starts with a 2-min incubation at 95 °C and then 25 cycles for 30 s at 94 °C, 30 sec at 55 °C, and 45 s at 72 °C; this is followed by a final incubation at 72 °C for 7 min. The forward primer for PCR amplification of GRP78/BiP is 5′-CTGGGTACATTTGATCTGACTGG-3′ and the reverse primer is 5′-GCATCCTGGTGGCTTTCCAGCCATTC-3′. The primers amplify a 397-bp mouse GRP78/BiP cDNA fragment.
Quantitative real-time RT-PCR analysis of GRP78/BiP transcripts
(1) A 2-μl aliquot of diluted cDNA template (12.5 ng) is mixed with 10 μl iQ SYBR Green Supermix (Bio-Rad), 150 nM forward and reverse real-time PCR primers for GRP78/BiP or β-actin (internal control), and nuclease-free water to a final volume of 20 μl. (2) The thermal cycling starts with a 10-min incubation at 95 °C, then 40 cycles for 15 s at 95 °C, and 1 min at 59 °C; this is followed by a final incubation at 4 °C to terminate the reaction. The primers used for GRP78/BiP quantitative real-time PCR assay are forward primer 5′-CATGGTTCTCACTAAAATGAAAGG-3′ and reverse primer 5′-GCTGGTACAGTAACAACTG-3′. Primers for the internal control β-actin transcript are the same as described in Section 3.1.2.
Measurement of GRP78/BiP protein level
The level of GRP78/BiP is elevated after prolonged stress treatment, although the fold of increase in protein level is usually less than that observed in mRNA levels due to the stability of the GRP78/BiP protein. An increase in the GRP78/BiP protein level is a good marker for UPR activation. However, this is not a very sensitive measure to detect low levels of ER stress and UPR activation. Therefore, it should not be concluded that the UPR is not activated if an increase in the GRP78/BiP protein is not detected. To evaluate ER stress and activation of the UPR, it is important to examine the changes in both GRP78/BiP mRNA and protein levels. Currently, monoclonal and polyclonal antibodies against GRP78/BiP are available commercially, and the level of GRP78/BiP protein in liver tissue or cultured cells can be determined through a standard Western blot analysis. (1) The liver protein lysate is prepared as described in Section 3.1.1. (2) Denatured liver proteins (50–80 μg) are separated by SDS-PAGE on a 10% Tris-glycine gel and transferred to a 45-μm PVDF nitrocellulose membrane in 0.19 M glycine, 25 mM Tris base, and 20% methanol. (3) Nonspecific binding sites on the membrane are blocked using 5% skimmed milk in PBST (PBS containing 0.1% Tween 20) for 1 h. (4) The membrane is incubated with a rabbit polyclonal anti-GRP78 antibody (SPA-826, StressGen) at a 1:1000 dilution for 3 h at room temperature or overnight at 4°. (5) The membrane is washed with PBST and then incubated with horseradish peroxidase (HRP)-conjugated secondary antibody for 2 h at room temperature. The membrane is washed three times with PBST and chemiluminescent signals are detected with the ECL detection reagent (RPN 2106, GE Healthcare).
3.1.3. Phosphorylation of eIF2α
Upon ER stress, PERK-mediated phosphorylation of translation initiation factor eIF2α inhibits general translation to reduce the protein-folding demand of the ER, while selectively stimulating translation of the ATF4 mRNA that encodes a transcription factor for the induction of proapoptotic factor CHOP under prolonged ER stress. Therefore, phosphorylated eIF2α is not only a reliable marker of PERK activation, but also a key transmitter of cell survival and death signals in response to ER stress (Rutkowski et al., 2006). Measurement of the levels of phosphorylated and total eIF2α protein is particularly informative for evaluating activation of the PERK/eIF2α UPR pathway and ER stress-induced apoptosis. However, it must be cautioned that eIF2α phosphorylation may not reflect PERK activation because there are at least three additional eIF2α kinases—general control of nitrogen metabolism kinase 2, heme-regulated inhibitor kinase, and double-stranded RNA-activated protein kinase—that phosphorylate the same site in response to amino acid deprivation, heme deficiency, or viral infection (Kaufman, 2004).
Measurement of phosphorylated eIF2α protein
(1) Liver tissue lysate is prepared by lysis in buffer containing 1% NP-40, 50 mM Tris-HCL [pH 7.5], 150 mM NaC1, 0.05% SDS, 0.5 mM Na vanadate, 100 mM NaF, 50 mM β-glycerophosphate, and 1 mM PMSF supplemented with protease inhibitors [EDTA-free, Roche] as described in Section 3.1.1. (2) Samples (50–80 μg) of denatured liver proteins are separated on a 10% Tris-glycine polyacrylamide gel and transferred to a nitrocellulose membrane in 0.19 M glycine, 25 mM Tris base, and 20% methanol. (3) Nonspecific-binding sites on the membrane are blocked by incubation with 5% (w/v) skimmed milk in PBST (PBS containing 0.1% Tween 20) for 1 h at room temperature. (4) The membrane is incubated with a rabbit anti-Ser51-phosphorylated eIF2α antibody (Biosource International) at a 1:1000 dilution for 3 h at room temperature or overnight at 4 °C. (5) The membrane is washed with PBST and then incubated with HRP-conjugated anti-rabbit secondary antibody for 2 h. (6) The membrane is washed with PBST three times and is then incubated with the ECL detection reagent to measure chemiluminescent signals (Fig. 20.3).
