

The crystal structure of dehaloperoxidase (DHP) isoenzyme B from the terebellid polychaete A. ornata, which exhibits both hemoglobin and peroxidase functions, has been determined at 1.58 Å resolution.
Keywords: dehaloperoxidases, globins, Amphitrite ornata, heme
Abstract
As members of the globin superfamily, dehaloperoxidase (DHP) isoenzymes A and B from the marine annelid Amphitrite ornata possess hemoglobin function, but they also exhibit a biologically relevant peroxidase activity that is capable of converting 2,4,6-trihalophenols to the corresponding 2,6-dihaloquinones in the presence of hydrogen peroxide. Here, a comprehensive structural study of recombinant DHP B, both by itself and cocrystallized with isoenzyme A, using X-ray diffraction is presented. The structure of DHP B refined to 1.58 Å resolution exhibits the same distal histidine (His55) conformational flexibility as that observed in isoenzyme A, as well as additional changes to the distal and proximal hydrogen-bonding networks. Furthermore, preliminary characterization of the DHP AB heterodimer is presented, which exhibits differences in the AB interface that are not observed in the A-only or B-only homodimers. These structural investigations of DHP B provide insights that may relate to the mechanistic details of the H2O2-dependent oxidative dehalogenation reaction catalyzed by dehaloperoxidase, present a clearer description of the function of specific residues in DHP at the molecular level and lead to a better understanding of the paradigms of globin structure–function relationships.
1. Introduction
Spanning all kingdoms of life, the globin superfamily is comprised of a vast number of proteins which exhibit a diverse array of functions. Although defined by the presence of a characteristic protein fold, namely the canonical 3/3 α-helical structure most commonly associated with myoglobin (Mb), the sequence homology between globins from different phyla may be as low as 10%. Thus, the combination of a highly conserved structural motif with a relatively low sequence homology, coupled with the diverse number of roles which globins fulfill, has been the driving force behind new research into understanding the nuances of globin structure–function relationships that enable specific discrete functions.
As an example of a globin found in marine organisms, structural and mechanistic investigations of the enzyme dehaloperoxidase (DHP) may reveal new insights into the structure–function paradigms of the globin superfamily. DHP is a bifunctional hemoprotein which acts as both the O2-transport protein and as a peroxidase in the terebellid polychaete Amphitrite ornata (LaCount et al., 2000 ▶; Chen et al., 1996 ▶), although how DHP performs these dual roles remains unclear at the present time. DHP has been shown to be a homodimer consisting of two identical subunits of ∼15.5 kDa in the asymmetric unit, each of which contains a heme protoporphyrin IX cofactor and eight α-helices (de Serrano et al., 2007 ▶; Zhang et al., 1996 ▶). These subunits are connected through an interface mainly comprised of salt bridges from acidic and basic amino-acid residues on the protein surface, specifically between Asp72 of one subunit and the side-chain groups of Arg122 and Asn126 of the other subunit (Lebioda et al., 1999 ▶; Chen et al., 2009 ▶). There is also a hydrophobic interaction between the Val74 residues of the two subunits (LaCount et al., 2000 ▶). The only cysteine present in DHP, Cys73, is located in close proximity to this interface. However, the distance between the S atoms within the dimer subunits is not sufficiently short to make a potential disulfide bond between the subunits likely.
Since the seminal work of Lebioda and coworkers revealed a small-molecule-binding pocket in close proximity to the heme active site (Lebioda et al., 1999 ▶), several studies have focused on the specifics of substrate localization in DHP (Davis et al., 2009 ▶; Smirnova et al., 2008 ▶; Nienhaus et al., 2008 ▶; Belyea et al., 2005 ▶), a feature that is unique to dehaloperoxidase among all known globins. More recently, it has been postulated that both external and internal small-molecule-binding sites may exist, with regulatory implications that may govern how DHP switches between its hemoglobin and peroxidase activities (Chen et al., 2009 ▶; Davis et al., 2009 ▶; Smirnova et al., 2008 ▶). One intriguing possibility is that the distal histidine His55, which has been observed in distinct ‘open’ and ‘closed’ conformations, mediates either H2O or O2 displacement from the heme in the presence of (tri)halophenol substrate, possibly serving as a trigger for peroxidase function (Chen et al., 2009 ▶). It has been hypothesized that the ‘open’ and ‘closed’ conformations are the consequence of an equilibrium between five-coordinate and six-coordinate (metaquo) forms of DHP at room temperature, respectively (Chen et al., 2009 ▶; Lebioda et al., 1999 ▶). Specifically, in the open conformation His55 is swung out of the distal pocket and solvent-exposed, with a heme Fe—NHis distance of ∼9 Å, and for this reason is unable to participate in hydrogen-bonding interactions that would stabilize the presence of a sixth axial heme ligand. The open conformation is not unique to DHP; it has been observed previously in myoglobin (Mb) at low pH (Tian et al., 1993 ▶; Zhu et al., 1992 ▶; Sage et al., 1991 ▶). In contrast, the ‘closed’ conformation of DHP, with His55 swung into the distal pocket, exhibits a heme Fe—NHis distance of 5.4 Å and represents a more reasonable distance for associating with the sixth heme ligand (i.e. water or dioxygen) via hydrogen-bonding interactions (de Serrano et al., 2007 ▶). This is clearly shown in the structure of oxyferrous DHP, in which a bent bonding geometry for the Fe—O(1)—O(2) adduct was observed, yielding a 2.8 Å hydrogen bond between O(2), the second O atom of molecular oxygen bound to the heme iron, and the N∊ atom of the distal histidine His55 (de Serrano et al., 2007 ▶). A similar closed conformation of the distal histidine in Mb has also been shown to exist under basic conditions (Tian et al., 1993 ▶; Zhu et al., 1992 ▶).
