

Abstract
Interleukin-31 (IL-31) is a recently described T cell-derived cytokine, mainly produced by T helper type 2 cells and related to the IL-6 cytokine family according to its structure and receptor. IL-31 is the ligand for a heterodimeric receptor composed of a gp130-like receptor (GPL) associated with the oncostatin M receptor (OSMR). A link between IL-31 and atopic dermatitis was shown by studying the phenotype of IL-31 transgenic mice and IL-31 gene haplotypes in patients suffering from dermatitis. In this study, we generated a potent IL-31 antagonist formed by external portions of OSMR and GPL fused with a linker. This fusion protein, OSMR-L-GPL, consisting of 720 amino acids, counteracted the binding of IL-31 to its membrane receptor complex and the subsequent signaling events involving the STATs and MAPK pathways. Neutralizing effects were found in IL-31-sensitive cell lines, including brain-derived cells and primary cultures of keratinocytes.
Keywords: Cytokines/Interleukins, Protein/Protein-Protein Interactions, Protein/Fusion, Receptors/Cytokine, Signal Transduction, Antagonist, Interleukin-31
Introduction
Interleukin-31 (IL-31)4 is a newly described T cell-derived cytokine, mostly produced by T helper type 2 cells, and involved in promoting skin disorders and regulating other allergic diseases such as asthma (1–5). This cytokine is related to the IL-6 cytokine family by its structure and receptor complex (6). In addition to IL-6 and IL-31, this family encompasses IL-11, IL-27, oncostatin M (OSM), leukemia inhibitory factor (LIF), ciliary neurotrophic factor, cardiotrophin-like cytokine, and neuropoietin. IL-31 is a four-helix bundle cytokine comprising two long crossover loops and one short loop (1). Its heterodimeric receptor is composed of gp130-like receptor (GPL, also known as GLM-R or IL-31RA (1, 7, 8)) and oncostatin M receptor (OSMR) (9, 10).
GPL was cloned as a member of the type I cytokine receptor family (7, 8, 11). Its name reflects its close relationship to gp130, the shared receptor subunit of the IL-6-type cytokines (12, 13). Both receptor genes are located in tandem on chromosome 5. GPL displays common structural motifs with the family of type I cytokine receptors, i.e. the cytokine-binding domain (CBD) with two pairs of conserved cysteine residues and a WSXWS box in the extracellular region. IL-31 binds to GPL before recruiting OSMR to form the high affinity receptor (14). On the other hand, OSMR can also heterodimerize with gp130 to form a functional type II OSM receptor (9, 10). The type I OSM receptor consists of the low affinity chain LIF receptor associated with gp130 (15, 16).
High expression levels of GPL have been found in tissues involved in reproduction, the myelomonocytic lineage, spleen, thymus, lung, skin, and trachea (7, 8). OSMR is also coexpressed in most of these tissues (10). The binding of IL-31 to its receptor leads to an intracellular activation via the recruitment of the Jak1, Jak2, STAT1, STAT3, and STAT5 signaling pathways, as well as the phosphatidylinositol 3-kinase/Akt cascade. SHP-2 and Shc adapter molecules are also recruited and contribute to an increased activation of the MAPK pathway in response to IL-31 (17–19).
An association of IL-31 with atopic dermatitis was found in the IL-31 transgenic mice phenotype that mimics the human pathology (20). Dillon et al. (1) showed that mice treated with intradermal injection of IL-31 or transgenic mice overexpressing IL-31 presented increased scratching behavior and developed severe dermatitis.
Several subsequent studies have demonstrated the involvement of IL-31 in atopic skin inflammation (3, 5, 21). Atopic individuals displayed an increased tendency to produce higher levels of IL-31 in response to external trigger factors, which may contribute to the development of pruritus. Moreover, IL-31 is overexpressed in pruritic atopic compared with nonpruritic psoriatic skin inflammation (5). Circulating cutaneous lymphocyte antigen-positive T cells produced IL-31 after activation and cutaneous lymphocyte antigen-positive skin-homing T cells, located in the epidermis of patients with atopic dermatitis, expressed IL-31 mRNA (3). Thus, IL-31 may contribute to the development of atopic dermatitis-induced skin inflammation and pruritus.
Soluble cytokine receptors, which are involved in the regulation of a number of physiological and pathological situations, can behave either as agonists or antagonists for a cytokine. The soluble counterparts of α-membrane chains, such as soluble IL-6 or ciliary neurotrophic factor receptors, are able to increase the functional responses to their respective ligands (22, 23). In contrast, β-chain-derived soluble receptors, such as soluble gp130 or soluble OSMR, can neutralize the responses to IL-6, IL-11, ciliary neurotrophic factor, OSM and IL-31 (24–26).
A new generation of cytokine antagonists is based on the fusion of soluble receptor fragments to trap their cognate ligand with a high affinity. To associate the external portions of receptor chains, two strategies were used: fusing soluble receptor(s) to the Fc domain of human IgG1 (27) or fusing two different ligand-binding domains together with a linker (28, 29).
In the present study, we generated a potent IL-31 antagonist consisting of the distal domains of OSMR associated through a linker to the GPL cytokine binding domain. This fusion protein, OSMR-L-GPL, specifically recognized IL-31 and displayed potent neutralizing activities on various IL-31 responsive models.