Figure 20.3.

Western blot analysis of phosphorylated and total eIF2α in murine liver. Wild-type C57BL/6J mice at 3 months of age were injected intraperitoneally with 2 μg/g body weight of tunicamycin in 150 mM dextrose. At 24 h after injection, liver protein samples were prepared as described in the text for Western blot analysis. Wild-type (+/+) and Perk-deleted (−/−) MEFs treated with Tm (5 μg/ml) for 8 h were included as controls. The phosphorylated and total eIF2α proteins were detected using different blots for the same liver protein samples.
Measurement of total eIF2α protein
(1) The same amounts of denatured liver protein are separated on a 10% Tris-glycine gel and transferred to a new PVDF membrane. (2) Nonspecific binding sites on the membrane are blocked using 5% skimmed milk in PBST for 1 h. (3) The membrane is incubated with a rabbit anti-total eIF2α antibody (Cell Signaling) at a 1:1000 dilution overnight at 4 °C. (4) The membrane is washed and incubated with the secondary antibody for ECL detection as described for the detection of phosphorylated eIF2α (Fig. 20.3).
Note: The use of serine-threonine phosphatase inhibitors in the lysis buffer such as β-glycerophosphate is critical in preserving the eIF2α phosphorylation status.
3.2. Experimental protocols for identification and characterization of ER stress-inducible proapoptotic factors in the liver
As discussed earlier, ATF4 and CHOP play a proapoptotic role in ER stress-induced cell death. Indeed, CHOP is the most well-studied and well-recognized indicator for ER stress-induced apoptosis both in vitro and in vivo. We have developed quantitative real-time RT-PCR to measure the induction of murine Atf4 and Chop mRNAs in the liver upon ER stress (Fig. 20.4). In addition, polyclonal anti-ATF4 and anti-CHOP antibodies are available commercially for the detection of ATF4 and CHOP protein levels in liver tissue through Western blot analysis.
Figure 20.4.

Measurement of Chop mRNA and protein in murine liver. (A) Western blot analysis of CHOP in murine liver. Wild-type C57BL/6J mice at 3 months of age were injected intraperitoneally with 2 μg/gram body weight of tunicamycin in 150 mM dextrose. At 24 h after injection, liver protein samples were prepared for Western blot analyses. Wild-type (+/+) and Chop-deleted (−/−) MEFs treated with Tm (5 μg/ml) for 30 h were included as controls. Levels of murine β-actin protein were determined as protein loading controls. (B) Quantitative real-time RT-PCR analysis of Chop mRNA in murine liver. The liver total RNA was prepared from the mice as described in A. The fold changes of mRNA levels were determined after normalization to internal control β-actin mRNA levels.
3.2.1. Quantitative real-time RT-PCR analysis of Atf4 and Chop mRNAs
Quantitative real-time RT-PCR analysis of Atf4 and Chop mRNAs
(1) The murine liver cDNA template is prepared from total liver RNA as described in Section 3.1.2. (2) A 2-μl aliquot of diluted cDNA template (12.5 ng) is mixed with 10 μl iQ SYBR Green Supermix, 150 nM forward and reverse real-time RT-PCR primers for Atf4, Chop, or β-actin, and nuclease-free water to a final volume of 20 μl. (3) The thermal cycling starts with a 10-min incubation at 95 °C, then 40 cycles for 15 s at 95 °C, and 1 min at 59 °C, followed by a final incubation at 4 °C. Primers used for Atf4 real-time PCR assay are forward primer 5′-ATGGCCGGCTATG-GATGAT-3′ and reverse primer 5′-CGAAGTCAAACTCTTTCAGATC-CATT-3′. The primers for Chop are forward primer 5′-CTGCCTTT CACCTTGGAGAC-3′ and reverse primer 5′-CGTTTCCTGGGGATGA-GATA-3′. The primers for internal control β-actin transcript are the same as described in Section 3.1.2.
3.2.2. Measurement of ATF4 and CHOP protein levels
Western blot analysis of ATF4 or CHOP protein in liver samples
(1) Liver protein sample preparation, SDS-PAGE, and PDVF membrane transfer are the same as described in Section 3.1.1. (2) Nonspecific-binding sites on the membrane are blocked by incubation with 5% skimmed milk in PBST for 1 h. (3) To detect ATF4, the membrane is incubated with rabbit polyclonal anti-CREB2/ATF4 antibody (SC-200, Santa Cruz Biotechnologies) at a 1:500 dilution for 3 h at room temperature. To detect CHOP, the membrane is incubated with rabbit polyclonal anti-GADD153/CHOP antibody (sc-793, Santa Cruz Biotechnologies) at a 1:200 dilution for 3 h at room temperature. (4) After incubation with anti-ATF4 or anti-CHOP primary antibody, the membrane is washed with PBST and then incubated with HRP-conjugated secondary antibody for 2 h at room temperature. (5) Chemiluminescent signals on the membrane are detected by the ECL detection reagent.