To date, X-ray diffraction studies of dehaloperoxidase have been limited to one isoenzyme (DHP A; Chen et al., 2009 ▶; de Serrano et al., 2007 ▶; LaCount et al., 2000 ▶; Zhang et al., 1996 ▶; Lebioda et al., 1999 ▶). However, two isoforms of dehaloperoxidase, termed DHP A and DHP B, occur in A. ornata and are encoded by two separate genes (dhpA and dhpB; Han et al., 2001 ▶). Both have been shown to catalyze the oxidative dehalogenation of 2,4,6-trihalogenated phenols to the corresponding 2,6-dihalo-1,4-benzoquinones in the presence of hydrogen peroxide (Feducia et al., 2009 ▶; D’Antonio et al., 2010 ▶). Although they share 96% sequence identity (DHP B differs from DHP A at five positions: I9L, R32K, Y34N, N81S and S91G), significant spectroscopic and mechanistic differences between dehaloperoxidase isoenzymes A and B have recently been elucidated (D’Antonio et al., 2010 ▶). These include the observations that DHP B is twofold to fourfold more active than DHP A (depending on the trihalophenol substrate), that it forms a Compound RH intermediate which exhibits different reactivity to the analogous species in DHP A and that DHP B exhibits a greater extent of substrate inhibition than DHP A. Furthermore, an interesting parallel may be drawn between DHP A and B and the α and β chains of Hb: two globin chains which are structurally homologous yet exhibit altered chemical reactivity (Feducia et al., 2009 ▶; D’Antonio et al., 2010 ▶). Given this, a number of interesting questions can be posed. For example, what are the structural features of DHP B and what are the differences when compared with DHP A? Will DHP B exhibit the same conformational flexibility of the distal histidine (His55) as DHP A? How does the S91G substitution in DHP B alter the proximal histidine (His89) binding? Is the small-molecule-binding pocket maintained in DHP B? Will DHP A and B form an αβ dimer or an α2β2 tetramer reminiscent of that found in hemoglobin (Hb) and will cooperative effects on O2-binding or peroxidase activity be observed? To begin to address these and other questions, here we present a structural study of recombinant DHP B both by itself and with isoenzyme A using X-ray diffraction. Such investigations of DHP B may provide critical insight that may help in understanding the mechanistic details of the H2O2-dependent oxidative dehalogenation reaction catalyzed by dehaloperoxidase and may advance our knowledge of how globins acquired such an enzymatic function. Our structural findings on DHP B are presented in the light of our recent mechanistic investigations on both isoenzymes (Feducia et al., 2009 ▶; D’Antonio et al., 2010 ▶) and together provide a clearer description of the function of DHP at the molecular level.
2. Materials and methods
2.1. Protein purification, characterization and crystallization
Buffer salts were purchased from Fisher Scientific. All other reagents and biochemicals, unless otherwise specified, were of the highest grade available from Sigma–Aldrich. The QIAprep Spin Miniprep Kit was from Qiagen Sciences (Valencia, California, USA) and the QuikChange II site-directed mutagenesis kit was purchased from Stratagene (La Jolla, California, USA). The required oligonucleotides were synthesized by IDT DNA Technologies Inc. Optical spectra were recorded using quartz microcuvettes (1 cm path length) on a Cary 50 UV–visible spectrophotometer equipped with thermostatted cell holders at 298 K.
For protein crystallization, the polyhistidine purification tag was removed from the 5′-end of the template plasmid pDHPB(6×His), which encodes the DHP B isoenzyme, using the QuikChange II site-directed mutagenesis kit. Mutagenesis [melting (368 K, 50 s), annealing (333 K, 50 s) and extension (341 K, 6 min)] was performed for 18 cycles using the mutagenic primers 5′-AGG AGA TAT ACC ATG GGG TTT AAA CAA GAT-3′ (sense) and 5′-ATC TTG TTT AAA CCC CAT GGT ATA TCT CCT-3′ (antisense). The plasmid was extracted using the QIAprep Spin Miniprep Kit and DNA sequencing of the resulting plasmid pDHPB(-6×-His) bearing the dhpB gene confirmed the success of the His-tag deletion and the absence of secondary mutations. Recombinant DHP B lacking the polyhistidine tag was subsequently overexpressed in Rosetta Escherichia coli cells (DE3 strain) and purified using the procedures established for the purification of DHP A (de Serrano et al., 2007 ▶). DHP A (non-His-tagged) was also expressed and purified as described previously (Davis et al., 2009 ▶) with only minor modification. The purification strategy (ammonium sulfate fractionation followed by ion-exchange and size-exclusion chromatography) resulted in protein that was of satisfactory purity for crystallization. As has previously been noted for dehaloperoxidase (Roach et al., 1997 ▶), the enzyme was initially isolated as a mixture of the ferric and oxyferrous forms. Enzyme in the purely ferric state was prepared by treatment with an excess of potassium ferricyanide as described elsewhere (de Serrano et al., 2007 ▶; D’Antonio et al., 2010 ▶). Only protein samples that exhibited Reinheitzahl values (R z) of greater than 4.0 were utilized in this study. The concentration of DHP B was determined spectrophotometrically (∊ 406 = 117.6 mM −1 cm−1; D’Antonio et al., 2010 ▶). Dehaloperoxidase activity assays of the enzyme preparations used here for crystallization confirmed proper enzymatic function (Feducia et al., 2009 ▶; D’Antonio et al., 2010 ▶).
The crystal structure of DHP B was determined for the protein in the ferric oxidation state and was confirmed by UV–visible spectroscopy, which showed the characteristic 406 nm Soret band absorption of the ferric protein (Feducia et al., 2009 ▶; D’Antonio et al., 2010 ▶). All crystallizations were carried out at 277 K using the hanging-drop vapor-diffusion method. For DHP B, the protein (9 mg ml−1) in 20 mM sodium cacodylate buffer pH 6.5 was mixed with an equal volume of crystallization solution and equilibrated against 600 µl crystallization solution [0.2 M ammonium sulfate and PEG 4000 in the concentration range 30–36%(w/v)]. Crystals initially grew in twinned clusters that appeared after 3 d of incubation and were subsequently improved by two rounds of microseeding. The crystals were allowed to grow for 7 d after microseeding and were harvested into mother liquor supplemented with 15% PEG 400 as a cryoprotectant and flash-frozen at 100 K for data collection. A diffraction data set was collected using Cu Kα radiation and was refined to a resolution of 1.58 Å with an R factor of 17.0% (R free = 21.4%). To grow crystals from a mixture of DHP A and B, the two proteins were combined in a 1:1 molar ratio (11 mg ml−1 final protein concentration), followed by mixing the protein solution with an equal volume of crystallization solution and incubating the mixture in a hanging drop over a 600 µl reservoir of crystallization solution as described above. The best diffraction-quality DHP AB crystals grew from 0.2 M ammonium sulfate and 32% PEG 4000.
