

Abstract
Drosophila Crumbs has been reported to attenuate Notch signaling by inhibition of γ-secretase cleavage at the wing margins. γ-Secretase is an intramembrane protease that is responsible for the generation of amyloid-β (Aβ) peptides from the β-amyloid precursor protein (APP). Here, we re-examined γ-secretase inhibition by human CRB2, which is the most abundant Crumbs ortholog in the brain. Transfected CRB2 inhibited proteolytic production of Aβ and APP intracellular domains from APP C-terminal fragments in HEK293 and SH-SY5Y cells. Conversely, knockdown of endogenous CRB2 increased γ-secretase cleavage products in SH-SY5Y cells. CRB2 inhibition of γ-cleavage was also detected in cell-free assays. CRB2 interacted with the γ-secretase complex, but was not a competitive substrate for γ-cleavage. The transmembrane domain of CRB2 was indispensable for inhibition of Aβ generation and mediated CRB2 binding with the γ-secretase complex. In addition, the cytoplasmic domain appeared to play a supportive role in γ-secretase inhibition, whereas mutational disruption of the two protein-binding motifs involved in the formation of cell adhesion complexes did not affect γ-secretase inhibition. Co-overexpression of presenilin-1 or APH-1 abrogated γ-secretase inhibition probably through prevention of the incorporation of CRB2 into the γ-secretase complex. Our results suggest that CRB2 functions as an inhibitory binding protein that is involved in the formation of a mature but inactive pool of the γ-secretase complex.
Keywords: Diseases/Alzheimer Disease, Diseases/Amyloid, Enzymes/Proteolytic, Membrane/Enzymes, Proteases/Secretases, Protein/Protein-Protein Interactions, Crumbs, Presenilin
Introduction
An emerging class of intramembrane proteases cleaves the transmembrane (TM)2 domains of proteins within the hydrophobic environment of the membrane, which releases polypeptides from the membrane into the extracellular milieu and the cytoplasm (reviewed in Refs. 1–3). The liberated peptides possess diverse biological activities including transcription factor and growth factor activities. These cleavage events are highly regulated and serve as key processes of signal transduction pathways. γ-Secretase is a representative intramembrane aspartyl protease and mediates cleavage of type I membrane proteins such as β-amyloid precursor protein (APP) and Notch receptors within their membrane-spanning segments (4). γ-Secretase cleavage exhibits no or relaxed sequence specificity, and, to date, more than 60 transmembrane proteins have been described as γ-secretase substrates. It is known that extracellular cleavage of the substrates at the juxtamembrane region is a prerequisite for γ-secretase proteolysis whether ectodomain shedding is constitutive or induced by ligand stimulation.
γ-Secretase cleavage of APP is subsequent to ectodomain shedding that is constitutively executed by α-secretase and β-secretase, and releases p3 and amyloid-β (Aβ) peptides into the extracellular space and APP intracellular domains (AICD) into the cytoplasm. The released AICD fragments translocate into the nucleus and regulate the transcription of specific target genes. On the other hand, the extracellular Aβ peptide is considered a cause of Alzheimer disease (AD). Thus, excessive Aβ peptides oligomerize to cause neuronal dysfunction and degeneration in the brains of AD patients resulting in the manifestation of severe dementia.
γ-Secretase is a membrane-embedded, multimeric protein complex composed of four membrane proteins: presenilin (PS), nicastrin (NCT), APH-1, and PEN-2. Presenilins (PS1 and PS2) have a catalytic center, although three other components are required for activity. Upon assembly and activation of the complex, PS is endoproteolyzed into an N-terminal fragment and a C-terminal fragment (CTF). The mechanism of intrinsic regulation of γ-secretase activity remains to be elucidated, although some regulatory proteins have recently been reported. To date, many of the reported regulatory proteins act as inhibitors of Aβ secretion by distinct mechanisms. An integral membrane component of coatomer-coated vesicles, termed p23 or TMP21, has dual inhibitory effects on Aβ generation both directly binding the γ-secretase complex to inhibit its activity and altering the trafficking of APP and/or γ-secretase complexes (5, 6). CD147, a member of the immunoglobulin superfamily, has also been reported as an inhibitory component of γ-secretase complexes (7); however, contradictory results showing that CD147 did not alter the proteolysis of APP have been reported (8). In contrast, retention in endoplasmic reticulum 1 (Rer1) decreases Aβ secretion through down-regulation of γ-secretase assembly by competing with APH-1 for NCT binding (9). Rer1 also binds to and retains unassembled PEN-2 in the endoplasmic reticulum (10). Phospholipase D1 has been reported to interrupt the incorporation of PEN-2 into γ-secretase complexes thereby inhibiting Aβ production (11).
Recently, Herranz et al. (12) reported that the Drosophila Crumbs transmembrane protein attenuates Notch signaling by inhibition of γ-secretase cleavage of the Notch receptor at the wing margins. A well known function of Crumbs orthologs is to organize a macromolecular protein scaffold at the intracellular face of the membrane, which is involved in the maintenance of apico-basal cell polarity and adherens junctions (reviewed in Ref. 13). Of three human Crumbs orthologs, CRB2 is expressed at a higher level in the brain, whereas CRB1 and CRB3 are mainly expressed in the retina and epithelia, respectively (14). In this study, we examined γ-secretase inhibition by human CRB2 in mammalian cells and attempted to explore its underlying mechanism. A previous report failed to detect any effect of truncated CRB2 on γ-secretase cleavage of APP and Notch in cultured HEK293 cells (15). However, using overexpression of full-length (FL) CRB2 as well as knockdown of endogenous CRB2 in mammalian cells we found that CRB2 bound the γ-secretase complex and inhibited proteolytic production of Aβ and AICD. The TM domain of CRB2 was indispensable for this inhibitory activity and for binding to PS1, whereas the cytoplasmic domain played a supportive role in the inhibition. CRB2 appears to be an inhibitory binding protein for the γ-secretase complex but is not a competitive substrate.
EXPERIMENTAL PROCEDURES
cDNA Constructs
A plasmid encoding FL human CRB2 cDNA was generated by subcloning the product obtained by reverse transcription-PCR of human retina mRNA into pBluescript II (Stratagene). For construction of a CRB2 expression vector without a tag, CRB2 cDNA was subcloned into pcDNA4 (Invitrogen). For construction of CRB2 with an internal FLAG tag (CRB2-iFLAG), the FLAG epitope tag sequence was inserted immediately downstream of the signal sequence (amino acid residues 1–36). CRB2 N-terminal truncation mutants (NT1, NT2, NT3, and NT4) were constructed by PCR using CRB2-iFLAG as a template. CRB2-NT5 was constructed by removing the FLAG tag from NT4. T-CRB2 that lacks the N-terminal 350 residues including the signal sequence but has an N-terminal FLAG tag was constructed by PCR. A TM domain chimera (NT5-TMC), in which the TM domain of CRB2-NT5 was replaced with the TM domain of the human TrkB receptor tyrosine kinase, was generated by PCR. CRB2 C-terminal truncation mutants (CT1 and CT2), a missense mutant at the FERM (protein band 4.1, ezrin, radixin, and moesin)-binding motif (Tyr1258 to Ala, Pro1260 to Ala, and Glu1264 to Ala; NT4-mFERM), and a PDZ (PSD-95, Discs Large and ZO-1)-binding motif (ERLI)-deleted mutant (NT4-ΔPDZ) were generated by PCR-based site-directed mutagenesis. The expression plasmids for CRB1 and CRB3 were obtained from Dr. Ben Margolis (University of Michigan Medical School). The plasmids encoding PS1 and NCT have been previously described (16). To construct APH-1b and PEN-2 expression plasmids, reverse transcription-PCR products were subcloned into pcDNA3-FLAG. Expression vectors for signal peptide peptidase-like protease 2b (SPPL2b) and human tumor necrosis factor-α (TNF-α) were constructed by subcloning their reverse transcription-PCR products into pcDNA4-Myc and pcDNA3-FLAG, respectively. The sequences of all constructs were confirmed by sequencing.
Antibodies and Reagents
Anti-human CRB2 polyclonal antibodies were raised in rabbits against synthetic polypeptides that were composed of the extracellular sequence between amino acid residues 276 and 292 (CLQRSDPALYGGVQAAF) for CRB2(N1) and the cytoplasmic sequence between residues 1248 and 1266 with an added N-terminal Cys residue (C+ARKRRQSEGTYSPSQQEVA) for CRB2(C). Each antibody was purified with a peptide-conjugated affinity column. The following antibodies were purchased from the respective manufacturer's: mouse anti-PS1 loop and rabbit anti-APP CTF (Chemicon); rabbit anti-NCT (N1660), mouse anti-FLAG tag (M2), and mouse anti-β-actin (Sigma); mouse anti-PS1 N-terminal fragment (IBL); rabbit anti-APH-1 (Covance), rabbit anti-PEN-2 (Zymed Laboratories Inc.), rabbit anti-Sec61α (Upstate), mouse anti-Flottilin-2 (BD Biosciences), mouse anti-KDEL (Stressgen); and mouse anti-Myc tag (9E10) (Santa Cruz Biotechnology). The γ-secretase inhibitor, {1S-benzyl-4R-[1S-carbamoyl-2-phenylethylcarbamoyl-1S-3-methylbutylcarbamoyl]-2R-hydroxy-5-phenylpentyl}carbamic acid tert-butyl ester (L685,458), was obtained from Calbiochem.