3.3. Immunohistochemical staining of ER stress-induced apoptotic markers in the liver
Immunohistochemical staining is an excellent method to visualize intensity, distribution, and localization of ER stress markers and apoptosis in liver tissue. We have successfully used modified immunohistochemical staining methods to amplify and visualize the CHOP signal and apoptotic events using paraffin-embedded liver tissue sections.
3.3.1. Immunohistochemical staining of CHOP in the liver
Tyramide signal amplification to detect CHOP
To stain and amplify the CHOP signal in liver tissue, we apply a method called tyramide signal amplification that utilizes the catalytic activity of HRP to generate high-density labeling of the CHOP protein (Speel et al., 1999; van Gijlswijk et al., 1997). The tyramide signal amplification staining process includes (1) binding of a rabbit anti-CHOP antibody to CHOP protein in the liver tissue section followed by secondary detection of the primary antibody with an HRP-labeled anti-rabbit antibody; (2) activation of multiple copies of a dye-labeled tyramide derivative by HRP; and (3) covalent coupling of the resulting highly reactive, short-lived tyramide radicals to nucleophilic residues in the vicinity of the HRP–CHOP interaction site, resulting in minimal diffusion-related loss of signal localization (Fig. 20.5A).
Figure 20.5.

Immunohistochemical staining of CHOP in the liver. (A) Schematic representation of the tyramide signal amplification process to detect CHOP protein in the liver. (B) CHOP staining in liver sections. Wild-type C57BL/6J mice at 3 months of age were injected intraperitoneally with 2 μg/gram body weight of tunicamycin in 150 mM dextrose. At 24 h after injection, fresh liver tissues were fixed in 4% PBS-buffered formalin, paraffin embedded, and 4-μm sections prepared. (Top) Liver sections from control mice. (Bottom) Liver sections from mice treated with Tm.
To stain and amplify the signal for CHOP protein in liver tissue sections, a commercial tyramide amplification kit (Invitrogen) is used. (1) Murine liver tissue fixation, sectioning, and deparaffination are carried out according to standard immunohistochemical staining protocols. (2) The liver tissue is permeabilized by incubating with 0.2% Triton X-100 for 10 min at room temperature followed by a rinse in PBS. (3) The endogenous peroxidase activity is quenched by incubating in 0.1% H2O2 in methanol for 1 h at room temperature. (4) The nonspecific-binding sites on the specimen are blocked by incubating the slide with 1% BSA in PBS for 1 h at room temperature. (5) The tissue section is labeled with a rabbit anti-CHOP primary antibody (sc-575, Santa Cruz Biotechnologies) at a 1:100 dilution in 1% BSA blocking reagent overnight at 4 °C. (6) After primary antibody incubation, the slide is rinsed with PBS three times and then incubated with HRP-conjugated antirabbit antibody for 60 min at room temperature. (7) The slide is rinsed with PBS three times and then incubated with the tyramide working solution (provided in the kit) for 7 min at room temperature. (8) The slide is rinsed with PBS three times and is then mounted with ProLong Gold antifade mounting medium containing 4′,6-diamidino-2-phenylindole (DAPI) to stain DNA (Molecular Probes, Invitrogen). The mounted slide is kept in the dark at room temperature for 24 h prior to analysis by fluorescence microscopy (Fig. 20.5B). Cells expressing CHOP exhibit red fluorescence when viewed at 556/573 nm (excitation/emission). DAPI-stained nuclei exhibit a blue color. The CHOP signal should be observed in nuclear, perinuclear, and cytosolic localizations.
Note: Because of the amplified detection inherent in the tyramide signal amplification process, background staining that was previously undetectable may become more prominent. Intense staining caused by nonspecific reaction of the tyramide with endogenous peroxidase in blood vessels may be observed. Because of sensitivity enhancements derived from the tyramide amplification process, dilution of the primary antibody may be increased 10-fold from the level used in conventional immunohistochemical staining of liver tissue sections.
3.3.2. Nuclear DNA fragmentation assay
During apoptosis, cells undergo many distinct morphological and biochemical changes. One striking apoptotic event involves cellular endonuclease cleavage of nuclear DNA between nucleosomes, thereby producing a mixture of DNA fragments. DNA fragmentation has been well recognized as a marker of the final stage of apoptosis (Schwartzman and Cidlowski, 1993). To detect ER stress-induced apoptosis in liver tissue sections, we have modified a DNA fragmentation assay by incorporating fluorescein-dUTP at the free 3′-hydroxyl ends of the fragmented DNA using the terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling (TUNEL) method. The fluorescein-labeled DNA can then be quantified as an indicator of apoptosis (Fig. 20.6).
Figure 20.6.