2.2. X-ray data collection and structure refinement
X-ray diffraction data were collected from the DHP B crystals using a Rigaku MicroMax007 HF copper rotating-anode generator (λ = 1.5418 Å) equipped with VariMax HF optics and a Rigaku R-AXIS IV++ image-plate detector (Biomolecular X-ray Crystallography Core Facility, University of North Carolina at Chapel Hill). The data set was collected from a single crystal at a crystal-to-detector distance of 85 mm using a 1° oscillation range and an exposure time of 3 min per image. The diffraction data for the DHP AB crystals were collected on the SER-CAT 22-BM beamline at the Advanced Photon Source (Argonne, Illinois, USA) using a wavelength of 0.91339 Å, a 1° oscillation range and a crystal-to-detector distance of 120 mm. The diffraction data were reduced and scaled using the HKL-2000 suite (Otwinowski & Minor, 1997 ▶). The crystals of both DHP B and DHP AB belonged to space group P212121, as do the crystals of DHP A, and had unit-cell parameters a = 60.65, b = 67.42, c = 67.48 Å and a = 60.165, b = 67.387, c = 67.653 Å, respectively, which are very similar to the unit-cell parameters of the DHP A crystals (de Serrano et al., 2007 ▶). The crystals of DHP B diffracted to a resolution of 1.58 Å and those of DHP AB diffracted to a resolution of 1.52 Å.
The structures of DHP B and DHP AB were determined by molecular replacement with Phaser (McCoy et al., 2005 ▶) using the metaquo structure of DHP A (PDB entry 2qfk; de Serrano et al., 2007 ▶) as a search model. Cycles of refinement and map calculation were carried out with REFMAC5 from the CCP4 suite of programs (Collaborative Computational Project, Number 4, 1994 ▶) and were iterated with model building using the Coot program (Emsley & Cowtan, 2004 ▶). All occupancies were refined manually. The final model of DHP B refined to an R factor of 17.0% (R free = 21.4%) and contained two protein molecules, with 94.0% of the residues in the most favored regions of the Ramachandran plot and the remaining 6.0% in allowed regions, whereas that of DHP AB refined to R work and R free values of 17.6% and 21.9%, respectively, with 93.6% of the residues in the most favored region and 6.4% of the residues in the allowed region of the Ramachandran plot. Under the conditions employed, DHP B crystallized with two molecules in the asymmetric unit; the main-chain atoms of the two molecules superposed with an average displacement of 0.289 Å. These findings are analogous to those for DHP A, which also was found to crystallize as a homodimer (de Serrano et al., 2007 ▶; LaCount et al., 2000 ▶). Data-collection and refinement statistics are summarized in Table 1 ▶. The structural coordinates for DHP B have been deposited in the Research Collaboratory for Structural Bioinformatics (RCSB) Protein Data Bank under accession code 3ixf.
Table 1. Data-collection and refinement statistics for DHP B.
Values in parentheses are for the highest resolution shell.
| DHP B | DHP AB complex† | |
|---|---|---|
| Data collection | ||
| Space group | P212121 | P212121 |
| Unit-cell parameters | ||
| a (Å) | 60.65 | 60.17 |
| b (Å) | 67.42 | 67.39 |
| c (Å) | 67.48 | 67.65 |
| α = β = γ (°) | 90 | 90, |
| Temperature (K) | 100 | 100 |
| Wavelength (Å) | 1.54 | 0.913 |
| Resolution (Å) | 30.17–1.58 (1.62–1.58) | 40.00–1.52 (1.56–1.52) |
| Rmerge‡ | 9.7 (54.7) | 7.9 (62.8) |
| I/σ(I) | 21.9 (2.5) | 19.3 (2.7) |
| Completeness (%) | 99.7 (99.9) | 97.0 (98.9) |
| Redundancy | 4.8 (4.8) | 4.9 (4.7) |
| Refinement | ||
| No. of reflections | 36546 (2665) | 39585 (2934) |
| Rwork/Rfree§ (%) | 17.0/21.4 | 17.6/21.9 |
| Average B factor (Å2) | ||
| All atoms | 13.82 | 10.13 |
| Protein | 12.52 | 9.07 |
| Water | 23.84 | 20.63 |
| No. of atoms | ||
| Protein | 3002 | 3139 |
| Water | 373 | 287 |
| R.m.s.d.¶ from ideal | ||
| Bond lengths (Å) | 0.024 | 0.011 |
| Bond angles (°) | 2.197 | 1.365 |
| Ramachandran plot†† (%) | ||
| Most favored | 94.0 | 93.6 |
| Allowed | 6.0 | 6.4 |
Protein crystallized using a 1:1 ratio of DHP A and DHP B.
R
merge = 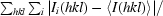
 , where I
i(hkl) is the ith measurement of I(hkl) and 〈I(hkl)〉 is the weighted mean of all measurements of I(hkl).
, where I
i(hkl) is the ith measurement of I(hkl) and 〈I(hkl)〉 is the weighted mean of all measurements of I(hkl).
R
work = 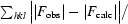
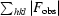 , where F
obs are the observed and F
calc are the calculated structure factors; R
free is the R factor for a subset (5%) of reflections selected before and not included in the refinement.
, where F
obs are the observed and F
calc are the calculated structure factors; R
free is the R factor for a subset (5%) of reflections selected before and not included in the refinement.
Root-mean-square deviation.
Calculated using PROCHECK (Laskowski et al., 1993 ▶).
3. Results and discussion
3.1. Overall DHP B protein structure
DHP B exhibits the canonical myoglobin fold consisting of eight helices (Phillips, 2001 ▶) identified by the letters A–H (of which the segments Pro29–Asn34, Lys36–Tyr38 and Lys87–His89 assume 310-helical conformations). As shown by the superposition of their backbone main-chain traces (Fig. 1 ▶), the structure of DHP B is nearly identical to that of isoenzyme A. Superposition of the main-chain atoms of the two dimeric molecules of DHP B with the equivalent chains of DHP A gives similar statistics for the coordinate r.m.s.d.: 0.257 Å for the A chains of DHP A and B and 0.236 Å for the B chains. The structural variations between DHP B and A are essentially limited to the amino-acid differences between the two isoenzymes, which cluster in the regions of the distal (R32K and Y34N; Fig. 2 ▶ a) and proximal (N81S and S91G; Fig. 2 ▶ b) sides of the heme cavity, with one additional substitution, I9L (Fig. 2 ▶ c), that is located in a hydrophobic region enclosed by residues Tyr16, Ile20, Phe115, Trp120, Phe60, Met64, Val104, Phe107 and Met108. However, it is apparent from backbone-superposition calculations that these amino-acid differences do not perturb the overall structural fold of DHP but are limited to hydrogen-bonding and nonbonding interactions within the molecule and with symmetry-related molecules.