Cell Lines and cDNA Transfection
HEK293 cells stably expressing wild-type APP (HEK/wtAPP) or APP-SC99 (HEK/C99), and SH-SY5Y neuroblastoma cells stably expressing wild-type APP (SY5Y/wtAPP), have been previously described (17, 18). Neuronal differentiation of SH-SY5Y cells was induced by treatment with 160 nm 12-O-tetradecanoylphorbol-13-acetate and 1 mm dibutyryl cAMP for 7 days. cDNA transfection was carried out using Lipofectamine 2000 reagent (Invitrogen) according to the manufacturer's protocol.
RNA Interference
For small interfering RNA (siRNA)-mediated knockdown of human CRB2, an siGENOME ON-TARGETplus duplex J-0180054-07 (CRB2-siRNA-3, 5′-CCUAAACGAUGGCCAUUGGUU-3′ (sense), 5′-CCAAUGGCCAUCGUUUAGGUU-3′ (antisense)) and a nontargeting control siRNA D-001810-05 were obtained from Dharmacon. SH-SY5Y cells in 6-well plates were transfected with 125 pmol of the siRNA duplexes using Lipofectamine RNAi MAX (Invitrogen) according to the manufacturer's protocol. Culture medium was changed 24 h after transfection, and cells were then cultured for another 24 h to prepare conditioned medium for Aβ measurement. Cells were harvested 48 h after siRNA transfection.
Immunoblotting and Immunoprecipitation
For immunoblotting, cells or membrane fractions were lysed in a lysis buffer (25 mm HEPES, pH 7.5, 150 mm NaCl, 2 mm EDTA, Complete protease inhibitor mixture (Roche Diagnostics) and 10% glycerol) containing 1% Nonidet P-40. Immunoblotting was performed as previously described (19). For immunoprecipitation, cells or brain homogenates were lysed in lysis buffer containing 1% CHAPSO. After preclearing with protein A-Sepharose CL-4B (Amersham Biosciences), lysates were incubated with antibody for 2 h, followed by overnight incubation with protein A-Sepharose at 4 °C. The immunoprecipitates were washed 3 times with 1% CHAPSO buffer and subjected to immunoblotting.
Blue Native (BN) Gel Electrophoresis
BN polyacrylamide gel electrophoresis was performed as previously described (20). In brief, microsomal membranes were prepared and then solubilized with ice-cold BN lysis buffer containing 0.7% n-dodecyl β-d-maltoside, 500 mm 6-aminocaproic acid, 50 mm BisTris, pH 7.0, plus Complete protease inhibitor mixture. The lysate was centrifuged at 100,000 × g for 20 min, and the supernatant was mixed at a ratio of 1:2 by volume with BN sample buffer containing 0.75% (w/v) Coomassie Brilliant Blue G-250, 500 mm 6-aminocaproic acid, 50 mm BisTris (pH 7.0), 30% glycerol, and 0.7% n-dodecyl β-d-maltoside. The samples were resolved in 6–13% BN gels, and transferred onto polyvinylidene difluoride membranes. Urease (hexamer 545 kDa, trimer 272 kDa, dimer 132 kDa) and bovine serum albumin (66 kDa) (Sigma) were used as standard proteins.
Cell-free AICD Generation and AICD Reporter Assays
A cell-free AICD generation assay was carried out based on a previous report (17). Briefly, crude membrane fractions obtained from HEK/C99 cells were solubilized in lysis buffer containing 0.5% CHAPSO and 5 mm 1,10-phenanthroline. Protein concentrations were determined with a protein DC assay kit (Bio-Rad) using bovine serum albumin as the standard, and adjusted to 1 mg/ml. After incubation for 0, 1, or 4 h at 37 °C, equal amounts of lysates were analyzed by immunoblotting with an anti-APP CTF antibody.
For the AICD reporter assay, the GAL4 DNA-binding domain/VP16, fused to the cytoplasmic tail of APP695 (APP-GV) or AICD (AICD-GV), was subcloned into pcDNA3 (Invitrogen). A GAL4 reporter plasmid encoding firefly luciferase (pG5luc) and a plasmid encoding Renilla luciferase (pRL-TK) were obtained from Promega. Subconfluent HEK293 cells in 6-well plates were transiently transfected with the indicated amount of DNA for each construct. To control for transfection efficiency, 20 ng of pRL-TK was added to each plasmid mixture. The cells were lysed 24 h after transfection, and firefly and Renilla luciferase activities were quantified using a dual luciferase reporter assay system (Promega) and a luminometer AB-2250 (Atto). Firefly luciferase values were standardized to the corresponding Renilla luciferase values. The Notch-ΔE reporter assay for Notch intracellular domain (NICD)-dependent transcriptional transactivation was described previously (21).
Cell-free Aβ Generation Assay
The generation of Aβ40 and Aβ42 peptides in CHAPSO-solubilized γ-secretase enzyme preparations using an exogenous recombinant APP-C99-FLAG substrate has been described previously (22). Essentially, the γ-secretase enzyme from HEK293 cell membranes was solubilized in a buffer containing 1% (w/v) CHAPSO, 50 mm HEPES-NaOH (pH 7.0), 150 mm NaCl, 5 mm CaCl2, 5 mm MgCl2, and Complete protease inhibitor mixture. The enzyme solution was then adjusted with the same buffer without CHAPSO to give a final detergent concentration of 0.25% CHAPSO (w/v). For the in vitro γ-secretase reaction, 10 μl of solubilized enzyme (0.6–0.7 mg/ml of protein) was incubated for 6 h at 37 °C with the recombinant APP-C99-FLAG substrate in the presence of 0.1% phosphatidylcholine in a final volume of 40 μl.
Aβ Measurement
The levels of Aβ40 and Aβ42 were measured using sandwich ELISA kits (WAKO Pure Chemical Industries). Each sample was assayed in triplicate and the values were averaged. The Aβ levels in the conditioned medium were normalized to the total protein concentrations of the cell lysates.
Lipid Raft Isolation
Lipid rafts were isolated as described previously (23) with some modifications. HEK293 or SH-SY5Y cells were washed three times with ice-cold PBS and lysed on ice for 20 min in 0.5% Lubrol lysis buffer (Serva) supplemented with Complete protease inhibitor mixture. The cell lysates were homogenized with 10 strokes of a Dounce homogenizer and centrifuged for 5 min at 1,000 × g at 4 °C in a microcentrifuge to remove insoluble material and nuclei. Lysates were adjusted to a final sucrose concentration of 45%, placed at the bottom of ultracentrifuge tubes, and overlaid with 6 ml of 35% and 3 ml of 5% sucrose. Lysates were ultracentrifuged at 4 °C in an SW41 rotor (Beckman) for 18 h at 38,000 × g. Twelve 1-ml fractions were collected from the top to the bottom and analyzed by immunoblotting.
RESULTS
Expression of CRB2 in Mammalian Cells and Brains
To assess protein expression of CRB2, affinity-purified polyclonal antipeptide antibodies, CRB2(N1) and CRB2(C), were generated against unique amino acid sequences at the extracellular and cytoplasmic regions, respectively, of human CRB2. Immunoblot analysis of CRB2-transfected HEK293 cells using either of these antibodies indicated that CRB2 migrated as a doublet of bands corresponding to molecular masses of 180 and 160 kDa (Fig. 1A). The electrophoretic mobility of CRB2 was increased by digestion with either endoglycosidase H (Endo H) or peptide-N-glycosidase F. Although the entire band of 160 kDa was completely shifted following either Endo H or peptide-N-glycosidase F digestion, the 180-kDa band was partially resistant to Endo H but completely sensitive to peptide-N-glycosidase F treatment (Fig. 1B). This result indicated that CRB2 is post-translationally modified by complex N-linked glycosylation.
FIGURE 1.

Expression and characterization of the human CRB2 protein. A, immunoblots of transfected human CRB2. Blots of HEK293 cells transfected with CRB2 or mock plasmids were probed with N-terminal, CRB2(N1) (left), or C-terminal, CRB2(C) (right), antibodies. B, glycosylation of CRB2. CRB2-transfected HEK293 cell lysates were treated with vehicle (Control), Endo H, or peptide-N-glycosidase F (PNGase F) for 1 h at 37 °C. The blot was probed with CRB2(N1). C, immunoblot of endogenous CRB2 in monkey brain. CRB2-transfected cell lysate was loaded as a size marker. The blot was probed with CRB2(N1) (left) or CRB2(N1) preabsorbed with the synthetic peptide immunogen (right). Asterisks in A–C indicate nonspecific bands.
To confirm endogenous expression of CRB2 in the mammalian brain, we immunoblotted cerebral cortex lysates of freshly prepared cynomolgus monkey brain with the CRB2(N1) antibody. This antibody recognized endogenous CRB2 that migrated at a molecular mass of ∼180 kDa (Fig. 1C). The specificity of this antibody was verified by the lack of immunoreactivity when antibody that had been preabsorbed with the synthetic peptide immunogen was used. These data indicated that the 180-kDa band represents the predominant species of endogenous CRB2 in mammalian brains and that the CRB2 expression plasmid can be used to analyze CRB2 cellular function.