EXPERIMENTAL PROCEDURES
Cells and Reagents
Cos-M6, HEK 293, Go-G-UVM, HsB2, CEM, Jurkat, KE37, and 1301 cell lines were cultured in RPMI medium 1640 supplemented with 10% fetal calf serum. Ba/F3 cells stably transfected with gp130, OSMR, and GPL were grown in the same culture medium supplement with 1 ng/ml murine IL-3. The human Th2 lymphocyte cell line derived from an atheroma infiltrate was maintained in culture with 10 ng/ml IL-2 (R&D Systems, Oxon, UK) and a stimulation through the CD3 and CD28 pathways. Human keratinocytes were purchased from PromoCell (Heidelberg, Germany) and maintained in keratinocyte growth medium-2 (PromoCell) following the manufacturer's instructions. Human IL-31 tagged with V5 and polyhistidine epitopes (V5-His) was produced in the laboratory as described previously (18). Human recombinant OSM was purchased from R&D Systems (Oxon, UK). The OSMR-Fc, GPL-Fc fusion proteins, monoclonal anti-OSMR (AN-V2) and IgG1 isotype control antibodies were produced in the laboratory. A polyclonal anti-GPL antibody (T3C15) was raised by immunizing rabbits with a 15-mer peptide chosen in the AB loop of the receptor as described previously (8). Polyclonal antibodies directed against phospho-STAT1 (Tyr701), phospho-STAT3 (Tyr705), phospho-MAPK (Thr202/Tyr204) and MAPK were bought from Upstate Technology (Lake Placid, NY). Polyclonal anti-STAT3 and anti-STAT1 antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Monoclonal anti-V5 antibodies coupled or not to peroxidase were purchased from Invitrogen. The anti-mouse and anti-rabbit peroxidase labeled antibodies were purchased from Clinisciences (Montrouge, France). Monoclonal anti-IL-31 antibodies AN-31C2 (IgG1), AN-31B71 (IgG2a), and AN-31A31 (IgG1) were generated in the laboratory according to conventional protocols. The polyclonal anti-human IL-31 antibody was purchased from R&D Systems (Oxon, UK).
Cloning of the Fusion Protein OSMR-L-GPL
The linker, corresponding to the short proximal part of the IL-6Rα extracellular domain (residues Ala323–Val362), was amplified by PCR (28). ClaI and EcoRI sites were introduced in the sense and antisense primers, respectively. The PCR product was cloned into pcDNA3.1/D TOPO V5-His vector. A fragment corresponding to the CBD regions and the Ig-like domain of OSMR (residues Met1–Ala428) was amplified by PCR. A HindIII and a ClaI restriction sites were introduced in the sense and antisense primers, respectively. The amplification product was cloned into the pcDNA3.1/D TOPO V5-His plasmid containing the linker. The fragment corresponding to the CBD of GPL (residues Leu21–Glu225) was amplified by PCR and introduced in 3′ side of the construct using EcoRI and NotI restriction sites. The construct was verified by DNA sequencing.
Quantification of IL-31 by ELISA
A 96-well microtiter plate was coated with two monoclonal antibodies (AN-31B71 and AN-31A31) at 5 μg/ml overnight at 4 °C. After saturation with 20% sucrose, 0.1 m Tris, pH 7.2, the wells were incubated with culture supernatants dilutions for 6 h at 37 °C. After three washing steps using 0.05% Tween 20 PBS, the wells were incubated with a biotinylated polyclonal anti-human IL-31 antibody (0.25 μg/ml) in 0.1% BSA, 0.01% Tween 20 PBS overnight at 4 °C. After washing steps, streptavidin-poly-horseradish peroxidase (Sanquin, Amsterdam, The Netherlands) (diluted 1:10,000 in 0.1% BSA, 0.01% Tween 20 PBS) was added in the wells for one hour at 37 °C. Binding of the streptavidin-horseradish peroxidase to the antibodies was visualized by the addition of 2,2′-azino-bis(3-ethylbenzthiazoline-6-sulphonic acid) substrate solution. The absorbance at 405 nm was determined using an ELISA reader.
Protein Expression and Purification
The HEK 293 cell line was stably transfected with pcDNA3.1D/TOPO OSMR-L-GPL V5-His using the Exgen transfection reagent (Euromedex, Souffelweyersheim, France). Cell supernatants were cleared by centrifugation and stored at −20 °C. They were submitted to an anion exchange column (GE Healthcare, Les Ullis, France) before an affinity purification step using nickel-Sepharose column chromatography (GE Healthcare). Purified fractions were submitted to SDS-PAGE, silver staining, and Western blot analyses. Some preliminary experiments were carried out by transient expression of the protein in Cos-M6 cells using the DEAE-dextran method. The chimeric cytokines (OSM-Fc and IL-31-Fc), consisting of the cytokines coupled to the Fc part of human IgG1, were expressed in Cos-M6 cells. After a 72-h transfection period, culture media were collected, and the protein was immunopurified on a protein A column. The native form of IL-31 was purified from the supernatant of a Th2 cell line. The supernatant was submitted to an affinity purification column using a monoclonal antibody directed against IL-31 (AN-31C2) coupled to Sepharose 4B beads (GE Healthcare). Purified fractions were submitted to SDS-PAGE and silver stained.
Endoglycosidase Treatment
OSMR-L-GPL was diluted in 1% Brij 96 lysis buffer and treated with 25 units/ml of N-glycosidase F (Roche, Mannheim, Germany) for 12 h at 37 °C before anti-V5 Western blot analyses.
Immunoprecipitation, Co-precipitation, and Western Blot Analyses
For immunoprecipitation experiments, samples were incubated overnight at 4 °C with indicated antibodies. Complexes were isolated using protein A beads, and the samples were then submitted to SDS-PAGE and transferred onto Immobilon membranes. The membranes were subsequently incubated overnight at 4 °C with the primary antibody, before being incubated with the appropriate peroxidase-labeled secondary antibody. The reaction was visualized by chemiluminescence according to the manufacturer's instructions (GE Healthcare). The density of immunodetected bands were integrated using the ImageJ program. Data analysis were performed by GraphPad Prism (GraphPad Software, San Diego, CA).
For co-precipitation experiments, chimeric cytokines fused with the human immunoglobulin Fc segment were incubated in the presence of the indicated soluble receptors overnight at 4 °C. Complexes were then isolated using protein A beads and submitted to Western blot analyses as described above.
Phosphorylation Assays
When indicated, OSMR-L-GPL was incubated for 2 h with the cytokines before being added for 10 min to the cells. Cells were lysed in SDS sample buffer (62.5 mm Tris-HCl, pH 6.8, 2% SDS, 10% glycerol, 50 mm dithiothreitol, 0.1% bromphenol blue), sonicated, and submitted to Western blot analyses as described above. Membranes were stripped in 0.1 m glycine, pH 2.5, for 2h, and neutralized in 1 m Tris-HCl, pH 7.6, before reblotting with the antibody recognizing all isoform proteins.