Figure 1.

Stereo ribbon diagram of the overlay of the Cα trace of DHP B (red) and DHP A (grey; PDB code 2qfk). The heme cofactor and the distal (His55; both conformations) and proximal (His89) histidines are shown in stick representation. Helices are labeled A–H and one (chain A) of the two subunits in the asymmetric unit is shown throughout.
Figure 2.

Superposition of the DHP isoenzymes showing the three regions of structural changes affected by the amino-acid substitutions. (a) Distal side of the heme cavity. Isoenzyme differences are noted at positions 32 (lysine in DHP B and arginine in DHP A) and 34 (asparagine in DHP B and tyrosine in DHP A). (b) Proximal side of the heme cavity. The relevant isoenzyme substitutions are at positions 81 (serine in DHP B and asparagine in DHP A) and 91 (glycine in DHP B and serine in DHP A). (c) Overlay of the region containing the residue at position 9 (leucine in DHP B and isoleucine in DHP A). The side chains are shown in red for DHP B and gray for DHP A. Residues are only labeled for DHP B.
Specifically, the amino-acid differences in the distal pocket (Fig. 2 ▶ a) introduce new hydrogen bonds, notably between the NH2 group of one of the Lys32 conformers and the backbone carbonyl O atom of Leu25 (2.80 Å) as well as between the NH2 group of one of the Asn34 conformers and the backbone carbonyl O atom of Glu31 (2.73 Å). The conformations of the intervening residues, as well as the residues participating in the formation of the distal pocket (e.g. phenylalanines 21, 24 and 35), are thus affected. In addition, the replacement of tyrosine in position 34 with asparagine affects the hydrogen-bonding interactions related to these two residues (Fig. 3 ▶). In DHP A Tyr34 forms only one weak hydrogen bond involving its hydroxyl group and the side chain of Asn96 (de Serrano et al., 2007 ▶). Upon the substitution of this tyrosine with asparagine in DHP B this hydrogen bond is lost, but Asn34 is capable of forming a much stronger hydrogen bond to the carbonyl O atom of Glu31 as well as a couple of very weak ionic interactions with the side chains of Glu31 and Asn96. Thus, since Tyr34 has been implicated as one possible site of radical formation in DHP B Compound ES (Feducia et al., 2009 ▶), its replacement by a redox-inactive asparagine residue may affect either the site of radical formation in DHP B Compound ES or may alter the pathway for the migration of this radical out of the active site. Furthermore, the substitution-induced differences in the distal pocket of DHP B versus DHP A directly affect both the conformations of critical active-site residues and the hydrogen-bonding networks that encompass the region putatively ascribed as the internal binding site, which may play a role in radical stabilization and radical migration and may also contribute to the differences in enzymatic activity (i.e. substrate specificity) observed between the two isoenzymes (D’Antonio et al., 2010 ▶).
Figure 3.

Stereoview of the proximal side of the heme cavity showing the 2F o − F c electron-density maps of (a) DHP B and (b) DHP A contoured at the 1.2σ level. Water molecules are represented as blue spheres. The differences in the region shown are limited to residue 34 (asparagine in DHP B and tyrosine in DHP A) and residue 91 (glycine in DHP B and serine in DHP A). The effect of the substitution of the tyrosine residue at position 34 by asparagine is most evident in the changes in the hydrogen-bonding interactions of these residues: Tyr34 (DHP A) makes a weak hydrogen-bonding contact (3.2 Å) to Asn96, whereas the Asn34 (DHP B) hydrogen-bonding interactions with Asn96 are mediated by a water molecule (distances of 2.78 and 2.81 Å, respectively) as illustrated.
On the proximal side of the heme pocket (Fig. 2 ▶ b), the differences between the two isoenzymes have a pronounced effect on the hydrogen-bonding network present and may also alter the bonding of the proximal histidine to the heme cofactor. The substitution of Asn81 in DHP A by a serine residue in DHP B places one of the Ser81 conformers in a position to establish a new hydrogen bond to one of the Gln85 NH2 conformers, thus possibly affecting the hydrogen bonding of Leu83 to His89 (the proximal histidine), as well as allowing the hydrogen bonding of Gln88 to the propionate A side chain of the heme cofactor. Additionally, the substitution of Ser91 by a glycine residue affords greater flexibility to the region surrounding the proximal histidine His89, possibly affecting the strength of the Fe—NHis bond and thus the electronics of the heme cofactor related to hydrogen peroxide activation. The increased mobility of Gly91 in DHP B is apparent from the resulting alternate backbone conformations of this residue (Fig. 2 ▶ b).
3.2. Heme–Fe ligation and conformations of the distal histidine
The active-site region, which includes the heme cofactor as well as the proximal and distal histidine residues, is identically located in the tertiary structure of the protein fold in both DHP isoenzymes (Figs. 1 ▶, 3 ▶ and 4 ▶). We have previously established that the binding of a ligand to the heme iron in DHP A is a crucial determinant of the conformation of the distal histidine His55 (Chen et al., 2009 ▶; Davis et al., 2009 ▶; Smirnova et al., 2008 ▶). For example, in the structure determined at 100 K a water molecule is bound to the heme iron as the sixth coordination ligand for DHP A in its ferric form (de Serrano et al., 2007 ▶). The presence of such a ligand stabilizes His55 in the ‘closed’ conformation by allowing the formation of a hydrogen-bonding interaction between the histidine N∊2 atom and the sixth ligand. This hydrogen bonding stabilizes both the ligand and the histidine conformation as being swung inside the distal pocket (i.e. ‘closed’). This pattern of ligand binding leading to a ‘closed’ conformation of the distal histidine also seems to occur in DHP B, as chain A exhibits a water molecule bound to the heme cofactor concomitant with His55 being swung inside the distal pocket, together with the presence of a hydrogen bond between the two at 3.12 Å (a virtually identical distance to that observed in DHP A; de Serrano et al., 2007 ▶). Moreover, the water molecule that is bound to heme iron as the sixth coordination ligand in DHP B is also located at a similar distance from the iron to that observed in the DHP A structure (Table 2 ▶ and Fig. 4 ▶; de Serrano et al., 2007 ▶).