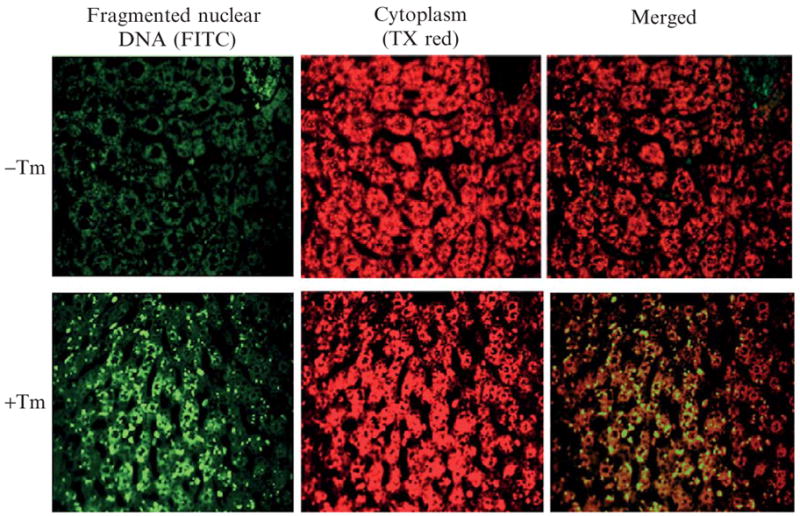
Immunohistochemical staining of nuclear DNA fragments in liver sections. Wild-type C57BL/6J mice at 3 months of age were injected intraperitoneally with 2 μg/gram body weight of tunicamycin in 150 mM dextrose. At 36 h after injection, fresh liver tissues were fixed in 4% PBS-buffered formalin, paraffin embedded, and 4-μm sections prepared. (Top) Liver sections from control mice. (Bottom) Liver sections from mice treated with Tm.
Analysis of DNA fragmentation in liver tissue sections
(1) Fixation, paraffin embedding, tissue sectioning, and deparaffination are performed according to standard immunohistochemical staining protocols. (2) The TUNEL labeling process is based on a commercial DNA fragmentation assay kit (Clontech). Pretreatment of tissue sections with NaCl and fixation by formaldehyde are carried out according to the product manual. (3) The liver tissue sections are treated with 20 μg/ml proteinase K in a Coplin jar for 10 min at room temperature. (4) The slide is washed with PBS and is fixed again by 4% formaldehyde/PBS for 5 min at room temperature. (5) The slide is washed by PBS and is then incubated in an equilibration buffer (200 nM potassium cacodylate, 25 mM Tris-HCl, 0.2 mM DD, 0.25 mg/ml BSA, and 2.5 mM cobalt chloride) for 10 min at room temperature. (6) For TUNEL labeling, the tissue section is incubated with TdT reaction buffer (1 μl TdT enzyme, 5 μl nucleotide mix, 45 μl equilibration buffer) in a dark, humidified 37 °C incubator for 2 h. (7) The reaction is terminated by immersing the slides in 2 × SSC (3 M NaCl, 300 mM Na3 citrate · H2O) for 15 min at room temperature. (8) The slide is washed with PBS twice and then stained with propidium iodide (PI) for 10 min at room temperature. (9) The slide is washed with deionized water and mounted with ProLong Gold antifade mount media (Molecular Probes, Invitrogen). (10) The slide is kept overnight at 4 °C in the dark prior to analysis by fluorescence microscopy. Apoptotic cells exhibit strong, nuclear green fluorescence using a standard fluorescein filter set (520 ± 20 nm). All cells stained with PI exhibit strong red cytoplasmic fluorescence when viewed at more than 620 nm.
Note: If tissue section permeabilization with Triton X-100 or incubation with proteinase K is insufficient, signals may be weak or not detected. However, if the tissue sections are overdigested with proteinase K, the sample may detach from the slides. The permeabilization and proteinase K incubation times need to be optimized to avoid these problems.
Acknowledgments
This work was supported by NIH Grants DK42394 (to RJK), HL52173 (to RJK), and PO1 HL057346 (to RJK) and American Heart Association Grant 0635423Z (to KZ). RJK is an Investigator of the Howard Hughes Medical Institute.