Luciferase Reporter Gene Assay
Cells were transfected, using kit V reagent (Amaxa, Gaithersburg, MD), with 2.5 μg of the SIEM-Luc reporter gene. This construct contains three consensus binding sites located upstream of a thymidine kinase minimal promoter (30). Twenty-four h after transfection, cells were serum-starved for 6 h and then incubated overnight with either medium alone or with the indicated combinations of cytokines and OSMR-L-GPL. Cells were washed with PBS, and 70 μl of lysis buffer was added to the wells (0.1 m KHPO4, pH 7.8, 0.1% Triton X-100) before adding luciferase. The luciferase activity was measured using a Packard TopCount luminometer (Meriden, CT).
Quantitative PCR
Total RNAs were extracted using the RNeasy micro kit (Qiagen), according to the manufacturer's protocol. Then, RNAs were reversed transcribed into cDNA using random hexamer primers and Superscript II reverse transcriptase (Invitrogen). Primers were designed using Primer3 software. PCR amplification reactions were carried out in duplicates with iQ-SYBR Green Supermix (Bio-Rad, Hercules, CA) in a 15 μl-reaction volume containing 200 nm of primers and 5 ng of cDNA, and using a Chromo4TM System (Bio-Rad). Thermal cycling was initiated with a 3-min incubation at 95 °C followed by 40 cycles of 95 °C for 10 s, 55 °C for 15 s, and 72 °C for 15 s. The ΔCt method was retained for quantification, and the GAPDH gene was used for normalization as described previously (31).
Proliferation Assays
The Ba/F3 stably expressing OSM receptor and IL-31 receptor chains (gp130, OSMRβ, and GPL chains) and Go-G-UVM cell lines were seeded in 96-well plates at a concentration of 5.103 cells/well in RPMI 5% fetal calf serum or 1.5.103 cells/well in RPMI 1% fetal calf serum, respectively. Serial dilutions of tested cytokines and fusion protein were performed in triplicate. After a 72-h incubation period, 0.5 μCi of [3H]Tdr (GE Healthcare, Les Ullis, France) was added to each well for the last 4 h of the culture, and the incorporated radioactivity was measured by scintillation counting (Packard Top count luminometer).
RESULTS
Design and Engineering of the Fusion Protein OSMR-L-GPL
OSMR-L-GPL fusion protein was designed to contain the minimal domains of OSMR and GPL required for high affinity IL-31 binding (Fig. 1A). The OSMR segment corresponding to the CBDs and the Ig-like domain, was placed at the N terminus side of the construct. It included the OSMR signal peptide to facilitate the secretion of the fusion protein. The CBD fragment of GPL was linked to the C terminus of the OSMR construction. A short flexible peptide corresponding to the membrane proximal part of IL-6Rα extracellular domain was added between both receptor parts. This linker was previously used for its weak immunogenicity and its flexibility, allowing the fusion protein to adopt an appropriate conformation (26). V5 and His tag epitopes were added at the C terminus of the OSMR-L-GPL fusion protein to facilitate affinity purification and Western blot detection. The resulting chimeric protein OSMR-L-GPL consisted of 720 amino acids (Fig. 1B).
FIGURE 1.

Schematic presentations of IL-31 receptor and OSMR-l-GPL fusion protein. A, schematic representations of the modular structure of IL-31 receptor components (OSMR and GPL) and of the fusion protein OSMR-L-GPL. The fusion protein contained the CBDs and the Ig-like domain of OSMR bound to the CBD of GPL. The linker consisted of a short flexible fragment of the extracellular proximal part of the IL-6Rα chain. Fn III, fibronectin III. B, schematic representation of the fusion protein OSMR-L-GPL. Numbers refer to the amino acid positions in the fusion protein. SP, signal peptide; N-ter, N-terminal; C-ter, C-terminal.
Expression and Characterization of OSMR-L-GPL
To produce the fusion protein, the cDNA encoding for OSMR-L-GPL was stably transfected in the HEK 293 cell line. Culture supernatants were submitted to a concentration step by an anion exchange column. OSMR-L-GPL was purified on a nickel column. The fractions obtained after elution were then submitted to SDS-PAGE before silver staining and Western blotting analyses. The soluble polypeptide displayed a molecular mass of 150 kDa corresponding to a mature protein of 145 kDa after subtracting molecular mass of the tags (Fig. 2A).
FIGURE 2.

Detection and biochemical analysis of OSMR-l-GPL. A, OSMR-L-GPL purified from a Ni+-agarose column was analyzed by SDS-PAGE. The gel was silver stained, and Western blotting analysis was performed using a monoclonal anti-V5 tag antibody. B and C, Cos-M6 cells were transfected with the mock vector or the vector-encoding OSMR-L-GPL. B, cell supernatants were immunoprecipitated with an isotype control monoclonal antibody (IgG1, 5 μg/ml) or with a monoclonal anti-OSMR antibody (AN-V2, 5 μg/ml). Immunorevelation was performed using an anti-GPL polyclonal antibody (T3C15, 1 μg/ml). IP, immunoprecipitation; WB, Western blot; BSA, bovine serum albumin. C, cell supernatants were treated for 12 h at 37 °C in the presence or not of 1 unit of N-glycosidase F (N-glyco F). Immunorevelation was performed using the monoclonal anti-V5 tag antibody.
To further characterize the fusion protein, we performed immunoprecipitation experiments using a monoclonal antibody directed against the extracellular part of OSMR (anti-OSMR, AN-V2). A signal corresponding to the expected molecular weight of OSMR-L-GPL was detected after an immunorevelation using antibody specifically recognizing GPL (anti-GPL, T3C15) (Fig. 2B). This result showed that OSMR and GPL domains were correctly encoded by the OSMR-L-GPL plasmid.
Deglycosylation experiments were then carried out using N-glycosidase F to remove N-linked polysaccharides from the protein backbone. A 25-kDa molecular mass shift (150 kDa versus 125 kDa) was observed (Fig. 2C). This result was in agreement with the predicted occupancy of 10 N-glycosylation sites, using the NetNGlyc 1.0 Prediction site.
IL-31 Directly Contacts OSMR-L-GPL
To analyze the ability and the specificity of the fusion protein to bind IL-31, immunoprecipitation experiments were carried out. Different concentrations of IL-31 fused to a Fc fragment, IL-31-Fc, were added to OSMR-L-GPL (Fig. 3A). Contact of IL-31 to the fusion protein was clearly demonstrated after precipitation and Western blotting analyses. A Kd of 2.4 nm was determined using GraphPad Prism software after signal intensity integration.