Figure 4.

Stereoview of the superposition of the distal pocket regions of DHP B (beige) and DHP A (green). Note the differences in the conformation of the distal histidine His55 in these two structures. A water molecule is coordinated to the heme iron of both structures. A hydrogen bond (3.12 Å) between the N∊2 atom of His55 (closed conformation) and the heme-bound water molecule is indicated.
Table 2. Comparison of heme-ligand parameters for DHP A and B.
Only the values for molecule A of the asymmetric unit are listed for both structures.
| DHP B | DHP A | |
|---|---|---|
| Fe—His89 N∊2 (Å) | 2.16 | 2.09 |
| Fe—His55 N∊2† (Å) | ||
| Inside conformation (Å) | 5.48 (5.41)‡ | 4.8 |
| Outside conformation (Å) | 9.8 (10.26) | — |
| Fe—water§ (Å) | 2.15 | 2.2 |
| Fe to pyrrole N plane (Å) | 0.21 | 0.04 |
| Fe–ligand tilt angle¶ (°) | 15.4 | 10.8 |
In the DHP B structure the distal histidine is present in conformations both inside and outside the pocket, whereas in the metaquo structure of DHP A (PDB entry 2qfk) it is only found in the conformation inside the pocket.
In the DHP B structure the distal histidine assumes two conformations when oriented both inside and outside the pocket.
Distances for water as the sixth coordination ligand of heme Fe are listed (see text).
The tilt angle is defined as the angle between the heme perpendicular and the Fe—O bond (Vangberg et al., 1997 ▶).
With respect to the heme active site, the structures and distances noted are very similar for the two isoenzymes of DHP regardless of the amino-acid substitutions that affect the distal and proximal heme environments. One noteworthy difference between DHP B and DHP A, however, is that the two proteins differ in the extent of ligand occupancy present in their respective crystal structures. Whereas the water molecule in DHP A is bound at a 1:1 ligand:protein ratio (taking into account a small percentage of molecular oxygen that is present; see below; de Serrano et al., 2007 ▶), the crystal structure of DHP B (chain A) exhibits only 45% ligand occupancy, with 34% of the ligand occupancy being water and 9% being molecular oxygen. Accordingly, His55 exhibits both the open and closed conformations in the same proportions as the sixth-ligand occupancy (Table 2 ▶). Specifically, the closed conformation (Fe—N∊2 distance of 5.48 Å) was observed with 45% occupancy, whereas the open conformation (Fe—N∊2 distance of ∼10 Å) was present with 55% occupancy.
The presence of only 45% occupancy of the sixth axial ligand in the structure of DHP B is possibly the consequence of partial photoreduction of the heme iron (de Serrano et al., 2007 ▶), leading to dissociation of the bound water molecule, which preferentially binds to heme in the FeIII oxidation state. This phenomenon of photoreduction of DHP B was more pronounced in chain B, in which the iron had undergone complete reduction and which resembled the structure of dithionite-reduced DHP A (PDB entry 3dr9; de Serrano et al., 2007 ▶), where the distal histidine is present only in the solvent-exposed open conformation owing to the absence of the sixth ligand with which to establish hydrogen bonds. Interestingly, in wild-type DHP A we did not observe any photoreduction of the heme iron in the metaquo form of the protein using Cu Kα X-ray radiation (de Serrano et al., 2007 ▶), whereas the DHP(C73S) mutant of this protein underwent complete reduction using this radiation source. The partially reduced iron in WT DHP B signifies that this form of the protein is reduced more easily than WT DHP A under these conditions and represents an apparent inconsistency as our spectroelectrochemical study of these two isoenzymes suggested that they had nearly identical reduction potentials (D’Antonio et al., 2010 ▶).
The displacement of the heme Fe relative to the heme pyrrole plane also supports the interpretation of partial ligand occupancy concomitant with the distal histidine His55 in both the open and closed conformations. In the hexacoordinate metaquo form of WT DHP A (de Serrano et al., 2007 ▶) the Fe is 0.04 Å below the heme plane (Table 2 ▶). In the structure of DHP B presented here the iron is located 0.21 Å below the pyrrole heme plane, which is more similar to the iron displacement observed in the reduced form of DHP A (de Serrano et al., 2007 ▶) and signifies that a major fraction of the heme iron is in the pentacoordinate state owing to dissociation of the water ligand upon reduction of the heme iron center.
3.3. Overall DHP AB cocrystal protein structure
The structure of the DHP AB heterodimer complex, which was crystallized at a 1:1 DHP A:DHP B molar ratio, was determined at 1.52 Å resolution and refined to an R factor of 17.6% (R free = 21.4%). The protein complex showed the symmetry of orthorhombic space group P212121, which is also the space group in which both DHP A (de Serrano et al., 2007 ▶) and DHP B crystallize under similar experimental conditions.
The results of molecular replacement with Phaser clearly demonstrate the presence of only two protomers in the asymmetric unit, with chain A mostly consisting of DHP A (approximately 80% DHP A and 20% DHP B, as assessed from the occupancies of the amino acids that differ between these two isoenzymes) and chain B predominantly derived from DHP B (approximately 70% of chain B is DHP B and 30% is DHP A). The main-chain atoms of chain A from the DHP AB structure superpose with those of chain A from the DHP A homodimer (average displacement of 0.159 Å), whereas the main-chain atoms of chain B in DHP AB superpose with chain B of the DHP B homodimer (average displacement of 0.158 Å).
As shown in Fig. 5 ▶, the superposition of the side chains is also very close. Specifically, the active sites are shown for chain A of the DHP AB heterodimer superposed with chain A of DHP A (Fig. 5 ▶ a) as well as for chain B of DHP AB superposed with chain B of DHP B (Fig. 5 ▶ b). Moreover, according to both its distance from the iron and the length of its hydrogen-bonding interaction with the distal histidine (His55), the heme-bound water molecule is positioned identically in the overlaid structures, further supporting the identities of the overlaid heme-pocket structures. However, one notable difference between the chains is the proximity of the Gln88 side chain to the carboxylate of heme propionate A (Fig. 5 ▶). The N∊2 atom of one of the conformers of Gln88 is at a hydrogen-bonding distance of 2.6 Å in both DHP B and chain B of DHP AB but is elongated to 3.5 Å in both DHP A (de Serrano et al., 2007 ▶) and chain A of DHP AB; it is not known at this time whether this difference in the hydrogen-bonding distance has a functional significance.