References
- Calfon M, Zeng H, Urano F, Till JH, Hubbard SR, Harding HP, Clark SG, Ron D. IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP-1 mRNA. Nature. 2002;415:92–96. doi: 10.1038/415092a. [DOI] [PubMed] [Google Scholar]
- Crompton M. The mitochondrial permeability transition pore and its role in cell death. Biochem J. 1999;341:233–249. [PMC free article] [PubMed] [Google Scholar]
- Davis RJ. Signal transduction by the JNK group of MAP kinases. Cell. 2000;103:239–252. doi: 10.1016/s0092-8674(00)00116-1. [DOI] [PubMed] [Google Scholar]
- Feng B, Yao PM, Li Y, Devlin CM, Zhang D, Harding HP, Sweeney M, Rong JX, Kuriakose G, Fisher EA, Marks AR, Ron D, et al. The endoplasmic reticulum is the site of cholesterol-induced cytotoxicity in macrophages. Nat Cell Biol. 2003;5:781–792. doi: 10.1038/ncb1035. [DOI] [PubMed] [Google Scholar]
- Fischer H, Koenig U, Eckhart L, Tschachler E. Human caspase 12 has acquired deleterious mutations. Biochem Biophys Res Commun. 2002;293:722–726. doi: 10.1016/S0006-291X(02)00289-9. [DOI] [PubMed] [Google Scholar]
- Gaut JR, Hendershot LM. The modification and assembly of proteins in the endoplasmic reticulum. Curr Opin Cell Biol. 1993;5:589–595. doi: 10.1016/0955-0674(93)90127-c. [DOI] [PubMed] [Google Scholar]
- Gunn KE, Gifford NM, Mori K, Brewer JW. A role for the unfolded protein response in optimizing antibody secretion. Mol Immunol. 2004;41:919–927. doi: 10.1016/j.molimm.2004.04.023. [DOI] [PubMed] [Google Scholar]
- Harding HP, Zeng H, Zhang Y, Jungries R, Chung P, Plesken H, Sabatini DD, Ron D. Diabetes mellitus and exocrine pancreatic dysfunction in perk−/− mice reveals a role for translational control in secretory cell survival. Mol Cell. 2001;7:1153–1163. doi: 10.1016/s1097-2765(01)00264-7. [DOI] [PubMed] [Google Scholar]
- Harding HP, Zhang Y, Bertolotti A, Zeng H, Ron D. Perk is essential for translational regulation and cell survival during the unfolded protein response. Mol Cell. 2000;5:897–904. doi: 10.1016/s1097-2765(00)80330-5. [DOI] [PubMed] [Google Scholar]
- Harding HP, Zhang Y, Zeng H, Novoa I, Lu PD, Calfon M, Sadri N, Yun C, Popko B, Paules R, Stojdl DF, Bell JC, et al. An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Mol Cell. 2003;11:619–633. doi: 10.1016/s1097-2765(03)00105-9. [DOI] [PubMed] [Google Scholar]
- Hetz C, Bernasconi P, Fisher J, Lee AH, Bassik MC, Antonsson B, Brandt GS, Iwakoshi NN, Schinzel A, Glimcher LH, Korsmeyer SJ. Proapoptotic BAX and BAK modulate the unfolded protein response by a direct interaction with IRE1alpha. Science. 2006;312:572–576. doi: 10.1126/science.1123480. [DOI] [PubMed] [Google Scholar]
- Hinnebusch A. Mechanism and Regulation of Initiator Methionyl-tRNA Binding Ribosomes. Cold Spring Harbor Press; Cold Spring Harbor, NY: 2000. [Google Scholar]
- Hitomi J, Katayama T, Eguchi Y, Kudo T, Taniguchi M, Koyama Y, Manabe T, Yamagishi S, Bando Y, Imaizumi K, Tsujimoto Y, Tohyama M. Involvement of caspase-4 in endoplasmic reticulum stress-induced apoptosis and Abeta-induced cell death. J Cell Biol. 2004;165:347–356. doi: 10.1083/jcb.200310015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufman RJ. Stress signaling from the lumen of the endoplasmic reticulum: Coordination of gene transcriptional and translational controls. Genes Dev. 1999;13:1211–1233. doi: 10.1101/gad.13.10.1211. [DOI] [PubMed] [Google Scholar]
- Kaufman RJ. Orchestrating the unfolded protein response in health and disease. J Clin Invest. 2002;110:1389–1398. doi: 10.1172/JCI16886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufman RJ. Regulation of mRNA translation by protein folding in the endoplasmic reticulum. Trends Biochem Sci. 2004;29:152–158. doi: 10.1016/j.tibs.2004.01.004. [DOI] [PubMed] [Google Scholar]
- Kaufman RJ, Scheuner D, Schröder M, Shen X, Lee K, Liu CY, Arnold SM. The unfolded protein response in nutrient sensing and differentiation. Nat Rev Mol Cell Biol. 2002;3:411–421. doi: 10.1038/nrm829. [DOI] [PubMed] [Google Scholar]
- Kim SJ, Zhang Z, Hitomi E, Lee YC, Mukherjee AB. Endoplasmic reticulum stress-induced caspase-4 activation mediates apoptosis and neurodegeneration in INCL. Hum Mol Genet. 2006;15:1826–1834. doi: 10.1093/hmg/ddl105. [DOI] [PubMed] [Google Scholar]