FIGURE 3.

Binding analysis of OSMR-l-GPL. Different concentrations of cytokines fused to the IgG1 Fc fragment were incubated in the presence of OSMR-L-GPL (4 nm) for 16 h at 4 °C before a pulldown with protein A beads. A, IL-31/OSMR-L-GPL association was detected using a monoclonal anti-V5 antibody coupled to peroxidase. An anti-IgG1 polyclonal antibody coupled to peroxidase was used to control the cytokines-Fc loading. Signals were integrated, and GraphPad software was used to plot the association curve to determine the Kd value. B, association with OSM. A.U., arbitrary units; WB, Western blot.
Because OSMR is a shared receptor subunit for both IL-31 and OSM, a putative association of OSM to the OSMR portion of the fusion protein was also investigated by adding OSM-Fc to OSMR-L-GPL (Fig. 3B). Under the same conditions, no association, or a very weak association between OSM and OSMR-L-GPL could be detected, underlying the specific recognition of OSMR-L-GPL for IL-31. These results are in agreement with previous studies demonstrating a direct association of OSM to gp130 but not to OSMR alone (10, 32, 33). Because IL-31 specifically bound to OSMR-L-GPL, we next analyzed the ability of the fusion protein to antagonize functional activities of IL-31.
Antagonistic Activity of OSMR-L-GPL on IL-31 Signaling
Previously, we reported that interaction of IL-31 with its membrane complex receptor on the glioblastoma cell line Go-G-UVM induced the recruitment of the following signaling pathways: Jak1, Jak2, STAT1, -3, -5, phosphatidylinositol 3-kinase/Akt, and MAPK (17, 18). Therefore, the ability of OSMR-L-GPL to neutralize the biological activity of IL-31 was studied by measuring the phosphorylation level of STAT1, STAT3, and MAPK in Go-G-UVM cells in response to IL-31 or IL-31 plus OSMR-L-GPL. Addition of IL-31 or OSM (2 nm) induced a robust tyrosine phosphorylation of the studied signaling proteins (Fig. 4A). The activation of the STATs and the MAPKs induced by IL-31 was entirely abrogated when the fusion protein OSMR-L-GPL was added (Fig. 4A). As expected, the addition of the soluble chimeric receptor had no effect on the phosphorylation levels of STAT1, STAT3, and MAPK in response to OSM. These findings indicated that OSMR-L-GPL was able to specifically antagonize IL-31 activity in signaling experiments.
FIGURE 4.

Antagonistic activity of OSMR-l-GPL on IL-31 signaling. A, 2 nm of OSM or IL-31 were preincubated with 4 nm of purified OSMR-L-GPL or with an equivalent volume of PBS for 2 h at 37 °C. Mixed reagents were added to the Go-G-UVM glioblastoma cell line for 10 min at 37 °C. STAT1, STAT3, and MAPK phosphorylation levels were determined by Western blot analyses. B, Go-G-UVM cells were transfected with a SIEM-Luc construct in order to analyze STAT3 transcriptional activity. OSM or IL-31 (4 nm) were mixed with a 10-fold molar excess of OSMR-L-GPL or with an equivalent volume of PBS as control. The proteins were added to Go-G-UVM cells for a 16-h culture period. The STAT3 activation level was measured by using a luminometer. S.D. are represented by error bars. C, increasing concentrations of OSMR-L-GPL were incubated for 2 h with IL-31 (0.4 nm) or added simultaneously with IL-31 to the cells for 10 min. The STAT3 tyrosine phosphorylation levels were determined by Western blot analysis.
The effect of OSMR-L-GPL on STAT3 transcriptional activity in response to IL-31 was analyzed in the same cell line (Fig. 4B). Go-G-UVM cells were transfected with a reporter construct containing three STAT3 consensus binding sites located upstream of a thymidine kinase minimal promoter (28). After a 48-h culture period, cells were stimulated for an additional 16 h with IL-31, OSM and the fusion protein. Luciferase transcription, which was induced by both cytokines, was specifically neutralized when IL-31 was added to the cells in combination with OSMR-L-GPL.
Subsequently, we performed a dose response analysis of OSMR-L-GPL using the same cells. Preincubation or simultaneous addition to the cells of both IL-31 and OSMR-L-GPL led to a similar inhibition of STAT3 phosphorylation (Fig. 4C). Results from Fig. 4C showed that a 10-fold molar excess of OSMR-L-GPL was sufficient to fully inhibit IL-31 biological activity.
Neutralization of IL-31 Native Form by OSMR-L-GPL
To determine the ability of OSMR-L-GPL to neutralize endogenous IL-31, we looked for a consistent source of native IL-31 (Fig. 5A). An important IL-31 secretion was detected in the culture supernatant of a human Th2 cell line. These cells were expanded, and the culture supernatant was harvested for IL-31 immunopurification (Fig. 5B). Similarly to that observed with recombinant glycosylated IL-31, several native IL-31 isoforms ranging from 24 to 33 kDa were observed. We then studied the ability of OSMR-L-GPL to neutralize the biological activity of the native form of IL-31 (Fig. 5C). STAT3 phosphorylation was analyzed in Go-G-UVM cell line following its contact with native, or mammalian recombinant IL-31, and OSMR-L-GPL. A similar STAT3 phosphorylation inhibition was observed in response to either recombinant glycosylated or native IL-31, when OSMR-L-GPL was added to the cells.
FIGURE 5.

Neutralization of IL-31 native form by OSMR-l-GPL. A, secretion of native IL-31 was measured in human cell lines by ELISA as indicated under “Experimental Procedures.” Error bar represents S.D. B, native IL-31 was immunopurified on an AN-C2 anti-IL-31 monoclonal antibody column and analyzed by SDS-PAGE and silver staining. C, after a 2-h contact between recombinant glycosylated IL-31 or native IL-31 (0.4 nm) with increasing concentrations of OSMR-L-GPL, the mixture was added to the Go-G-UVM cell line for 10 min. The STAT3 tyrosine phosphorylation levels were determined by Western blot analysis.