Figure 5.

Superposition of the active site of DHP AB with the active sites of DHP A and DHP B. (a) Chain A of the DHP AB complex (blue) best superposes with the active site of chain A of DHP A (PDB entry 2qfk; yellow). (b) Chain B of the DHP AB complex (blue) best matches the active site of chain B of DHP B (yellow). Distances for the hydrogen bond between the distal histidine (His55) and the water coordinated to the heme iron and for that between Gln88 and the heme propionate A are also indicated.
Interestingly, PISA analysis (Protein Interfaces, Surfaces and Assemblies; Krissinel & Henrick, 2007 ▶) of the state of assembly and interfaces of the two chains of the DHP AB heterodimer suggests that they are most likely to be present as monomers in the asymmetric unit and hence are not predicted to form a complex in solution. However, examination of the chain interfaces for all three structures (DHP A, DHP B and DHP AB) reveals differences in the distances of relevant residues involved in these interfacial interactions which may have ramifications beyond the PISA analysis. In particular, one of the major interfaces involving residues Asp72 and Arg122 from both chains in DHP AB is shown in Fig. 6 ▶(c) and is contrasted with those of the two chains of DHP A (Fig. 6 ▶ a) and DHP B (Fig. 6 ▶ b). The interfacial hydrogen-bonding distances of 2.4 Å between these two pairs of residues in the DHP AB heterodimer is indicative of a very strong interaction between the side chains involved. These hydrogen bonds are weaker in the DHP B homodimer (2.5–3.1 Å; Fig. 6 ▶ b) and are further elongated to 3.2–3.4 Å in the DHP A homodimeric structure (Fig. 6 ▶ a; de Serrano et al., 2007 ▶). Therefore, there appears to be a trend in the strength of the interaction between the two chains of this interface, with DHP AB being the strongest, DHP B somewhat weaker and the weakest interaction being that found in the DHP A structure. As a whole, there may be a preference in chain assembly for the asymmetric unit of the DHP AB complex over that of either the DHP A or DHP B homodimers, as well as a difference in the strength of the interaction of the residues involved in the chain interfaces, but these interactions and their significance require further investigation by other means.
Figure 6.

Interface of chain A (bottom chain) and chain B (top chain) located in the asymmetric unit of DHP. The ribbon diagram of the Cα trace is shown for (a) DHP A (PDB entry 2qfk; dark blue), (b) DHP B (cyan) and (c) DHP AB at a 1:1 protein ratio (pink). The residues involved in the interface interactions, Asp72, Arg122, Asn126 and Val74 of both chains, are displayed in stick representation and the relevant hydrogen-bonding distances are indicated. The E, F and H helices involved in these interactions are also indicated.
3.4. Structure–function relationship of DHP
Across the superfamily, globins have been shown to perform a diverse number of roles that include O2 transport, small-molecule sensing or scavenging, redox chemistry and, as discussed here, peroxidase activity. This range of biomolecular processes is remarkable given the highly conserved structural motif that defines globins despite their relatively low sequence homologies. In light of the structural constraints of the canonical 3/3 globin fold, understanding the nuances of globin structure that enable their specific and functionally unique activities is paramount to establishing the paradigms of globin structure–function relationships. Within this context, our recent focus has been the elucidation of the molecular details that enable both O2-transport and peroxidase functions in the globin dehaloperoxidase from the terebellid polychaete A. ornata. In addition to being the coelomic hemoglobin of this marine worm (Weber et al., 1977 ▶), DHP possesses a biologically relevant peroxidase activity in that it catalyzes the oxidative degradation of trihalophenols to dihaloquinones (Chen et al., 1996 ▶). Whereas A. ornata has been shown to possess two genes, dhpA and dhpB (Han et al., 2001 ▶), that encode dehaloperoxidase isoenzymes A and B, respectively, only isoenzyme A has been characterized structurally. Thus, in order to support recent and ongoing detailed mechanistic investigations (Feducia et al., 2009 ▶; D’Antonio et al., 2010 ▶; Osborne et al., 2009 ▶) and to provide further insight into the molecular details of the protein environment that support a bifunctional heme active site, we present here the structural characterization of DHP B both as a homodimer and as a heterodimer crystallized with isoenzyme A.
