

Abstract
Neurotransmitter transporters are responsible for removal of biogenic amine neurotransmitters after release into the synapse. These transporters are the targets for many clinically relevant drugs, such as antidepressants and psychostimulants. A high resolution crystal structure for the monoamine transporters has yet to be solved. We have developed a homology model for the serotonin transporter (SERT) based on the crystal structure of the leucine transporter (LeuTAa) from Aquifex aeolicus. The objective of the present studies is to identify the structural determinants forming the entrance to the substrate permeation pathway based on predictions from the SERT homology model. Using the substituted cysteine accessibility method, we identified residues predicted to reside at the entrance to the substrate permeation pathway that were reactive with methanethiosulfonate (MTS) reagents. Of these residues, Gln332 in transmembrane helix (TMH) VI was protected against MTS inactivation in the presence of serotonin. Surprisingly, the reactivity of Gln332 to MTS reagents was enhanced in the presence of cocaine. Bifunctional MTS cross-linkers also were used to examine the distances between helices predicted to form the entrance into the substrate and ion permeation pathway. Our studies suggest that substrate and ligand binding may induce conformational shifts in TMH I and/or VI, providing new opportunities to refine existing homology models of SERT and related monoamine transporters.
Keywords: Cysteine-mediated Cross-linking, Neurotransmitter Transport, Neurotransmitters, Protein Structure, Transport Drugs, Cocaine
Introduction
The serotonin (5-hydroxytryptamine (5-HT)2) transporter (SERT) is a member of the neurotransmitter sodium symporters (NSSs) (also referred as Na+/Cl−-dependent transporters), whose function is to return 5-HT into presynaptic neurons after release into the synapse (1–3). Other family members include the transporters for dopamine (DAT), norepinephrine, γ-aminobutyric acid, and glycine (1–4). These transporters are of particular interest as targets for many drugs, including antidepressants, anticonvulsants, and abused psychostimulants, such as cocaine and amphetamines (3). Although these transporters may share a common topology of 12 transmembrane α-helices (TMHs), intracellular N and C termini, and a large extracellular loop between TMHs III and IV with putative glycosylation sites, actual structural information for these proteins is limited.
A high resolution crystal structure for members of the human neurotransmitter transporter family has yet to be solved, impeding the study of these important drug targets. A breakthrough came when the crystal structure of a bacterial homologue of Na+/Cl−-dependent neurotransmitter transporters was determined (5). The leucine transporter (LeuTAa) from the bacteria Aquifex aeolicus was crystallized with the substrate and two Na+ ions occluded in the structure (5). It shares 20–25% of overall sequence identity with the eukaryotic neurotransmitter transporters, including clusters of high sequence conservation (5). Our group and others have supported the use of LeuTAa as an appropriate template for homology modeling of SERT and other biogenic amine transporters (6–10).
Homology models of mammalian NSS members based on the LeuTAa structure provide new opportunities to investigate the arrangement of the TMHs, the mechanisms of substrate transport, and the sites for antagonist binding. The substituted cysteine accessibility method (SCAM) approach has been used extensively to study the topology of membrane transport proteins (11–16). The technique involves the reactivity of an introduced cysteine to sulfhydryl-specific reagents (either membrane-impermeant or permeant), thus defining protein topology. By using this analysis, TMHs I and III of SERT had been implicated as part of the substrate permeation pathway (12, 15). Cysteine-scanning mutagenesis has also been applied to extracellular loop 4 (EL4) of SERT. Strong inhibition by MTS reagents and changes in MTS accessibility by substrate translocation are consistent with a role for the loop in conformational changes associated with substrate transport (16). Engineered Zn2+ binding sites and bifunctional MTS reagents have also been used to confirm the proximity and orientation of TMHs I and III of the human SERT (hSERT) (10). Disulfide cross-linking studies have provided information regarding orientation and proximity of important residues to protein function (17, 18). These molecular approaches are useful in validating structural predictions that are based on NSS homology models generated from the LeuTAa structure. Indeed, several models for NSS members have been generated and now can provide the basis for rational structure-function studies exploring these important transporters (6, 10, 19–21).
In the current study, we used our SERT homology model to explore the extracellular surface of the substrate permeation pathway. Our SERT homology model predicts that the extracellular entrance to the permeation pathway is formed by TMHs I, III, VI, X, and XI as well as EL4. To identify structural components of SERT involved in the substrate permeation pathway, residues predicted to be at the entrance to the permeation pore were subjected to the SCAM approach using the membrane-impermeant thiol-reactive 2-(trimethylammonium)ethyl methanethiosulfonate bromide (MTSET) (11–13, 15, 16). Further examination was performed by determining the rate of inactivation at those positions where ligand binding affected MTS reactivity. We also defined the proximities of these residues by cross-linking with bifunctional MTS reagents of different lengths. Our studies suggest that, in accordance with our model, TMHs I and VI and TMHs VI and XI are in close proximity to each other. Our data are consistent with the homology model of SERT, thus providing the opportunity to further extend SERT characterization and the possibility of developing more refined models of SERT and other members of the neurotransmitter transporter family.
EXPERIMENTAL PROCEDURES
Site-directed Mutagenesis and Construction of hSERT Cys Mutants
The QuikChange® mutagenesis kit (Stratagene, La Jolla, CA) was used to generate the following hSERT Cys mutants: TMH I, I108C; TMH VI, A330C, A331C, and Q332C; TMH X, T497C and G498C; EL4, E396C, V397C, and A398C. All mutations were generated in hSERT C109A background, a conserved MTS-sensitive cysteine in the first extracellular loop of SERT, thus preventing the inhibition of transport at that position by MTS reagents (22). The following hSERT Cys double mutants were constructed using existing restriction endonuclease sites allowing exchange of sequence encoding for the Cys mutants: I108C/A330C, I108C/A331C, I108C/Q332C, I108C/E396C, F556C/A330C, F556C/A331C, and F556C/Q332C. TMHs I–VI, I-EL4, and VI–XI double mutants were constructed by excising the BsiWI-EcoNI fragment from the TMH VI or EL4 Cys mutant cDNAs and ligating this fragment into the corresponding site in the TMH I or TMH XI Cys mutant cDNA, respectively. The remaining hSERT Cys double mutants (F556C/T497C and F556C/G498C) were generated using F556C as a template to introduce the second mutation. All mutations were confirmed by DNA sequencing (University of Michigan Sequencing Core, Ann Arbor, MI) and subcloned into pcDNA3.1+ vector.