Comparison of OSMR-L-GPL with Other IL-31 Inhibitors
To compare the inhibitory activities of soluble receptors and OSMR-L-GPL, we studied the ability for the different receptor forms to inhibit STAT3 phosphorylation induced by IL-31 (Fig. 6A). Soluble forms of OSMR and GPL, separately or in combination, were not able to decrease the STAT3 phosphorylation level observed in response to IL-31. In contrast, the same amount of OSMR-L-GPL fully suppressed the phosphorylation of STAT3 induced by IL-31. These results established that the fusion protein displayed a potent inhibitory effect on the IL-31 response, compared with separately expressed sOSMR and sGPL.
FIGURE 6.

Comparison of OSMR-L-GPL with other inhibitors. A, IL-31 (2 nm) was preincubated with 15 nm of the soluble receptors sGPL or sOSMR, or a combination of both, or OSMR-L-GPL for 2 h at 37 °C. Mixed reagents were added to the Go-G-UVM cell line for 10 min at 37 °C. The STAT3 tyrosine phosphorylation levels were determined by Western blot analysis. B, following a preincubation of IL-31 (0.4 nm) with increasing concentrations of the purified OSMR-L-GPL or a polyclonal neutralizing anti-IL-31 antibody for 2 h at 37 °C, the Go-G-UVM cell line was activated for 10 min at 37 °C. The STAT3 phosphorylation levels were determined by Western blot analysis.
In the following experiment, we compared the inhibitory activities of OSMR-L-GPL to a neutralizing polyclonal antibody on the IL-31-induced STAT3 activation in the Go-G-UVM cells (Fig. 6B). The cells were stimulated with a constant amount of IL-31 preincubated with increasing concentrations of the antibody. 20 nm of the antibody could completely inhibit the phosphorylation of STAT3 induced by IL-31. In comparison, a 10-fold lower concentration of OSMR-L-GPL (2 nm) was sufficient to fully inhibit the response. Obtained results indicated that the OSMR-L-GPL antagonist was 10-fold more potent than the neutralizing antibody.
OSMR-L-GPL Is a Potent IL-31 Antagonist in Cell Proliferation Assays
We then studied the ability of the fusion protein to antagonize IL-31 in a proliferation assay using the murine Ba/F3 cell line transfected with gp130, OSMR, and GPL. This cell line proliferates in response to IL-31 or OSM (18). Increasing concentrations of OSMR-L-GPL were added to fixed amounts of IL-31 or OSM (80 and 8 pm, respectively, corresponding to concentrations leading to a 50% of maximum signal in the assay), and then a tritiated thymidine incorporation was carried out. OSMR-L-GPL neutralized the activity of IL-31 in a dose-dependent manner. Half-inhibition was observed for a concentration of 0.67 nm corresponding to an 8-fold molar excess of the fusion protein compared with IL-31 (Fig. 7). In contrast, OSMR-L-GPL had no significant effect on OSM-induced cell proliferation, underlining the specificity of the fusion protein for IL-31.
FIGURE 7.

Proliferation response of transfected Ba/F3 cell line upon stimulation with OSM, IL-31 and OSMR-L-GPL. Ba/F3 cells stably transfected with OSM and IL-31 receptor components were cultured in triplicates with 2-fold dilutions of purified OSMR-L-GPL, or an equivalent volume of PBS, and fixed amounts of OSM (8 ρm) or IL-31 (80 ρm). After a 48-h culture period, a [3H]thymidine pulse was carried out for 4 h, and the amount of incorporated radioactivity was determined.
OSMR-L-GPL Suppresses Cytostatic Effect, Morphological Changes, and Gene Induction upon Stimulation with IL-31
A second functional assay was developed to assess the specificity of the OSMR-L-GPL neutralizing effect. This functional assay was based on the cytostatic activity of OSM toward tumoral cell lines (34, 35). Similarly to OSM, we observed that addition of IL-31 to glioblastoma-derived cell lines led to an inhibition of cell proliferation (Fig. 8A) and an accumulation of the cells in the G0 phase.5 In the absence of cytokines, Go-G-UVM cells presented an elongated appearance with cytoplasmic extensions and intercellular contacts. After a 5-day cytokine exposure, Go-G-UVM cells underwent morphological changes and became rounder, and the cellular extensions wore off (Fig. 8B). Addition of the fusion protein to the cultures prevented, in a specific manner, the inhibition of cell proliferation and the morphological changes observed in the presence of IL-31 (Fig. 8).
FIGURE 8.

Inhibition of cell morphological changes and cytostatic effect induced by IL-31. A, Go-G-UVM cells were cultured in triplicates with 2-fold dilutions of purified OSMR-L-GPL or with an equivalent volume of PBS and with IL-31 or OSM (1.67 nm). After a 5-day culture period, a [3H]thymidine pulse was carried out, and the incorporated radioactivity was determined. B, phase contrast micrographs of Go-G-UVM cells after a 5-day culture period in the presence of indicated cytokines (1.67 nm) with purified OSMR-L-GPL or PBS. c.p.m., counts per min.
We then studied the effect of the fusion protein on proinflammatory genes induced by IL-31 in Go-G-UVM cells. These genes were previously identified from a screening array experiment.6 We tested the ability of OSMR-L-GPL to modulate their expressions in response to IL-31 or OSM in the Go-G-UVM cells (Fig. 9). Interestingly, the proinflammatory gene signature (IL-6, VEGF-A, CXCL16) and also SOCS3 were induced by both OSM and IL-31, but the OSMR-L-GPL fusion protein specifically reversed the IL-31 effect.
FIGURE 9.

Analysis of OSMR-l-GPL activity on IL-31 gene induction. OSM or IL-31 (2 nm) were mixed with 4 nm of purified OSMR-L-GPL or with an equivalent volume of PBS. After a 2-h contact at 37 °C, proteins were added to Go-G-UVM cell cultures for 6 h. After RNA extraction and cDNA synthesis, quantitative reverse transcription-PCR analyses of IL-6, vascular endothelial growth factor A (VEGFA), CXCL16, and SOCS3 genes expression were carried out (*, p < 0.05, Student's t test). Error bars represent S.D.
