

Abstract
Many tumor suppressor proteins act to blunt the effects of mitogenic signaling pathways. Loss of function mutations in the merlin tumor suppressor underlie neurofibromatosis type 2 (NF2), a familial autosomal dominant cancer syndrome. Studies of Drosophila suggest that Hippo (hpo) is required for inhibition of cell proliferation mediated by dMer, the orthologue of human merlin. Mammalian sterile 20-like kinase-2 (Mst2) is a mammalian Hpo orthologue, and numerous studies implicate Mst2 as a tumor suppressor. Mst2 is negatively regulated by the proto-oncoprotein Raf-1 in a manner independent of the kinase activity of Raf-1. We sought to determine whether, in mammalian cells, merlin could positively regulate Mst2. We also sought to determine whether Mst2, in addition to being negatively regulated by Raf-1, might itself reciprocally regulate Raf-1. In contrast to findings from Drosophila, we find no evidence that mammalian merlin positively regulates mammalian Mst2. Instead, surprisingly, RNA interference silencing of Mst2 leads to elevated inhibitory phosphorylation of Raf-1 at Ser-259 and impaired Raf-1 kinase activity. Consequent to this, ERK pathway activation and cell proliferation are attenuated. Phosphatase-2A (PP2A) dephosphorylates Raf-1 Ser-259 in response to mitogens. Interestingly RNA interference silencing of Mst2 triggers a striking proteasome-dependent decrease in the levels of the catalytic subunit of PP2A (PP2A-C). A similar effect is achieved upon silencing of large tumor suppressor (LATS)-1 and LATS2, direct substrates of Mst2. Our studies reveal a more complex role for Mst2 than previously thought. The Mst2 → LATS1/2 pathway, by maintaining PP2A-C levels, may, in some situations, positively affect mitogenic signaling.
Keywords: ERK, MAP Kinases (MAPKs), Raf, Tumor, Tumor Suppressor, Hippo Pathway, Mst2, Neurofibromatosis-2 (NF2)
Introduction
Controlling the intensity and duration of mitogen-regulated signaling pathways exerts a profound impact on cell proliferation and survival. The integration of proliferative and tumor suppressor signals is central to normal cellular function and to proliferative diseases such as cancer. Mitogen-activated protein kinase (MAPK)4 signal transduction pathways are present in all eukaryotic cells and regulate a diverse array of cellular functions, including cell differentiation, proliferation, migration, apoptosis, and inflammation. MAPKs are crucial to the integration of proliferative and survival signals in cancer. MAPK signal transduction pathways include a central MAPK-kinase kinase (MAP3K) → MAPK-kinase (MAP2K) → MAPK core module (1, 2). In mammals, mitogen activation of the extracellular signal-regulated kinase (ERK) MAPK group requires Ras, a monomeric G protein recruited by receptor and non-receptor tyrosine kinases (3). Active Ras (Ras-GTP) recruits downstream MAP3Ks of the Raf family (Raf-1, B-Raf, and A-Raf) (3). The mechanism of activation of Raf-1, consequent to Ras binding, is quite complex and not yet fully understood.
In resting cells, Raf-1 is restrained in an inactive state through phosphorylation at Ser-259 (possibly catalyzed by Akt or protein kinase A) (4, 5). The interaction of Raf-1 phospho-Ser-259 with 14-3-3 proteins helps to keep Raf-1 in an inhibitory conformation (6, 7). Upon Ras binding, Raf-1 activation requires dephosphorylation of Ser-259 and consequent dissociation of 14-3-3 proteins (8–10). In addition, a number of Ras-dependent phosphorylation events, notably at Tyr-341 (catalyzed by Src), Ser-338 (possibly catalyzed by p21-activated kinases (PAKs)), Thr-491, and Ser-494 (by unknown kinase (s)), are required (11). Under some circumstances, Raf-1 can either homo-oligomerize or hetero-oligomerize with B-Raf. The latter complex can enable B-Raf trans activation of Raf-1, a process that may contribute further to Raf-1 activation (12, 13).
Several tumor-suppressive pathways directly counter the pro-mitogenic actions of Ras-dependent signaling. For example, neurofibromin, mutations of which underlie type-1 NF, is a Ras-GTPase-activating protein that functions to promote Ras-GTPase activity and consequent Ras inactivation (14). Merlin, through mechanisms that are not fully understood, also suppresses mitogenic signaling including activation of ERK and PAKs (reviewed in Ref. 15). Still other tumor suppressors recruit signaling pathways that act to induce genes that inhibit cell proliferation or promote apoptosis.
The mammalian Ste20-like kinases 1 and 2 (Mst1 and Mst2) are members of the germinal center kinase family of Ser/Thr kinases (16). Drosophila hippo (hpo) and Caenorhabditis elegans CST-1 are orthologues of Mst1/2 (16, 17). Functionally, hpo is a bona fide tumor suppressor in Drosophila (18, 19). Hpo and mammalian Mst1/2 regulate a highly conserved signaling cascade, wherein Hpo/Mst1/2 activate a protein complex consisting of the Ser/Thr kinase Warts (large tumor suppressor 1 and large tumor suppressor 2, LATS1/2, in mammals) and the adapter proteins Salvador (WW domain-containing adapter 45, WW45, in mammals) and Mob as tumor suppressor (Mats-Mps-one binder-1, MOB1, in mammals). Hpo, in complex with Salvador, phosphorylates and activates Warts, a process that also requires Mats. Activated Warts then phosphorylates and inhibits Yorkie (Yes-associated protein, YAP, in mammals), a transcription factor that, when active, triggers a pro-proliferative, antiapoptotic program of gene expression (reviewed in Refs. 20 and 21). Genetic studies of Drosophila indicate that Hpo is an effector for dMerlin and dExpanded Drosophila orthologues of mammalian merlin (22). However, it is still unclear whether Mst1, Mst2, or both are merlin targets in mammalian cells.
Most studies suggest that mammalian Mst1 and Mst2, like Hpo, are also tumor suppressors (20, 21, 23, 24). Studies of cultured cells document conservation of the Mst1/2 → LATS → YAP pathway (20, 21, 25). Mst2 can also associate with members of the RASSF (Ras association domain family) family of tumor suppressors. This association activates Mst2 and serves to promote Ras-mediated apoptosis (26–29). Moreover, disruption of Mst1 causes significant lymphoid hyperproliferation (30). Lastly, liver-specific overexpression of YAP causes the spontaneous development of hepatocellular carcinoma (31).
However, the cellular functions of Mst1 and Mst2 and their lower metazoan orthologues may be more complex. Thus, in contrast to Drosophila, RNA interference (RNAi) knockdown studies in C. elegans point to a pro-survival rather than a pro-apoptotic function for CST-1 (32). Moreover, disruption of mst1, although leading to enhanced lymphocyte proliferation, apparently does not affect LATS1/2 function (30). Disruption of mst1 and mst2 in liver leads to hepatocellular carcinoma via a YAP-dependent but LATS1/2-independent process (33). In addition, recent studies of RASSF function indicate that although RASSF6 can induce Mst2-dependent apoptosis, RASSF6 dissociates from Mst2 after Mst2 activation, and this free RASSF6 can trigger Mst2-independent apoptosis (34).
In situ, Mst2 can also interact with Raf-1 in a manner independent of the kinase activity of Raf-1 (24). In complex with Raf-1, the pro-apoptotic kinase activity of Mst2 is blunted (24). Genetic deletion of Raf-1 or stimulation with apoptogenic agents liberates Mst2 and fosters Mst2 apoptogenic signaling (24). Collectively however, the relative contribution of the Raf-1-Mst2 or RASSFs-Mst2 complexes to antiproliferative or apoptogenic signaling is still unclear.
In the studies described herein, we sought to determine whether merlin could regulate Mst2, in part, through destabilization of the Raf-1-Mst2 complex, thereby promoting activation of Mst2. Alternatively, although it is well documented that the interaction of Mst2 with Raf-1 serves to restrain the pro-apoptotic activation of Mst2 (24), a role for Mst2 in regulation of Raf-1 or MAPK signaling has not been investigated. We therefore also sought to determine whether Mst2 could influence Raf-1 activity. Our results reveal an unexpected positive role for Mst2 in Raf-1/ERK pathway regulation and function.
