

Abstract
Background
Identifying developmental processes regulated by Notch1 can be addressed in part by characterizing mice with graded levels of Notch1 signaling strength. Here we examine development in embryos expressing various combinations of Notch1 mutant alleles. Mice homozygous for the hypomorphic Notch112f allele, which removes the single O-fucose glycan in epidermal growth factor-like repeat 12 (EGF12) of the Notch1 ligand binding domain (lbd), exhibit reduced growth after weaning and defective T cell development. Mice homozygous for the inactive Notch1lbd allele express Notch1 missing an ~20 kDa internal segment including the canonical Notch1 ligand binding domain, and die at embryonic day ~E9.5. The embryonic and vascular phenotypes of compound heterozygous Notch112f/lbd embryos were compared with Notch1+/12f, Notch112f/12f, and Notch1lbd/lbd embryos. Embryonic stem (ES) cells derived from these embryos were also examined in Notch signaling assays. While Notch1 signaling was stronger in Notch112f/lbd compound heterozygotes compared to Notch1lbd/lbd embryos and ES cells, Notch1 signaling was even stronger in embryos carrying Notch112f and a null Notch1 allele.
Results
Mouse embryos expressing the hypomorphic Notch112f allele, in combination with the inactive Notch1lbd allele which lacks the Notch1 ligand binding domain, died at ~E11.5-12.5. Notch112f/lbd ES cells signaled less well than Notch112f/12f ES cells but more strongly than Notch1lbd/lbd ES cells. However, vascular defects in Notch112f/lbd yolk sac were severe and similar to Notch1lbd/lbd yolk sac. By contrast, vascular disorganization was milder in Notch112f/lbd compared to Notch1lbd/lbd embryos. The expression of Notch1 target genes was low in Notch112f/lbd yolk sac and embryo head, whereas Vegf and Vegfr2 transcripts were increased. The severity of the compound heterozygous Notch112f/lbd yolk sac phenotype suggested that the allelic products may functionally interact. By contrast, compound heterozygotes with Notch112f in combination with a Notch1 null allele (Notch1tm1Con) were capable of surviving to birth.
Conclusions
Notch1 signaling in Notch112f/lbd compound heterozygous embryos is more defective than in compound heterozygotes expressing a hypomorphic Notch112f allele and a Notch1 null allele. The data suggest that the gene products Notch1lbd and Notch112f interact to reduce the activity of Notch112f.
Background
Notch transmembrane receptors are important regulators of cell fate determination in numerous cell types [1-3]. Notch receptors in Drosophila and mammals are covalently modified with O-fucose on many epidermal growth factor-like (EGF) repeats of the extracellular domain [4]. An important O-fucose site resides in epidermal growth factor-like repeat 12 (EGF12) which, together with EGF11, is required for canonical Notch ligand binding to Drosophila Notch [5-7] and to mammalian Notch1 [8]. A point mutation that precludes the addition of fucose to EGF12 in Drosophila Notch results in enhanced binding of both Delta and Serrate Notch ligands, and a hyperactive Notch that is refractory to Fringe [9]. However, the same mutation (Notch112f) in cultured mammalian cells gives markedly reduced signaling in a Notch reporter signaling assay [10,11], predicting a Notch1 null phenotype in vivo. Surprisingly however, homozygous Notch112f/12f mice are viable and fertile, but exhibit retarded growth and mild defects in T cell development in the thymus [12], consistent with weak Notch1 signaling. Notch1+/12f heterozygotes are indistinguishable from wild type in terms of growth and T cell development. However, compound heterozygotes carrying Notch112f and the inactive Notch1lbd allele, which lacks the ligand binding domain and generates an inactive ~280 kDa Notch1 receptor at the cell surface, are not born [12]. Therefore Notch112f is a hypomorphic allele in mammals and the O-fucose glycan in the ligand binding domain is required for optimal Notch1 signaling. Homozygous Notch1lbd/lbd embryos die at ~E9.5 [8,12] with an indistinguishable phenotype compared to Notch1 null embryos (Notch1in32/in32 and Notch1tm1Con/tm1Con) described by others [13,14]. Heterozygous Notch1+/lbd and Notch1+/tm1Con mice are viable and fertile whereas Notch112f/lbd compound heterozygotes die between E11.5 and E12.5, significantly later than either Notch1lbd/lbd [12] or Notch1 null embryos [13,14] that do not express Notch1 [15-17].