[3H]5-HT Uptake Assays of hSERT Mutants
The ability of each Cys mutant to transport 5-HT was determined. Human embryonic kidney-293 (HEK-293) cells were maintained in Dulbecco's modified Eagle's medium supplemented with 1% penicillin, streptomycin (10,000 units/ml), 2 mm glutamine, and 10% dialyzed fetal bovine serum at 37 °C in a humidified CO2 incubator. The cells (1 × 105 cells/well) were plated in tissue culture plates coated with poly-d-lysine 18 h prior to transfection. The cells were transiently transfected with each mutant cDNA using Lipofectamine 2000 per the manufacturer's protocol (Invitrogen). Forty-eight hours after transfection, the uptake assays were performed. First, cells were washed once with KRH buffer (120 mm NaCl, 4.7 mm KCl, 2.2 mm CaCl2, 10 mm Hepes, 1.2 mm KH2PO4, 1.2 mm MgSO4, and 1.8 g/liter d-glucose at pH 7.4) and then incubated in KRH buffer with or without 10 μm fluoxetine (for nonspecific or total uptake, respectively) for 10 min at 37 °C. Next, 20 nm [3H]5-HT (∼110 Ci/mmol) supplemented with 100 μm ascorbic acid and 100 μm pargyline was added to each well and incubated for another 10 min at 37 °C. The assay was terminated by washing the cells three times with KRH buffer, solubilizing with Microscint-20, and shaken overnight, and radioactivity was determined using a Packard Topcount NXT microplate scintillation counter. Data were analyzed using GraphPad Prism version 3.0 (GraphPad Software, San Diego, CA).
Sensitivity of Cys Mutants to MTS Reagents and Protection Studies Using SERT Ligands
HEK-293 cells were plated, transfected, and maintained as described above. Forty-eight hours after transfection, the uptake assays were performed. Cells were washed with PBS/CM buffer (137 mm NaCl, 2.7 mm KCl, 1.5 mm KH2PO4, 8.1 mm Na2HPO4, 0.1 mm CaCl2, and 1.0 mm MgCl2) and then incubated in KRH buffer containing 1 mm MTSET, 10 μm fluoxetine (nonspecific uptake), or KRH buffer (total uptake) for 10 min at room temperature. The MTS reagents were made fresh in deionized water at the time of the assay. Following MTSET treatment, cells were washed twice with PBS/CM, and the [3H]5-HT uptake assay was performed as described above. For protection studies, HEK-293 cells (2 × 104 cells/well) were transiently transfected with the mutant cDNA of interest and incubated for 48 h at 37 °C in a humidified CO2 incubator. At the time of the assay, the cells were preincubated with buffer, 20 μm unlabeled 5-HT, 20 μm MDMA, 200 μm MPP+, or 50 μm cocaine for 5 min at room temperature, followed by incubation in the presence or absence of MTSET for 10 min. Saturating concentrations of the ligands were used to ensure full occupancy of the transporters. Next, [3H]5-HT uptake assays were performed as described above with nonspecific uptake determined using 10 μm fluoxetine. Data were analyzed using GraphPad Prism version 3.0 (GraphPad Software).
Rate of Inactivation Studies
HEK-293 cells (2 × 104 cells/well) were plated, transfected, and maintained at 37 °C before the assay. Forty-eight hours after transfection, cells were washed once with PBS/CM buffer and incubated with ligand (cocaine or RTI-55) for 5 min at room temperature. The cells were then coincubated with increasing concentrations of MTSET in the presence or absence of ligand for 10 min at room temperature. The cells were washed twice with PBS/CM, and the [3H]5-HT uptake assay was performed as previously described. Nonspecific uptake was determined using 10 μm fluoxetine. The rate of inactivation was calculated after determining the concentration of MTSET that inhibited 50% of maximal inactivation (23). Data analysis was performed using GraphPad Prism version 3.0 (GraphPad Software).
Proximity Studies Using Bifunctional MTS Reagents
To characterize the proximity of our double cysteine mutants, we performed cross-linking studies using the bifunctional MTS reagents: 1,3-propanediyl bismethanethiosulfonate (MTS-3-MTS) and 1,4-butanediyl bismethanethiosulfonate (MTS-4-MTS). Briefly, 48 h after transient transfection, HEK-293 cells (2 × 104 cells/well) expressing the double mutant of interest were washed once with PBS/CM buffer and then incubated with 100 μm MTS-3-MTS, 100 μm MTS-4-MTS, 100 μm propyl methanethiosulfonate (PMTS) (a monofunctional MTS reagent as a control), or buffer alone (total uptake) for 10 min at room temperature. The cells were washed twice with PBS/CM, and [3H]5-HT uptake assays were performed as described above. The nonspecific uptake was determined using 10 μm fluoxetine. Data analysis was performed using GraphPad Prism version 3.0 (GraphPad Software).
Immunoblot and Surface Expression Analysis of hSERT Mutants
HEK-293 cells were plated (5 × 105 cells/well) 18 h prior to transfection as described above. Forty-eight hours after transient transfection with each mutant cDNA, the cells were washed twice with PBS and incubated with 300 μl of radioimmune precipitation solubilization buffer (10 mm Tris-base, pH 7.4, 150 mm NaCl, 1 mm EDTA, 0.1% SDS, 1% Triton X-100, 1% sodium deoxycholate) for 30 min while shaking at 4 °C. Solubilized cells were transferred to prechilled microcentrifuge tubes, and nonsolubilized material was pelleted at 20,000 × g for 10 min at 4 °C. BCA assay (Bio-Rad) was performed to determine protein concentration using an aliquot of the supernatant. Protein extracts were separated by SDS-PAGE, followed by transfer of proteins to polyvinylidene difluoride membrane (Bio-Rad). The blots were blocked with 5% nonfat dried milk in PBS containing 0.1% Tween 20 and probed with an hSERT monoclonal antibody (mAB Technologies, Inc.) for 1 h. The dilution of the antibody used was 1:1000, followed by three washes in PBS/Tween solution. The blots were then incubated with a secondary antibody (goat anti-mouse horseradish peroxidase) for 1 h at a dilution of 1:5000, followed by three washes in PBS/Tween solution. Blots were developed using enhanced chemiluminescence (ECL, Amersham Biosciences) and exposed to film.
Surface expression binding experiments were performed in 24-well plates (1 × 105 cells/well) precoated with poly-d-lysine as described previously (9). The day after plating, cells were washed once with KRH buffer. A saturating concentration (20 nm) of [3H]citalopram was used as a radiolabeled ligand for hSERT. Radiolabeled ligand and cells were incubated at 4 °C for 1 h. After incubation, cells were rapidly washed twice with KRH buffer, solubilized with a 10% SDS solution, and transferred into a scintillation vial. Bound [3H]citalopram was determined using a Beckman LS 1801 liquid scintillation counter. Total binding was established in the presence of KRH buffer. Nonspecific binding was defined as the binding of radiolabeled ligand in the presence of fluoxetine (10 μm). Internal binding was determined in the presence of MPP+ (400 μm).