EXPERIMENTAL PROCEDURES
Cell Lines and Cell Culture
HEI-193 cells are an immortalized, patient-derived NF2 schwannoma line (35). akt2/3−/−;akt-1flox/flox primary mouse embryonic fibroblast (MEF) cells, kindly provided by Philip Tsichlis, are a mouse embryo fibroblast cell line in which both akt2 and akt3 have been disrupted and endogenous akt1 has been replaced with a floxed allele. akt1 can be disrupted with adenoviral Cre recombinase (below), resulting in cells depleted of all three Akt isoforms. SKOV3 is a human ovarian cancer cell line (36). For transient transfection experiments, HEK293 cells were used. These cell lines were cultured in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum (Atlanta Biologicals), 2.5 mm l-glutamine, and 1% penicillin/streptomycin. The RT4 NF2.17 is a rat schwannoma cell line engineered to stably express wild type merlin from a doxycycline-inducible (Tet-On) promoter (37). These cells were cultured in the above mentioned medium further supplemented with 1 μg/ml puromycin and 500 μg/ml G418. NIH3T3 cells were purchased from ATCC and cultured in Dulbecco's modified Eagle's medium supplemented with 10% calf serum 2.5 mm l-glutamine, 2.5 mm pyruvate and 1% penicillin/streptomycin. Cells were grown and maintained at 37 °C and 5% CO2 in a humidified incubator. Where indicated, cells were serum-starved for 18–20 h in medium supplemented with 0.5% serum. pBabe-puro retroviral constructs encoding constitutively active S218D/S221D MEK1 (MEK-DD) and constitutively active Raf-1 (Raf-22W, a C-terminal construct expressing amino acids 321–552 and missing Ser-259) were obtained from Addgene. To generate MEK-DD or Raf-22W retroviruses, each construct or control pBabe-puro was co-transfected with constructs expressing retroviral essential proteins; pMDLgpRRE, pRSVrev, and pMD2.VSVG into 293 cells in 10-cm dishes. After 48 h, cells were grown in only 5 ml of complete growth medium for further 24 h, after which the supernatant was collected, spun, and filtered through a 0.45-μm polyvinylidene difluoride syringe filter. To prepare stably overexpressing HEI-193 or SKOV3 cells, the relevant retroviruses were added to subconfluent cultures of either in six-well plates at a 1:2 dilution. 72 h later, cells were grown in selection medium containing 2 μg/ml puromycin for a further 72 h and then maintained in medium supplemented with 0.5 μg/ml puromycin thereafter.
To determine whether constitutively active C-Raf-1 (C-Raf 22W) or MEK1 (MEK1DD) could rescue the inhibition of ERK and cell proliferation in Mst2 knockdown cells, cells were plated in 6-well plates and treated with either Mst2 RNAi (oligo-1) or control RNAi (GFP below). For proliferation assays, cells were treated as described previously. For Western blotting, cells were lysed 72 h after transfection in RIPA buffer without any form of stimulation, subjected to SDS-PAGE analysis, and immunoblotted with the indicated antibodies.
Plasmid DNA Transfections
We routinely used Lipofectamine 2000 (Invitrogen) for HEK-293 cell transfection. We used the following plasmids: pCMV5-FLAG-MST2, pMT3-HA-Raf-1, and pCMV5-Myc-merlin. Transfections were performed in 10-cm tissue culture dishes at 70–80% confluency. Cells were lysed 48 h after transfection in Lysis buffer A (20 mm Hepes, pH 7.4, 150 mm NaCl, 2 mm EGTA, 1 mm EDTA, 50 mm β-glycerophosphate, 50 mm NaF, 1 mm dithiothreitol, 1 mm Na3VO4, 1% Triton X-100, 10% glycerol, 10 μm aprotinin, and 0.5 μm phenylmethylsulfonyl fluoride) for immunoprecipitations.
RNAi
We used previously described methods for RNAi (38). The following small interfering RNA oligos (Dharmacon) were used: Mst2-human oligo-1, sense, 5′-uccggucaaguugucgcaauu-3′, antisense, 5′-uugcgacaacuugaccgga-3′; Mst2-human oligo-2, sense, 5′-cagcccauauguuguaaagua-3′, antisense, 5′-uacuuuacaacauaugggcug-3′; Mst2-human 3′-untranslated region targeted, sense, 5′-acuaggaauugaaggaaua-3′, antisense, 5′-uauuccuucaauuccuagu-3′; murine control Mst2 sequences, sense, 5′-ucuggucaagugguugcaauu-3′, antisense, 5′-aauugcaaccacuugaccagauu-3′. To knock down murine NIH3T3 cell Mst2, we used the following two mouse-specific Mst2 oligos simultaneously: mouse Mst2-oligo-1, sense, 5′-cguuucagacauaauuaga-3′, antisense, 5′-ucuaauuuaugucugaaacguu-3′, and mouse Mst2 oligo-2, sense, 5′-ggaauauucuccucaauac-3′, antisense, 5′-guauugaggagaauauucc-3′. For control human, Mst2 oligo-1 was used. To silence Raf-1 in HEI-193 cells, the following oligos were used: sense, 5′-caucagacaacucuuauug-3′, antisense, 5′-caauaagaguugucugaug-3′. For silencing human LATS1/2, the following oligonucleotides were used: LATS1, sense, 5′-cggcaagauagcauggauu-3′, antisense, 5′-aauccaugcuaucuugccg-3′; LATS2, sense, 5′-ggaaaucagauauucuug-3′, antisense, 5′-caaggaauaucugauuucc-3′. GFP-specific RNAi oligonucleotides were: sense, 5′-gacguaaacggccacaagu-3′, antisense, 5′-acuuguggccguuuacgta-3′. For negative controls, the murine control oligo (for human cell lines), human Mst2 oligo-1 (for murine cell lines), or GFP oligos were used, as indicated in the figure legends.
Immunoprecipitation and Immunoblotting
Immunoprecipitation and immunoblotting were performed as described previously (38). Where indicated, cells were serum-starved for 18–20 h prior to treatment with 50 ng/ml EGF for various times.
For immunoprecipitation, cells were lysed in Lysis buffer A. Extracts were cleared by centrifugation (5000 × g). Protein concentrations in the cleared lysates were determined using the Bradford assay. For co-immunoprecipitation of the endogenous Raf-1-Mst2 complex from NIH3T3 cells or HEI-193, we used 500 μg of total lysate protein per condition. For immunoprecipitation of the Raf-1-B-Raf complex, we used 250 μg of cell extract/condition.
We used 1× RIPA buffer (50 mm Tris-HCl, pH 7.4, 150 mm NaCl, 1 mm phenylmethylsulfonyl fluoride, 1 mm EDTA, 5 μg/ml aprotinin, 50 mm β-glycerophosphate, 50 mm NaF, 1 mm Na3VO4, 1% Triton X-100, 0.25% sodium deoxycholic acid, and 0.5% SDS) to prepare cell extracts for direct immunoblotting. Equal amounts of lysate protein/condition (15–25 μg of lysate/lane) were used.
Antibodies and Reagents
Anti-merlin, anti-Akt anti-phospho-Akt Ser-473, anti-phospho-Akt Thr-308, anti-phospho-Raf-1 Ser-259, anti-phospho-S6 ribosomal protein Ser-235/236, anti-phospho-Raf-1 Ser-338, anti-phospho-MEK1/2 Ser-221/227, anti-phospho-ERK Thr-202/Tyr-204, anti-phospho-tyrosine, anti-caspase-3, anti-poly(ADP-ribose) polymerase, anti-phosphatase-2A-C, anti-phosphatase-2A-A, anti-LATS1, LY294002, and platelet-derived growth factor were from Cell Signaling Technologies. Anti-LATS2 was from Bethyl laboratories, anti-Raf-1 and anti-B-Raf (for kinase assay immunoprecipitation and detection of C-Raf 22W), purified MEK1 K97R, and Raf-1 kinase assay kit were from Upstate Biotechnology. Anti-14-3-3, anti-MEK1, anti-MEK1 C-terminal (for detection of MEK1DD) anti-ERK2, horseradish peroxidase-conjugated mouse IgG, and rabbit IgG were from Santa Cruz Biotechnology. Anti-β-actin, anti-FLAG tag, staurosporine, myelin basic protein, and doxycycline were from Sigma-Aldrich. Z-VAD-FMK and doxorubicin were from R&D systems. Recombinant human EGF and lactacystin were from Calbiochem. Anti-Mst2 was from Epitomics. Phospho-Mst2 antibody was a gift from Dr. Joseph Avruch. Anti-Raf-1 mouse monoclonal antibody was from BD Biosciences, and [γ-32P-ATP] was from PerkinElmer Life Sciences. Horseradish peroxidase-conjugated anti-mouse light chain-specific IgG and anti-rabbit light chain-specific IgG were from Jackson ImmunoResearch Laboratories.
RNA Extraction and RT-PCR
For RNA extraction, cells were seeded and treated with RNAi oligos as explained previously. 72 h later, cells were lysed and subjected to total RNA extraction using the TRI Reagent kit (Ambion) according to manufacturer's recommendations. Following extraction, 0.5 μg of total RNA for each was separately subjected to concurrent RT-PCR and PCR using the Titan One Tube RT-PCR kit (Roche Applied Science). The following primer sets were used in PCR reactions to generate a 243-bp product of human protein phosphatase-2A catalytic subunit (PP2A-C): 5′ primer, catgctgaagcgacattgtt; 3′ primer, aagtagaagggaggccttgg. β-Actin primers were part of the PCR kit.
Cre Recombinase and Merlin (NF2) Adenovirus
Wild type merlin and Cre recombinase adenoviruses were gifts from, respectively, Andrea I. McClatchey and Michael Mendelsohn. Viral concentration was determined based on A260. For infection of cells, viruses were used at a multiplicity of infection of 50–250, depending on experimental requirements.
Cell Proliferation Assays
HEI-193, SKOV3, and NIH3T3 cell proliferation assays were performed as described (38).
In Vitro Kinase Assay
We used the Raf-1 kinase assay kit (Upstate Biotechnology) to assay Raf-1. Anti-B-Raf was from Santa Cruz Biotechnology, and anti-phospho-MEK1/2 Ser-217/221 was from Cell Signaling Technologies. Raf-1 immunoprecipitation and kinase assay were performed as described (38). The reactions were stopped with the addition of EDTA to a final concentration of 50 mm to each sample. Raf-1 beads were removed by centrifugation, and the supernatant was transferred to a clean microcentrifuge tube. 40 μl of 3× Laemmli SDS-PAGE sample loading buffer was added to both pellet and supernatant and boiled for 5 min. For detection of phospho-MEK using chemiluminescence, 20 μl of supernatant was separated on SDS-PAGE and subjected to immunoblotting with either phospho-specific or total MEK Ser-217/221 antibody.