- Krajewski S, Tanaka S, Takayama S, Schibler MJ, Fenton W, Reed JC. Investigation of the subcellular distribution of the bcl-2 oncoprotein: Residence in the nuclear envelope, endoplasmic reticulum, and outer mitochondrial membranes. Cancer Res. 1993;53:4701–4714. [PubMed] [Google Scholar]
- Lee AH, Iwakoshi NN, Glimcher LH. XBP-1 regulates a subset of endoplasmic reticulum resident chaperone genes in the unfolded protein response. Mol Cell Biol. 2003;23:7448–7459. doi: 10.1128/MCB.23.21.7448-7459.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipson KL, Fonseca SG, Urano F. Endoplasmic reticulum stress-induced apoptosis and auto-immunity in diabetes. Curr Mol Med. 2006;6:71–77. doi: 10.2174/156652406775574613. [DOI] [PubMed] [Google Scholar]
- Marciniak SJ, Yun CY, Oyadomari S, Novoa I, Zhang Y, Jungreis R, Nagata K, Harding HP, Ron D. CHOP induces death by promoting protein synthesis and oxidation in the stressed endoplasmic reticulum. Genes Dev. 2004;18:3066–3077. doi: 10.1101/gad.1250704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumoto M, Minami M, Takeda K, Sakao Y, Akira S. Ectopic expression of CHOP (GADD153) induces apoptosis in M1 myeloblastic leukemia cells. FEBS Lett. 1996;395:143–147. doi: 10.1016/0014-5793(96)01016-2. [DOI] [PubMed] [Google Scholar]
- McCullough KD, Martindale JL, Klotz LO, Aw TY, Holbrook NJ. Gadd153 sensitizes cells to endoplasmic reticulum stress by down-regulating Bcl2 and perturbing the cellular redox state. Mol Cell Biol. 2001;21:1249–1259. doi: 10.1128/MCB.21.4.1249-1259.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mori K. Tripartite management of unfolded proteins in the endoplasmic reticulum. Cell. 2000;101:451–454. doi: 10.1016/s0092-8674(00)80855-7. [DOI] [PubMed] [Google Scholar]
- Nadanaka S, Yoshida H, Kano F, Murata M, Mori K. Activation of mammalian unfolded protein response is compatible with the quality control system operating in the endoplasmic reticulum. Mol Biol Cell. 2004;15:2537–2548. doi: 10.1091/mbc.E03-09-0693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagawa T, Yuan J. Cross-talk between two cysteine protease families: Activation of caspase-12 by calpain in apoptosis. J Cell Biol. 2000;150:887–894. doi: 10.1083/jcb.150.4.887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagawa T, Zhu H, Morishima N, Li E, Xu J, Yankner BA, Yuan J. Caspase-12 mediates endoplasmic-reticulum-specific apoptosis and cytotoxicity by amyloid-beta. Nature. 2000;403:98–103. doi: 10.1038/47513. [DOI] [PubMed] [Google Scholar]
- Nishitoh H, Matsuzawa A, Tobiume K, Saegusa K, Takeda K, Inoue K, Hori S, Kakizuka A, Ichijo H. ASK1 is essential for endoplasmic reticulum stress-induced neuronal cell death triggered by expanded polyglutamine repeats. Genes Dev. 2002;16:1345–1355. doi: 10.1101/gad.992302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishitoh H, Saitoh M, Mochida Y, Takeda K, Nakano H, Rothe M, Miyazono K, Ichijo H. ASK1 is essential for JNK/SAPK activation by TRAF2. Mol Cell. 1998;2:389–395. doi: 10.1016/s1097-2765(00)80283-x. [DOI] [PubMed] [Google Scholar]
- Ohoka N, Yoshii S, Hattori T, Onozaki K, Hayashi H. TRB3, a novel ER stress-inducible gene, is induced via ATF4-CHOP pathway and is involved in cell death. EMBO J. 2005;24:1243–1255. doi: 10.1038/sj.emboj.7600596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pugazhenthi S, Nesterova A, Sable C, Heidenreich KA, Boxer LM, Heasley LE, Reusch JE. Akt/protein kinase B up-regulates Bcl-2 expression through cAMP-response element-binding protein. J Biol Chem. 2000;275:10761–10766. doi: 10.1074/jbc.275.15.10761. [DOI] [PubMed] [Google Scholar]
- Puthalakath H, O’Reilly LA, Gunn P, Lee L, Kelly PN, Huntington ND, Hughes PD, Michalak EM, McKimm-Breschkin J, Motoyama N, Gotoh T, Akira S, et al. ER stress triggers apoptosis by activating BH3-only protein Bim. Cell. 2007;129:1337–1349. doi: 10.1016/j.cell.2007.04.027. [DOI] [PubMed] [Google Scholar]
- Rao RV, Hermel E, Castro-Obregon S, del Rio G, Ellerby LM, Ellerby HM, Bredesen DE. Coupling endoplasmic reticulum stress to the cell death program: Mechanism of caspase activation. J Biol Chem. 2001;276:33869–33874. doi: 10.1074/jbc.M102225200. [DOI] [PubMed] [Google Scholar]
- Ron D, Habener JF. CHOP, a novel developmentally regulated nuclear protein that dimerizes with transcription factors C/EBP and LAP and functions as a dominant-negative inhibitor of gene transcription. Genes Dev. 1992;6:439–453. doi: 10.1101/gad.6.3.439. [DOI] [PubMed] [Google Scholar]