CRB2 Suppresses Aβ Secretion and AICD Generation in Cultured Cell-based Assays
To determine the effect of CRB2 on γ-secretase function we first assayed the effect of CRB2 expression on γ-secretase cleavage activity. γ-Secretase cleaves APP and Notch at multiple sites; C-terminal cleavage releases AICD and NICD, respectively, into the cytoplasm, whereas N-terminal cleavage yields several Aβ and Notch-β species, respectively. We first confirmed that CRB2 suppresses Notch-mediated transcriptional activation as previously reported (12) using a Notch-ΔE luciferase reporter assay (Fig. 2A). Notch-ΔE lacks the ectodomain of Notch receptor and is constitutively cleaved by γ-secretase. The specificity of this cell-based assay for γ-secretase cleavage was demonstrated by treatment with a potent γ-secretase inhibitor, L685,458, which attenuated luminescence in a dose-dependent manner. Co-expressed CRB2 clearly decreased γ-secretase-dependent emission of luciferase luminescence but did not suppress the γ-secretase-independent luminescence emission evoked by NICD transfection. Similar results were obtained in an APP luciferase reporter assay (Fig. 2B). These results suggested that CRB2 inhibits γ-secretase cleavage and thereby proteolytic production of NICD and AICD.
FIGURE 2.

CRB2 suppresses Aβ secretion and AICD generation. A and B, luciferase reporter assays of γ-secretase cleavage of Notch-ΔE (A) and APP (B). HEK293 cells in 6-well plates were transiently transfected with the indicated combinations of expression (+) and mock (m) plasmids. Eight hours after transfection, the culture medium was replaced by Dulbecco's modified Eagle's medium, 10% fetal bovine serum containing the indicated concentration of the γ-secretase inhibitor L685,458. Luciferase activity was analyzed 24 h post-transfection as described under ”Experimental Procedures.“ Relative luciferase activity is expressed as fold-activation (relative to the level of a reporter gene in the presence of Notch-ΔE or APP-GV after normalization with co-transfected Renilla luciferase activity). The values shown are the mean ± S.D. for three experiments. C, Aβ secretion from CRB2-transfected cells. Mock- or CRB2-expression plasmids were transiently transfected into HEK/wtAPP cells, and Aβ40 and Aβ42 levels in the conditioned medium were measured by ELISA. *, p < 0.01 versus the mock control by an unpaired, two-tailed Student's t test. The expression level of CRB2 was determined by immunoblotting of the cell lysates (right). D, Aβ secretion from CRB2-knockdown cells. Differentiated SY5Y/wtAPP cells were transfected with a control siRNA duplex or with a CRB2-targeted siRNA duplex (CRB2 k/d), and Aβ levels in the conditioned medium were measured by ELISA. *, p < 0.01 versus the control. The RNA interference-mediated knockdown for CRB2 was confirmed by immunoblotting of the cell lysates (right). E, a cell-free assay of Aβ generation. Membrane fractions from HEK293 cells transfected with mock or CRB2 plasmids were solubilized in CHAPSO-lysis buffer, and then incubated with a recombinant C99-FLAG substrate for 6 h at 37 °C in the presence or absence of L685,458. Aβ levels were measured by a sandwich ELISA. *, p < 0.01 versus the mock control. F, a cell-free AICD generation assay. Crude membrane fractions obtained from HEK/C99 cells were solubilized in 0.5% CHAPSO-lysis buffer containing 5 mm 1,10-phenanthroline. Protein concentrations were determined and adjusted to 1 mg/ml. After incubation for 0, 1, or 4 h at 37 °C, equivalent amounts of lysates were subjected to immunoblotting and probed with the anti-APP CTF antibody. The graph shows the relative density of the AICD bands (mean ± S.D. from three experiments).
We next investigated the effect of CRB2 on Aβ secretion using cultured mammalian cells. When transfected into HEK/wtAPP cells, CRB2 decreased the level of secreted Aβ40 and Aβ42 (Fig. 2C). No difference in the degree of inhibition of Aβ40 and Aβ42 was observed. A similar decrease was induced following transfection of CRB2 into human neuroblastoma SY5Y/wtAPP cells and HEK/C99 cells. The latter cell line stably expressed β-secretase-cleaved APP-C99, the direct substrate of γ-secretase. These results suggested that CRB2 inhibition is observed in non-neuronal and neuronal cell lines and occurs at the level of γ-secretase cleavage but not at the preceding β-secretase cleavage.
To confirm the role of CRB2 in γ-secretase cleavage, we assayed the effect of reducing the cellular level of CRB2 by siRNA on Aβ secretion (Fig. 2D). Expression of endogenous CRB2 in differentiated SH-SY5Y cells, and its knockdown by siRNA, was confirmed by immunoblotting. Knockdown of CRB2 in differentiated SY5Y/wtAPP cells resulted in a 1.3-fold increase in secretion of Aβ40 and Aβ42 (Fig. 2D). To rule out the possibility that the increase in Aβ secretion by CRB2 knockdown in the differentiated SY5Y/wtAPP cells might be somehow related to artificial overexpression of APP, we confirmed that a similar increase in Aβ occurred in differentiated native SH-SY5Y cells when treated with the same siRNA (data not shown).
These results are in contradiction to a previous report that described that overexpression of CRBs did not affect Aβ production in HEK293 cells (15). To address this discrepancy, we examined CRB1, T-CRB2, and CRB3 expressing plasmids that are the same or similar to plasmids used in the previous study (supplemental Fig. S1). Transient transfection with these plasmids into HEK/wtAPP cells resulted in a significant decrease in secreted Aβ. When transfected into SH-SY5Y cells, all constructs but T-CRB2 significantly decreased Aβ secretion. However, when transfected into HEK293 cells, the Aβ decrease was not statistically significant as reported (15). The secreted Aβ levels in native HEK293 cells were approximately 10 times and four times less than those in HEK/wtAPP cells and SH-SY5Y cells, respectively. We presumed that the previous study failed to detect a significant decrease in Aβ due to the low level of secreted Aβ in HEK293 cells. In addition, signal peptide-deleted T-CRB2 decreased the Aβ levels less efficiently than CRB2-iFLAG and N-terminal truncated but signal peptide-bearing CRB2-NT4. This suggested that deletion of the signal peptide perturbed the inhibitory activity. Our result also suggested that CRB1 and CRB3 inhibit Aβ generation. But we could not evaluate the inhibition by the endogenous proteins, because a specific antibody recognizing endogenous CRB1 or CRB3 is not available at present.
CRB2 Inhibits γ-Secretase Cleavage of APP in Cell-free Assays
We next investigated whether CRB2 suppression of Aβ secretion is indeed caused by direct inhibition of γ-secretase activity, using an in vitro γ-secretase assay (Fig. 2E). In this assay, a CHAPSO-solubilized membrane fraction of HEK293 cells, transiently transfected with CRB2 or mock plasmids, was incubated with recombinant APP-C99 at 37 °C for 6 h. This assay allowed evaluation of Aβ generation in cell-free conditions, under which Aβ generation could be specifically inhibited by the γ-secretase inhibitor L685,458. CRB2 transfection significantly inhibited Aβ production in this assay.
To assess the inhibitory effect of CRB2 on cell-free production of AICD fragments, the amount of AICD generated from membrane fractions of mock- or CRB2-transfected HEK/C99 cells was compared by immunoblotting (Fig. 2F). Less AICD fragments were generated from membranes of CRB2-transfected cells than from membranes of mock-transfected cells. These results suggested that CRB2 indeed inhibits γ-secretase cleavage of APP-C99.
CRB2 Binds with the PS1·γ-Secretase Complex
Exogenous expression of CRB2 did not alter the expression level of the PS1·γ-secretase complex as previously reported (15). To determine whether CRB2 directly interacts with the PS1 complex, we analyzed the binding between CRB2 and each component of the complex by co-immunoprecipitation assays (Fig. 3A). CRB2 was immunoprecipitated with the CRB2(C) antibody from lysates of HEK293 cells that were co-transfected with CRB2 and four components of the complex. In the transfected cells, the immature complex containing PS1-FL and immature NCT was accumulated. PS1-FL, APH-1, and immature NCT did co-precipitate with CRB2, whereas proteolyzed PS1, PEN-2, and mature NCT showed weak co-precipitation. This result suggested that CRB2 preferentially binds to a premature PS1 subcomplex composed of PS1-FL, immature NCT, and APH-1 in an overexpression condition. Under physiological conditions, the major pool of endogenous PS1 is endoproteolyzed and incorporated into the mature γ-secretase complex. We therefore investigated binding between endogenous CRB2 and γ-secretase complexes in a cynomolgus monkey brain lysate by co-immunoprecipitation. Immunoprecipitation of endogenous CRB2 with the CRB2(N1) antibody co-precipitated four components of the γ-secretase complex (Fig. 3B). Both FL and proteolyzed fragments of PS1 were co-immunoprecipitated with CRB2. A similar result was obtained with an SH-SY5Y cell lysate (data not shown). These results suggested that endogenous CRB2 interacts with mature as well as immature γ-secretase complex under physiological conditions.
FIGURE 3.