RESULTS
Construction and Transport Activity of hSERT Cys Mutant
The generation of a human serotonin transporter homology model based on the crystal structure of the A. aeolicus leucine transporter (LeuTAa), a bacterial homologue of the Na+/Cl−-dependent transporters, provided us with the opportunity to develop a rational hypothesis for the identity of residues involved with substrate transport (5). Molecular structure/function studies examining residues modeled at the entrance to the putative permeation pathway can provide evidence that validates directly our homology model. This model proposes that the entrance to the pore is formed by the participation of several TMHs, along with EL4. Residues modeled at the entrance to the permeation pore for hSERT were as follows: TMH I (Ile108), TMH VI (Ala330, Ala331, and Gln332), TMH X (Thr497 and Gly498), and EL4 (Glu396, Val397, and Ala398) (Fig. 1). These residues reside above the putative extracellular gate and the substrate binding site (Fig. 1A).
FIGURE 1.

Homology model of the SERT demonstrating the presence of residues predicted to be at the entrance to the permeation pore. The SERT TMHs predicted to form the entrance to the permeation pathway (TMHs I, VI, X, XI, and EL4) are shown. A, homology model of SERT based on the closed-closed conformation of LeuTAa (5) showing the predicted location of the 5-HT substrate binding site. The protein backbone is colored light blue, and three residues in the substrate pocket (Tyr176, Phe335, and Asp98) are colored green. The residues above this pocket, in the extracellular facing vestibule, are colored pink and include Ala330, Ala331, Gln332, Ile108, and the gating residues Arg104 and Glu493. B, SERT homology model with the residues (labeled in white) predicted to be positioned at the extracellular surface of the transporter. Each residue was computationally mutated to cysteine, and the approximate distance (in Å) between the Cys pair (S–S) was determined using PyMOL. PyMOL was allowed to position the -SH side chain in the default orientation with no steric clashes observed. C, overlay of different conformations of LeuTAa. Closed-closed conformation is shown in blue, whereas the tryptophan-bound LeuTAa structure is shown in orange (30). Residues corresponding to hSERT Ile108, Ala330, Ala331, and Gln332 are shown in stick form on TMH I and VI, respectively. The structures reveal significant movement of TMHs I and VI as a result of the ligand (tryptophan) binding.
To probe the contribution of these residues in the substrate permeation pathway, we employed the SCAM (11–16). This approach allows us to study the aqueous accessibility and sensitivity of introduced cysteines to thiol-specific reagents. We individually mutated each amino acid to a cysteine in the hSERT C109A background. This native cysteine in the first extracellular loop of SERT is known to be reactive with MTS reagents (15). Our homology model suggested that no other native cysteines were in proximity (within 10 Å) to the mutated residues, suggesting the lack of influence by these other Cys residues. All of the Cys mutants were functional for 5-HT uptake, although I108C, Q332C, V397C, T497C, and G498C exhibited reduced function (Fig. 2). Of these, the most marked loss of function due to Cys replacement was seen at Q332C. The Q332C mutant displayed 14 ± 3% transport activity compared with hSERT C109A (Fig. 2). The sensitivity of our assay system allowed us to test mutants reliably that retained at least 5% of transport function.
FIGURE 2.

Transport activity of hSERT Cys mutants. C109A and hSERT Cys mutant cDNAs were transiently transfected in HEK-293 cells as described under “Experimental Procedures.” Forty-eight hours post-transfection, the cells were assayed for [3H]5-HT transport activity. Nonspecific uptake was determined using 10 μm fluoxetine and typically represented between 3 and 5% of total uptake. In the case of the Q332C mutant, which demonstrated the lowest specific uptake, total uptake was 12,560 ± 210 cpm, and nonspecific uptake was 650 ± 20 cpm. The results represent means ± S.E. (error bars) from at least three separate experiments performed in triplicate.
Immunoblot Analysis of hSERT Cys Mutants Protein Expression
To examine the protein expression of each Cys mutant, we performed immunoblot analysis using an hSERT-specific monoclonal antibody. HEK-293 cells were transiently transfected with the mutant of interest as described under “Experimental Procedures.” A 65 kDa band represented hSERT (15) (Fig. 3A). The more mature glycosylated forms of the protein migrate around 75–80 kDa (Fig. 3A). Also, SERT is thought to form higher order oligomeric structures (24). The high molecular weight species could represent dimers and oligomers (Fig. 3A). The Cys replacement at positions 108 and 397 reduced the amount of the highly glycosylated forms of hSERT, which could provide one possible explanation for the loss of transport observed with these mutants (Figs. 2 and 3A). Surprisingly, Q332C was expressed at levels comparable with hSERT C109A (Fig. 3A). This glutamine is conserved among members of the Na+/Cl−-dependent family of transporters, suggesting its importance in transporter function. Replacement with a different residue could account for the loss of transport function observed. The remaining transporter mutants expressed amounts of the various forms of the protein comparable with C109A (Fig. 3A). These data suggest that loss of transporter function observed for Q332C, T497C, and G498C does not reflect disruption in the overall biosynthesis of the protein. Furthermore, radioligand binding experiments revealed that all mutant transporters were expressed on the cell surface at a level of 20–40% of total transporters (Fig. 3B). This finding would suggest that the marked reductions in transport activity observed for some mutants are not associated with defects in trafficking to the cell surface.
FIGURE 3.

Evaluation of hSERT Cys mutant protein expression. A, whole cell lysates from HEK-293 cells transiently transfected with the hSERT C109A mutant, hSERT Cys single mutants, and hSERT Cys double mutants were prepared as described under “Experimental Procedures.” Proteins were separated by SDS-PAGE, and the transporter was detected by immunoblotting using an hSERT monoclonal antibody. The arrows denote hSERT migration at 65 kDa. A more highly glycosylated form migrates at ∼75–80 kDa. The blots are representative of three independent experiments. B, cell surface radioligand binding for wild-type or hSERT mutants. Assays were performed in 24-well plates coated with poly-d-lysine as described under “Experimental Procedures.” A saturating concentration of [3H]citalopram (20 nm) was used for hSERT and mutants. Nonspecific binding was defined as the binding of radiolabeled ligand in the presence of 10 μm fluoxetine. Internal binding was determined as the binding in the presence of 400 μm MPP+. Specific surface binding was calculated as (total binding − binding in the presence fluoxetine) − (binding in the presence of MPP+ − binding in the presence of fluoxetine). Transporter molecules were calculated assuming one citalopram bound per transporter. Bars, the mean of three independent experiments ± S.E. (error bars).
Sensitivity of hSERT Cys Mutants to MTS Reagents
To examine the reactivity of each Cys mutant to MTS reagents, SCAM studies were performed in HEK-293 cells transiently transfected with the C109A mutant or each Cys mutant cDNA. The aqueous accessibility and functional importance of the residues were assessed after incubating with 1 mm MTSET. This membrane-impermeant compound covalently modifies cysteine residues that are accessible to the aqueous environment of the protein. Disruption in transporter function by MTS reagents implicates the residue as being present in or near the substrate pore. The results were analyzed using normalized data for each mutant in order to examine MTS reactivity across different activity levels of the individual mutants. We observed significant (50–75%) MTSET-induced inactivation of transport at the I108C, A331C, Q332C, V397C, T497C, and G498C mutants (Fig. 4A). The decrease in [3H]5-HT uptake following treatment with MTSET suggested the presence of these residues in a hydrophilic environment of the protein that when modified disrupts transport activity.