The availability of these Notch1 mutant alleles suggested a genetic approach to determining effects of varying Notch1 signaling strength. The Notch1lbd mutation generates a non-functional but cell surface-expressed Notch1 that cannot signal [8,12]. Notch1tm1Con lacks Notch1 on the cell surface due to the absence of its transmembrane domain [14]. Notch1in32 homozygous embryos have no Notch1 transcripts [13] and an indistinguishable phenotype from Notch1tm1Con homozygotes which lack Notch1 based on western analyses [15,18]. Notch1+/- heterozygotes carrying either of the Notch1 null alleles exhibit Notch1 signaling defects in certain cell types, an effect attributed to Notch1 haploinsufficiency rather than to a dominant negative effect in Notch1tm1Con [18-21]. In this paper we compare embryogenesis and vasculogenesis in compound heterozygotes expressing the hypomorphic Notch112f allele with either the inactive Notch1lbd allele [8,12] or the Notch1tm1Con null allele [14].
Results
Notch signaling in Notch112f/lbd compound heterozygous ES cells
The Notch112f and Notchlbd alleles investigated here are diagrammed in Fig. 1A and 1B and their identification by PCR genotyping is shown in Fig. 1C. Previous studies showed that Notch112f/lbd compound heterozygotes die by ~E12.5 [12]. To examine Notch ligand binding and the strength of Notch signaling in more detail, ES cells were derived from Notch112f/lbd compound heterozygous blastocysts and compared to ES cells derived from Notch112f/12f and Notch1lbd/lbd homozygous blastocysts and wild type ES cells (Fig. 2). All cell lines bound the anti-Notch1 extracellular domain mAb 8G10 equivalently, and therefore expressed the various Notch1 molecules similarly at the cell surface (Fig. 2A). Each mutant line exhibited a decrease in the low level of soluble Delta1 binding observed with wild type ES cells (Fig. 2B). Binding of Delta1 is not reduced to zero even in Notch1 null ES cells because of the presence of Notch2, Notch3 and Notch4 [17]. Notch signaling was analysed in co-culture assays with L cells or L cells expressing full length Delta1 or Jagged1 ligand. This reporter assay revealed a graded reduction in Notch signaling with Notch112f/12f >Notch112f/lbd >Notch1lbd/lbd ES cells (Fig. 2C-D). This graded response was also observed by western analysis using Notch1 antibody Val1744 [15] which detects the ~110 kDa Notch1 fragment generated by γ-secretase complex cleavage of Notch1. The level of activated Notch1 in Notch112f/lbd ES cells was less than in Notch112f/12f ES cells, which was lower than in control ES cells, while Notch1lbd/lbd ES cells had undetectable levels of activated Notch1 (Fig. 2E). Nevertheless, all ES cell populations, including Notch1lbd/lbd ES cells, expressed equivalent levels of full-length Notch1 (Fig. 2E). Taken together, these data indicate that Notch112f and Notch1lbd expression and transit to the cell surface were similar to wild type Notch1, but Notch1 signaling was reduced in mutant cells: Notch112f signaling was sightly less than wild type; signaling from the combination of Notch112f and Notch1lbd was further reduced, and signaling by Notch1lbd alone was essentially absent. Previous experiments have shown that Notch1lbd/lbd and Notch1in32/in32 ES cells which lack Notch1 [13,15,16], are equally deficient in Delta1-Fc binding and Notch1 signaling [12].
Figure 1.
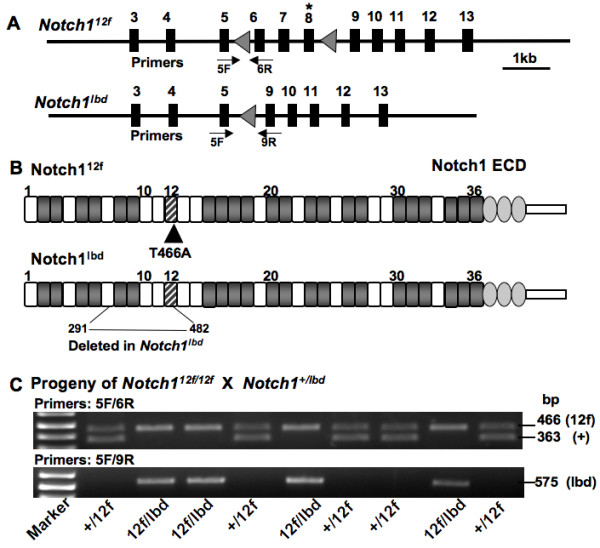
Generation of Notch112f/lbd embryos. (A) Diagram of the Notch112f and Notch1lbd alleles. (B) Diagram of mouse Notch1 EGF repeats in Notch112f and Notch1lbd extracellular domains. The EGF repeats with putative O-fucosylation sites are shaded in gray and the mutation in EGF12 is shown. (C) Genotyping by PCR from E9.5 yolk sac DNA of a litter from a Notch112f/12f × Notch1+/lbd cross. Primers 5F and 6R detect the Notch112f and Notch1+ alleles, primers 5F and 9R detect the Notch1lbd allele.