To assess the ability of merlin to activate Mst2, we expressed in HEK293 cells 1 μg of Myc-tagged merlin and 300 ng of FLAG-tagged Mst2. After 18 h, FLAG-MST2 was immunoprecipitated and assayed as described with myelin basic protein (MBP) as a substrate.
Statistical Analysis
All cell proliferation values are presented as -fold change over controls (time 0) ± S.E. Control values were, therefore, set to 1 and not subjected to statistical analysis. Unpaired t tests were routinely performed to evaluate whether the difference between samples was significant (p < 0.05). Densitometry of immunoblot band intensity was performed using ImageJ version 1.24 software (National Institutes of Health) and expressed as means of three independent experiments ± S.E.
RESULTS
Merlin Does Not Activate Schwannoma Cell Mst2
HEI-193 cells are immortalized patient-derived vestibular schwannoma cells bearing a loss of function splice-site mutation in nf2 (35). We reasoned that if merlin were an upstream regulator of Mst2, then 1) RNAi silencing of Mst2 would abrogate the cellular effects of merlin reintroduction and 2) reintroduction of merlin would activate endogenous Mst2. Thus, we exploited RNAi to silence HEI-193 cell Mst2. After 24 h, cells were infected with an adenovirus expressing either a wild type nf2 construct or GFP (control). Clearly, overexpression of merlin led, in a dose-dependent manner, to apoptotic cell death as indicated by increased cleavage of caspase-3. RNAi silencing of Mst2 did not inhibit or alter the level of merlin-stimulated caspase-3 cleavage, indicating that Mst2 is not required for merlin-induced cell death (Fig. 1A).
FIGURE 1.

Merlin does not function upstream of or positively regulate MST2. A, knockdown of Mst2 does not inhibit merlin-meditated cell death. HEI-193 cells were transfected with either human Mst2-specific RNAi or control (Ctr) (mouse-specific Mst2 RNAi) and infected with a LacZ (LAZ) or NF2-overexpressing adenovirus (Ad) at a 50 or 250 multiplicity of infection (MOI) 24 h later. Cells were lysed in RIPA buffer 48 h after infection and subjected to immunoblotting with the indicated antibodies. B, merlin does not promote disruption of the Raf-1-Mst2 complex. co-immunoprecipitations (IP) in HEI-193 cells (left) and NIH3T3 cells (right). Cells were lysed in Lysis buffer A, and 500 μg of total lysate was used. WB, Western blot. C, merlin does not promote phosphorylation of Mst2 on Thr-180. Total RIPA buffer lysates from HEI-193 cells infected with NF2-overexpressing adenovirus for the indicated amount of time were subjected to immunoblot analysis with phospho-Mst2 (Phosp-Mst2) Thr-180 (T180) antibody. STR = staurosporine D, merlin overexpression does not affect Mst2 kinase activity. 0.3 μg of FLAG-Mst2 was co-transfected with 1 μg of Myc-merlin into HEK293 cells, and 18 h later, cells were lysed, and 500 μg of total lysate was subjected to immunoprecipitation with FLAG antibody. 50% of immunoprecipitated beads-FLAG-Mst2 complex was used in an in vitro kinase assay with myelin basic protein as substrate.
Raf-1 can interact with and inhibit Mst2 (24). Dissociation of Mst2 from Raf-1 coincides with increased Mst2 kinase activity and cell death (24). We therefore sought to determine whether merlin could disrupt the Raf-1-Mst2 complex. As is evident in Fig. 1B, infection of either HEI-193 or NIH3T3 cells with the merlin-overexpressing adenovirus did not detectably alter the levels of Mst2 present in Raf-1 immunoprecipitates.
Still, despite this, it is possible that Raf-1-bound Mst2 is not accessible to merlin. The process of Mst2 activation in response to apoptotic stimuli requires homo-oligomerization and autophosphorylation on Thr-180 followed by caspase-3 cleavage (20, 21, 24). We therefore asked whether merlin could promote the kinase activity (either autophosphorylation or activity toward an exogenous substrate) of Mst2. To ensure that the phosphorylated Mst2 was not lost to caspase-3 cleavage, we included a control sample treated with the pan caspase inhibitor Z-VAD (OMe)-FMK. HEI-193 cells were infected with NF2-overexpressing adenovirus, and samples were analyzed at various time points thereafter (Fig. 1C). From Fig. 1C, it is clear that merlin did not promote the activating phosphorylation at Thr-180. Staurosporine (STR), a known activator of Mst2, was used as a positive control. Merlin also did not reduce the level of intact Mst2 polypeptide, consistent with the idea that merlin did not induce cleavage of Mst2 (Fig. 1C). To examine more directly merlin regulation of Mst2 kinase, we co-transfected 293 cells with FLAG-Mst2 and either vector or Myc-merlin. FLAG-Mst2 was immunoprecipitated and subjected to in vitro kinase assay with MBP as a substrate. Despite considerable amounts of Mst2 in the immunoprecipitate, little or no kinase activity toward MBP could be detected in the presence or absence of merlin. Staurosporine (STR), a known activator of Mst2, did result in an increase in Mst2-catalyzed MBP phosphorylation (Fig. 1D). Taken together, these results suggest that either merlin and Mst2 function separately in mammalian cells to regulate apoptotic cell death or that their relationship is more complex than that implied by the epistatic relationship observed in Drosophila studies.
Mst2 Is Required for Optimal Activation of the Ras/Raf/MEK/ERK Mitogen-activated Pathway and Cell Proliferation
It was first reported that hpo was the principal component of a pro-apoptotic/antiproliferative pathway in Drosophila (reviewed in Ref. 39). Studies of Mst2, a mammalian orthologue of hpo, suggest a similar conclusion (reviewed in Refs. 20 and 21). Given that Raf-1 functions to suppress Mst2 function (24), we wondered whether Mst2 might have a reciprocal role in Raf-1 regulation. We silenced HEI-193 cell Mst2, and to our surprise, depletion of Mst2 coincided with a decrease in serum-stimulated cell proliferation (Fig. 2A, left). Similar results were observed for SKOV3 cells (Fig. 2A, right) and, more modestly, for NIH3T3 Cells (supplemental Fig. 1). Of particular note, EGF-stimulated phosphorylation of the Raf-1 substrate MEK and its downstream target ERK was impaired upon silencing of Mst2 (supplemental Fig. 5A). Taken together, these data indicate that Mst2 may, unexpectedly, be required for optimal mitogen-mediated activation of the Raf/MEK/ERK pathway and cell proliferation.
FIGURE 2.

Mst2 is required for optimal activation of the Ras/Raf/MEK/ERK mitogen-activated pathway and cell proliferation. A, RNAi silencing of Mst2 reduces cell proliferation. Left, HEI-193; right, SKOV3. Cells were transfected with 100 nm human-specific Mst2 or control (Ctr) (mouse) RNAi. 24 h later, cells were serum-starved overnight and trypsinized, and equal numbers were plated in triplicate per condition. At the indicted time points, cells were counted with a hemocytometer. Parallel samples were lysed at each time point for immunoblot analysis with the indicated antibodies. Numbers represent -fold change ± S.E. relative to T0. (# indicates p < 0.05, n = 3. Control values (time 0) were set to 1 and not subjected to statistical analysis. B, Mst2 RNAi impairs MAPK signaling in response to EGF stimulation. HEI-193 cells (left) and SKOV3 cells (right) were treated with human-specific Mst2 RNAi or control (mouse) RNAi (100 nm). After 48 h, cells were serum-starved for 18–20 h and then treated with 50 ng/ml EGF (+EGF) or vehicle (−EGF) for 10 min. Lysates were subjected to immunoblotting with the indicated antibodies. Phosp-MEK-1/2, phospho-MEK1/2; Tot-Mek-1, total Mek1.
Mitogens Enhance Assembly of the Raf-1-Mst2 Complex
O'Neill et al. (24) demonstrated that serum starvation favors formation of the Raf-1-Mst2 complex, whereas treatment of cells with serum promoted dissociation of this complex. In light of our findings that Mst2 was required for optimal activation of the Raf/MEK/ERK MAPK cascade and cell proliferation, we wondered whether EGF could promote assembly or disassembly of the Raf-1-Mst2 complex. NIH3T3 cells were serum-starved overnight and then treated with 50 ng/ml EGF for various times after which endogenous Raf-1 or Mst2 was subjected to reciprocal immunoprecipitation/immunoblotting with the indicated antibodies (Fig. 3A). Interestingly, in our hands, EGF treatment resulted in enhanced assembly of the endogenous Raf-1-Mst2 complex (Fig. 3A). Similar results were obtained for HEK-293 cells transfected with wild type HA-Raf-1 and FLAG-Mst2 and stimulated with 10% fetal bovine serum (supplemental Fig. 2). EGF enhanced neither the polypeptide levels of Mst2 nor its phosphorylation on Thr-180 (supplemental Fig. 3). These data indicate that Raf-1 and Mst2 may exist as part of a dynamic, signal-dependent complex that functions to promote cell proliferation and survival. However, in the absence of Raf-1 (i.e. Raf-1−/− cells) or when cells are treated with pro-apoptotic agonists such as FasL or staurosporine, Mst2 homo-oligomerizes and fosters apoptotic cell death.