FIGURE 4.
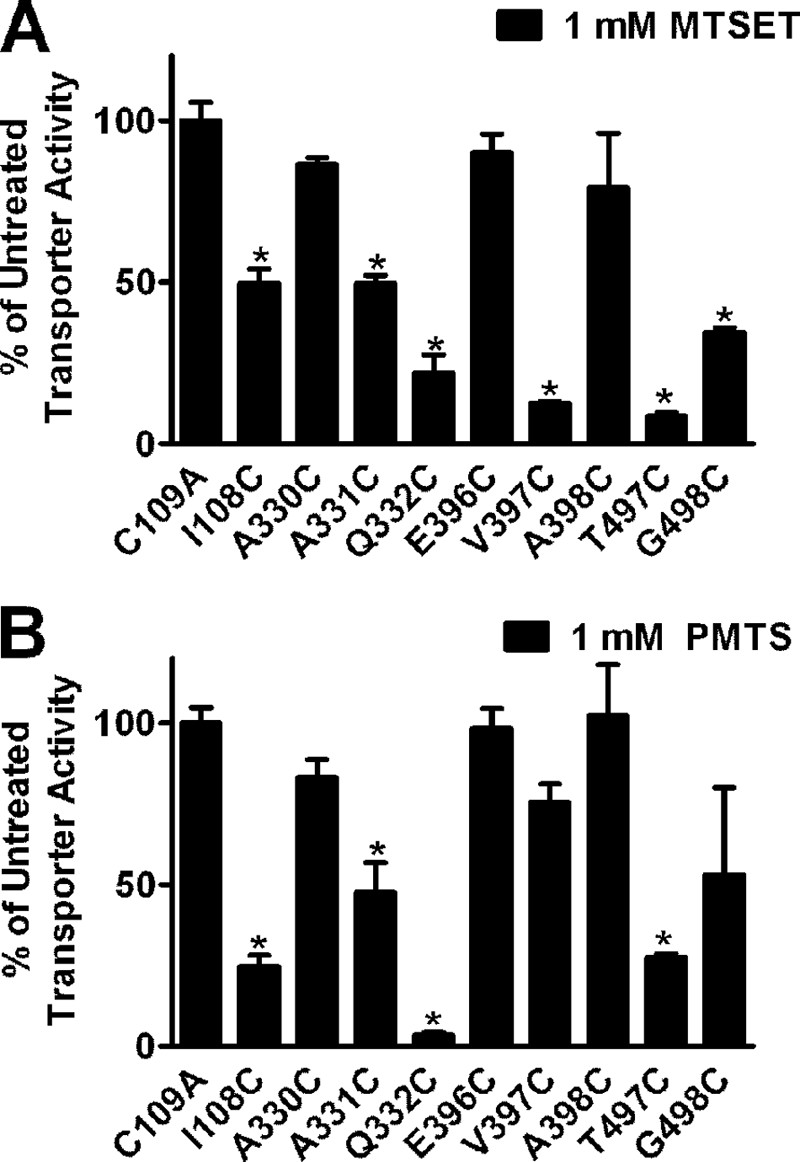
Sensitivity of residues predicted to reside at the opening of the pore to MTS reagents. HEK-293 cells transiently transfected with C109A and hSERT Cys mutant cDNAs were incubated with 1 mm MTSET (A) or 1 mm PMTS (B) for 10 min at room temperature, followed by [3H]5-HT uptake as described under “Experimental Procedures.” Bars, means ± S.E. (error bars) from three independent experiments performed in triplicate. Normalized data are presented for each mutant. *, p < 0.001 compared with C109A using a one-way analysis of variance with a post hoc Dunnett's test.
Potential electrostatic effects that could influence accessibility and reactivity associated with MTSET, which is positively charged, were ruled out by examining the sensitivity of each mutant to the neutral MTS compound PMTS. Reactivity of the mutants with PMTS paralleled the results obtained with MTSET with the exception that sensitivity of the V397C and G498C mutants did not reach significance for PMTS. In general, the results with PMTS alleviated concerns that electrostatic effects were impacting inactivation of the mutants by MTSET (Fig. 4B).
Protection against MTS Reactivity by SERT Ligands
The SCAM approach identified residues that are sensitive and accessible to MTS reagents. In order to understand better the proximity of these residues to critical sites for ligand binding, we examined the ability of different SERT substrates and antagonists to protect against reactivity with MTSET. HEK-293 cells expressing the Cys mutants were coincubated with MTSET in the presence or absence of a particular ligand, as described under “Experimental Procedures.” The ability of a ligand to alter sensitivity was revealed by a change in [3H]5-HT uptake compared with the activity levels in the absence of ligand for each individual mutant.
Among the mutants tested, TMH VI residues displayed the most marked changes in MTSET reactivity in the presence of 5-HT. Coincubation with unlabeled 5-HT fully protected Q332C from MTSET inactivation, possibly suggesting that the residue resides near a binding site for 5-HT and is altered conformationally upon 5-HT binding (Fig. 5A). We also tested the ability of MDMA and MPP+ to protect from MTSET inactivation, but neither affected reactivity with the reagent (Fig. 5, B and C).
FIGURE 5.

Protection against MTS reactivity by SERT substrates. The ability of 5-HT (A), MDMA (B), and MPP+ (C) to protect the mutants from MTS reactivity was evaluated in HEK-293 cells transiently transfected with hSERT Cys mutants. Briefly, the cells were incubated with buffer, 20 μm 5-HT, 20 μm MDMA, or 200 μm MPP+ for 5 min at room temperature, followed by incubation in the presence or absence of 1 mm MTSET for 10 min. After washing, [3H]5-HT uptake assays were performed as described under “Experimental Procedures.” Bars, mean ± S.E. (error bars) from three independent experiments performed in triplicate. The data were normalized to the total uptake of each individual mutant. *, p < 0.01 versus MTSET treatment using a one-way analysis of variance with Bonferroni's post hoc test.
Effect of Cocaine on MTS Reactivity
We extended our studies by testing the ability of cocaine to alter MTSET sensitivity. Coincubation with cocaine enhanced reactivity of Q332C to MTSET (Fig. 6A). Notably, cocaine and unlabeled 5-HT had opposite effects on the inactivation process at Q332C (Figs. 5A and 6A). One possibility is that cocaine binding induces a conformational change in the transporter, making Q332C more accessible to MTSET, thus enhancing transporter inactivation and inhibition of [3H]5-HT uptake.