Neutralization of IL-31 Activity by OSMR-L-GPL in Human Epidermal Keratinocytes
We recently reported that OSM is a potent keratinocyte activator and plays an important role in cutaneous inflammatory responses (36). Moreover, IL-31 induced release of chemokines by human epidermal keratinocytes, and after IL-31 intradermal injection, a neutrophil infiltration as well as a thickening of epidermis were observed (1). Given these observations, we studied the neutralizing potential of OSMR-L-GPL in primary cultures of human keratinocytes. Both IL-31 and OSM induced STAT3 recruitment, but the fusion protein specifically inhibited STAT3 phosphorylation induced by IL-31 (Fig. 10).
FIGURE 10.
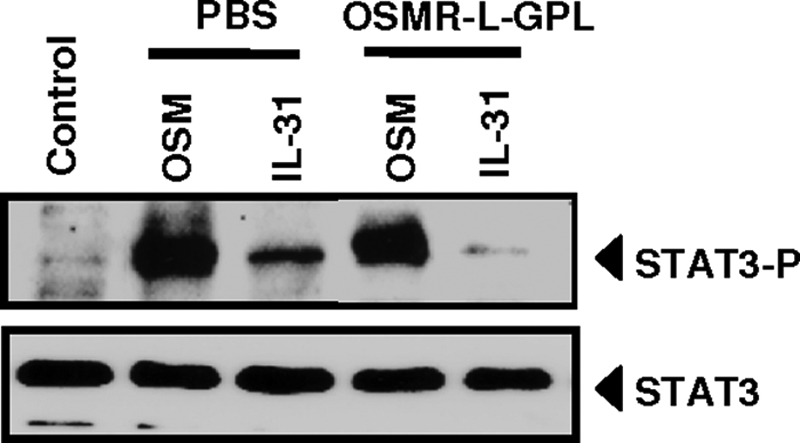
Neutralization of IL-31 activity by OSMR-l-GPL in human epidermal keratinocytes. OSM or IL-31 (2 nm) were mixed with 4 nm of purified OSMR-L-GPL or with an equivalent volume of PBS for 2h at 37 °C, before being added to primary cultures of human keratinocytes for 10 min at 37 °C. Phospho-STAT3 and STAT3 protein levels were measured by western-blot analyses.
DISCUSSION
Recent studies indicate that IL-31 is a key trigger of atopic dermatitis and possibly other allergic diseases such as asthma (1–5). An agent that neutralizes this cytokine may have a beneficial role in these diseases.
In the present study, we developed an approach to neutralize IL-31 by fusing the extracellular portions of its two receptor chains. The first five extracellular modules of OSM receptor, composed of a half CBD followed by an Ig-like domain and a second complete CBD, were associated to GPL/IL-31R CBD. We had previously observed that the CBD of GPL was involved in direct binding of IL-31, through a site 2 motif (14). Conversely, IL-31 interaction with the OSMR chain was based on contact between the site 3 of the cytokine and the Ig-like domain of OSMR (26).
OSMR is also engaged in the formation of a second cytokine receptor by heterodimerizing with gp130 in the presence of OSM (9, 10). We previously reported that OSM first binds to the CBD of gp130 via its site 2 and then to the Ig-like domain of OSMR via its site 3 (33, 37). OSM displays numerous functional properties, such as cytostatic activities toward solid tumor-derived cell lines, proliferation as well as activation of endothelial cells, induction of acute phase protein synthesis by liver and induction of proinflammatory responses in in vivo models. Strikingly, inactivation of OSMR in the mouse led to a nearly normal phenotype with only a slight decrease in platelet number (38), suggesting that this pathway is likely recruited in pathological situations rather than in healthy animals.
In the present work, we particularly focused on the functional properties of OSMR-L-GPL, using human cells that were sensitive to both IL-31 and OSM. This focus allowed us to precisely determine the specificity of the developed fusion protein. In all tested systems, OSMR-L-GPL only neutralized IL-31 without affecting its neighboring cytokine, OSM.
IL-31 and OSM belong to a larger family of cytokines that encompasses IL-6, IL-11, LIF, ciliary neurotrophic factor, cardiotrophin-like cytokine, and IL-27 (39). These cytokines bind to their receptors through different binding sites referred to as sites 1, 2 and 3. These mediators can be subdivided in two subgroups: those recruiting three receptor components via sites 1, 2 and 3, and a second subgroup (IL-31, OSM, LIF, IL-27), requiring the involvement of only two receptor subunits recognized through interactions with the sites 2 and 3 (14). A recent report described the possibility of neutralizing the LIF response by using a mouse construction inhibiting interactions between the receptor subunits and sites 2 and 3 of the cytokine (40). Our findings extend this approach to the human IL-31 receptor and emphasize the specificity of the observed neutralizing response.
Obtained results indicate that OSMR-L-GPL is a potent inhibitor for IL-31 functional responses. Determination of its affinity led to a 2.4 nm Kd value, which is in agreement with the OSMR-L-GPL amount required to neutralize the functional activity of IL-31 in the different bioassays we did use. As previously described for IL-6, we demonstrated the superior activity of the fusion protein compared with the separate soluble receptors sOSMR and sGPL. Nevertheless, high concentrations of soluble GPL and soluble OSMR can display a low level inhibitory action as we previously observed I a different cell background (26). We also showed that the inhibitory activity of our antagonist is more potent than a neutralizing polyclonal antibody directed against IL-31. Recently, similar results were also observed concerning an IL-6 inhibitor (41). Thus, our results confirm that, to trap a cytokine and antagonize its effects, a fusion of the receptor subunit fragments might be an interesting strategy with promising applications.
Interestingly, we also found that IL-31 displayed cytostatic activity toward glioblastoma cell lines, similar to OSM. IL-31 inhibited the proliferation of these cells, induced morphological modifications (cells became rounder and lost their cytoplasmic extensions), and increased the transcription of proinflammatory genes. OSMR-L-GPL suppressed these properties of IL-31 on glioblastoma-derived cells.
Until today, the main functional properties or deleterious activities of IL-31 were reported in skin models and cutaneous pathologies (1, 2, 4, 42). Overexpression of IL-31 or its subcutaneous injection in mice led to increased scratching behavior and a development of severe dermatitis. In line with these observations, IL-31 was increased in an animal model of atopic dermatitis (NC/Nga mice) and in patients suffering from atopic dermatitis (3–5). Recently, an IL-31 single nucleotide polymorphism was associated with this pathology (43). Here, we showed that OSMR-L-GPL was also able to neutralize IL-31 mediated signaling in primary cultures of human keratinocytes.