FIGURE 3.

Mitogens enhance assembly of the Raf-1-Mst2 complex; Mst2 is required for optimal doxorubicin-induced cell death. A, EGF promotes assembly of the Raf-1-Mst2 complex. NIH3T3 cells were serum-starved overnight and then treated with 50 ng/ml EGF for the indicated time points before lysis and immunoprecipitation (IP) with Raf-1 antibody. Immunoprecipitates were then immunoblotted (IB) with anti-Mst2 antibody. Phospho-ERK (Pho-ERK) immunoblotting was used as a positive control readout for activation of the Ras-Raf/ERK pathway by EGF. WB, Western blot. Tot ERK, total ERK. B, in response to genotoxic stress, knockdown of Mst2 promotes cell survival. SKOV3 or HEI-193 cells were transfected with human-specific Mst2 RNAi or control RNAi (Ctr RNAi, mouse). 48 h later, cells were treated with either 10 μm doxorubicin or vehicle (DMSO) for 8 h and then lysed. Equal amounts were immunoblotted with antibodies to the indicated apoptotic markers. FL, full length. PARP, poly(ADP-ribose) polymerase; casp-3, caspase-3.
Consistent with this latter idea, silencing of Mst2 in Raf-1−/− MEFs abolishes their sensitivity to apoptotic agonists (24, 40). Inasmuch as our findings (Fig. 2) suggested a positive role for Mst2 in mitogen signaling, we wanted to determine whether Mst2 could also function as a pro-apoptotic enzyme in response to apoptogenic stimuli. We used the DNA-intercalating cancer chemotherapeutic drug doxorubicin as a pro-apoptotic agonist. We silenced Mst2 in either SKOV3 or HEI-193 cells and treated cells with either vehicle or 10 μm doxorubicin. Cell lysates were prepared and subjected to immunoblot analysis with anti-cleaved poly(ADP-ribose) polymerase, anti-cleaved capase-3 (apoptosis markers), or anti-Mst2. Consistent with previous studies, silencing of Mst2 impaired doxorubicin-induced apoptosis, as indicated by reduced SKOV3 and HEI-193 cell poly(ADP-ribose) polymerase and caspase-3 cleavage as well as cleavage of Mst2 into a 36-kDa fragment (Fig. 3B and supplemental Fig. 4). Thus, Mst2 may be a critical component in doxorubicin-induced cell death. This finding raises the interesting idea that Mst2 may have both a cell proliferative function and an apoptogenic function depending on the stimulus and cell context.
Mst2 Is Not Required for EGF Receptor Activation or Integrity of the Raf-1-B-Raf Complex
We wished to determine the molecular mechanisms underlying the impaired MEK phosphorylation and cell proliferation observed upon silencing of Mst2. From Fig. 2B, it is clear that silencing of Mst2, by reducing agonist-stimulated MEK phosphorylation, impairs a step upstream of MEK. The binding of EGF to its receptor induces receptor dimerization and phosphorylation on specific tyrosine residues. Anti-phosphotyrosine immunoblot analysis of lysates prepared from EGF-treated cells indicates that phosphorylation of the EGF receptor is not affected by Mst2 RNAi (Fig. 4A). Thus, Mst2 is required for the optimal function of a step between EGF receptor and MEK.
FIGURE 4.

Mst2 is not required for optimal EGF receptor activation or for the integrity of the Raf-1-B-Raf complex. A, Mst2 RNAi does not affect EGF receptor phosphorylation. Left, HEI-193; Right, SKOV3. (Please note that these are the same total lysates saved from the samples used to immunoprecipitate Raf-1 for the kinase assay in Fig. 5A, also used for supplemental Fig. 5A; hence the phospho-MEK (phosp-MEK), total MEK (Tot-MEK), and Mst2 blots have been reproduced in these figures for the convenience of the reader). Cells were treated as indicated in Fig. 5A below and subjected to immunoblot analysis with the indicated antibodies. Total ERK (Tot-ERK) immunoblotting was performed as a gel loading control. Ctr RNAi, control RNAi. B, integrity of the Raf-1-B-Raf complex is not affected by Mst2 RNAi. HEI-193 cells were grown and transfected with 100 nm human-specific Mst2 or control (mouse) RNAi. After 48 h cells, were serum-starved for 18–20 h and then treated with EGF or vehicle for 10 min. Raf-1 was then immunoprecipitated (IP) and subjected to SDS-PAGE and immunoblot analysis with the indicated antibodies. WB, Western blot.
The heteromerization of Raf-1 and B-Raf is thought to be required for optimal mitogenic activation of ERK (12, 13, 41). Mst2 can interact in situ with Raf-1 but not B-Raf (24). With these two observations in mind, we wondered whether the Mst2 indirectly supported maintenance of the Raf-1-B-Raf complex. Fig. 4B shows that silencing Mst2 has no effect on the level of B-Raf associated with Raf-1, indicating that the integrity of the Raf-1-B-Raf complex does not require Mst2.
Mst2 Positively Regulates Raf-1 Activation via Suppression of the Inhibitory Phosphorylation of Raf-1 on Ser-259; Mst2 Is Not Required for B-Raf Kinase Activation
We next wanted to determine whether the kinase activity of Raf-1 or B-Raf was affected by Mst2 silencing. Raf-1 and B-Raf were separately immunoprecipitated from cells subjected to control or Mst2 RNAi and treated with either vehicle or 50 ng/ml EGF. The Raf immunoprecipitates were then subjected to in vitro kinase assays with purified MEK1 K97R as substrate. Raf-1 immunoprecipitated from cells subjected to Mst2 RNAi manifested reduced mitogen-induced kinase activity when compared with Raf-1 from cells subjected to control RNAi (Fig. 5A). Surprisingly, silencing of Mst2 had no detectable effect on B-Raf kinase activity (Fig. 5B). Collectively, these results indicate that Mst2 is required for optimal mitogen activation of Raf-1 but not B-Raf.
FIGURE 5.

Mst2 positively regulates Raf-1 activation via suppression of the inhibitory phosphorylation of Raf-1 on Ser-259; Mst2 is not required for B-Raf activation. A, Raf-1 kinase activity is impaired upon Mst2 RNAi. SKOV3 cells were grown and transfected with 100 nm human-specific Mst2 RNAi or control (Ctr) (mouse) RNAi. After 48 h, cells were serum-starved for 18–20 h and then treated with EGF for the indicated time points before lysis in Lysis buffer A. Raf-1 was then immunoprecipitated (IP) and assayed using purified MEK1 K97R as a substrate. A part of the total lysate was saved and separately subjected to immunoblotting with phospho-MEK1/2 Ser-217/221, total MEK, or Mst2 antibodies. (Note: These are the same lysates used in Fig. 4A (SKOV3 blot) and supplemental Fig. 5A; hence the phospho-MEK (Phosp-MEK-1), total MEK, and Mst2 blots have been reproduced for the convenience of the reader.) For the in vitro kinase assay, MEK phosphorylation was quantitated with ImageJ as shown in the bar graphs. Data shown are means ± S.E. Replicates were analyzed by Student's t test (n = 3, # indicates p = 0.02, Dens. Units indicates densitometric units). WB, Western blot. B, the kinase activity of B-Raf is not affected by Mst2 knockdown. Cell handling, B-Raf immunoprecipitation, and assaying were performed as for Raf-1 in A above. C, Ser-259, an inhibitory phosphoacceptor site on Raf-1, is hyperphosphorylated upon silencing of Mst2. HEI-193 cells were treated either with 100 nm Mst2 targeting RNAi (human Mst2-RNAi) or with control RNAi (mouse). 48 h later, cells were serum-starved and then stimulated with 50 ng/ml EGF for the indicated time points before lysis. Lysates were immunoblotted with the indicated antibodies. D and E, densitometric analysis of the effect of Mst2 RNAi on Raf-1 Ser-259 phosphorylation and MEK1/2 217/221 phosphorylation. The experiment in C was repeated three times, and phospho-MEK1/2 Ser-217/221 (D) and phospho-Raf-1 Ser-259 (E) were quantified with ImageJ software. Each data point represents the average of three independent experimental densitometry values of that particular time point ± S.E. Data were analyzed by Student's t test. # indicates p < 0.05 for control versus Mst2 RNAi samples indicated. F, similar results of hyperphosphorylation of Raf-1 Ser-259 upon Mst2 silencing were obtained using a second human-specific Mst2 oligonucleotide targeting a different part of the Mst2 transcript.