FIGURE 6.

Effect of cocaine on reactivity of hSERT Q332C mutant with MTSET. A, the ability of the hSERT antagonist cocaine to alter the reactivity of hSERT Cys mutants to MTSET was assessed in HEK-293 cells transiently transfected with each Cys mutant. The cells were incubated with buffer or 50 μm cocaine for 5 min at room temperature, followed by incubation in the presence or absence of 1 mm MTSET for 10 min. After washing, [3H]5-HT uptake assays were performed as described under “Experimental Procedures.” Bars, mean ± S.E. (error bars) from three independent experiments performed in triplicate. The data were normalized to the total uptake of each individual mutant. #, p < 0.001 versus MTSET treatment using a one-way analysis of variance with Bonferroni's post hoc test. The effect of cocaine on the rate of MTSET inactivation was determined in HEK-293 cells transiently transfected with the cDNA for the Q332C mutant. The cells were incubated with increasing concentrations of MTSET in the presence (filled squares) or absence (filled triangles) of 50 μm cocaine (B) or 300 nm RTI-55 (C), a cocaine analog, followed by determining [3H]5-HT uptake, as described under “Experimental Procedures.” The nonspecific uptake was determined using 10 μm fluoxetine. MTSET inactivated Q332C at an estimated rate of ∼700 m −1 min−1 versus ∼140 m−1 min−1 in the presence or absence of cocaine, respectively. MTSET inactivated Q332C at a calculated rate of ∼2600 m−1 min−1 in the presence of RTI-55, which is estimated to be significantly greater than a rate that causes complete inactivation at ∼125 μm under the conditions used. Data represent the mean from three independent experiments performed in triplicate.
To characterize further the cocaine effect on MTSET reactivity of Q332C, we examined the rate of MTSET-induced inactivation. The study involved determining the amount of transporter activity remaining after incubating with increasing concentrations of the MTS reagent. The rate is determined from the concentration of reagent that results in half-maximal inactivation (23, 25). Our studies revealed an ∼5-fold increase in the rate of MTSET inactivation at the Q332C mutant in the presence of 50 μm cocaine versus the absence of cocaine (Fig. 6B). The cocaine analog, RTI-55, also enhanced the rate of inactivation over that observed in the absence of ligand (Fig. 6C).
Construction, Transport Activity, and Protein Expression of hSERT Cys Double Mutants
Our SERT model provides the opportunity to make predictions about the proximity relationships of the helices implicated in forming the entrance to the permeation pathway. To examine the distance between residues identified at the entrance to the pore, we took advantage of the available Cys single mutants and generated Cys double mutants in the C109A background (Table 1). Our laboratory has previously observed that hSERT F556C is sensitive to inactivation by MTSET (9). In the present study, F556C as well as the Cys single mutants described above were used to generate a series of hSERT Cys double mutants: I108C/A330C, I108C/A331C, I108C/Q332C, I108C/E396C, F556C/A330C, F556C/A331C, F556C/Q332C, F556C/T497C, and F556C/G498C.
TABLE 1.
Predictions of distances at the hSERT Cys double mutants
The homology model of SERT based on the LeuTAa was used to measure the approximate S–S distances between residues. Each residue was computationally mutated to cysteine, and the distance between the Cys pair (S–S) was determined (PyMOL; DeLano Scientific, LLC, San Carlos, CA). Approximate distances were based on an orientation of the -SH side chains, which were positioned by the default setting in PyMOL with no steric clashes observed.
| Mutant pair | Predicted distance (Cys–Cys) |
|---|---|
| Å | |
| TMH XI-X | |
| F556C-T497C | 12.1 |
| F556C-G498C | 8.0 |
| TMH XI-VI | |
| F556C-A330C | 7.7 |
| F556C-A331C | 5.5 |
| F556C-Q332C | 12.9 |
| TMH I-VI | |
| I108C-A330C | Inaccessible |
| I108C-A331C | 5.0 |
| I108C-Q332C | 3.5 |
| I108C-E396C | Inaccessible |
Prior to exploring the approximate distances among these residues, all mutants were analyzed for [3H]5-HT transport. All of the double mutants showed detectable [3H]5-HT uptake compared with C109A in HEK-293 cells transiently transfected with each mutant cDNA (Fig. 7). Although significant loss of function was evident for I108C/Q332C, I108C/E396C, and F556C/Q332C, sufficient functional activity was observed to allow further characterization (Fig. 7).
FIGURE 7.

Transport activity of hSERT Cys double mutants. hSERT C109A and Cys double mutant cDNAs were transiently transfected in HEK-293 cells as described under “Experimental Procedures.” After 48 h, the cells were assayed for [3H]5-HT transport activity. Nonspecific uptake was determined using 10 μm fluoxetine. The results represent means ± S.E. (error bars) from at least three separate experiments performed in triplicate.
To evaluate protein expression of the double mutants, immunoblot analysis was performed in whole cell lysates from transfected cells (Fig. 3). The apparent loss in the highly glycosylated form of the protein at I108C/E396C could explain the significant decrease in transport activity observed (Figs. 3 and 7). At I108C/A330C, modest loss of the highly glycosylated form of the protein also could explain the reduced uptake (Figs. 3 and 7). The remaining mutants showed amounts of protein expression similar to C109A (Fig. 3).
Cross-linking Studies Using Bifunctional MTS Reagents
To characterize the proximity of the Cys double mutants, we performed cross-linking studies using the bifunctional MTS reagent MTS-3-MTS (10). This compound is capable of cross-linking Cys residues within a distance of 5–6.5 Å (17). Our SERT model predicted that I108C/A331C were at an approximate distance of 5 Å (Table 1). The premise of the assay is dependent upon the cross-linking reagent reacting to a greater extent with the double mutant as compared with the parental single Cys mutants. In the case of I108C/A331C, MTS-3-MTS cross-linked and significantly inhibited [3H]5-HT uptake compared with the single mutants, suggesting that these two residues are accessible and in proximity to each another (Fig. 8A). The transport activity remaining after cross-linker treatment at both single and double mutants is displayed to permit visualization of cross-linker impact at the double mutants. Results were analyzed using normalized data for each mutant, where activity remaining in the absence of reagent was normalized to total uptake for each mutant. Surprisingly, MTS-3-MTS significantly inhibited [3H]5-HT uptake at I108C/A330C (Fig. 8A). Our model, however, suggested that I108C and A330C were not accessible to each other (Fig. 1B and Table 1). Therefore, we did not expect to observe inhibition of [3H]5-HT uptake with the bifunctional MTS reagent.
FIGURE 8.