- Ron D, Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol. 2007;8:519–529. doi: 10.1038/nrm2199. [DOI] [PubMed] [Google Scholar]
- Rutkowski DT, Arnold SM, Miller CN, Wu J, Li J, Gunnison KM, Mori K, Sadighi Akha AA, Raden D, Kaufman RJ. Adaptation to ER stress is mediated by differential stabilities of pro-survival and pro-apoptotic mRNAs and proteins. PLoS Biol. 2006;4:e374. doi: 10.1371/journal.pbio.0040374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saleh M, Mathison JC, Wolinski MK, Bensinger SJ, Fitzgerald P, Droin N, Ulevitch RJ, Green DR, Nicholson DW. Enhanced bacterial clearance and sepsis resistance in caspase-12-deficient mice. Nature. 2006;440:1064–1068. doi: 10.1038/nature04656. [DOI] [PubMed] [Google Scholar]
- Sanges D, Marigo V. Cross-talk between two apoptotic pathways activated by endoplasmic reticulum stress: Differential contribution of caspase-12 and AIF. Apoptosis. 2006;11:1629–1641. doi: 10.1007/s10495-006-9006-2. [DOI] [PubMed] [Google Scholar]
- Scheuner D, Song B, McEwen E, Lui C, Laybutt R, Gillespie P, Saunders T, Bonner-Weir S, Kaufman RJ. Translational control is required for the unfolded protein response and in vivo glucose homeostasis. Mol Cell. 2001;7:1165–1176. doi: 10.1016/s1097-2765(01)00265-9. [DOI] [PubMed] [Google Scholar]
- Schroder M, Kaufman RJ. The mammalian unfolded protein response. Annu Rev Biochem. 2005;74:739–789. doi: 10.1146/annurev.biochem.73.011303.074134. [DOI] [PubMed] [Google Scholar]
- Schwartzman RA, Cidlowski JA. Apoptosis: The biochemistry and molecular biology of programmed cell death. Endocr Rev. 1993;14:133–151. doi: 10.1210/edrv-14-2-133. [DOI] [PubMed] [Google Scholar]
- Scorrano L, Oakes SA, Opferman JT, Cheng EH, Sorcinelli MD, Pozzan T, Korsmeyer SJ. BAX and BAK regulation of endoplasmic reticulum Ca2+: A control point for apoptosis. Science. 2003;300:135–139. doi: 10.1126/science.1081208. [DOI] [PubMed] [Google Scholar]
- Shen X, Ellis RE, Lee K, Liu CY, Yang K, Solomon A, Yoshida H, Morimoto R, Kurnit DM, Mori K, Kaufman RJ. Complementary signaling pathways regulate the unfolded protein response and are required for C. elegans development. Cell. 2001;107:893–903. doi: 10.1016/s0092-8674(01)00612-2. [DOI] [PubMed] [Google Scholar]
- Shi Y, An J, Liang J, Hayes SE, Sandusky GE, Stramm LE, Yang NN. Characterization of a mutant pancreatic eIF-2alpha kinase, PEK, and co-localization with somatostatin in islet delta cells. J Biol Chem. 1999;274:5723–5730. doi: 10.1074/jbc.274.9.5723. [DOI] [PubMed] [Google Scholar]
- Shi Y, Vattem KM, Sood R, An J, Liang J, Stramm L, Wek RC. Identification and characterization of pancreatic eukaryotic initiation factor 2 alpha-subunit kinase, PEK, involved in translational control. Mol Cell Biol. 1998;18:7499–7509. doi: 10.1128/mcb.18.12.7499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sok J, Wang XZ, Batchvarova N, Kuroda M, Harding H, Ron D. CHOP-dependent stress-inducible expression of a novel form of carbonic anhydrase VI. Mol Cell Biol. 1999;19:495–504. doi: 10.1128/mcb.19.1.495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Speel EJ, Hopman AH, Komminoth P. Amplification methods to increase the sensitivity of in situ hybridization: Play card(s) J Histochem Cytochem. 1999;47:281–288. doi: 10.1177/002215549904700302. [DOI] [PubMed] [Google Scholar]
- Tan Y, Dourdin N, Wu C, De Veyra T, Elce JS, Greer PA. Ubiquitous calpains promote caspase-12 and JNK activation during endoplasmic reticulum stress-induced apoptosis. J Biol Chem. 2006;281:16016–16024. doi: 10.1074/jbc.M601299200. [DOI] [PubMed] [Google Scholar]
- Tirasophon W, Lee K, Callaghan B, Welihinda A, Kaufman RJ. The endoribonuclease activity of mammalian IRE1 autoregulates its mRNA and is required for the unfolded protein response. Genes Dev. 2000;14:2725–2736. doi: 10.1101/gad.839400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tirasophon W, Welihinda AA, Kaufman RJ. A stress response pathway from the endoplasmic reticulum to the nucleus requires a novel bifunctional protein kinase/endoribonuclease (Ire1p) in mammalian cells. Genes Dev. 1998;12:1812–1824. doi: 10.1101/gad.12.12.1812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urano F, Wang X, Bertolotti A, Zhang Y, Chung P, Harding HP, Ron D. Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science. 2000;287:664–666. doi: 10.1126/science.287.5453.664. [DOI] [PubMed] [Google Scholar]