Raf-1 signaling is tightly regulated through phosphorylation and dephosphorylation at specific target serine, threonine, and tyrosine residues, (reviewed in Refs. 42 and 43). Ser-259 is an inhibitory phosphorylation thought to be a target of both Akt (protein kinase B) and the cAMP-dependent protein kinase (4, 5). Conversely, phosphorylation of Ser-338 by the PAKs and Tyr-341 by Src is required for optimal Raf-1 kinase activity (44, 45). Several additional sites require dephosphorylation or phosphorylation during Raf-1 activation, and these remain partially or poorly characterized (11). In resting cells, Ser-259 is hyperphosphorylated, whereas phosphorylation on Ser-338 is at a basal level. Growth factor/mitogen stimulation induces rapid dephosphorylation of Ser-259 and a rapid increase in Ser-338 phosphorylation coincident with a marked increase in the overall kinase activity of Raf-1 (8). To determine whether silencing of Mst2 resulted in increased phosphorylation of Raf-1 on Ser-259, HEI-193 cells were treated with Mst2 or control small interfering RNA and then with either vehicle or EGF for various times. As evident from Fig. 5C, second panel from top, silencing Mst2 enhances basal and EGF-stimulated Raf-1 phosphorylation at multiple sites. Thus, Raf-1 Ser-259 phosphorylation is high in resting cells but decreases with EGF treatment, returning to basal levels by 30 min. Overall, Raf-1 Ser-259 phosphorylation is enhanced by Mst2 RNAi. Paradoxically, the basal and EGF-stimulated activating phosphorylation at Ser-338 is also enhanced upon Mst2 silencing (Fig. 5A, third panel from the top). Despite this, Raf-1 immunoprecipitated from cells subjected to Mst2 RNAi manifests reduced kinase activity (Fig. 5A), coincident with reduced in situ phosphorylation of MEK1 Ser-217/221, the phosphoacceptor sites on MEK1 targeted by Raf-1 and B-Rafs (Fig. 5C, second panel from the bottom). The experiment in Fig. 5A was repeated three times, and the correlation between Mst2 RNAi and changes in Raf-1 phosphorylation and activity were indirectly quantified by ImageJ densitometry software (Fig. 5, D and E).
To rule out off target RNAi effects, we acquired a second Mst2 oligonucleotide specific to a different site on the Mst2 transcript and repeated this experiment. Again, silencing of Mst2 resulted in increased Raf-1 Ser-259 and Ser-338 phosphorylation and impaired MEK phosphorylation in response to EGF stimulation (Fig. 5F). Similar results, including Mst2 RNAi-incurred reduced ERK phosphorylation, were obtained in studies of SKOV3 cells (supplemental Fig. 5A) and in NIH3T3 cells using a mouse-specific Mst2 RNAi oligonucleotide (supplemental Fig. 5B). These findings indicate that Mst2 is a positive regulator of Raf-1 activation in response to mitogen stimulation and that Mst2 supports Raf-1 activation via suppression of the inhibitory phosphorylation of Raf-1 on Ser-259.
To confirm that the reduced cell proliferation and MEK activation observed upon Mst2 RNAi were due to suppression of Raf-1 activation, we first wished to determine whether the proliferation of HEI-193 cells, which is impaired upon Mst2 silencing, also required Raf-1. Silencing of Raf-1 substantially reduces HEI-193 cell proliferation (supplemental Fig. 7). We next performed an epistasis study. Thus, we tested whether or not the effect of Mst2 RNAi on cell proliferation could be reversed upon contemporaneous expression of constitutively active MEK1 (MEK-DD) or constitutively active Raf-1 (Raf-22W, missing Ser-259, see “Experimental Procedures”). Fig. 6 shows that this is indeed the case. Thus, ectopic expression of MEK-DD or Raf-22W does not impair the ability of Mst2 RNAi to elevate endogenous Raf-1 Ser-259 phosphorylation (Fig. 6A). Of note, the cells in Fig. 6A were treated briefly with mitogen, without serum starvation, so as to produce an acute and robust activation of ERK. Under these circumstances, expression of either constitutively active construct does modestly restore the HEI-193 cell ERK phosphorylation seen in cells growing in serum but lost upon Mst2 RNAi. Most importantly, expression of either constitutively active construct completely restores the proliferation defect incurred upon silencing of Mst2 (Fig. 6B).
FIGURE 6.

Ectopic, stable expression of either constitutively active MEK1 or constitutively active Raf-1 rescues the proliferation defect in Mst2 knockdown Cells. A, expression of MEK1DD (MEK1-S218D/S221D) or C-Raf 22W (a C-terminal construct expressing amino acids 321–552 and thus missing Ser-259) has no effect on the elevation in endogenous Raf-1 Ser-259 phosphorylation (Phosp-Raf-1S259) incurred upon silencing of Mst2 but can partially reverse the resulting decrease in ERK phosphorylation in cells maintained in 10% serum. HEI-193 cells stably overexpressing MEK1DD, C-Raf 22W, or empty pBabe-puro vector were treated with human-specific Mst2 RNAi (oligo-1) or control RNAi (Ctr RNAi) (GFP). 72 h later, cell extracts were prepared and immunoblotted with the indicated antibodies. B, constitutively active MEK-DD or Raf-22W can rescue HEI-193 cells from the reduction in cell proliferation incurred upon Mst2 silencing. Top panel, the stable HEI-193-pBabe-puro, MEK1DD, or C-Raf 22W cell lines were treated with either control RNAi (+GFP) or Mst2 RNAi (+Mst2) as described in A above. A cell proliferation assay was performed as described in the legend for Fig. 2A. An unpaired Student's t test was performed to ascertain that overexpression of MEK1DD or C-Raf 22W resulted in significant rescue of cell proliferation upon Mst2 knockdown relative to the pBabe-puro Mst2 knockdown cell line (control cell line). The bottom panel documents the level of Mst2 RNAi and MEK-DD or Raf-22W expression during the time course of the study. Data shown are means ± S.E.
Loss of Expression Levels of Phosphatase-2A Subunit C and Not Hyperactivation of the Akt/PI3K Pathway Is Responsible for the Inhibitory Hyperphosphorylation of Raf-1 Ser-259 in Mst2 Knockdown Cells
Both Mst1 and Mst2 have been shown to directly interact with and negatively regulate Akt phosphorylation and kinase activity (46). Akt, along with cAMP-dependent protein kinase, are the two well characterized kinases thought to phosphorylate Raf-1 Ser-259 (4, 5). We therefore wondered whether silencing Mst2 resulted in increased Akt activity, thereby enhancing Raf-1 Ser-259 phosphorylation. Indeed, Mst2 RNAi substantially increased Akt phosphorylation at the activating Thr-380 phosphoacceptor site (Fig. 7A). Silencing of Mst2 also elevates EGF-stimulated phosphorylation of ribosomal protein S6 (supplemental Fig. 6). Phosphatase and tensin homologue deleted on chromosome 10 (PTEN) is a phosphatidylinositol 3′ (PtdIns-3′) phosphatase that functions to deactivate the PtdIns-3-kinase pathway. We did not observe any changes in the expression levels of the phosphatidylinositol phosphate phosphatase PTEN upon silencing Mst2 (Fig. 7B), indicating that the increased phosphorylation of Akt was not due to elevation in PtdIns-3-kinase activity.
FIGURE 7.

Mst2 functions to maintain levels of the catalytic subunit of PP2A (PP2A-C); although silencing of Mst2 enhances Akt activation, Akt is not required for phosphorylation of Raf-1 at Ser-259. A, Mst2 RNAi leads to hyperactivation of the PI3K/Akt pathway. HEI-193 cell RNAi transfection, handling, cell lysis, and immunoblotting were performed as described in the legend for Fig. 5A. Ctr RNAi, control RNAi; Phosp-AKT S473, phospho-AKT Ser-473. Data shown are means ± S.E. B, PTEN levels are not affected by silencing of Mst2 in both HEI-193 and SKOV3 cells. C and D, neither PI3K inhibitor LY294002 (C) nor deletion of all three Akt isoforms (D) could affect the Raf-1 Ser-259 hyperphosphorylation observed upon silencing Mst2. For C, HEI-193 cells were treated with vehicle (−) (DMSO) or 50 μm LY294002 (+) as indicated. Cells were then treated with EGF for the indicated time points. Equal amounts of total lysates were then separated by SDS-PAGE and immunoblotted with the indicated antibodies. For D, akt2/3−/−;akt-1flox/flox MEFs were treated with Cre recombinase-expressing adenovirus (Cre Aden) to obtain Akt MEFs expressing no Akt isoform. Control MEFs were treated with Lac-Z (Laz in the figure)-expressing adenovirus. MEFs were then transfected with mouse-specific Mst2 RNAi. Human Mst2 RNAi was used as control. After 48 h, MEFs were treated with EGF or vehicle (0) as indicated. MEFs were then lysed in RIPA buffer, and equal amounts of lysate were subjected to SDS-PAGE and immunoblot analysis with the indicated antibodies. E–G, levels of PP2A-C are diminished upon Mst2 RNAi. HEI-193 (E) or SKOV3 (G) cells were treated with human-specific Mst2 or control RNAi (mouse). NIH3T3 cells (F) were treated with the different mouse-specific oligo set described under “Experimental Procedures” and the human Mst2 RNAi (oligo-1) as control. Lysates were then subjected to immunoblot analysis for the expression levels of PP2A-C, PP2A-A, 14-3-3-β, or PTEN. Only the expression levels of PP2A-C were found to be significantly affected by Mst2 silencing. E, in the bar graph, # indicates p < 0.001 for control versus Mst2 RNAi samples. Data shown are means ± S.E.