CRB2 binds the PS1 complex. A, co-immunoprecipitation (IP) assays of transfected CRB2. CRB2-iFLAG was co-transfected with PS1, NCT, APH-1, and PEN-2 into HEK293 cells, and the expressed CRB2 was then precipitated from cell lysates with the CRB2(C) antibody. Precipitation with rabbit IgG was used as a negative control. The precipitates were subjected to immunoblotting with anti-FLAG, anti-PS1 loop, anti-NCT, anti-APH-1, or anti-PEN-2 antibodies. Images of long exposed films are also shown for PS1-CTF, NCT, and PEN-2 (long). B, co-immunoprecipitation assays of endogenous CRB2. CRB2 was precipitated from lysates of monkey brain with CRB2(N1) antibody. The precipitate was analyzed by immunoblotting with anti-PS1 loop, anti-NCT, anti-APH-1, or anti-PEN-2 antibodies. Precipitation with rabbit IgG and immunoblotting with antibody against Sec61α, an unrelated membrane protein, were used as negative controls. C, BN gel electrophoresis for CRB2. Microsomal membranes prepared from SH-SY5Y cells (lane 1), non-transfected HEK293 cells (lane 2), CRB2-NT4-transfected HEK293 cells (lanes 3 and 5–8), and HEK293 cells co-transfected with CRB2-NT4 and γ-secretase components (lane 4) were solubilized with BN-lysis buffer containing 0.7% n-dodecyl β-d-maltoside. The cleared lysate was analyzed by BN electrophoresis, and the blots were probed with CRB2(N1), anti-PS1 N-terminal fragment, anti-NCT, anti-APH-1, or anti-PEN-2 antibodies. Asterisk and the number sign indicate the mature form of the γ-secretase complex and the NCT-APH-1 subcomplex, respectively.
Reverse co-immunoprecipitation with an anti-PS1 loop, anti-NCT, anti-APH-1, or anti-PEN-2 antibody, followed by immunoblotting with the CRB2(C) or CRB2(N1) antibody gave a background signal that was too high to allow for evaluation of specific signals. Therefore, as a second method, to investigate whether the γ-secretase complex contains CRB2, we analyzed microsomal membrane fractions of cultured cells by BN gel electrophoresis in which protein complexes are separated according to their molecular size (Fig. 3C). In this analysis the mature form of the γ-secretase complex migrated at a molecular mass of ∼500 kDa as indicated by PS1-, NCT-, APH-1-, and PEN-2-immunoreactive bands. Immunoblotting for CRB2 indicated that transfected CRB2 in HEK293 cells was contained in complexes of a similar molecular weight to the γ-secretase complex (Fig. 3C, lane 3). Additional incorporation of CRB2 into higher molecular weight complexes is consistent with previous studies in which CRB2 was shown to be a component of macromolecular cell-adhesion complexes (13). BN analysis using native SH-SY5Y cells also revealed endogenous CRB2-containing complexes at the similar molecular weight, although this band was weak (Fig. 3C, lane 1). When co-transfected with CRB2 and four components of the γ-secretase complex, CRB2(N1) antibody detected dense bands at the same molecular masses as the mature complex and the NCT-APH-1 subcomplex (Fig. 3C, lane 4), suggesting that CRB2 bound with the immature subcomplex as well as the mature complex.
The N-terminal Extracellular Domain of CRB2 Is Dispensable for Inhibition of Aβ Secretion
To define the domain within CRB2 that is required for its inhibitory activity, we prepared a series of N-terminal deletion (NT series) and C-terminal truncation (CT series) CRB2 mutants (Fig. 4A). To avoid potential functional perturbation that might arise due to deletion of the signal sequence or to fusion of a tag peptide to either terminus, a FLAG epitope tag was inserted immediately after the signal sequence of all mutants as well as of CRB2-FL. Individual constructs were transfected into HEK/C99 cells and their equivalent protein expression was confirmed by immunoblotting using an anti-FLAG tag antibody (Fig. 4B). The Aβ concentrations in the conditioned medium were then measured by a sandwich ELISA (Fig. 4C). Transfection of the CRB2-FL construct, or of any of the NT series of mutants, suppressed the level of secreted Aβ40 and Aβ42. However, cells transfected with the CT1 mutant, which lacks the TM and cytoplasmic domains of CRB2, secreted Aβ40 and Aβ42 at levels that were equivalent to those observed in the mock-transfected control. The CT2 mutant that lacked the cytoplasmic domain caused weak but significant attenuation of Aβ secretion. These results demonstrated that the N-terminal extracellular domain is dispensable for the inhibitory activity of CRB2 against Aβ production.
FIGURE 4.

CRB2 extracellular domain is dispensable for inhibition of Aβ secretion. A, scheme of the CRB2 mutants. The large extracellular region of FL CRB2 contains epidermal growth factor-like (EGF) domains and laminin A globular domain-like (LAG) repeats. SS and TMD represent the signal sequence and transmembrane domain, respectively. FL and all mutant CRB2 constructs have the N-terminal signal sequence followed by an inserted FLAG epitope tag. B, the expression of transfected CRB2 constructs. The cell lysates were immunoblotted with anti-FLAG antibody (upper panel). The blot was reprobed with anti-β-actin antibody as a loading control (lower panel). C, Aβ secretion from mutant CRB2-transfected cells in B. Mock or the indicated mutant CRB2 plasmid was transiently transfected into HEK/C99 cells, and Aβ levels in the conditioned medium were measured by a sandwich ELISA specific for Aβ40 and Aβ42. The error bars represent the S.D. *, p < 0.01 versus the mock control by an unpaired t test.
The CRB2 TM Domain Is Indispensable for γ-Secretase Inhibition and Mediates CRB2 Binding with the PS1 Complex
The CRB2-NT4 mutant was more effective in suppression of Aβ secretion than CRB2-FL. To further delineate the inhibitory domain of CRB2, we tested additional mutants that were constructed by removing the FLAG epitope tag from CRB2-NT4 (named CRB2-NT5) and by replacing the TM domain of CRB2-NT5 with the TM domain of the human TrkB receptor tyrosine kinase, a protein that is not cleaved by γ-secretase (named NT5-TMC) (Fig. 5A). Inhibition of Aβ secretion by CRB2-NT5 was equivalent to that by CRB2-NT4, indicating that the FLAG tag did not affect CRB2-NT4 function. In contrast, NT5-TMC showed no inhibitory activity when transfected into HEK/C99 cells, further suggesting the importance of the CRB2 TM domain for inhibition. Inhibition of Aβ secretion by CRB2-FL, NT4, and NT5 was accompanied by intracellular accumulation of APP-C99 and APP-C83, which are direct substrates of γ-secretase (Fig. 5B). To further confirm the PS1 complex-binding domain of CRB2, we performed coimmunoprecipitation assays using lysates of the transfected cells (Fig. 5C). Proteolyzed PS1 co-precipitated with CRB2-FL, NT4, and NT5 but not with CT1 or NT5-TMC. These data implied that the CRB2 TM domain is essential for binding to the PS1 complex and that CRB2·PS1 complex binding correlates with γ-secretase inhibition.
FIGURE 5.
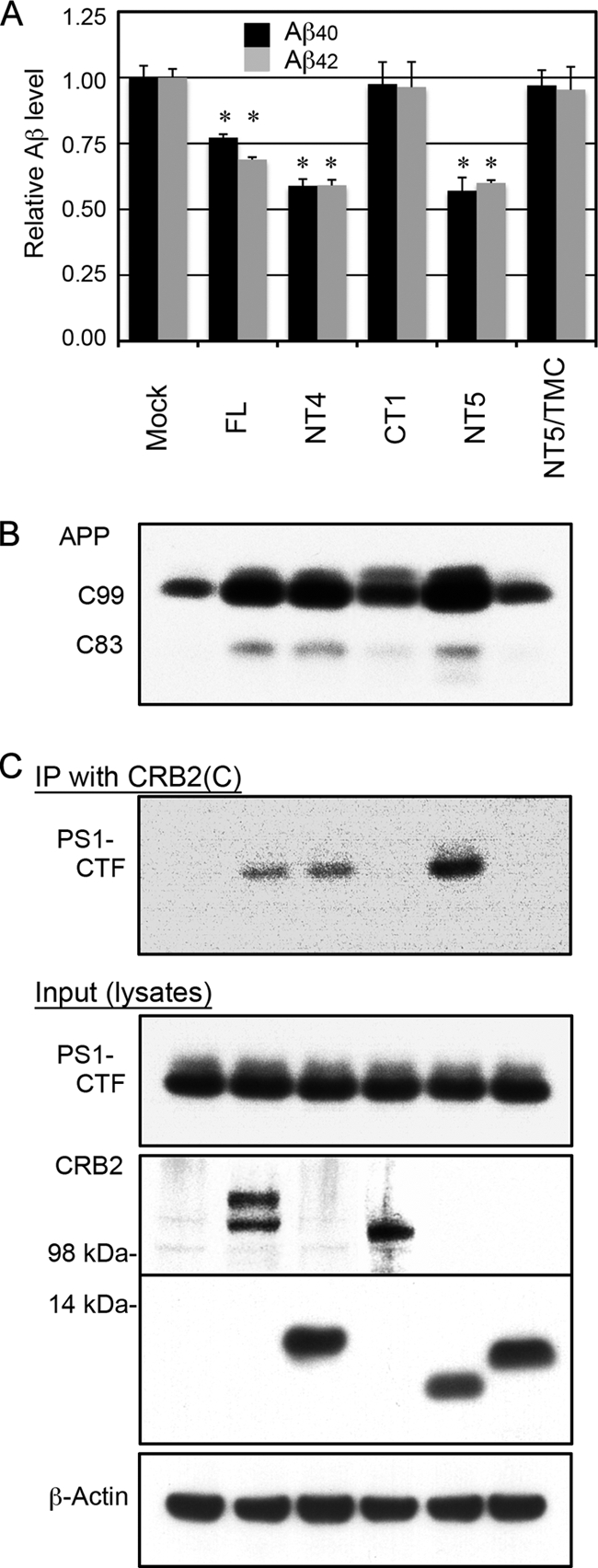
CRB2 TM domain mediates CRB2-inhibition of Aβ secretion and CRB2-PS1 binding. A, Aβ secretion from mutant CRB2-transfected cells. Mock or the indicated mutant CRB2 plasmid were transiently transfected into HEK/C99 cells, and Aβ40 and Aβ42 levels in the conditioned medium were measured by ELISA. CRB2-NT5 corresponds to FLAG tag-deleted CRB2-NT4, and NT5-TMC is a TM domain chimera in which the TM domain of CRB2-NT5 was replaced with the TM domain of the human TrkB receptor tyrosine kinase. Values are the mean ± S.D. *, p < 0.01 versus the mock control by an unpaired t test. B, the cell lysates in A were immunoblotted with an anti-APP CTF antibody. The APP cleavage products C99 and C83 are indicated. C, the cell lysates in A were immunoprecipitated (IP) with the CRB2(C) antibody, and the precipitates were immunoblotted with the anti-PS1 loop antibody (top panel). The same cell lysates (Input) were immunoblotted with anti-PS1 loop (second panel), CRB2(N1) (third panel), CRB2(C) (fourth panel), and anti-β-actin antibody (fifth panel). The sample order in B and C is the same as in A.