- van Gijlswijk RP, Zijlmans HJ, Wiegant J, Bobrow MN, Erickson TJ, Adler KE, Tanke HJ, Raap AK. Fluorochrome-labeled tyramides: Use in immunocytochemistry and fluorescence in situ hybridization. J Histochem Cytochem. 1997;45:375–382. doi: 10.1177/002215549704500305. [DOI] [PubMed] [Google Scholar]
- Wang XZ, Harding HP, Zhang Y, Jolicoeur EM, Kuroda M, Ron D. Cloning of mammalian Ire1 reveals diversity in the ER stress responses. EMBO J. 1998;17:5708–5717. doi: 10.1093/emboj/17.19.5708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Rutkowski DT, Dubois M, Swathirajan J, Saunders T, Wang J, Song B, Yau GD, Kaufman RJ. ATF6alpha optimizes long-term endoplasmic reticulum function to protect cells from chronic stress. Dev Cell. 2007;13:351–364. doi: 10.1016/j.devcel.2007.07.005. [DOI] [PubMed] [Google Scholar]
- Yamaguchi H, Wang HG. CHOP is involved in endoplasmic reticulum stress-induced apoptosis by enhancing DR5 expression in human carcinoma cells. J Biol Chem. 2004;279:45495–45502. doi: 10.1074/jbc.M406933200. [DOI] [PubMed] [Google Scholar]
- Yamamoto K, Sato T, Matsui T, Sato M, Okada T, Yoshida H, Harada A, Mori K. Transcriptional induction of mammalian ER quality control proteins is mediated by single or combined action of ATF6alpha and XBP1. Dev Cell. 2007;13:365–376. doi: 10.1016/j.devcel.2007.07.018. [DOI] [PubMed] [Google Scholar]
- Yaman I, Fernandez J, Liu H, Caprara M, Komar AA, Koromilas AE, Zhou L, Snider MD, Scheuner D, Kaufman RJ, Hatzoglou M. The zipper model of translational control: A small upstream ORF is the switch that controls structural remodeling of an mRNA leader. Cell. 2003;113:519–531. doi: 10.1016/s0092-8674(03)00345-3. [DOI] [PubMed] [Google Scholar]
- Ye J, Rawson RB, Komuro R, Chen X, Davé UP, Prywes R, Brown MS, Goldstein JL. ER stress induces cleavage of membrane-bound ATF6 by the same proteases that process SREBPs. Mol Cell. 2000;6:1355–1364. doi: 10.1016/s1097-2765(00)00133-7. [DOI] [PubMed] [Google Scholar]
- Yoneda T, Imaizumi K, Oono K, Yui D, Gomi F, Katayama T, Tohyama M. Activation of caspase-12, an endoplastic reticulum (ER) resident caspase, through tumor necrosis factor receptor-associated factor 2- dependent mechanism in response to the ER stress. J Biol Chem. 2001;276:13935–13940. doi: 10.1074/jbc.M010677200. [DOI] [PubMed] [Google Scholar]
- Yoshida H, Matsui T, Yamamoto A, Okada T, Mori K. XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell. 2001;107:881–891. doi: 10.1016/s0092-8674(01)00611-0. [DOI] [PubMed] [Google Scholar]
- Zeng L, Lu M, Mori K, Luo S, Lee AS, Zhu Y, Shyy JY. ATF6 modulates SREBP2-mediated lipogenesis. EMBO J. 2004;23:950–958. doi: 10.1038/sj.emboj.7600106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang K, Shen X, Wu J, Sakaki K, Saunders T, Rutkowski DT, Back SH, Kaufman RJ. Endoplasmic reticulum stress activates cleavage of CREBH to induce a systemic inflammatory response. Cell. 2006;24:587–599. doi: 10.1016/j.cell.2005.11.040. [DOI] [PubMed] [Google Scholar]
- Zhang K, Wong HN, Song B, Miller CN, Scheuner D, Kaufman RJ. The unfolded protein response sensor IRE1alpha is required at 2 distinct steps in B cell lymphopoiesis. J Clin Invest. 2005;115:268–281. doi: 10.1172/JCI21848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J, Lhoták S, Hilditch BA, Austin RC. Activation of the unfolded protein response occurs at all stages of atherosclerotic lesion development in apolipoprotein E-deficient mice. Circulation. 2005;111:1814–1821. doi: 10.1161/01.CIR.0000160864.31351.C1. [DOI] [PubMed] [Google Scholar]
- Zinszner H, Kuroda M, Wang X, Batchvarova N, Lightfoot RT, Remotti H, Stevens JL, Ron D. CHOP is implicated in programmed cell death in response to impaired function of the endoplasmic reticulum. Genes Dev. 1998;12:982–995. doi: 10.1101/gad.12.7.982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zong WX, Li C, Hatzivassiliou G, Lindsten T, Yu QC, Yuan J, Thompson CB. Bax and Bak can localize to the endoplasmic reticulum to initiate apoptosis. J Cell Biol. 2003;162:59–69. doi: 10.1083/jcb.200302084. [DOI] [PMC free article] [PubMed] [Google Scholar]