Figure 2.
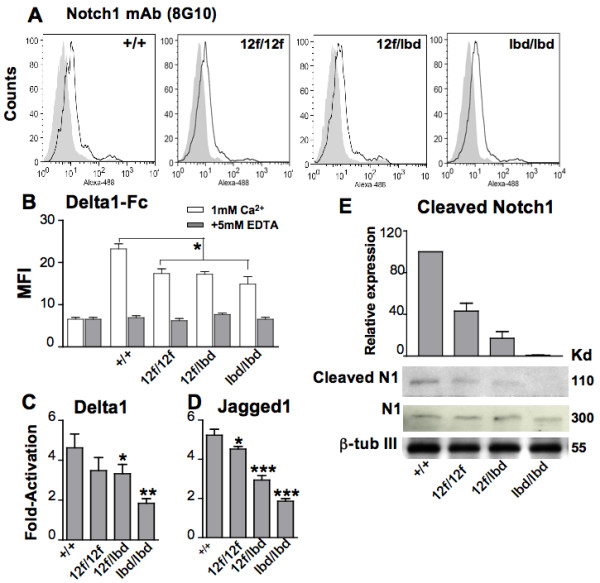
A graded reduction in Notch1 signaling in Notch112f/lbd ES cells. (A) Notch1 expression on the surface of ES cells (Notch1+/+, Notch112f/12f, Notch112f/lbd and Notch1lbd/lbd) was analyzed by flow cytometry using anti-Notch1 mAb 8G10 (solid line). Shaded profiles are secondary Ab only. (B) Delta1-Fc binding to ES cells. Control is secondary antibody alone. 5 mM EDTA inhibited ligand binding to control levels (gray). Data are mean ± SEM (n = 4), * p < 0.05 between Notch1+/+ and all mutant lines. (C) Delta1-induced Notch signaling and (D) Jagged1-induced Notch1 signaling were determined by co-culturing ES cells with Delta1/L or Jagged1/L cells compared to control L cells after transfection of a Notch reporter construct. Bars represent fold-activation ± SEM (n = 4), * p < 0.05; ** p < 0.01, *** p < 0.001 based on the two-tailed Student's t test; (E) Whole cell lysates from ES cells were subjected to western analysis using the Val1744 antibody for activated Notch1 and the 8G10 antibody for full length Notch1. The histogram shows the relative expression of activated Notch1 after normalization to β-tubulin III (mean ± SEM from 4 experiments).
Embryogenesis in Notch112f/lbd compound heterozygous embryos
Embryonic development was compared between Notch112f/12f, Notch112f/lbd and Notch1lbd/lbd embryos. At E9.5 Notch112f/lbd embryos formed 17-21 somites compared to 23-26 somites in Notch112f/12f embryos, the same as Notch1+/+ embryos, and 13-17 somites in Notch1lbd/lbd embryos [8], the same as Notch1tm1Con null embryos [14] (Table 1). Compared to Notch112f/12f and Notch1+/12f embryos, Notch112f/lbd embryos also showed severely defective vasculogenesis in yolk sac at E9.5, similar to Notch1lbd/lbd yolk sac. By contrast, Notch112f/lbd embryos at E9.5 and E10.5 exhibited milder defects in development than Notch1lbd/lbd embryos [12] (Fig. 3), although the ballooning of the pericardial sac and defective heart development were severe, and similar to mutants globally defective in Notch signaling such as mutants lacking Pofut1 [22], RBPJk [23] or presenilins 1 and 2 [24]. Taken together, these data indicate that two copies of Notch112f do not noticeably affect mouse embryogenesis at a gross level, whereas a single copy of Notch112f with Notch1lbd support embryonic development ~2.0-2.5 days longer than embryos with two copies of Notch1lbd.