We next asked whether the elevation in Akt phosphorylation, and presumably activation, contributed to increased Raf-1 Ser-259 phosphorylation consequent to Mst2 RNAi. To determine this, we tested whether pharmacologic inhibition of PtdIns-3-kinase or genetic ablation of Akt could blunt the increase in Raf-1 Ser-259 phosphorylation observed upon Mst2 RNAi. We silenced HEI-193 cell Mst2 and then treated the cells with the PtdIns-3-kinase inhibitor LY294002 prior to treatment with EGF or vehicle (Fig. 7C). LY294002 blocked Akt, activating phosphorylation almost completely, as indicated by immunoblotting with phosphospecific antibodies that detect Akt phosphorylated at Thr-308 and Ser-473 or with antibodies that detect phosphorylated ribosomal S6 protein, another target of PtdIns-3-kinase. Under these conditions, LY294002 had no effect on the Raf-1 Ser-259 hyperphosphorylation induced by Mst2 RNAi (Fig. 7C). akt2/3−/−;akt-1flox/flox MEFs have undergone targeted disruption of akt-2 and akt-3 and bear an akt-1 gene as a floxed allele. Expression of Cre recombinase in these cells results in MEFs null for all three Akt isoforms. Disruption of all three Akt isoforms did not impair Raf-1 Ser-259 hyperphosphorylation incurred by silencing Mst2 (Fig. 7D.) Thus, although of Mst2 RNAi may result in an increase in the activating phosphorylation of Akt, the hyperphosphorylation of Raf-1 Ser-259 seen upon silencing Mst2 is likely not mediated by Akt or the PtdIns-3-kinase pathway.
The phosphorylation/dephosphorylation status of Raf-1 Ser-259 is also determined by a combination of other factors including Ras-GTP, 14-3-3 binding, and dephosphorylation by the protein serine/threonine PP2A (reviewed in Refs. (42 and 43). PP2A is a trimeric enzyme composed of a dimeric core consisting of a structural A subunit, a catalytic C subunit, and a variable B subunit that confers targeting specificity (43, 47, 48). Mst2 can interact with Raf-1 in situ. In addition, ectopically expressed Raf-1 co-immunoprecipitates with a phosphatase that can dephosphorylate Mst2 in vitro (24). Moreover, PP2A is likely the phosphatase responsible for dephosphorylation of Raf-1 Ser-259 (9, 10, 49, 50) and Akt-Thr-308 (51, 52), both of which exhibit hyperphosphorylation upon Mst2 RNAi. We therefore asked whether Mst2 could affect PP2A levels. We used the human-specific Mst2 RNAi oligo-1 to silence HEI-193 or SKOV3 cell Mst2. We used the murine-specific Mst2 oligo set to silence NIH3T3 cell Mst2.
To our surprise, silencing of Mst2 resulted in a significant decrease in the polypeptide levels of PP2A-C. This effect was specific to the PP2A C subunit inasmuch as Mst2 RNAi had no significant effect on PP2A-A subunit, 14-3-3, or PTEN levels (Fig. 7E). Similar results were obtained in SKOV3 Cells and NIH3T3 cells (Fig. 7, F and G, respectively). These results indicate that Mst2 indirectly regulates Raf-1 Ser-259 phosphorylation, and potentially, the phosphorylation of other kinases through maintenance of the expression levels of PP2A-C. Moreover it points to the possibility of the existence of a different regulatory mechanisms for the various PP2A subunits insofar as knockdown of Mst2 affected the expression levels of PP2A-C and not PP2A-A.
We next sought to begin to determine the mechanism of Mst2-mediated PP2A-C stabilization. Silencing of Mst2 has no effect on either HEI-193 or SKOV3 cell PP2A-C mRNA, as judged by reverse transcription-PCR (Fig. 8A). By contrast, treatment with the specific proteasome inhibitor lactacystin completely reverses the loss of HEI-193 cell PP2A-C incurred upon Mst2 RNAi (Fig. 8B). Of particular note, and consistent with the idea of a role for Mst2-dependent PP2A-C stabilization in reducing Raf-1 Ser-259 phosphorylation, lactacystin also produces a reduction in Raf-1 Ser-259 phosphorylation, even in the presence of Mst2 RNAi. Thus, Mst2-dependent stabilization of PP2A-C is a post-translational process likely involving a reduction in proteasome-dependent PP2A-C degradation.
FIGURE 8.
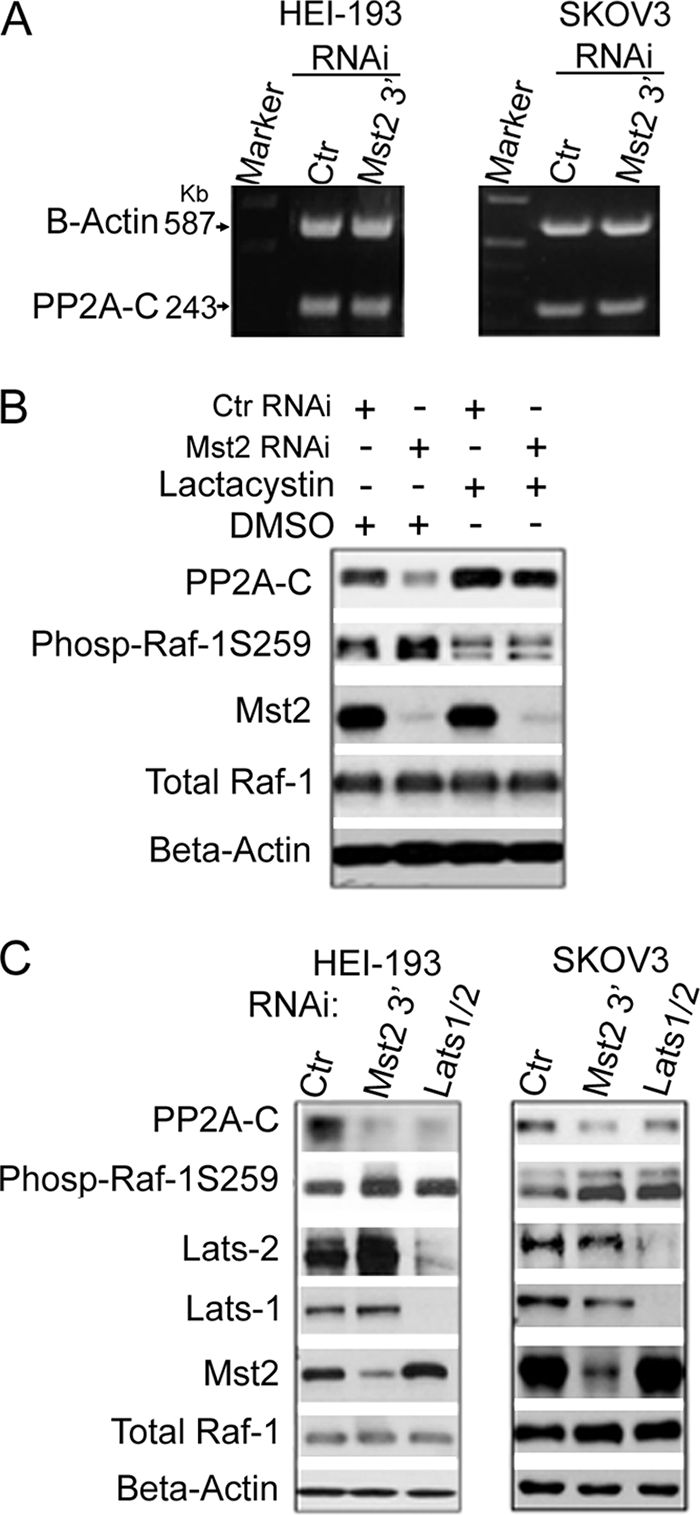
Mst2 stabilizes PP2A-C through a post-translational mechanism involving stabilization of the PP2A polypeptide. PP2A-C stabilization requires the Mst2 substrate kinases LATS1/2. A, Mst2 RNAi has no effect on PP2A-C transcript levels. HEI-193 or SKOV3 cells were subjected to Mst2 RNAi targeting the 3′-untranslated region of human Mst2 transcript (the RNA samples for this study were prepared in parallel with cells used in panel C, which shows the efficacy of Mst2 RNAi). For control (Ctr), GFP-specific RNAi was used. RNA was prepared and subjected to reverse transcription-PCR to assess PP2A-C levels. β-Actin reverse transcription-PCR served as a loading control. B, the proteasome inhibitor lactacystin restores PP2A-C levels depleted upon Mst2 RNAi, coincident with a decrease in Raf-1 Ser-259 phosphorylation. HEI-193 cells were treated with human-specific Mst2 RNAi (oligo-1) and 10 μm lactacystin 48 h later for 18 h as indicated. GFP RNAi was used as control. Extracts were prepared and immunoblotted with the indicated antibodies. C, RNAi silencing of LATS1/2 results in destabilization of PP2A-C and coincident increased Raf-1 Ser-259 phosphorylation. As indicated, either HEI-193 or SKOV3 cells were subjected to the 3′-untranslated region targeting the Mst2 RNAi oligo described in A above, which is different from the Mst2 oligo-1 used in Fig. 7, E and F, or the murine oligo set used for NIH3T3 cells (Fig. 7G). A sample, contemporaneously treated with human-specific LATS1 and LATS2 RNAi oligonucleotides, was also included. GFP RNAi was used as control. Cell extracts were prepared and subjected to immunoblotting with the indicated antibodies. Phos-Raf-1S259, phosho-Raf-1 Ser-259.
Mst2 is an orthologue of Drosophila Hippo. As such, in some, but not all cells (33), Mst2 directly phosphorylates and activates the mammalian LATS1/2, orthologues of Drosophila Warts. We silenced either HEI-193 or SKOV3 cell LATS1 and LATS2 contemporaneously. In both instances, this resulted in a decrease in PP2A-C levels comparable with that observed upon silencing of Mst2 (Fig. 8C). As in Fig. 6A, the cells used in Fig. 8C were not serum-starved and were treated acutely with mitogen. Nevertheless, silencing of LATS1/2, as with Mst2 RNAi, also elevates Raf-1 phosphorylation at Ser-259, albeit to a degree more modest than that observed for Mst2 RNAi (likely due to the technical difficulties of contemporaneously silencing both LATS1 and LATS2). Thus, at least two established elements of the mammalian Mst2 pathway are necessary to stabilize the PP2A-C polypeptide in situ.