Efficient Inhibition by CRB2 Requires Its Cytoplasmic Domain but Not Its FERM- or PDZ-binding Motif
The cytoplasmic domain-truncation mutant CT2 also attenuated Aβ secretion but did so less efficiently than the FL protein. This result suggested that the cytoplasmic domain is required for efficient inhibition of γ-secretase activity. The CRB2 cytoplasmic domain contains two conserved protein-binding motifs: FERM- and PDZ-binding motifs. These motifs are required for its function in cell adhesion (24). To clarify whether these motifs are also involved in the inhibition of γ-secretase activity, we generated CRB2 constructs mutated at these motifs by modifying the CRB2-NT4 mutant. The resulting mutants were: NT4-CT2, which lacks the entire cytoplasmic and extracellular domains, NT4-mFERM, which harbors critical amino acid substitutions of three residues in the FERM-binding motif (Tyr1258 to Ala, Pro1260 to Ala, and Glu1264 to Ala), and NT4-ΔPDZ, which harbors a deletion of the PDZ-binding motif. The same mutations in CRB3 reportedly led to a defect in tight junction development (24). These mutants were transiently transfected into HEK/C99 cells, following which secreted Aβ levels were evaluated by ELISA (Fig. 6). NT4-CT2 significantly inhibited Aβ secretion, but the degree of inhibition was less pronounced than that by NT4. In contrast, the inhibition of Aβ secretion by NT4-mFERM and NT4-ΔPDZ was equivalent to that of NT4. The differences in the degree of Aβ suppression were not attributable to differences in the expression levels of CRB2 mutants. These results suggested that, in addition to the TM domain, the cytoplasmic domain is also required for efficient inhibition of Aβ secretion, and that the two conserved protein-binding motifs are not involved in this inhibition.
FIGURE 6.
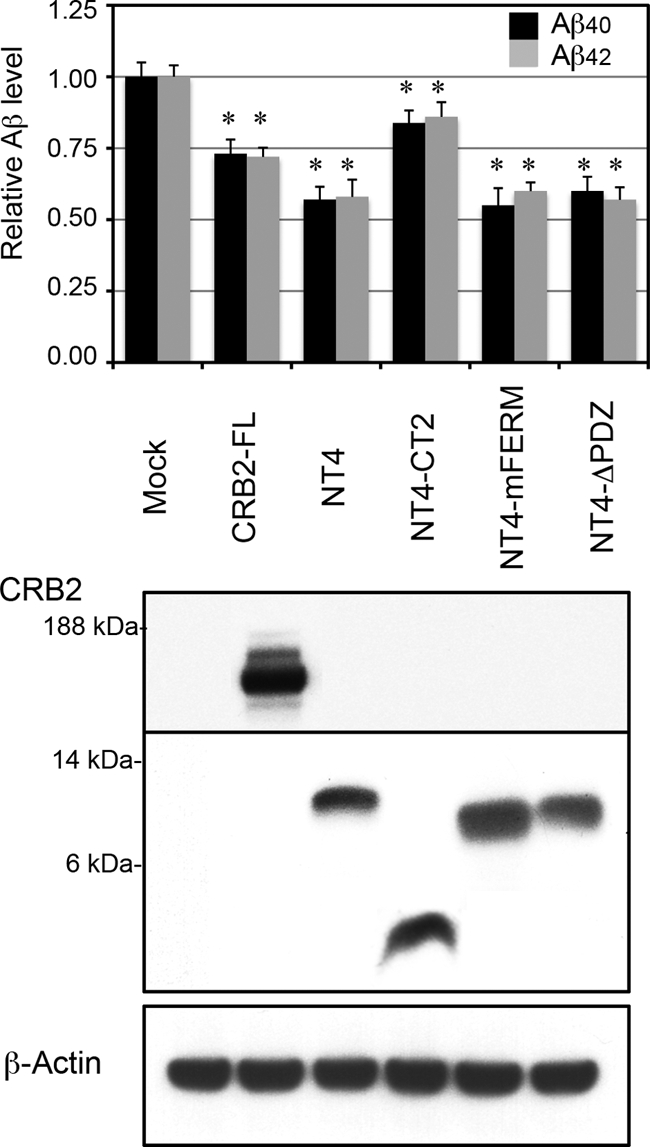
Efficient inhibition of Aβ secretion requires the CRB2-cytoplasmic domain, but not the FERM- or PDZ-binding motifs. Aβ secretion from mutant CRB2-transfected cells. Mock and the indicated mutant CRB2 plasmids were transiently transfected into HEK/C99 cells, and Aβ40 and Aβ42 levels in the conditioned medium were measured by ELISA. NT4-CT2 lacks the entire cytoplasmic and extracellular domain. NT4-mFERM harbors amino acid substitutions of three critical residues in the FERM-binding motif, and NT4-ΔPDZ harbors a deletion of the PDZ-binding motif. Values are the mean ± S.D. *, p < 0.01 versus the mock control by an unpaired t test. The protein expression was confirmed by immunoblotting the total cell lysates with anti-FLAG antibody (upper and middle panels). The blot was reprobed with anti-β-actin antibody as a loading control (lower panel).
CRB2 Is Not a Competitive Substrate for γ-Secretase
It is still controversial whether γ-secretase substrates competitively inhibit each others cleavage (25–27). CRB2 is a potential substrate for γ-secretase, because CRB2 is a type I TM protein. However, CRB2(N1) antibody did not detect any cross-reactive band in the conditioned medium of cultured CRB2-transfected HEK293 cells (supplemental Fig. S2A), suggesting that the ectodomain of CRB2 is not released into the culture medium. The NT4 mutant more effectively inhibits Aβ secretion than CRB2-FL and has a short extracellular domain composed of the putative signal sequence (36 amino acids) and the FLAG tag (6 amino acids). To determine whether CRB2-FL and the NT4 mutant are substrates for γ-secretase, we assayed the effect of treatment with the γ-secretase inhibitor L685,458 on the cellular level and degradation of CRB2 and NT4 (supplemental Fig. S2B). L685,458 treatment of HEK293 cells transfected with APP caused accumulation of APP-CTFs, which are direct substrates for γ-secretase. In contrast, it did not lead to any change in the level and degradation of CRB2-FL or NT4. These results suggested that CRB2 and NT4 are not substrates for γ-secretase.
It has previously been shown for Notch and APP that proteolyzed cytoplasmic fragments (NICD and AICD) are too unstable to be detected by immunoblotting of cell lysates. However, AICD generated by γ-secretase cleavage in a cell-free assay can be easily detected by immunoblotting. To confirm that CRB2-FL and NT4 were not cleaved by γ-secretase into unstable fragments that could not be detected by immunoblotting, we further assayed potential cleavage of CRB2-FL and NT4 by γ-secretase in a similar cell-free assay to that used for assay of AICD generation. Thus, CHAPSO-solubilized microsomal membrane fractions were prepared from CRB2-FL- or NT4-transfected cells, which were then incubated in a buffer containing a protease inhibitor mixture at 37 °C for 3 h. Immunoblotting of these reaction mixtures did not reveal any proteolyzed fragments (supplemental Fig. S2C), again suggesting that CRB2 and the NT4 mutant are not cleaved by γ-secretase. In addition, we further tested whether Notch-ΔE transfection could augment the inhibitory effect of CRB2 on Aβ secretion. Measurement of secreted Aβ levels in HEK/C99 cells transfected with CRB2 and/or Notch-ΔE showed no additive inhibitory effect on Aβ secretion following co-transfection of Notch-ΔE with CRB2 (supplemental Fig. S2D). These results indicated that CRB2 and NT4 mutants are not competitive substrates for γ-secretase.
CRB2 Transfection Does Not Detectably Alter the Subcellular Localization of PS1
One possible mechanism underlying the inhibitory activity of CRB2 is that CRB2 might sequester the γ-secretase complex away from the cellular sites or microdomains at which the complex meets and cleaves APP-C99. We investigated the effect of CRB2 transfection on the localization of PS1 in cholesterol-rich lipid raft microdomains, which have been implicated in γ-secretase cleavage of APP (23). The hydrophilic nonionic detergent Lubrol WX-based lysates were prepared from HEK293 cells transfected with mock or CRB2 plasmid, followed by fractionation in a discontinuous sucrose density gradient and immunoblotting analysis of the fractions for PS1 and CRB2 (supplemental Fig. S3). Flottilin-2, an established marker of lipid rafts, was indeed predominantly concentrated in the detergent-insoluble fractions, whereas the non-raft marker BiP was localized in the detergent-soluble fractions. Transfected CRB2 was distributed in the detergent-soluble fractions. The distribution of PS1 was not altered by CRB2 cotransfection. Thus, PS1 was equivalently enriched in the detergent-insoluble fractions of cell lysates transfected with mock or CRB2 plasmid. A similar result was obtained with SH-SY5Y cells (data not shown).