Table 1.
Somite Numbers in Notch1 Mutants
| Genotype | Stage | No. Embryos | No. Somites |
|---|---|---|---|
| +/12f or +/+ | E9.5 | 8 | 23,23,24,24,24,25,25,26 |
| 12f/12f | E9.5 | 4 | 23,24,25,26 |
| 12f/lbd | E9.5 | 6 | 17,17,18,18,19,21 |
| 12f/tm1Con | E9.5 | 7 | 18,19,21,21,23,24,26 |
| lbd/lbd | E9.5 | 6 | 13,14,14,16,17,17, |
| tm1Con/tm1Con | E9.5 | * | ≤ 14 |
| +/12f or +/+ | E10.5 | 3 | 33,34,35 |
| 12f/12f | E10.5 | 3 | 32,32,34 |
| 12f/lbd | E10.5 | 5 | 18,18,21,22,23 |
Somites were counted in embryos of Notch1+/+, Notch1+/12f, Notch112f/12f, Notch112f/lbd, Notch112f/tm1Con and Notch1lbd/lbd at E9.5. *Somite numbers in Notch1tm1Con/tm1Con embryos are from Conlon et al. [14].
Figure 3.
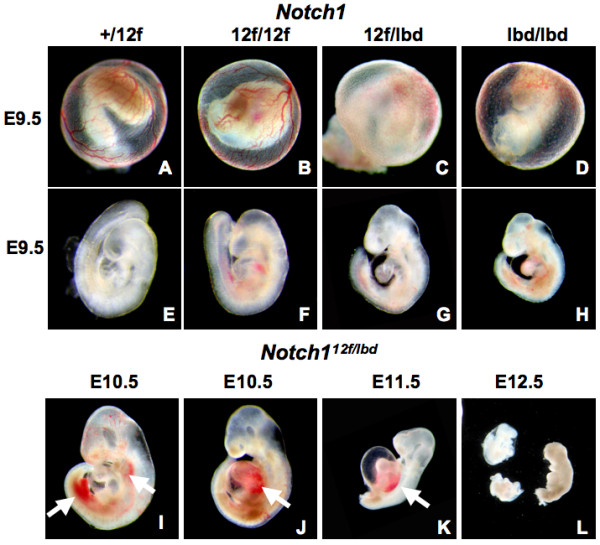
Embryogenesis in Notch112f/lbd embryos. (A-D) Vascularization of yolk sac in Notch1+/12f, Notch112f/12f, Notch112f/lbd and Notchllbd/lbd embryos at E9.5. Large vitelline blood vessels were present in Notch1+/12f and Notch112f/12f yolk sacs, but absent in the Notch112f/lbd and Notch1lbd/lbd mutants. (E-H) Morphology of embryos at E9.5. Notch112f/12f are similiar to Notch1+/12f, Notch112f/lbd are markedly underdeveloped, and Notch1lbd/lbd are severely underdeveloped. (I-L) Notch112f/lbd embryos from E10.5-E12.5. White arrows show hemorrhaging in E10.5 and E11.5 embryos; most E12.5 embryos were resorbing. The number of embryos examined at each stage is given in Table 1.
Vasculogenesis in yolk sac appears to require stronger Notch1 signaling than in the embryo
Notch1 signaling is critical for vasculogenesis during mouse embryogenesis [25]. Loss of Notch1 in embryos [26] or in endothelial cells [27] causes embryonic lethality with severe vascularization defects in yolk sac, placenta and embryo. Blood that had leaked from the heart and blood vessels was apparent in Notch112f/lbd embryos (Fig. 3I-K; arrows). Vascular organization in embryos was examined by staining with anti-Pecam1 (endothelial marker platelet/endothelial cell adhesion molecule-1). Notch112f/12f embryos (Fig. 4B, F, J, N) did not exhibit any apparent defects in brain, heart or intersomitic vascularization compared to Notch1+/12f embryos. Notch112f/lbd embryos exhibited somewhat disorganized vascularization in embryos, especially in the main trunk of the anterior cardinal vein, the vascular network of the head and heart, and in intersomitic vessels (Fig. 4C, G, K, O). Notch1lbd/lbd embryos exhibited severe defects in vascularization (Fig. 4D, H, L, P). Therefore, the extensive vascularization in E9.5 and older Notch112f/lbd embryos appears to be well supported by the level of Notch1 signaling provided by the Notch112f allele. Considering that the vascular defects in yolk sac of compound heterozygous Notch112f/lbd and homozygous Notch1lbd/lbd embryos were similarly severe, the comparatively milder defects in Notch112f/lbd embryos indicated that Notch1 signaling from a single copy of Notch112f, while not sufficient to support vascularization in yolk sac at E9.5, is able to support a high level of vascularization in E9.5 embryos. It seems that vascularization in yolk sac requires stronger Notch1 signaling than in the embryo.