DISCUSSION
Mammalian Mst2 has been best characterized as a tumor suppressor protein recruited by pharmacologic (e.g. staurosporine) or physiological (e.g. FasL) apoptogenic stimuli (23, 24, 27, 29). A positive role of Mst2 in mitogen signaling has not been reported. We have shown that in multiple cell types, Mst2 can promote either pro-apoptotic or proliferative responses depending on the nature of the stimulus. In response to EGF, Mst2 is required for optimal Raf-1 → ERK signaling and cell proliferation. By contrast, Mst2 is required in these cell types for maximal doxorubicin-induced apoptotic cell death. Mst2 apparently functions to maintain the expression of the PP2A-C subunit, which in turn reduces Raf-1 Ser-259 inhibitory phosphorylation. Although Mst2 RNAi apparently enhances Akt activity, we see no role for Akt in mediating Raf-1 Ser-259 phosphorylation regardless of Mst2 status. Raf-1 Ser-259 and Akt Thr-308 are known PP2A targets (49–52). Given that (i) knockdown of Mst2 also coincides with a significant decrease in PP2A-C levels coincident with increased Raf-1 Ser-259 phosphorylation and (ii) stabilization of PP2A-C by lactacystin leads to decreased Raf-1 Ser-259 phosphorylation in the presence of Mst2 RNAi, it is reasonable to propose that Mst2 regulates Raf-1 activation at least in part through the maintenance of optimal expression levels of PP2A-C.
We do not yet have a complete picture of how Mst2 regulates PP2A-C levels. Our results suggest that this process is post-translational and may involve the ubiquitin proteasome system inasmuch as PP2A-C transcript levels are unaffected by Mst2 RNAi, whereas lactacystin, a specific proteasome inhibitor, restores PP2A-C levels depleted by Mst2 RNAi. Moreover, our results suggest that an intact Mst2 signaling pathway is necessary for maintenance of PP2A-C. Thus, contemporaneous silencing of the Mst2 substrate kinases LATS1 and LATS2 also reduces PP2A-C. However, further studies will be necessary to determine how LATS1/2 couple to mechanisms affecting PP2A-C stability. Although overexpression studies have suggested that PP2A-C and PP2A-A exist in a complex with Raf-1, we could not detect endogenous PP2A-C or PP2A-A in endogenous Mst2 or Raf-1 immunoprecipitates. If such an endogenous complex exists to stabilize PP2A, the stoichiometry of the endogenous complex may be low, or the complex may be unstable. Others have found that the expression of PP2A-C is tightly controlled by an as yet unknown autoregulatory translational mechanism that ensures constant amounts of PP2A-C protein in the cell (53). Our results, by contrast, would seem to indicate that PP2A-C protein levels can be affected by the Mst2 pathway and are therefore not necessarily constant. Indeed, it is emerging that PP2A-C levels can be quite dynamic.
Clues to the mechanism of PP2A-C stabilization may come from recent studies of lipopolysaccharide-stimulated macrophages. Lipopolysaccharide treatment of macrophages renders cells refractory to repeated exposure in part through an agonist-induced stabilization of PP2A-C. This is mediated in part by valosin-containing protein/p97, which is required to maintain elevated PP2A-C levels, consequent to lipopolysaccharide treatment, through a process involving Tyr nitration of the PP2A-C polypeptide (54). Whether or not Mst2 operates in a similar manner to maintain PP2A-C remains to be determined.
PP2A can dephosphorylate Akt Thr-308 (51, 52). Mst2 also negatively regulates Akt through a direct interaction that correlates with decreased phosphorylation of Akt Thr-308 and Ser-473 (46). We find that silencing Mst2 increases Akt phosphorylation on Thr-308. Whether the Akt-Mst2 interaction, in conjunction with decreased PP2A-C levels, contributes to decreased Akt-Thr-308 phosphorylation is unclear. That Mst2, by maintaining PP2A levels, can exert an impact on Raf-1 by fostering the relief of Ser-259-mediated inhibition, while suppressing activating phosphorylation of Raf-1 Ser-338 and Akt 308, is indicative of the complexity of Raf-1 regulation. In addition, the findings suggest that Mst2, through PP2A, can have both positive and negative effects on mitogenic pathways.
Hpo, the Drosophila orthologue of Mst1/2, is also a bona fide tumor suppressor. Genetic epistasis studies suggest that dMerlin, the orthologue of hMerlin, exerts its antiproliferative effects in part through Hpo (22). Despite the apparent relationship between dMerlin and Hpo, our findings indicate that human Mst2 is unlikely to be a rate-limiting effector for mammalian merlin. It is possible, however, that Mst1 and Mst2 function redundantly as Hpo orthologues downstream of merlin in mammals. Alternatively, in mammals, Mst1 may be the sole Hpo orthologue that serves as a merlin effector.
To conclude, our results suggest a complex role for Mst2 in the regulation of mitogenic signaling and cell proliferation. Through direct signaling to LATS and YAP, Mst2 likely functions as a tumor suppressor. However, Mst2-dependent YAP inactivation occurs independently of LATS kinases in hepatocellular carcinoma; while in fibroblasts, LATS activation occurs at high cell density even if both Mst1 and Mst2 are disrupted (33). In addition to the LATS/YAP pathway, Mst2 exists in a complex with Raf-1. Release of Mst2 from this complex, in response to apoptogenic stress, coincides with Mst2 activation, including activation of the LATS/YAP pro-apoptotic pathway. We find that mitogens stimulate the association between Mst2 and Raf-1. We do not yet know whether this complex or free Mst2 mediates the stabilization of PP2A-C. However, inasmuch as Raf-1-associated Mst2 is thought to be inactive, free Mst2 may mediate the effects on Raf-1, which apparently involve the Mst2-substrates LATS1/2 and suppression of proteasomal degradation of PP2A-C. Insofar as the cell proliferation defect mediated by Mst2 RNAi is reversed upon expression of active MEK or Raf-1 mutants, this pathway is likely relevant to the promotion of cell proliferation by Mst2. However, we cannot rule out other mechanisms yet to be identified. Indeed, we also show here that Mst2 RNAi exerts a paradoxical and complicated effect on the phosphorylation status of key signaling proteins, promoting both activating and inhibitory phosphorylation of Raf-1 on Ser-338 and Ser-259, respectively. In addition, we show that phosphorylation of Akt on Thr-308 as well as Ser-235/236 of the S6 ribosomal protein (supplemental Fig. 6) in response to EGF stimulation is up-regulated in Mst2 knockdown cells.
Acknowledgments
We thank Philip Tsichlis (Tufts Medical Center) for the akt2/3−/−;akt-1flox/flox cells, Andrea McClatchey (Massachusetts General Hospital) for merlin-expressing recombinant adenovirus, and Joseph Avruch (Massachusetts General Hospital) for the phospho-specific Mst2 antibody. We thank Michael Mendelsohn (Tufts Medical Center) for the Cre recombinase adenovirus and for continued support.
This work was supported, in whole or in part, by National Institutes of Health Grant CA112399 (to J. M. K.).
This article was selected as a Paper of the Week.

The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. 1–7.
- MAPK
- mitogen-activated protein kinase
- MAP2K
- MAPK-kinase
- MAP3K
- MAPK-kinase-kinase
- ERK
- extracellular signal-regulated kinase
- MEK
- MAPK/ERK kinase
- MEF
- mouse embryonic fibroblast
- Mst
- mammalian sterile-20-like
- LATS
- large tumor suppressor
- NF
- neurofibromatosis
- PAK
- p21-activated kinase
- PP2A
- protein phosphatase-2A
- PP2A-A
- PP2A A scaffolding subunit
- PP2A-C
- PP2A catalytic subunit
- PtdIns
- phosphatidylinositol
- RNAi
- RNA interference
- oligo
- oligonucleotide
- GFP
- green fluorescent protein
- RIPA
- radioimmune precipitation
- MBP
- myelin basic protein
- EGF
- epidermal growth factor
- PTEN
- phosphatase and tensin homolog
- RASFF
- Ras association domain family
- YAP
- Yes-associated protein
- DMSO
- dimethyl sulfoxide
- Z
- benzyloxycarbonyl
- FMK
- fluoromethyl ketone
- PI3K
- phosphatidylinositol 3-kinase
- d
- Drosophila
- h
- human.