Cross-linking studies using bifunctional MTS reagents at hSERT Cys double mutants. HEK-293 cells transiently expressing the hSERT C109A, Cys single mutants, or Cys double mutants were incubated with 100 μm MTS-3-MTS (A and B) or 100 μm MTS-4-MTS (C and D) for 10 min at room temperature. Following incubation, the cells were washed twice and assayed for [3H]5-HT uptake as described under “Experimental Procedures.” The data were normalized to the total uptake of each individual mutant. Data represent the mean ± S.E. (error bars) from three independent experiments performed in triplicate. #, p < 0.001 versus C109A and each single mutant forming the double mutant using a one-way analysis of variance with Bonferroni's post hoc test.
Our SERT model suggested that I108C and Q332C were approximately ∼3.5 Å from each other. Unless the bifunctional cross-linker is able to fold onto itself, we predicted this distance to be incompatible with efficient cross-linking. We also predicted inaccessibility between I108C and E396C. Overall, no effect was expected at these pairs of mutants. In agreement with our expectations, no significant changes in uptake were observed at those positions after MTS-3-MTS treatment compared with the single mutants (Fig. 8A).
Our model predicted larger distances between residues in TMHs VI and XI and TMHs X and XI (Table 1). To examine these predictions, we determined the ability of MTS-3-MTS to cross-link and inactivate Cys double mutants in those helices. As predicted, none of the double mutants from these domains displayed significant inhibition of [3H]5-HT uptake after MTS-3-MTS treatment as compared with each single mutant (Fig. 8B). Next, we evaluated the sensitivity of the double mutants to a longer bifunctional MTS reagent MTS-4-MTS. This compound is capable of spanning the distance between Cys residues ∼7.8 Å apart (17). Among the double mutants tested, our model predicted that F556C and A330C were at a distance of ∼7.7 Å (Table 1). MTS-4-MTS significantly inhibited [3H]5-HT uptake at F556C/A330C consistent with the model (Fig. 8D). Moreover, no significant changes in the remaining pairs of mutants were detectable because the modeled distances separating these residues were too large to be cross-linked by the bifunctional reagents used in this study. However, MTS-4-MTS was able to reduce significantly [3H]5-HT uptake at I108C/A330C and I108C/A331C (Fig. 8C). As described above, MTS-3-MTS also inhibited 5-HT transport at the I108C/A330C double mutant, although these residues were predicted to be inaccessible to each other (Fig. 8A and Table 1). To rule out the possibility that the inhibition of [3H]5-HT uptake observed by both reagents may be due to both cysteines being individually alkylated but not cross-linked, we examined the sensitivity of the mutants to the small size monofunctional MTS reagent PMTS (26). Pretreatment with PMTS did not inhibit 5-HT transport at I108C/A330C (data not shown) as compared with both of the bifunctional MTS reagents. It is possible, however, that the bifunctional reagents were able to inhibit transport as a result of the bulk added by modification. The PMTS results are consistent with the conclusion that the bifunctional MTS reagents are in fact cross-linking these two Cys mutants. Due to the flexibility of the bifunctional MTS cross-linkers providing a range of distances and allowing their spans to overlap, we concluded that the bifunctional MTS sensitivity observed for I108C/A330C supports an experimentally determined distance between these residues of 6–7 Å.
One model of substrate translocation suggests that TMHs I, II, VI, and VII tilt inward to promote a cytoplasm-facing conformation (27). The movement of these helices during transport is predicted to alter the distance between TMHs VI and XI. In the presence of 5-HT, F556C and A330C/F556C demonstrated enhanced reactivity with MTS-3-MTS (Fig. 9A). The enhanced reactivity of F556C in the presence of 5-HT most likely contributed to increased reactivity of the other double mutants (A331C/F556C and Q332C/F556C) to MTS-3-MTS (data not shown). Interestingly, cocaine increased reactivity of the A330C/F556C mutant to MTS-3-MTS compared with the respective single mutants (Fig. 9B). Reactivity of the other TMH VI/XI double mutants with MTS-3-MTS was unaffected by the presence of cocaine (data not shown).
FIGURE 9.

Effects of SERT ligands on reactivity of bifunctional MTS reagents at hSERT TMH VI and XI Cys double mutant. HEK-293 cells transiently expressing the hSERT Cys single mutants or Cys double mutant were incubated with 100 μm MTS-3-MTS for 10 min at room temperature in the presence or absence of 20 μm 5-HT (A) or 50 μm cocaine (B) (5-min preincubation). Following incubation, the cells were washed twice and assayed for [3H]5-HT uptake as described under “Experimental Procedures.” The data were normalized to the specific uptake of the untreated control for each individual mutant. Data represent the mean ± S.E. (error bars) from three independent experiments performed in triplicate. *, p < 0.05 using a t test compared with MTS-3-MTS alone.
DISCUSSION
In this study, a homology model of SERT based on the crystal structure of a member of the neurotransmitter sodium symporter family was used to obtain insights into the extracellular entrance to the substrate permeation pathway (9, 10). Our model suggests that the entrance to the pore is lined by TMHs I, III, VI, X, and XI as well as EL4 (Fig. 1, A and B). In the SERT, TMHs I and III have been shown to contain residues associated with substrate and antagonist binding (12, 15). In separate studies on SERT, extracellular loop 4 and a four-helix bundle composed of TMHs I, II, VI, and VII have been associated with conformational changes coupled to substrate transport (16).
We employed the SCAM approach that has been used extensively to probe the accessibility of introduced cysteines to sulfhydryl-specific reagents to learn about topological location and functional importance of residues in membrane proteins. The cysteine mutants were well tolerated at the introduced positions, with the exception of Gln332, a polar residue conserved among members of the NSSs. In HEK-293 cells transiently transfected with Q332C, reduced 5-HT transport activity was observed, although immunoblot analysis revealed that protein expression for this mutant was not altered. The pronounced reduction in transport activity observed with the Q332C mutant may reflect its importance for transporter function. By using MTSET, an impermeant thiol-reactive compound, we identified residues in TMHs I, VI, X, and EL4 that were exposed to the extracellular environment and possibly the putative entrance to the substrate permeation pathway. Specifically, I108C, A331C, Q332C, V397C, T497C, and G498C demonstrated a significant decrease in 5-HT uptake following MTSET treatment. Also, our previous work in TMH XI had identified hSERT Phe556 at the entrance to the permeation pore (9).
Our studies identified residues that are accessible to the external medium. However, using ligands to protect residues from inactivation by MTS reagents can be used as evidence that a residue may be part of or near a specific binding site (12, 14, 15). Interestingly, studies with Q332C revealed protection by unlabeled 5-HT. Conversely, coincubation with cocaine enhanced reactivity of the mutant to the MTS reagent. One explanation would be that the residue is in or near the binding site for 5-HT; alternatively, the transporter could undergo a conformational change associated with substrate binding or transport occluding the residue from the extracellular medium. The fact that cocaine, a nontransported inhibitor, enhances reactivity to MTSET at the same position suggests that what we observed could be indirect protection by substrate transport. Moreover, cocaine increased the rate of inactivation, suggesting that the antagonist may stabilize the protein in an external facing conformation, making the Cys more accessible. Therefore, hSERT/Gln332 may be part of a conformation-sensitive area that is occluded when substrate is transported but is accessible when cocaine binds. Consistent with this conclusion was the finding that both the cocaine and 5-HT effect on Q332C sensitivity to MTSET was sodium-dependent (data not shown), consistent with previous findings that ligand-induced conformational changes in SERT require sodium (28). Similar effects were observed for I155C in the norepinephrine transporter and Y403C in the glutamate transporter GLT-1, where substrate transport and antagonist binding inhibited and stimulated inactivation, respectively (14, 29).