Figure 4.
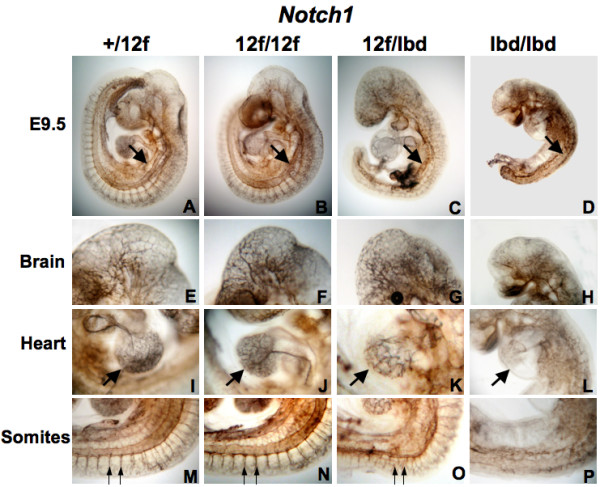
Defects in vascular remodeling in Notch112f/lbd E9.5 embryos. All whole mount embryos were stained with Ab to Pecam1. (A-D) Morphogenesis of the main trunk of the anterior cardinal vein (arrow) in Notch112f/lbd and Notch1lbd/lbd mutant embryos is defective compared to Notch112f/12f and control Notch1+/12f embryos. (E-H) Vascular remodeling in brain in Notch1+/12f and Notch112f/12f is similar but is defective in Notch112f/lbd and severely defective in Notch1lbd/lbd embryos. (I-L) Vascular remodeling in heart is defective in Notch112f/lbd and more severely affected in Notch1lbd/lbd embryos. (M-P) Intersomitic vessels (arrows) were well-organized in Notch1+/12f and Notch112f/12f embryos but were mildly disorganized in Notch112f/lbd and essentially absent from Notch1lbd/lbd embryos. The number of embryos examined was 3 - 4 of each genotype.
Notch1 target gene expression in E9.5 yolk sac versus embryo
Whereas vascularization was severly affected in both yolk sac and embryo of Notch1lbd/lbd embryos, only the yolk sac of Notch112f/lbd compound heterozygous embryos exhibited extremely defective vascularization. To investigate further, the expression of vasculogensis-related and Notch1 target genes was examined by real-time PCR using total RNA isolated from E10.5 Notch112f/lbd and Notch1+/12f yolk sacs and embryo heads. The relative expression levels of Pecam1 and Vegf were increased in Notch112f/lbd yolk sacs and embryos, and Vegfr2 expression was increased in Notch112f/lbd embryo heads (Fig. 5A-C). Therefore loss of Notch1 signaling upregulated transcription of the Pecam1, Vegf and Vegfr2 genes. Interestingly, the increased expression of Vegf and Vegfr2 was greater in Notch112f/lbd embryos, consistent with the relative strength of Notch1 signaling being greater in yolk sac. Expression of the Notch1 target genes Hes5, Hey1 and Hey2 was reduced in Notch112f/lbd yolk sac (Fig. 5D-F), but the level of Hes1 transcripts was not changed (data not shown). In embryos, only the expression of Hes5 was significantly reduced compared to control. The expression of Ang1, Tie2 and Ephrin-B2 which are involved in angiogenesis, as well the expression of Notch1 itself, were not changed in Notch112f/lbd yolk sac or embryos (data not shown). The fact that the increase in Vegf and Vegfr2 transcripts was more in embryo head than yolk sac (418% vs 170% for Vegf; 227% vs. 148% for Vegfr2; Fig. 5B and 6C), and the fact that the reduction in Notch target gene expression was greater in yolk sac than embryo head, correlated generally with Notch1 signal strength and the greater severity of vascularization defects in yolk sac versus embryo head.