Interestingly, a recent study reported the ability for a neutralizing IL-31 monoclonal antibody to ameliorate scratching behavior in a mouse model of dermatitis (44). Additional work will be required to develop a murine IL-31 antagonist based on a similar approach and to validate its ability to neutralize IL-31 in animal models of skin diseases. The present work and further studies will contribute to the design of new approaches to treat cutaneous pathologies.
Acknowledgments
We thank Plate forme Automatisée de Developpement d'Anticorps Monoclonaux (PADAM) for monoclonal antibodies and Catherine Guillet for careful review of the manuscript. We also thank David Perret for initial technical advice.
This work was supported by Grant 5176 from the Association pour la Recherche contre le Cancer and by the Ciblage Moléculaire et Applications Thérapeutiques (CIMATH) Program of the Région Pays de la Loire.
C. Diveu, unpublished observation.
L. Preisser, unpublished data.
- IL-31
- interleukin-31
- GPL
- gp130-like receptor
- OSMR
- oncostatin M receptor
- LIF
- leukemia inhibitory factor
- ELISA
- enzyme-linked immunosorbent assay
- PBS
- phosphate-buffered saline
- MAPK
- mitogen-activated protein kinase
- CBD
- cytokine-binding domain.
REFERENCES
- 1.Dillon S. R., Sprecher C., Hammond A., Bilsborough J., Rosenfeld-Franklin M., Presnell S. R., Haugen H. S., Maurer M., Harder B., Johnston J., Bort S., Mudri S., Kuijper J. L., Bukowski T., Shea P., Dong D. L., Dasovich M., Grant F. J., Lockwood L., Levin S. D., LeCiel C., Waggie K., Day H., Topouzis S., Kramer J., Kuestner R., Chen Z., Foster D., Parrish-Novak J., Gross J. A. (2004) Nat. Immunol. 5, 752–760 [DOI] [PubMed] [Google Scholar]
- 2.Takaoka A., Arai I., Sugimoto M., Yamaguchi A., Tanaka M., Nakaike S. (2005) Eur. J. Pharmacol. 516, 180–181 [DOI] [PubMed] [Google Scholar]
- 3.Bilsborough J., Leung D. Y., Maurer M., Howell M., Boguniewicz M., Yao L., Storey H., LeCiel C., Harder B., Gross J. A. (2006) J. Allergy Clin. Immunol. 117, 418–425 [DOI] [PubMed] [Google Scholar]
- 4.Takaoka A., Arai I., Sugimoto M., Honma Y., Futaki N., Nakamura A., Nakaike S. (2006) Exp. Dermatol. 15, 161–167 [DOI] [PubMed] [Google Scholar]
- 5.Sonkoly E., Muller A., Lauerma A. I., Pivarcsi A., Soto H., Kemeny L., Alenius H., Dieu-Nosjean M. C., Meller S., Rieker J., Steinhoff M., Hoffmann T. K., Ruzicka T., Zlotnik A., Homey B. (2006) J. Allergy Clin. Immunol. 117, 411–417 [DOI] [PubMed] [Google Scholar]
- 6.Heinrich P. C., Behrmann I., Haan S., Hermanns H. M., Müller-Newen G., Schaper F. (2003) Biochem. J. 374, 1–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ghilardi N., Li J., Hongo J. A., Yi S., Gurney A., de Sauvage F. J. (2002) J. Biol. Chem. 277, 16831–16836 [DOI] [PubMed] [Google Scholar]
- 8.Diveu C., Lelièvre E., Perret D., Lak-Hal A. H., Froger J., Guillet C., Chevalier S., Rousseau F., Wesa A., Preisser L., Chabbert M., Gauchat J. F., Galy A., Gascan H., Morel A. (2003) J. Biol. Chem. 278, 49850–49859 [DOI] [PubMed] [Google Scholar]
- 9.Thoma B., Bird T. A., Friend D. J., Gearing D. P., Dower S. K. (1994) J. Biol. Chem. 269, 6215–6222 [PubMed] [Google Scholar]
- 10.Mosley B., De Imus C., Friend D., Boiani N., Thoma B., Park L. S., Cosman D. (1996) J. Biol. Chem. 271, 32635–32643 [DOI] [PubMed] [Google Scholar]
- 11.Yamaoka K., Okayama Y., Kaminuma O., Katayama K., Mori A., Tatsumi H., Nemoto S., Hiroi T. (2009) Int. Arch. Allergy Immunol. 149, 73–76 [DOI] [PubMed] [Google Scholar]
- 12.Hibi M., Murakami M., Saito M., Hirano T., Taga T., Kishimoto T. (1990) Cell 63, 1149–1157 [DOI] [PubMed] [Google Scholar]
- 13.Bravo J., Heath J. K. (2000) EMBO J. 19, 2399–2411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Le Saux S., Rousseau F., Barbier F., Ravon E., Grimaud L., Danger Y., Froger J., Chevalier S., Gascan H. (2010) J. Biol. Chem. 285, 3470–3477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gearing D. P., Thut C. J., VandeBos T., Gimpel S. D., Delaney P. B., King J., Price V., Cosman D., Beckmann M. P. (1991) EMBO J. 10, 2839–2848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gearing D. P., Comeau M. R., Friend D. J., Gimpel S. D., Thut C. J., McGourty J., Brasher K. K., King J. A., Gillis S., Mosley B., et al. (1992) Science 255, 1434–1437 [DOI] [PubMed] [Google Scholar]
- 17.Dreuw A., Radtke S., Pflanz S., Lippok B. E., Heinrich P. C., Hermanns H. M. (2004) J. Biol. Chem. 279, 36112–36120 [DOI] [PubMed] [Google Scholar]
- 18.Diveu C., Lak-Hal A. H., Froger J., Ravon E., Grimaud L., Barbier F., Hermann J., Gascan H., Chevalier S. (2004) Eur. Cytokine Netw. 15, 291–302 [PubMed] [Google Scholar]