A crystal structure of LeuTAa with the amino acid tryptophan bound has been reported (30). Tryptophan binds to LeuTAa but is not transported, locking the transporter in an open-to-out conformation. Examination of the tryptophan-bound LeuTAa structure may reveal the impact of ligand binding on the movement of TMH I and VI. In this structure, TMH VI shifts significantly upward and inward toward the pore. TMH I shifts toward EL4, moving Gln34 (the equivalent of hSERT Ile108) toward the pore entrance (Fig. 1C). If cocaine induces a similar conformational shift, the increased sensitivity of Q332C to MTSET in the presence of cocaine could be explained. A comprehensive model of substrate and inhibitor binding for DAT in the context of the LeuTAa structure predicts that cocaine and cocaine analogs bind between several segments of the transporter, including TMH VI (19). The inhibitor binding site would overlap with the substrate binding site for DAT, suggesting a competitive inhibition of transport (19). Our data also support a model where residues in TMH VI are in contact or near the substrate and cocaine binding sites for SERT. In fact, our data also suggest that cocaine and its analogs stabilize the outward facing conformation of SERT, in agreement with the DAT model (19). Furthermore, cocaine has been shown to reduce the accessibility of residues near the cytoplasmic surface of the transporter to membrane-permeant MTS reagents (27). This finding provides further support that cocaine may stabilize an outward facing conformation consistent with our observation for increased reactivity of Q332C in the presence of cocaine.
Our SERT homology model also provides the opportunity to make predictions about the distances between and orientation of the helices forming the entrance to the permeation pathway. Previous studies confirmed the proximity of hSERT TMHs I and III by successfully generating Zn2+-binding sites and through cross-linking studies (10, 31). In the present study, cross-linking experiments using MTS-3-MTS supported the orientation of residues Val102 (TMH I) and Met180 (TMH III) toward each other. Our SERT model supports this proximity relationship previously identified for TMHs I and III (10).
We constructed additional Cys double mutants to confirm the proximity relationships of the helices forming the entrance to the permeation pathway. Some of the mutations induced significant loss of transport activity, particularly the two mutants that contained Q332C. However, due to the sensitivity of the uptake assay, all of the mutants were further evaluated if they retained at least 5% of transporter function. We examined the effect of two bifunctional MTS reagents in the inactivation of transporter function at the double mutants. In the presence of MTS-3-MTS, a significant decrease in [3H]5-HT uptake was observed at I108C/A330C and I108C/A331C, thus defining the orientation of residues in TMH I and VI. Accordingly, the predicted distance between I108C and A331C was ∼5 Å, but a larger distance was expected for I108C/A330C (Table 1).
Our model predicted that I108C and A330C would be inaccessible to each other because they would reside on opposite sides of their respective helices. The helical breaks that are predicted to exist halfway through the membrane bilayer for these helices may provide for conformational flexibility that allows for this accessibility. An alternative explanation for the reactivity of these residues with the cross-linking reagents could be related to the flexibility of the compounds as opposed to conformational changes in the protein. Again, based on the tryptophan-bound LeuTAa structure (Fig. 1C), we propose that the conformation of TMH VI may be dynamic and possibly allow for the helix to rotate clockwise. This alternative conformation may be associated with translocation steps and could place Gln34 and Val248 (hSERT Ile108 and Ala330) in closer proximity to each other when the transporter is in the open-to-out conformation. This rotation also would provide for another intermediate conformation whereby I108C and A331C as well as F556C and A330C could be cross-linked. Experimental results and homology modeling have suggested that part of the conformational change associated with substrate translocation includes a tilting of TMHs I, II, VI, and VII (27). A shift in these helices would be supported by our results. For example, as TMH VI tilts inward during transport, F556C on TMH XI would be predicted to become more exposed. The increased reactivity of F556C and A330C/F556C to the bifunctional MTS reagent in the presence of 5-HT is consistent with the proposed conformational shifts in TMHs I, II, VI, and VII associated with an inward facing conformation (27). Interestingly, a recent report identifying a potential second substrate binding site on the dopamine transporter suggests a similar conformational shift for TMHs I and V (32).
Our model suggests that TMHs VI and XI as well as X and XI are separated by larger distances that would be inaccessible for the MTS-3-MTS compound (Table 1). Our data revealed no significant changes in transporter function for the double mutants tested in those helices. Consequently, we decided to use a longer bifunctional cross-linker, MTS-4-MTS, to measure the distance of those residues predicted to reside further away from each other. This particular reagent was capable of cross-linking residues ∼7.8 Å apart. MTS-4-MTS inhibited 5-HT transport at F556C/A330C, whereas MTS-3-MTS did not. Accordingly, our model proposed that F556C was separated from A330C by approximately 7.7 Å, a span too great for MTS-3-MTS. Interestingly, the conformation of these helices induced by the presence of cocaine appears to not only increase accessibility of Q332C but also shorten the distance between A330C and F556C (TMHs VI and XI, respectively) from ∼7.7 Å predicted in our closed-closed state model (Table 1) to the 5–6.5 Å indicated by MTS-3-MTS reactivity of the double mutant. A straightforward explanation of these results is that cocaine binding promotes a conformation where Ala330 and Phe556 are in closer proximity to each other. This could be a result of an open-to-out conformation similar to that observed in tryptophan-bound LeuTAa (Fig. 1C) or the occluded-out structure proposed in the modeling of cocaine binding to DAT (19). The model of cocaine binding to DAT does not explicitly address potential changes in the distances between TMH VI and XI, but our data would suggest that this distance is narrowed in the presence of cocaine. Thus, if TMHs I, II, VI, and VII move as a bundle in response to cocaine binding (27), this outward facing conformation of SERT would leave TMHs VI and XI in close proximity to each other. With regard to the other double mutants generated between these helices, most pairs exhibited larger predicted distances (>8 Å) than the spans of either MTS-3-MTS or MTS-4-MTS (Table 1). Although we attempted to use longer bifunctional reagents, one limitation encountered was their restricted solubility, thus impairing their use for our studies.