Co-overexpression of PS1 and APH-1 Abrogates γ-Secretase Inhibition by CRB2
Based on the above results, we assumed that CRB2 is incorporated into the PS1·γ-secretase complex as an inhibitory binding protein. PS1 complex assembly is considered to be a highly regulated and ordered process. It is currently accepted that NCT and APH-1 first form a subcomplex prior to sequential incorporation of PS1 and PEN-2. Association of PEN-2 results in a proteolytically active complex, which contains endoproteolyzed PS1 fragments and is transported from the endoplasmic reticulum to the trans-Golgi apparatus. It is not yet known which component of this complex is a direct binding partner of CRB2, although CRB2 appears to bind preferentially to premature forms of the PS1 complex (Fig. 3A). To clarify whether overexpression of components of the complex might influence the inhibitory effect of CRB2, we assayed the effect of co-overexpression of individual core component proteins with CRB2 on CRB2 inhibition of γ-secretase activity. We used the CRB2-NT4 in this assay, because this construct was more effective in inhibition of Aβ secretion than CRB2-FL. Thus, following transient co-transfection of NT4 with individual protein components of the complex into HEK/C99 cells, Aβ levels in the conditioned medium were measured (Fig. 7A). Neither co-expression of NCT nor PEN-2 with NT4 caused any alteration in secreted Aβ levels compared with those observed following co-transfection with the mock control. However, co-expression of PS1 or APH-1 with NT4 almost completely abrogated inhibition of Aβ secretion by CRB2-NT4. Substitution of CRB2-FL for CRB2-NT4 gave similar results but less prominent differences in Aβ levels (data not shown).
FIGURE 7.
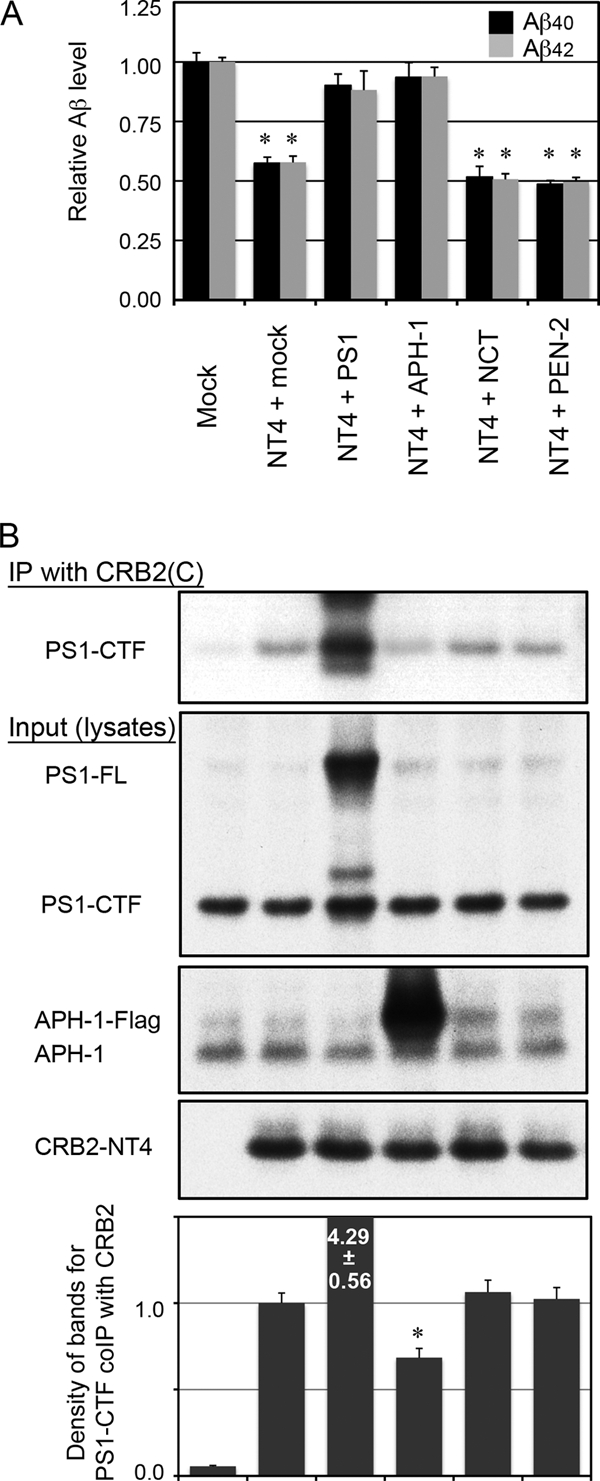
Co-overexpression of PS1 or APH-1 abrogates γ-secretase inhibition by CRB2. A, Aβ secretion from HEK/C99 cells co-transfected with CRB2-NT4 and individual components of the γ-secretase complex. CRB2-NT4 was transiently co-transfected with mock, PS1, APH-1, NCT, or PEN-2 plasmids into HEK/C99 cells, and the culture medium was changed 24 h post-transfection. The cells were then cultured for a further 24 h, and Aβ40 and Aβ42 levels in the conditioned medium was measured by ELISA. Values are the mean ± S.D. *, p < 0.01 versus the mock-transfected control by an unpaired t test. B, the cell lysates in A were immunoprecipitated (IP) with the CRB2(C) antibody and the precipitate was immunoblotted with the anti-PS1 loop antibody (top panel). Cell lysates were immunoblotted with an anti-PS1 loop (second panel), anti-APH-1 (third panel), or CRB2(C) antibody (fourth panel). The graph shows the relative density of the bands for PS1-CTF co-precipitated with CRB2 (mean ± S.D. from three experiments). *, p < 0.01 versus the NT4 + mock-transfected control by an unpaired t test. The sample order in B is the same as in A.
We reasoned that simultaneous overexpression of PS1 or APH-1 might have somehow prevented the incorporation of CRB2 into the γ-secretase complex. To test this possibility, we cotransfected HEK/C99 cells with CRB2-NT4 and individual components of the γ-secretase complex, and determined the amount of PS1, a catalytic component of the γ-secretase complex, which co-precipitated with CRB2 (Fig. 7B). Co-expression of NT4 with PS1 did indeed increase the binding between these two proteins, although a large part of exogenous PS1 remained uncleaved and was not incorporated into the active γ-secretase complex. Co-expression of NT4 with APH-1, but not with NCT or PEN-2, reduced its co-precipitation with endogenous PS1. This result suggested that co-overexpression of APH-1 prevented the incorporation of CRB2-NT4 into the PS1 complex.
CRB2 Does Not Inhibit SPPL2b Cleavage of TNF-α in a Cultured Cell-based Assay
Signal peptide peptidases (SPP) and SPP-like proteases (SPPL) belonging to an intramembrane aspartyl protease family, share a common consensus motif, GXGD, at the catalytic center with γ-secretase. Therefore, despite their opposite membrane topology, some γ-secretase inhibitors such as L685,458 are also effective for inhibition of SPP or SPPL cleavage (28). There is a possibility that CRB2 is a common inhibitor for GXGD-type proteases. SPPL2b mediates intramembrane proteolysis of TNF-α (29, 30). TNF-α is first cleaved by the TNF-α converting enzyme, and the resultant N-terminal, membrane-bound fragment (15 kDa) is further cleaved at an intramembrane site by SPPL2b. To determine whether CRB2 inhibits SPPL2b cleavage of TNF-α, we transiently transfected HEK293 cells with SPPL2b, TNF-α, and/or CRB2 and analyzed proteolyzed fragments of TNF-α by immunoblotting (supplemental Fig. S4). In the absence of cotransfected SPPL2b, TNF-α converting enzyme-cleaved N-terminal fragments of TNF-α accumulated, whereas co-transfection of SPPL2b markedly decreased accumulation of these 15-kDa fragments. As previously reported (30), treatment with L685,458 enhanced accumulation of these fragments by inhibition of SPPL2b cleavage of TNF-α. In contrast, co-expression of CRB2 did not increase the accumulation of the TNF-α converting enzyme-cleaved fragments, suggesting that CRB2 does not inhibit SPPL2b activity.
DISCUSSION
In this study, we described the inhibition of γ-secretase cleavage of APP by human CRB2 in cultured mammalian cells. Our data show that transfected CRB2 inhibited proteolytic production of Aβ and AICD from APP CTFs. Conversely, knockdown of endogenous CRB2 increased the amount of γ-secretase cleavage products in SH-SY5Y cells. Inhibition of Aβ and AICD generation could be detected by either cell-based or cell-free assays. CRB2 bound with the γ-secretase complex through the TM domain, which was required for the inhibitory activity. In addition to the TM domain, the cytoplasmic domain appeared to play a supportive role in γ-secretase inhibition, but the FERM- and PDZ-binding motifs were not involved in this inhibition. CRB2 is a γ-secretase complex-interacting protein, but is not a competitive substrate for γ-cleavage. Overexpressed FL CRB2 decreased secreted Aβ levels to 70–80% of the control level, suggesting that CRB2 is a negative modulator of γ-secretase activity rather than a potent inhibitor.