Figure 5.
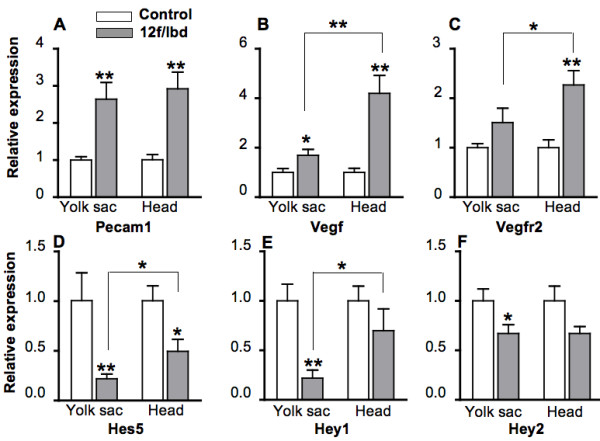
Real-time PCR of vasculogenic and Notch target genes in Notch112f/lbd yolk sac and embryo. Total RNA extracted from E10.5 yolk sac or embryonic head was reverse-transcibed and subjected to real-time PCR. Numbers of transcripts were normalized to β-actin, and the average relative expression of Notch1+/12f samples was set to 1. (A-F) Relative expression of Pecam1, Vegf, Vegfr2, Hes5, Hey1, and Hey2 as marked. Bars represent SEM (n = 6). The two-tailed Student's t test was used in control versus mutant yolk sac and embryo head comparisons; a one-tailed Student's t test was used in mutant yolk sac versus mutant embryo head comparisons; * p < 0.05; ** p < 0.01
Figure 6.
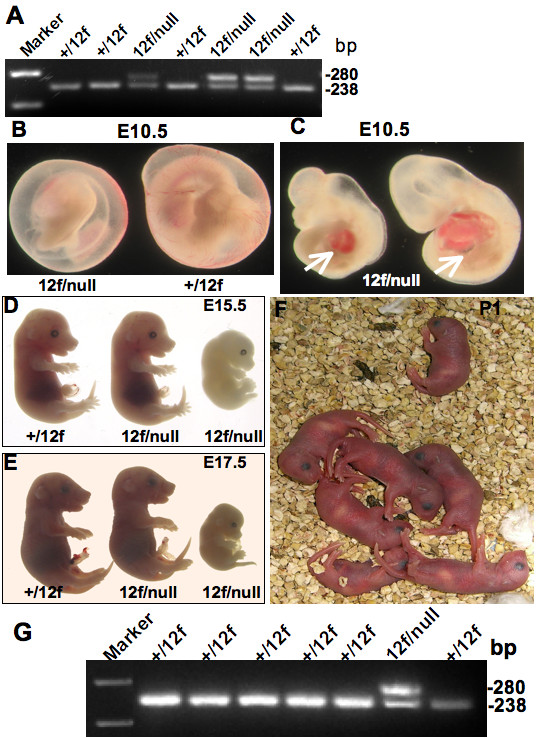
Notch112f/tm1Con embryos survive longer than Notch112f/lbd embryos. (A) PCR genotype of an E9.5 litter showed the 280 bp PCR product from Notch1tm1Con allele and the 238 bp product from the Notch112f allele. (B) Yolk sac vascularization of E10.5 Notch112f/tm1Con and Notch1+/12f embryos. (C) Notch112f/tm1Con embryos at E10.5 exhibit heamorrhaging around the heart (arrows). (D) Notch112f//tm1Con and control embryos at E15.5. One Notch112f/tm1Con embryo was defective but the other had no obvious defects. (E) Notch112f/tm1Con and control embryos at E17.5. One Notch112f/tm1Con embryo was defective but the other had no obvious defects. (F) Photo of a litter on postnatal day 1 (P1) which included one pup identified as Notch112f/tm1Con by PCR genotyping below. The pup was indistingishable but died within a few days. (G) PCR genotype of the P1 litter in panel F.