- 19.Chattopadhyay S., Tracy E., Liang P., Robledo O., Rose-John S., Baumann H. (2007) J. Biol. Chem. 282, 3014–3026 [DOI] [PubMed] [Google Scholar]
- 20.Leung D. Y., Bieber T. (2003) Lancet 361, 151–160 [DOI] [PubMed] [Google Scholar]
- 21.Neis M. M., Peters B., Dreuw A., Wenzel J., Bieber T., Mauch C., Krieg T., Stanzel S., Heinrich P. C., Merk H. F., Bosio A., Baron J. M., Hermanns H. M. (2006) J. Allergy Clin. Immunol. 118, 930–937 [DOI] [PubMed] [Google Scholar]
- 22.Davis S., Aldrich T. H., Stahl N., Pan L., Taga T., Kishimoto T., Ip N. Y., Yancopoulos G. D. (1993) Science 260, 1805–1808 [DOI] [PubMed] [Google Scholar]
- 23.Honda M., Yamamoto S., Cheng M., Yasukawa K., Suzuki H., Saito T., Osugi Y., Tokunaga T., Kishimoto T. (1992) J. Immunol. 148, 2175–2180 [PubMed] [Google Scholar]
- 24.Narazaki M., Yasukawa K., Saito T., Ohsugi Y., Fukui H., Koishihara Y., Yancopoulos G. D., Taga T., Kishimoto T. (1993) Blood 82, 1120–1126 [PubMed] [Google Scholar]
- 25.Sporeno E., Paonessa G., Salvati A. L., Graziani R., Delmastro P., Ciliberto G., Toniatti C. (1994) J. Biol. Chem. 269, 10991–10995 [PubMed] [Google Scholar]
- 26.Diveu C., Venereau E., Froger J., Ravon E., Grimaud L., Rousseau F., Chevalier S., Gascan H. (2006) J. Biol. Chem. 281, 36673–36682 [DOI] [PubMed] [Google Scholar]
- 27.Economides A. N., Carpenter L. R., Rudge J. S., Wong V., Koehler-Stec E. M., Hartnett C., Pyles E. A., Xu X., Daly T. J., Young M. R., Fandl J. P., Lee F., Carver S., McNay J., Bailey K., Ramakanth S., Hutabarat R., Huang T. T., Radziejewski C., Yancopoulos G. D., Stahl N. (2003) Nat. Med. 9, 47–52 [DOI] [PubMed] [Google Scholar]
- 28.Ancey C., Küster A., Haan S., Herrmann A., Heinrich P. C., Müller-Newen G. (2003) J. Biol. Chem. 278, 16968–16972 [DOI] [PubMed] [Google Scholar]
- 29.Renné C., Kallen K. J., Müllberg J., Jostock T., Grötzinger J., Rose-John S. (1998) J. Biol. Chem. 273, 27213–27219 [DOI] [PubMed] [Google Scholar]
- 30.Lelièvre E., Plun-Favreau H., Chevalier S., Froger J., Guillet C., Elson G. C., Gauchat J. F., Gascan H. (2001) J. Biol. Chem. 276, 22476–22484 [DOI] [PubMed] [Google Scholar]
- 31.Vandesompele J., De Preter K., Pattyn F., Poppe B., Van Roy N., De Paepe A., Speleman F. (2002) Genome Biol. 3, RESEARCH0034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liu J., Modrell B., Aruffo A., Marken J. S., Taga T., Yasukawa K., Murakami M., Kishimoto T., Shoyab M. (1992) J. Biol. Chem. 267, 16763–16766 [PubMed] [Google Scholar]
- 33.Olivier C., Auguste P., Chabbert M., Lelièvre E., Chevalier S., Gascan H. (2000) J. Biol. Chem. 275, 5648–5656 [DOI] [PubMed] [Google Scholar]
- 34.Zarling J. M., Shoyab M., Marquardt H., Hanson M. B., Lioubin M. N., Todaro G. J. (1986) Proc. Natl. Acad. Sci. U.S.A. 83, 9739–9743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Grant S. L., Begley C. G. (1999) Mol. Med. Today 5, 406–412 [DOI] [PubMed] [Google Scholar]
- 36.Boniface K., Diveu C., Morel F., Pedretti N., Froger J., Ravon E., Garcia M., Venereau E., Preisser L., Guignouard E., Guillet G., Dagregorio G., Pène J., Moles J. P., Yssel H., Chevalier S., Bernard F. X., Gascan H., Lecron J. C. (2007) J. Immunol. 178, 4615–4622 [DOI] [PubMed] [Google Scholar]
- 37.Plun-Favreau H., Perret D., Diveu C., Froger J., Chevalier S., Lelièvre E., Gascan H., Chabbert M. (2003) J. Biol. Chem. 278, 27169–27179 [DOI] [PubMed] [Google Scholar]
- 38.Tanaka M., Hirabayashi Y., Sekiguchi T., Inoue T., Katsuki M., Miyajima A. (2003) Blood 102, 3154–3162 [DOI] [PubMed] [Google Scholar]
- 39.Murakami M., Kamimura D., Hirano T. (2004) Growth Factors 22, 75–77 [DOI] [PubMed] [Google Scholar]
- 40.Metz S., Naeth G., Heinrich P. C., Müller-Newen G. (2008) J. Biol. Chem. 283, 5985–5995 [DOI] [PubMed] [Google Scholar]
- 41.Wiesinger M. Y., Haan S., Wüller S., Kauffmann M. E., Recker T., Küster A., Heinrich P. C., Müller-Newen G. (2009) Chem. Biol. 16, 783–794 [DOI] [PubMed] [Google Scholar]
- 42.Heise R., Neis M. M., Marquardt Y., Joussen S., Heinrich P. C., Merk H. F., Hermanns H. M., Baron J. M. (2009) J. Invest Dermatol. 129, 240–243 [DOI] [PubMed] [Google Scholar]
- 43.Schulz F., Marenholz I., Fölster-Holst R., Chen C., Sternjak A., Baumgrass R., Esparza-Gordillo J., Grüber C., Nickel R., Schreiber S., Stoll M., Kurek M., Rüschendorf F., Hubner N., Wahn U., Lee Y. A. (2007) J. Allergy Clin. Immunol. 120, 1097–1102 [DOI] [PubMed] [Google Scholar]
- 44.Grimstad O., Sawanobori Y., Vestergaard C., Bilsborough J., Olsen U. B., Grønhøj-Larsen C., Matsushima K. (2009) Exp. Dermatol. 18, 35–43 [DOI] [PubMed] [Google Scholar]