Several previous studies failed to identify CRB2 as a component of the γ-secretase complex (reviewed in Ref. 31). We speculate that one or more of the following reasons may explain this failure. 1) Because CRB2 is only expressed in specific organs or cell lines, γ-secretase complexes isolated from another source would not contain the CRB2 protein. 2) Because CRB2 is probably not incorporated into 100% of the γ-secretase complexes, the level of CRB2 co-purified with the complexes may not have been high enough for detection. 3) It is sometimes difficult to detect and identify high molecular weight proteins such as CRB2. 4) Because CRB2 is a negative regulator of γ-secretase activity, activity-dependent isolation of the γ-secretase complex might result in failure to co-purify CRB2.
The major cellular pool of CRB family proteins resides at cell-cell contact sites at which it forms macromolecular complexes with Pals1 and PATJ through PDZ-binding motifs, with EPB41L5 and MPP5 through FERM-binding motifs, and where it also further interacts with other proteins (13). The conserved C-terminal region harboring the two protein-binding motifs is essential for CRB function during tight junction development (24, 32). Similarly, we found that the C-terminal, TM, and cytoplasmic domains of CRB2 were required for inhibition of γ-secretase cleavage of APP. However, our results suggest that the FERM- and PDZ-binding motifs are not involved in its inhibitory activity. Therefore, the γ-secretase inhibitory activity of CRB2 is considered to be independent of its well studied function in cell adhesion and polarity determination.
Our data suggested that CRB2 neither sequestered the γ-secretase complex away from subcellular domains rich in its APP-CTF substrates nor functioned as a competitive substrate for γ-secretase. Instead our results support the hypothesis that CRB2 interacts with the PS1·γ-secretase complex as an inhibitory binding protein. Co-overexpression of CRB2 with either PS1 or APH-1 attenuated CRB2 suppression of Aβ secretion. Furthermore, co-immunoprecipitation assays showed that co-expressed APH-1 reduced the amount of CRB2 bound to endogenous PS1. One possible explanation of these data is that CRB2 binds with PS1 and APH-1 and that heterodimer formation between CRB2 and co-overexpressed PS1 or APH-1 precludes incorporation of CRB2 into the maturing γ-secretase complex, thereby preventing CRB2 from acting as an inhibitory binding protein. In contrast, when cotransfected with CRB2 and NCT, the CRB2-NCT dimers could be incorporated into the complexes to exert the inhibitory activity. Because the γ-secretase complex assembly is considered to be a highly regulated and ordered process, it can be envisaged that the CRB2-PS1 and CRB2-APH-1 dimers may preclude incorporation of CRB2 into the complexes but that the CRB2-NCT dimer may not. A second possibility is that CRB2 binds with NCT and that excessive PS1 or APH-1 mitigated CRB2 inhibition by competing with CRB2 for binding with NCT in the maturing γ-complex. Because NCT has been reported to directly bind with both PS1 and APH-1, it is possible that CRB2 could compete with PS1 and APH-1 for binding with NCT in the maturing γ-secretase complex (10, 33–35).
It remains to be elucidated whether CRB2 exhibits a constitutive or at least partially inducible expression in mammalian brains. Herranz et al. (12) showed that Crumbs expression is induced by Notch activation and provides a negative feedback for Notch signaling at Drosophila wing margins. However, we could not reproduce CRB2 induction by exogenously expressed Notch-ΔE or by NICD in cultured SH-SY5Y neuroblastoma cells (data not shown). A recent report showed that γ-secretase activity in brains is altered during aging, and this change could confer a risk for sporadic AD in aged subjects (36). A reduction of CRB2 expression might lead to insufficient inhibition of γ-secretase activity, which could enhance Aβ generation in human brains and play a causative role in the development of AD. It is a future issue to investigate whether the inhibition by CRB2 is involved in a variable fine-tuning of cellular γ-secretase activity in vivo and whether CRB2 expression is reduced in aged brains and/or brains of patients with AD.
Several lines of data suggest that APP-C99 is processed by γ-secretase activity mainly at the plasma membrane or in endocytotic compartments, although a major pool of the γ-secretase complex resides at early secretory compartments (37, 38). On the other hand, previous reports (39, 40) estimated that only a small portion of the total proteolyzed PS1 fragments in cellular membranes is engaged in active γ-secretase complexes. The results in these reports indicate that the major cellular pool of the PS complex is the mature proteolyzed form but remains catalytically inactive. Inhibitory binding proteins such as CRB2 may contribute to the formation of an inactive pool of γ-secretase complexes by binding with a subset of the complex. Previous studies have shown that Aβ levels in brains can be reduced by decreasing γ-secretase activity (reviewed in Ref. 41). Therefore, an exact understanding of the mechanism by which cellular γ-secretase activity is negatively regulated might provide the basis for a novel therapeutic strategy for AD other than development of direct inhibitors of γ-secretase.
γ-Secretase·PS complexes interact with many cell-cell and cell-matrix adhesion proteins and are involved in synapse formation/remodeling in brains. PS complexes bind to adhesion proteins such as telencephalin and CRB2, whereas γ-secretase cleaves many membrane proteins including nectin-1α and ephrin receptors (reviewed in Ref. 42). In particular, N- and E-cadherins serve as both binding partners and protease substrates of γ-secretase (43, 44). Although it remains unresolved how synaptic plasticity is orchestrated, dysfunction of γ-secretase·PS complexes could cause synaptic dysregulation, eventually leading to neurodegeneration (45). Additionally, γ-secretase complex-interacting adhesion proteins such as N-cadherin and CRB2 are simultaneously involved in the regulation of cellular Aβ secretion (this article and see Ref. 46).
Many clinical mutations of CRB1 cause retinal degeneration including autosomal recessive retinitis pigmentosa, Leber congenital amaurosis, and autosomal dominant-pigmented paravenous chorioretinal atrophy, although the exact underlying pathological mechanism remains unresolved (47). Future studies are required to determine whether a dysfunction of CRB2 plays a role in the pathogenesis of AD.
Acknowledgments
We thank Drs. Raphael Kopan, Ben Margolis, Ryosuke Takahashi, Shinichiro Nakamura, and Takashi Uehara for Notch constructs, CRB1 and CRB3 constructs, pcDNA3-FLAG vectors, monkey brain tissues, and SH-SY5Y cells, respectively. We acknowledge the Central Laboratory of the Shiga University of Medical Science for assistance with DNA sequencing.
This work was supported in part by a grant-in-aid for scientific research on Priority Area-Research on Pathomechanisms of Brain Disorders from the Ministry of Education, Culture, Sports, Science and Technology, Japan (to M. N.), and a grant from the Program for the Promotion of Fundamental Studies in Health Sciences of the National Institute of Biomedical Innovation (05-26) (to M. T., M. O., S. T., and M. N.).

The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1–S4.
- TM
- transmembrane
- AD
- Alzheimer disease
- APP
- β-amyloid precursor protein
- Aβ
- amyloid-β
- AICD
- APP intracellular domain
- PS
- presenilin
- NCT
- nicastrin
- NICD
- Notch intracellular domain
- SPPL2b
- signal peptide peptidase-like protease 2b
- TNF-α
- tumor necrosis factor-α
- CTF
- C-terminal fragment
- FL
- full-length
- PDZ
- PSD-95/Discs Large/ZO-1
- FERM
- protein band 4.1/ezrin/radixin/moesin
- ELISA
- enzyme-linked immunosorbent assay
- BN
- blue native
- siRNA
- small interfering RNA
- CHAPSO
- 3-[(3-cholamidopropyl)dimethylammonio]-2-hydroxy-1-propanesulfonic acid
- Endo H
- endoglycosidase H
- BisTris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol
- wt
- wild-type.