Notch112f may function to birth in the absence of Notch1lbd
The severity of the Notch112f/lbd phenotype suggested an interaction between Notch112f and Notch1lbd that interfered with signaling by Notch112f. In this case, compound heterozygous embryos expressing a Notch112f allele and a Notch1 null allele might be expected to have a milder phenotype than Notch112f/lbd embryos. Notch112f/12f mice were crossed with Notch1+/tm1Con heterozygotes and embryos were examined at E9.5 and later (Fig. 6, Table 2). Some Notch112f/tm1Con embryos died between E11.5 and E12.5 with similar defects to Notch112f/lbd embryos. However, this is ~1.5 days later than observed with Notch1tm1Con/tm1Con homozygous embryos who were mostly dead by E10 [14]. However, nearly one third of the Notch112f/tm1Con embryos developed beyond E12.5 and died at various times during embryogenesis, including after birth (Table 2). Two Notch112f/tm1Con pups were found after birth, but none were observed after postnatal day 7 (Fig. 6, Table 2). Somite numbers in Notch112f/tm1Con embryos varied from as low as Notch112f/lbd embryos to as high as wild type embryos (Table 1). Taken together, these results indicate that Notch112f receptors present at a 50% dose in vivo, generate stronger Notch1 signaling than Notch112f in combination with Notch1lbd. This provides genetic evidence that Notch112f and Notch1lbd may functionally interact.
Table 2.
Notch112f/tm1Con pups may survive to birth
| Stage | 12f/12f × +/tm1Con | 12f/12f × +/lbd | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Litters | Pups | +/12f | 12f/tm1Con | Litters | Pups | +/12f | 12f/lbd | ||||
| E9.5 | 3 | 23 | 11 | 12 | 5 | 38 | 18 | 20 | |||
| E10.5 | 3 | 23 | 13 | 10 | 8 | 70 | 37 | 33 | |||
| E11.5 | 4 | 24 | 16 | 8 | 7 | 48 | 28 | 20 | |||
| E12.5 | 5 | 25 | 21 | 4 | 8 | 33 | 28 | 5 | |||
| E13.5 | 5 | 27 | 21 | 6 | 5 | 23 | 23 | 0 | |||
| E15.5 | 3 | 13 | 9 | 4 | - | - | - | - | |||
| E17.5 | 5 | 19 | 17 | 2 | - | - | - | - | |||
| P1 | 6 | 25 | 23 | 2 | - | - | - | - | |||
| Wean | 13 | 50 | 50 | 0 | 8 | 30 | 30 | 0 | |||
Embryos (E9.5 -- E17.5), post-natal pups (P1) and mice after weaning were genotyped from crosses of Notch112f/12f female × Notch1+/tm1Con male or crosses of Notch112f/12f female × Notch1+/lbd male. The data from Notch1+/12f × Notch112f/lbd crosses were previously published [12] but are included for ease of comparison.
Discussion and Conclusions
In this paper we show that Notch1 signaling is greatly reduced in Notch112f/lbd ES cells and compound heterozygous embryos, but is significantly greater than in Notch1lbd/lbd ES cells or homozygous embryos. The presence of the hypomorphic Notch112f allele allows vasculogenesis to proceed further and embryos to survive ~1.5-2 days longer. The vascular system develops early during mammalian embryogenesis. Initially, endothelial cell precursors differentiate and coalesce into a primitive network of undifferentiated blood vessels (the primary vascular plexus) in both the mammalian embryo and its extraembryonic membrane the yolk sac, in a process termed vasculogenesis [28]. Subsequently, the primary vascular plexus is remodeled into a highly organized and functionally competent vascular network in a process termed angiogenesis [29,30]. These processes are controlled by several signaling molecules, including vascular endothelial growth factor (VEGF) and its receptors [31], angiopoeitin 1 and its receptor Tie2 [32], Ephrin-B ligands and EphB receptors [33], TGFβ and its receptors [34], and Notch receptors and their ligands Delta and Jagged [25,26,35-38]. Defects in vasculogenesis are one of the major reasons that Notch1 null embryos die at mid-gestation [13,26]. Conditional mutation of Notch1 in vascular endothelial cells using the Tie2-Cre transgene showed that embryos lacking endothelial cell Notch1 die at ~E10.5 with profound vascular defects in placenta, yolk sac, and the embryo proper [27]. The Notch1 target genes Hey1 and Hey2 are also essential for embryonic vascular development [39]. A requirement for Notch signaling in the maintenance of vascular homestasis and the repression of endothelial cell proliferation is also indicated in adult mice by conditional deletion of RBP-Jκ in endothelial cells [40].