The single-crystal X-ray diffraction study of the DHP B homodimer at 1.58 Å resolution revealed a protein structure that was similar overall to that previously observed for the homodimer of DHP A (LaCount et al., 2000 ▶; Lebioda et al., 1999 ▶; de Serrano et al., 2007 ▶). The superposition of the backbone trace of isoenzyme B with that of isoenzyme A showed that the primary differences between the two structures were limited to regions surrounding the five amino-acid substitutions that distinguish these two isoenzymes and can be clustered into three distinct regions: (i) the distal cavity (R32K and Y34N), (ii) the proximal cavity (N81S and S91G) and (iii) the hydrophobic region in proximity to I9L. While the effects of the latter are mitigated by the relatively small structural difference between isoleucine and leucine, the former two regions lead to differences that are much more significant. Specifically, both the N81S and S91G substitutions impact on the bonding character of the proximal histidine His89. Compared with DHP A, the Fe—NHis bond is elongated by 0.07 Å in DHP B. Based on the Fe—NHis vibrational frequency, previous resonance Raman spectroscopic studies have shown that DHP exhibits an imidazolate character that is weaker than in peroxidases but stronger than in globins (Belyea et al., 2006 ▶). In the monofunctional peroxidases (e.g. cytochrome c peroxidase and horseradish peroxidase), the strength of this bond has been implicated as a contributing factor in the ‘push–pull’ effect, which has been hypothesized to play an essential role in facilitating the requisite O—O bond-cleavage step needed to generate Compound I (or ES), the catalytically active species of the Poulos–Kraut mechanism (Poulos & Kraut, 1980 ▶). Generally, the Asp–His–Fe catalytic triad typical of peroxidases consists of a series of electronic interactions that results in a charge relay to the imidazole ring of the proximal histidine (Goodin & McRee, 1993 ▶; Poulos & Kraut, 1980 ▶). This localization of charge is responsible for an electronic ‘push’ of electron density into the O–O bond which aids its cleavage. Interestingly, DHP does not have an Asp–His–Fe catalytic triad but instead contains a Leu83–His89–Fe triad, in which there is a strong hydrogen-bond interaction between the Nδ atom of His89 and the backbone carbonyl of Leu83. This interaction is weak when compared with the Asp–His interaction in peroxidases, but has been shown to provide polarization of the proximal histidine that is stronger than in typical globins (Franzen, 2001 ▶). The Ser→Gly substitution at position 91 affords greater flexibility to His89 and may be one of the contributing factors behind the elongation of the Fe—NHis bond in DHP B compared with isoenzyme A. Similarly, the Asn→Ser substitution at position 81 alters the proximal hydrogen-bonding network, which includes Gln85, Leu83, the proximal histidine His89 and Gln88 as well as the heme propionate A and may either contribute to this bond elongation or potentially affect (de)protonation events during catalytic turnover. It has also been hypothesized that the lone pairs of the S atom of Met86, one of which is in contact with the proximal His89, could aid the ‘push’ effect through charge transfer (Belyea et al., 2006 ▶). The weakening of the Fe—NHis bond compared with typical monofunctional peroxidases may be related to the fact that DHP functions both as a hemoglobin and as a peroxidase. However, further studies of the proximal cavity, coupled with additional investigations on the distal residues involved in the ‘pull’ effect, will be necessary to deconvolute the factors that contribute to this observation.
The distal cavity substitutions R32K and Y34N comprise the other major region in which structural differences are observed between DHP B and DHP A. As Tyr34 has been implicated as one of the possible sites of radical formation in DHP A Compound ES (Feducia et al., 2009 ▶), its substitution by asparagine in DHP B rules out radical formation at this position in this case. Tyr34 also participates in a weak hydrogen bond between its hydroxyl group and the side chain of Asn96. This hydrogen bond is lost on the substitution of tyrosine by asparagine, but Asn34 forms a much stronger hydrogen bond to the carbonyl O atom of Glu31, as well as a couple of very weak ionic interactions with the Glu31 and Asn96 side chains. The Arg→Lys substitution at position 32 also introduces new hydrogen bonds, notably between the NH2 group of one of the Lys32 conformers and the backbone carbonyl O atom of Leu25. Interestingly, the findings from our previous EPR spectroscopic studies (Feducia et al., 2009 ▶; D’Antonio et al., 2010 ▶) of the radical(s) present in the Compound ES intermediates of DHP A and B may be related to substitution-induced hydrogen-bonding differences present at the sites of radical formation between these two isoenzymes. The EPR spectroscopic study (D’Antonio et al., 2010 ▶) of the Compound ES intermediate of DHP B at pH 7 showed that this signal was strikingly similar to that observed for DHP A at pH 5 (Feducia et al., 2009 ▶) but dissimilar to that observed at pH 7. Furthermore, we noted time-dependent changes in the DHP B Compound ES radical signal (but not in that of DHP A; Feducia et al., 2009 ▶; D’Antonio et al., 2010 ▶) and hypothesized a pathway-dependent radical migration out of the active site that was pH-specific. Thus, the structural evidence here supports the hypothesis that the DHP B amino-acid substitutions may affect the hydrogen-bonding networks that originate at these two critical positions in the distal pocket, which may play an important role in the formation, stabilization and/or migration of the Compound ES radical formed during catalytic turnover.
Although some chain heterogeneity was observed, the structure of the DHP AB heterodimer also demonstrates how the DHP A and B monomers may preferentially interact with each other. Examination of the chain interfaces for all three structures (DHP A, DHP B and DHP AB) reveals differences in the distances of residues involved in interfacial hydrogen bonding, specifically between residues Asp72 and Arg122, which may suggest a preference for a heterodimeric complex over a purely homodimeric one should the A and B monomers interact in solution. However, outside the interfacial regions the overall structures of the two chains in DHP AB closely resemble those of the homodimers of DHP A and DHP B for both main-chain and side-chain superposition. The one notable exception is that the hydrogen-bonding interaction between the N∊2 atom of one of the conformers of Gln88 and the heme propionate A differs between the two chains in DHP AB, with that in chain B being 2.6 Å whereas that in chain A is 3.5 Å. The consequence, if any, of this elongation of nearly 1 Å is not known at this time. One possibility, however, is that these interfacial interactions lead to the formation of A. ornata erythrocruorin, a giant hemoglobin found in the vascular system of the marine worm (Chiancone et al., 1980 ▶). Although speculation at this point, future study of both DHP A and DHP B in the presence of conditions known to stabilize erythrocruorin may lead to its formation and is currently under investigation (Chiancone et al., 1981 ▶).
4. Conclusion
We have been able to show that although dehaloperoxidase B is structurally very similar to DHP A there are distinct differences between the two that may manifest themselves in the different catalytic activities exhibited by these isoenzymes. Subtle alterations in the active-site hydrogen-bonding network as well as the substitution of a critical active-site tyrosine residue are likely to be the structural factors behind the spectroscopic differences noted for the radical in DHP B Compound ES when compared with that of isoenzyme A (D’Antonio et al., 2010 ▶) and suggest that ‘fine-tuning’ of the active site can lead to profound differences in the mechanism of action between these two isoenzymes. Furthermore, despite having been observed in other globins, the extent of the conformational flexibility of the distal histidine appears to be a hallmark of dehaloperoxidases and may function as a molecular trigger that serves to discriminate between peroxidase and hemoglobin function that is dependent upon the type of halophenol (monohalophenol versus trihalophenol) present. In light of the structural findings presented here, a firmer understanding of the structure–function relationship in dehaloperoxidase is rapidly emerging and may have potential implications for future studies involving globin protein engineering or directed evolution of DHP for bioremediation.