Taken together, we identified portions of helices that contribute to the entrance into the substrate permeation pathway. Our data suggest that TMHs I and VI as well as VI and XI are in close proximity, confirming the orientation of residues toward each other as predicted from our model (Fig. 1). We propose that TMHs I or VI possess flexible conformations more consistent with the tryptophan-bound LeuTAa structure and may be closer than what the original closed-closed state model predicted. A better understanding of the contribution of these central helices in forming the permeation pathway will guide the development of more refined models of SERT and other members of the neurotransmitter transporter family.
Acknowledgments
We thank Cody Wenthur and Natalie Sealover for assistance with data collection.
This work was supported, in whole or in part, by National Institutes of Health Grant DA018682 (to E. L. B. and D. E. N.).
- 5-HT
- 5-hydroxytryptamine
- SERT
- serotonin transporter
- hSERT
- human SERT
- DAT
- dopamine transporter
- TMH
- transmembrane helix
- SCAM
- substituted cysteine accessibility method
- MTS
- methanethiosulfonate
- MTSET
- 2-(trimethylammonium)ethyl methanethiosulfonate bromide
- MTS-3-MTS
- 1,3-propanediyl bismethanethiosulfonate
- MTS-4-MTS
- 1,4-butanediyl bismethanethiosulfonate
- NSS
- neurotransmitter sodium symporter
- EL4
- extracellular loop 4
- PBS
- phosphate-buffered saline
- PMTS
- propyl methanethiosulfonate
- CM
- calcium and magnesium.
REFERENCES
- 1.Gether U., Andersen P. H., Larsson O. M., Schousboe A. (2006) Trends Pharmacol. Sci. 27, 375–383 [DOI] [PubMed] [Google Scholar]
- 2.Henry L. K., Field J. R., Adkins E. M., Parnas M. L., Vaughan R. A., Zou M. F., Newman A. H., Blakely R. D. (2006) J. Biol. Chem. 281, 2012–2023 [DOI] [PubMed] [Google Scholar]
- 3.Torres G. E., Gainetdinov R. R., Caron M. G. (2003) Nat. Rev. Neurosci. 4, 13–25 [DOI] [PubMed] [Google Scholar]
- 4.Henry L. K., Meiler J., Blakely R. D. (2007) Mol. Interv. 7, 306–309 [DOI] [PubMed] [Google Scholar]
- 5.Yamashita A., Singh S. K., Kawate T., Jin Y., Gouaux E. (2005) Nature 437, 215–223 [DOI] [PubMed] [Google Scholar]
- 6.Beuming T., Shi L., Javitch J. A., Weinstein H. (2006) Mol. Pharmacol. 70, 1630–1642 [DOI] [PubMed] [Google Scholar]
- 7.Henry L. K., Defelice L. J., Blakely R. D. (2006) Neuron 49, 791–796 [DOI] [PubMed] [Google Scholar]
- 8.Ravna A. W., Jaronczyk M., Sylte I. (2006) Bioorg. Med. Chem. Lett. 16, 5594–5597 [DOI] [PubMed] [Google Scholar]
- 9.Torres-Altoro M. I., White K. J., Rodríguez G. J., Nichols D. E., Barker E. L. (2008) Protein Sci. 17, 1761–1770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.White K. J., Kiser P. D., Nichols D. E., Barker E. L. (2006) Protein Sci. 15, 2411–2422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Akabas M. H., Stauffer D. A., Xu M., Karlin A. (1992) Science 258, 307–310 [DOI] [PubMed] [Google Scholar]
- 12.Chen J. G., Sachpatzidis A., Rudnick G. (1997) J. Biol. Chem. 272, 28321–28327 [DOI] [PubMed] [Google Scholar]
- 13.Chen J. G., Liu-Chen S., Rudnick G. (1998) J. Biol. Chem. 273, 12675–12681 [DOI] [PubMed] [Google Scholar]
- 14.Chen J. G., Rudnick G. (2000) Proc. Natl. Acad. Sci. U.S.A. 97, 1044–1049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Henry L. K., Adkins E. M., Han Q., Blakely R. D. (2003) J. Biol. Chem. 278, 37052–37063 [DOI] [PubMed] [Google Scholar]
- 16.Mitchell S. M., Lee E., Garcia M. L., Stephan M. M. (2004) J. Biol. Chem. 279, 24089–24099 [DOI] [PubMed] [Google Scholar]
- 17.Loo T. W., Clarke D. M. (2001) J. Biol. Chem. 276, 36877–36880 [DOI] [PubMed] [Google Scholar]
- 18.Wang J. B., Moriwaki A., Uhl G. R. (1995) J. Neurochem. 64, 1416–1419 [DOI] [PubMed] [Google Scholar]
- 19.Beuming T., Kniazeff J., Bergmann M. L., Shi L., Gracia L., Raniszewska K., Newman A. H., Javitch J. A., Weinstein H., Gether U., Loland C. J. (2008) Nat. Neurosci. 11, 780–789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kaufmann K. W., Dawson E. S., Henry L. K., Field J. R., Blakely R. D., Meiler J. (2009) Proteins 74, 630–642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Indarte M., Madura J. D., Surratt C. K. (2008) Proteins 70, 1033–1046 [DOI] [PubMed] [Google Scholar]
- 22.Chen J. G., Liu-Chen S., Rudnick G. (1997) Biochemistry 36, 1479–1486 [DOI] [PubMed] [Google Scholar]
- 23.Rudnick G. (2002) Transmembrane Transporters, pp. 125–141, Wiley-Liss, Inc., Hoboken, NJ [Google Scholar]
- 24.Kilic F., Rudnick G. (2000) Proc. Natl. Acad. Sci. U.S.A. 97, 3106–3111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Keller P. C., 2nd, Stephan M., Glomska H., Rudnick G. (2004) Biochemistry 43, 8510–8516 [DOI] [PubMed] [Google Scholar]
- 26.Mascia M. P., Trudell J. R., Harris R. A. (2000) Proc. Natl. Acad. Sci. U. S. A. 97, 9305–9310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Forrest L. R., Zhang Y. W., Jacobs M. T., Gesmonde J., Xie L., Honig B. H., Rudnick G. (2008) Proc. Natl. Acad. Sci. U.S.A. 105, 10338–10343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Androutsellis-Theotokis A., Rudnick G. (2002) J. Neurosci. 22, 8370–8378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zarbiv R., Grunewald M., Kavanaugh M. P., Kanner B. I. (1998) J. Biol. Chem. 273, 14231–14237 [DOI] [PubMed] [Google Scholar]
- 30.Singh S. K., Piscitelli C. L., Yamashita A., Gouaux E. (2008) Science 322, 1655–1661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tao Z., Zhang Y. W., Agyiri A., Rudnick G. (2009) J. Biol. Chem. 284, 33807–33814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Quick M., Winther A. M., Shi L., Nissen P., Weinstein H., Javitch J. A. (2009) Proc. Natl. Acad. Sci. U.S.A. 106, 5563–5568 [DOI] [PMC free article] [PubMed] [Google Scholar]