REFERENCES
- 1.Brown M. S., Ye J., Rawson R. B., Goldstein J. L. (2000) Cell 100, 391–398 [DOI] [PubMed] [Google Scholar]
- 2.Beel A. J., Sanders C. R. (2008) Cell Mol. Life Sci. 65, 1311–1334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wolfe M. S. (2009) Chem. Rev. 109, 1599–1612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hass M. R., Sato C., Kopan R., Zhao G. (2009) Semin. Cell Dev. Biol. 20, 201–210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen F., Hasegawa H., Schmitt-Ulms G., Kawarai T., Bohm C., Katayama T., Gu Y., Sanjo N., Glista M., Rogaeva E., Wakutani Y., Pardossi-Piquard R., Ruan X., Tandon A., Checler F., Marambaud P., Hansen K., Westaway D., St. George-Hyslop P., Fraser P. (2006) Nature 440, 1208–1212 [DOI] [PubMed] [Google Scholar]
- 6.Vetrivel K. S., Gong P., Bowen J. W., Cheng H., Chen Y., Carter M., Nguyen P. D., Placanica L., Wieland F. T., Li Y. M., Kounnas M. Z., Thinakaran G. (2007) Mol. Neurodegener. 2, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhou S., Zhou H., Walian P. J., Jap B. K. (2005) Proc. Natl. Acad. Sci. U.S.A. 102, 7499–7504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vetrivel K. S., Zhang X., Meckler X., Cheng H., Lee S., Gong P., Lopes K. O., Chen Y., Iwata N., Yin K. J., Lee J. M., Parent A. T., Saido T. C., Li Y. M., Sisodia S. S., Thinakaran G. (2008) J. Biol. Chem. 283, 19489–19498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Spasic D., Raemaekers T., Dillen K., Declerck I., Baert V., Serneels L., Füllekrug J., Annaert W. (2007) J. Cell Biol. 176, 629–640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kaether C., Scheuermann J., Fassler M., Zilow S., Shirotani K., Valkova C., Novak B., Kacmar S., Steiner H., Haass C. (2007) EMBO Rep. 8, 743–748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cai D., Netzer W. J., Zhong M., Lin Y., Du G., Frohman M., Foster D. A., Sisodia S. S., Xu H., Gorelick F. S., Greengard P. (2006) Proc. Natl. Acad. Sci. U.S.A. 103, 1941–1946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Herranz H., Stamataki E., Feiguin F., Milán M. (2006) EMBO Rep. 7, 297–302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gosens I., den Hollander A. I., Cremers F. P., Roepman R. (2008) Exp. Eye Res. 86, 713–726 [DOI] [PubMed] [Google Scholar]
- 14.van den Hurk J. A., Rashbass P., Roepman R., Davis J., Voesenek K. E., Arends M. L., Zonneveld M. N., van Roekel M. H., Cameron K., Rohrschneider K., Heckenlively J. R., Koenekoop R. K., Hoyng C. B., Cremers F. P., den Hollander A. I. (2005) Mol. Vis. 11, 263–273 [PubMed] [Google Scholar]
- 15.Pardossi-Piquard R., Chen F., Silva-Gagliardi N. F., Szego M., McInnes R., McGlade C. J., St. George-Hyslop P., Fraser P. E. (2007) Biochemistry 46, 13704–13710 [DOI] [PubMed] [Google Scholar]
- 16.Yu G., Nishimura M., Arawaka S., Levitan D., Zhang L., Tandon A., Song Y. Q., Rogaeva E., Chen F., Kawarai T., Supala A., Levesque L., Yu H., Yang D. S., Holmes E., Milman P., Liang Y., Zhang D. M., Xu D. H., Sato C., Rogaev E., Smith M., Janus C., Zhang Y., Aebersold R., Farrer L. S., Sorbi S., Bruni A., Fraser P., St. George-Hyslop P. (2000) Nature 407, 48–54 [DOI] [PubMed] [Google Scholar]
- 17.Nakaya Y., Yamane T., Shiraishi H., Wang H. Q., Matsubara E., Sato T., Dolios G., Wang R., De Strooper B., Shoji M., Komano H., Yanagisawa K., Ihara Y., Fraser P., St. George-Hyslop P., Nishimura M. (2005) J. Biol. Chem. 280, 19070–19077 [DOI] [PubMed] [Google Scholar]
- 18.Takeda K., Araki W., Tabira T. (2004) Eur. J. Neurosci. 19, 258–264 [DOI] [PubMed] [Google Scholar]
- 19.Wang H. Q., Nakaya Y., Du Z., Yamane T., Shirane M., Kudo T., Takeda M., Takebayashi K., Noda Y., Nakayama K. I., Nishimura M. (2005) Hum. Mol. Genet. 14, 1889–1902 [DOI] [PubMed] [Google Scholar]
- 20.Schägger H., von Jagow G. (1991) Anal. Biochem. 199, 223–231 [DOI] [PubMed] [Google Scholar]
- 21.Tagami S., Okochi M., Yanagida K., Ikuta A., Fukumori A., Matsumoto N., Ishizuka-Katsura Y., Nakayama T., Itoh N., Jiang J., Nishitomi K., Kamino K., Morihara T., Hashimoto R., Tanaka T., Kudo T., Chiba S., Takeda M. (2008) Mol. Cell. Biol. 28, 165–176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li Y. M., Lai M. T., Xu M., Huang Q., DiMuzio-Mower J., Sardana M. K., Shi X. P., Yin K. C., Shafer J. A., Gardell S. J. (2000) Proc. Natl. Acad. Sci. U.S.A. 97, 6138–6143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vetrivel K. S., Cheng H., Lin W., Sakurai T., Li T., Nukina N., Wong P. C., Xu H., Thinakaran G. (2004) J. Biol. Chem. 279, 44945–44954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fogg V. C., Liu C. J., Margolis B. (2005) J. Cell Sci. 118, 2859–2869 [DOI] [PubMed] [Google Scholar]
- 25.Chen F., Yu G., Arawaka S., Nishimura M., Kawarai T., Yu H., Tandon A., Supala A., Song Y. Q., Rogaeva E., Milman P., Sato C., Yu C., Janus C., Lee J., Song L., Zhang L., Fraser P. E., St. George-Hyslop P. H. (2001) Nat. Cell Biol. 3, 751–754 [DOI] [PubMed] [Google Scholar]
- 26.Berezovska O., Jack C., Deng A., Gastineau N., Rebeck G. W., Hyman B. T. (2001) J. Biol. Chem. 276, 30018–30023 [DOI] [PubMed] [Google Scholar]
- 27.Lleó A., Waldron E., von Arnim C. A., Herl L., Tangredi M. M., Peltan I. D., Strickland D. K., Koo E. H., Hyman B. T., Pietrzik C. U., Berezovska O. (2005) J. Biol. Chem. 280, 27303–27309 [DOI] [PubMed] [Google Scholar]
- 28.Weihofen A., Lemberg M. K., Friedmann E., Rueeger H., Schmitz A., Paganetti P., Rovelli G., Martoglio B. (2003) J. Biol. Chem. 278, 16528–16533 [DOI] [PubMed] [Google Scholar]
- 29.Fluhrer R., Grammer G., Israel L., Condron M. M., Haffner C., Friedmann E., Böhland C., Imhof A., Martoglio B., Teplow D. B., Haass C. (2006) Nat. Cell Biol. 8, 894–896 [DOI] [PubMed] [Google Scholar]
- 30.Friedmann E., Hauben E., Maylandt K., Schleeger S., Vreugde S., Lichtenthaler S. F., Kuhn P. H., Stauffer D., Rovelli G., Martoglio B. (2006) Nat. Cell Biol. 8, 843–848 [DOI] [PubMed] [Google Scholar]
- 31.Dries D. R., Yu G. (2008) Curr. Alzheimer Res. 5, 132–146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.den Hollander A. I., Johnson K., de Kok Y. J., Klebes A., Brunner H. G., Knust E., Cremers F. P. (2001) Hum. Mol. Genet. 10, 2767–2773 [DOI] [PubMed] [Google Scholar]
- 33.Kaether C., Capell A., Edbauer D., Winkler E., Novak B., Steiner H., Haass C. (2004) EMBO J. 23, 4738–4748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lee S. F., Shah S., Li H., Yu C., Han W., Yu G. (2002) J. Biol. Chem. 277, 45013–45019 [DOI] [PubMed] [Google Scholar]
- 35.Gu Y., Chen F., Sanjo N., Kawarai T., Hasegawa H., Duthie M., Li W., Ruan X., Luthra A., Mount H. T., Tandon A., Fraser P. E., St. George-Hyslop P. (2003) J. Biol. Chem. 278, 7374–7380 [DOI] [PubMed] [Google Scholar]
- 36.Placanica L., Zhu L., Li Y. M. (2009) PLoS ONE 4, e5088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cupers P., Bentahir M., Craessaerts K., Orlans I., Vanderstichele H., Saftig P., De Strooper B., Annaert W. (2001) J. Cell Biol. 154, 731–740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Maltese W. A., Wilson S., Tan Y., Suomensaari S., Sinha S., Barbour R., McConlogue L. (2001) J. Biol. Chem. 276, 20267–20279 [DOI] [PubMed] [Google Scholar]
- 39.Lai M. T., Chen E., Crouthamel M. C., DiMuzio-Mower J., Xu M., Huang Q., Price E., Register R. B., Shi X. P., Donoviel D. B., Bernstein A., Hazuda D., Gardell S. J., Li Y. M. (2003) J. Biol. Chem. 278, 22475–22481 [DOI] [PubMed] [Google Scholar]
- 40.Beher D., Fricker M., Nadin A., Clarke E. E., Wrigley J. D., Li Y. M., Culvenor J. G., Masters C. L., Harrison T., Shearman M. S. (2003) Biochemistry 42, 8133–8142 [DOI] [PubMed] [Google Scholar]
- 41.Wolfe M. S. (2008) Neurotherapeutics 5, 391–398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Parks A. L., Curtis D. (2007) Trends Genet. 23, 140–150 [DOI] [PubMed] [Google Scholar]
- 43.Georgakopoulos A., Marambaud P., Efthimiopoulos S., Shioi J., Cui W., Li H. C., Schütte M., Gordon R., Holstein G. R., Martinelli G., Mehta P., Friedrich V. L., Jr., Robakis N. K. (1999) Mol. Cell 4, 893–902 [DOI] [PubMed] [Google Scholar]
- 44.Marambaud P., Wen P. H., Dutt A., Shioi J., Takashima A., Siman R., Robakis N. K. (2003) Cell 114, 635–645 [DOI] [PubMed] [Google Scholar]
- 45.Saura C. A., Choi S. Y., Beglopoulos V., Malkani S., Zhang D., Shankaranarayana Rao B. S., Chattarji S., Kelleher R. J., 3rd, Kandel E. R., Duff K., Kirkwood A., Shen J. (2004) Neuron 42, 23–36 [DOI] [PubMed] [Google Scholar]
- 46.Uemura K., Lill C. M., Banks M., Asada M., Aoyagi N., Ando K., Kubota M., Kihara T., Nishimoto T., Sugimoto H., Takahashi R., Hyman B. T., Shimohama S., Berezovska O., Kinoshita A. (2009) J. Neurochem. 108, 350–360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Richard M., Roepman R., Aartsen W. M., van Rossum A. G., den Hollander A. I., Knust E., Wijnholds J., Cremers F. P. (2006) Hum. Mol. Genet. 15, R235–243 [DOI] [PubMed] [Google Scholar]