Supplementary Material
PDB reference: dehaloperoxidase B, 3ixf
Acknowledgments
We would like to thank the NCSU Molecular Biotechnology Training Program for an NIH T32 Biotechnology Traineeship grant (JD), the Army Research Office for grant 52278-LS (SF) and the North Carolina State University (RG) for their financial support of this research. We would also like to thank Dr Zhongmin Jin at SER-CAT (Advanced Photon Source) for synchrotron data collection. The Advanced Photon Source is supported by the US Department of Energy, Office of Science, Office of Basic Energy Sciences under Contract No. W-31-109-Eng-38. We acknowledge Dr Robert Rose (NC State University) for his insight into the interpretation of the structural data.
References
- Belyea, J., Belyea, C. M., Lappi, S. & Franzen, S. (2006). Biochemistry, 45, 14275–14284. [DOI] [PubMed]
- Belyea, J., Gilvey, L. B., Davis, M. F., Godek, M., Sit, T. L., Lommel, S. A. & Franzen, S. (2005). Biochemistry, 44, 15637–15644. [DOI] [PubMed]
- Chen, Y. P., Woodin, S. A., Lincoln, D. E. & Lovell, C. R. (1996). J. Biol. Chem.271, 4609–4612. [PubMed]
- Chen, Z., de Serrano, V., Betts, L. & Franzen, S. (2009). Acta Cryst. D65, 34–40. [DOI] [PubMed]
- Chiancone, E., Brenowitz, M., Ascoli, F., Bonaventura, C. & Bonaventura, J. (1980). Biochim. Biophys. Acta, 623, 146–162. [DOI] [PubMed]
- Chiancone, E., Ferruzzi, G., Bonaventura, C. & Bonaventura, J. (1981). Biochim. Biophys. Acta, 670, 84–92. [DOI] [PubMed]
- Collaborative Computational Project, Number 4 (1994). Acta Cryst. D50, 760–763.
- D’Antonio, J., D’Antonio, E. L., Bowden, E. F., Smirnova, T., Franzen, S. & Ghiladi, R. A. (2010). Submitted. [DOI] [PMC free article] [PubMed]
- Davis, M. F., Gracz, H., Vendeix, F. A., de Serrano, V., Somasundaram, A., Decatur, S. M. & Franzen, S. (2009). Biochemistry, 48, 2164–2172. [DOI] [PubMed]
- Emsley, P. & Cowtan, K. (2004). Acta Cryst. D60, 2126–2132. [DOI] [PubMed]
- Feducia, J., Dumarieh, R., Gilvey, L. B., Smirnova, T., Franzen, S. & Ghiladi, R. A. (2009). Biochemistry, 48, 995–1005. [DOI] [PubMed]
- Franzen, S. (2001). J. Am. Chem. Soc.123, 12578–12589. [DOI] [PubMed]
- Goodin, D. B. & McRee, D. E. (1993). Biochemistry, 32, 3313-3324. [PubMed]
- Han, K., Woodin, S. A., Lincoln, D. E., Fielman, K. T. & Ely, B. (2001). Mar. Biotechnol.3, 287–292. [DOI] [PubMed]
- Krissinel, E. & Henrick, K. (2007). J. Mol. Biol.372, 774–797. [DOI] [PubMed]
- LaCount, M. W., Zhang, E., Chen, Y. P., Han, K., Whitton, M. M., Lincoln, D. E., Woodin, S. A. & Lebioda, L. (2000). J. Biol. Chem.275, 18712–18716. [DOI] [PubMed]
- Laskowski, R. A., MacArthur, M. W., Moss, D. S. & Thornton, J. M. (1993). J. Appl. Cryst.26, 283–291.
- Lebioda, L., LaCount, M. W., Zhang, E., Chen, Y. P., Han, K., Whitton, M. M., Lincoln, D. E. & Woodin, S. A. (1999). Nature (London), 401, 445. [DOI] [PubMed]
- McCoy, A. J., Grosse-Kunstleve, R. W., Storoni, L. C. & Read, R. J. (2005). Acta Cryst. D61, 458–464. [DOI] [PubMed]
- Nienhaus, K., Nickel, E., Davis, M. F., Franzen, S. & Nienhaus, G. U. (2008). Biochemistry, 47, 12985–12994. [DOI] [PubMed]
- Osborne, R. L., Coggins, M. K., Raner, G. M., Walla, M. & Dawson, J. H. (2009). Biochemistry, 48, 4231–4238. [DOI] [PubMed]
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol.276, 307–326. [DOI] [PubMed]
- Phillips, G. N. (2001). Handbook of Metalloproteins, edited by A. Messerschmidt, R. Huber, T. L. Poulos & K. Weighardt, vol. 1, pp. 5–15. Chichester: John Wiley & Sons.
- Poulos, T. L. & Kraut, J. (1980). J. Biol. Chem.255, 8199–8205. [PubMed]
- Roach, M. P., Chen, Y. P., Woodin, S. A., Lincoln, D. E., Lovell, C. R. & Dawson, J. H. (1997). Biochemistry, 36, 2197–2202. [DOI] [PubMed]
- Sage, J. T., Morikis, D. & Champion, P. M. (1991). Biochemistry, 30, 1227–1237. [DOI] [PubMed]
- Serrano, V. de, Chen, Z., Davis, M. F. & Franzen, S. (2007). Acta Cryst. D63, 1094–1101. [DOI] [PubMed]
- Smirnova, T. I., Weber, R. T., Davis, M. F. & Franzen, S. (2008). J. Am. Chem. Soc.130, 2128–2129. [DOI] [PubMed]
- Tian, W. D., Sage, J. T. & Champion, P. M. (1993). J. Mol. Biol.233, 155–166. [DOI] [PubMed]
- Vangberg, T., Bocian, D. F. & Ghosh, A. (1997). J. Biol. Inorg. Chem.2, 526–530.
- Weber, R. E., Mangum, C., Steinman, H., Bonaventura, C., Sullivan, B. & Bonaventura, J. (1977). Comp. Biochem. Physiol. A Comp. Physiol.56, 179–187. [DOI] [PubMed]
- Zhang, E., Chen, Y. P., Roach, M. P., Lincoln, D. E., Lovell, C. R., Woodin, S. A., Dawson, J. H. & Lebioda, L. (1996). Acta Cryst. D52, 1191–1193. [DOI] [PubMed]
- Zhu, L., Sage, J. T., Rigos, A. A., Morikis, D. & Champion, P. M. (1992). J. Mol. Biol.224, 207–215. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: dehaloperoxidase B, 3ixf
PDB reference: dehaloperoxidase B, 3ixf