REFERENCES
- 1.Brown M. D., Sacks D. B. (2009) Cell. Signal. 21, 462–469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Widmann C., Gibson S., Jarpe M. B., Johnson G. L. (1999) Physiol. Rev. 79, 143–180 [DOI] [PubMed] [Google Scholar]
- 3.Lowy D. R., Willumsen B. M. (1993) Annu. Rev. Biochem. 62, 851–891 [DOI] [PubMed] [Google Scholar]
- 4.Dhillon A. S., Pollock C., Steen H., Shaw P. E., Mischak H., Kolch W. (2002) Mol. Cell. Biol. 22, 3237–3246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zimmermann S., Moelling K. (1999) Science 286, 1741–1744 [DOI] [PubMed] [Google Scholar]
- 6.Clark G. J., Drugan J. K., Rossman K. L., Carpenter J. W., Rogers-Graham K., Fu H., Der C. J., Campbell S. L. (1997) J. Biol. Chem. 272, 20990–20993 [DOI] [PubMed] [Google Scholar]
- 7.Tzivion G., Luo Z., Avruch J. (1998) Nature 394, 88–92 [DOI] [PubMed] [Google Scholar]
- 8.Dhillon A. S., Meikle S., Yazici Z., Eulitz M., Kolch W. (2002) EMBO J. 21, 64–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jaumot M., Hancock J. F. (2001) Oncogene 20, 3949–3958 [DOI] [PubMed] [Google Scholar]
- 10.Kubicek M., Pacher M., Abraham D., Podar K., Eulitz M., Baccarini M. (2002) J. Biol. Chem. 277, 7913–7919 [DOI] [PubMed] [Google Scholar]
- 11.Kyriakis J. M. (2007) Biochim. Biophys. Acta 1773, 1238–1247 [DOI] [PubMed] [Google Scholar]
- 12.Rushworth L. K., Hindley A. D., O'Neill E., Kolch W. (2006) Mol. Cell. Biol. 26, 2262–2272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Garnett M. J., Rana S., Paterson H., Barford D., Marais R. (2005) Mol. Cell 20, 963–969 [DOI] [PubMed] [Google Scholar]
- 14.Xu G. F., Lin B., Tanaka K., Dunn D., Wood D., Gesteland R., White R., Weiss R., Tamanoi F. (1990) Cell 63, 835–841 [DOI] [PubMed] [Google Scholar]
- 15.Scoles D. R. (2008) Biochim. Biophys. Acta 1785, 32–54 [DOI] [PubMed] [Google Scholar]
- 16.Pombo C. M., Force T., Kyriakis J., Nogueira E., Fidalgo M., Zalvide J. (2007) Front. Biosci. 12, 850–859 [DOI] [PubMed] [Google Scholar]
- 17.Dan I., Watanabe N. M., Kusumi A. (2001) Trends Cell Biol. 11, 220–230 [DOI] [PubMed] [Google Scholar]
- 18.Harvey K. F., Pfleger C. M., Hariharan I. K. (2003) Cell 114, 457–467 [DOI] [PubMed] [Google Scholar]
- 19.Pantalacci S., Tapon N., Léopold P. (2003) Nat. Cell Biol. 5, 921–927 [DOI] [PubMed] [Google Scholar]
- 20.Ling P., Lu T. J., Yuan C. J., Lai M. D. (2008) Cell. Signal. 20, 1237–1247 [DOI] [PubMed] [Google Scholar]
- 21.Radu M., Chernoff J. (2009) Curr. Biol. 19, R421–R425 [DOI] [PubMed] [Google Scholar]
- 22.Hamaratoglu F., Willecke M., Kango-Singh M., Nolo R., Hyun E., Tao C., Jafar-Nejad H., Halder G. (2006) Nat. Cell Biol. 8, 27–36 [DOI] [PubMed] [Google Scholar]
- 23.Graves J. D., Gotoh Y., Draves K. E., Ambrose D., Han D. K., Wright M., Chernoff J., Clark E. A., Krebs E. G. (1998) EMBO J. 17, 2224–2234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.O'Neill E., Rushworth L., Baccarini M., Kolch W. (2004) Science 306, 2267–2270 [DOI] [PubMed] [Google Scholar]
- 25.Oka T., Mazack V., Sudol M. (2008) J. Biol. Chem. 283, 27534–27546 [DOI] [PubMed] [Google Scholar]
- 26.Avruch J., Praskova M., Ortiz-Vega S., Liu M., Zhang X. F. (2006) Methods Enzymol. 407, 290–310 [DOI] [PubMed] [Google Scholar]
- 27.Cooper W. N., Hesson L. B., Matallanas D., Dallol A., von Kriegsheim A., Ward R., Kolch W., Latif F. (2009) Oncogene 28, 2988–2998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cox A. D., Der C. J. (2003) Oncogene 22, 8999–9006 [DOI] [PubMed] [Google Scholar]
- 29.Matallanas D., Romano D., Yee K., Meissl K., Kucerova L., Piazzolla D., Baccarini M., Vass J. K., Kolch W., O'Neill E. (2007) Mol. Cell 27, 962–975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhou D., Medoff B. D., Chen L., Li L., Zhang X. F., Praskova M., Liu M., Landry A., Blumberg R. S., Boussiotis V. A., Xavier R., Avruch J. (2008) Proc. Natl. Acad. Sci. U.S.A. 105, 20321–20326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dong J., Feldmann G., Huang J., Wu S., Zhang N., Comerford S. A., Gayyed M. F., Anders R. A., Maitra A., Pan D. (2007) Cell 130, 1120–1133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lehtinen M. K., Yuan Z., Boag P. R., Yang Y., Villén J., Becker E. B., DiBacco S., de la Iglesia N., Gygi S., Blackwell T. K., Bonni A. (2006) Cell 125, 987–1001 [DOI] [PubMed] [Google Scholar]
- 33.Zhou D., Conrad C., Xia F., Park J. S., Payer B., Yin Y., Lauwers G. Y., Thasler W., Lee J. T., Avruch J., Bardeesy N. (2009) Cancer Cell 16, 425–438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ikeda M., Kawata A., Nishikawa M., Tateishi Y., Yamaguchi M., Nakagawa K., Hirabayashi S., Bao Y., Hidaka S., Hirata Y., Hata Y. (2009) Sci. Signal. 2, ra59. [DOI] [PubMed] [Google Scholar]
- 35.Lepont P., Stickney J. T., Foster L. A., Meng J. J., Hennigan R. F., Ip W. (2008) Mutat. Res. 637, 142–151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.King B. L., Carter D., Foellmer H. G., Kacinski B. M. (1992) Am. J. Pathol. 140, 23–31 [PMC free article] [PubMed] [Google Scholar]
- 37.Sun C. X., Haipek C., Scoles D. R., Pulst S. M., Giovannini M., Komada M., Gutmann D. H. (2002) Hum. Mol. Genet. 11, 3167–3178 [DOI] [PubMed] [Google Scholar]
- 38.Chadee D. N., Kyriakis J. M. (2004) Nat. Cell Biol. 6, 770–776 [DOI] [PubMed] [Google Scholar]
- 39.Saucedo L. J., Edgar B. A. (2007) Nat. Rev. Mol. Cell Biol. 8, 613–621 [DOI] [PubMed] [Google Scholar]
- 40.Mikula M., Schreiber M., Husak Z., Kucerova L., Rüth J., Wieser R., Zatloukal K., Beug H., Wagner E. F., Baccarini M. (2001) EMBO J. 20, 1952–1962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chadee D. N., Xu D., Hung G., Andalibi A., Lim D. J., Luo Z., Gutmann D. H., Kyriakis J. M. (2006) Proc. Natl. Acad. Sci. U.S.A. 103, 4463–4468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Baccarini M. (2005) FEBS Lett. 579, 3271–3277 [DOI] [PubMed] [Google Scholar]
- 43.Dhillon A. S., von Kriegsheim A., Grindlay J., Kolch W. (2007) Cell Cycle 6, 3–7 [DOI] [PubMed] [Google Scholar]
- 44.Chaudhary A., King W. G., Mattaliano M. D., Frost J. A., Diaz B., Morrison D. K., Cobb M. H., Marshall M. S., Brugge J. S. (2000) Curr. Biol. 10, 551–554 [DOI] [PubMed] [Google Scholar]
- 45.King A. J., Sun H., Diaz B., Barnard D., Miao W., Bagrodia S., Marshall M. S. (1998) Nature 396, 180–183 [DOI] [PubMed] [Google Scholar]
- 46.Cinar B., Fang P. K., Lutchman M., Di Vizio D., Adam R. M., Pavlova N., Rubin M. A., Yelick P. C., Freeman M. R. (2007) EMBO J. 26, 4523–4534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Eichhorn P. J., Creyghton M. P., Bernards R. (2009) Biochim. Biophys. Acta 1795, 1–15 [DOI] [PubMed] [Google Scholar]
- 48.Westermarck J., Hahn W. C. (2008) Trends Mol. Med. 14, 152–160 [DOI] [PubMed] [Google Scholar]
- 49.Abraham D., Podar K., Pacher M., Kubicek M., Welzel N., Hemmings B. A., Dilworth S. M., Mischak H., Kolch W., Baccarini M. (2000) J. Biol. Chem. 275, 22300–22304 [DOI] [PubMed] [Google Scholar]
- 50.Adams D. G., Coffee R. L., Jr., Zhang H., Pelech S., Strack S., Wadzinski B. E. (2005) J. Biol. Chem. 280, 42644–42654 [DOI] [PubMed] [Google Scholar]
- 51.Andrabi S., Gjoerup O. V., Kean J. A., Roberts T. M., Schaffhausen B. (2007) Proc. Natl. Acad. Sci. U.S.A. 104, 19011–19016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Millward T. A., Zolnierowicz S., Hemmings B. A. (1999) Trends Biochem. Sci 24, 186–191 [DOI] [PubMed] [Google Scholar]
- 53.Baharians Z., Schönthal A. H. (1998) J. Biol. Chem. 273, 19019–19024 [DOI] [PubMed] [Google Scholar]
- 54.Ohama T., Brautigan D. L. (2010) J. Biol. Chem. 285, 8711–8718 [DOI] [PMC free article] [PubMed] [Google Scholar]