Interestingly, Notch112f/lbd embryos allowed us to observe that vasculogenesis is regulated to different extents in yolk sac and embryo by Notch1 signaling. Thus, vascular defects in Notch112f/lbd yolk sac were as severe as in Notch1lbd/lbd yolk sac, but vascular defects in Notch112f/lbd embryo heads were comparatively mild. The vasculogenic phenotype of Notch112f/lbd embryos was also milder than reported for Jagged1 or Notch1 or Notch1/4 null embryos [13,26,37], reflecting the presence of a low level of Notch1 signaling in Notch112f/lbd compound heterozygotes. The reduced strength of Notch1 signaling was responsible for defective artery development in Delta-like 4 (Dll4) heterozygous embryos [38]. Hes5 and Hey1 are Notch1 target genes, and Notch1 downregulates expression of Hesr-1/Hey1 thereby enhancing expression of its target gene Vegfr2 in endothial cells [41]. In addition, Vegf is upregulated six-fold in Hey1/2 double knock-out embryos [39]. Notch1 has also been proposed to regulate vasculogenesis and angiogenesis via induction of Ephrin-B2 [42,43] and Ang1 [44,45], and suppression of Vegfr-2/Kdr [41,46]. Consistent with this, we observed enhanced suppression of Vegfr2 and Vegf in Notch112f/lbd yolk sac and embryo. However, we observed no change in the expression of Ang1, Tie2, Ephrin-B2 or Notch1 itself, although experiments in human endothelial cells indicate that Ang1 and Tie2 are Notch1 target genes [44,45]. Ephrin-B2 was reported to respond to Notch4, but not to Notch1, through Delta-like 4 in differentiating HUVEC cells [43], so it was perhaps not surprising that Ephrin-B2 expression was unchanged in Notch112f/lbd yolk sac or embryo. Thus, decreased Notch1 signaling may inhibit vascular development in yolk sac more than in embryos by inducing more Vegf and Vegfr2 through generating less Hes5 and Hey1 mRNA in yolk sac.
The prolonged embryonic development supported by the hypomorphic Notch112f allele was only ~1.5-2 days for Notch112f/lbd embryos compared to Notch1lbd/lbd [8,12], or Notch1 null embryos [13,14]. By contrast Notch112f/12f, Notch1+/lbd or Notch1+/- heterozygotes are viable and fertile [12-14,20]. This suggests that Notch1lbd may interfere with Notch112f in a process termed negative complementation for Abruptex Notch mutants in Drosophila [47,48]. The basis of negative complementation is most commonly attributed to the products of the mutant alleles interacting physically [47]. Thus Notch1lbd may either be dominant negative and inhibit Notch112f activity, or may not form a functional dimer or higher oligomer with Notch112f, if that is required for Notch1 to function. We prefer the latter hypothesis because there is no evidence to date that Notch1lbd behaves as a dominant negative in Notch1+/lbd heterozygotes [8,12]. Unfortunately, attempts to prove the existence of dimers or higher oligomers of Notch1 expressed at endogenous levels have so far been unsuccessful and previous attempts came to opposite conclusions. While two groups found that overexpressed Notch1 transfected into cultured cells may form dimers through the transmembrane domain or the extracellular domain EGF repeats, one group concluded that dimerization is necessary for Notch1 to signal [49], while the other concluded that Notch1 signals without the need for dimerization, and is present mainly as a monomer on the cell surface [50]. Both studies characterized transiently-transfected Notch1 expressed at much higher levels than endogenous Notch1, which might induce anomolous interactions.
If Notch1lbd reduces the effective amount of Notch112f to a level insufficient to sustain development, we reasoned that Notch112f expressed in the context of a Notch1 null background may function better. In fact, we found that a significant proportion of Notch112f/tm1Con embryos survived beyond E12.5 and that some survived to birth. On the other hand, some compound heterozygous Notch112f/tm1Con embryos died at ~E11.5 with similar defects to Notch112f/lbd. This indicates that Notch112f at a dose of 50% functions at a threshold of Notch1 signaling strength that variably sustains embryogenesis through to birth - a stochastic effect or perhaps a genetic background effect, since Notch1+/12f and Notch1+/lbd mice were not extensively backcrossed to C57Bl/6. Nevertheless, the Notch1 signal strength generated by a single copy of Notch112f was intermediate between Notch112f/12f and Notch112f/lbd, revealing the importance of maintaining a certain level of Notch1 signaling for mouse embryogenesis to proceed. Fig. 7 summarizes these findings in a diagram which describes a mini-allelic series of available Notch1 mutants. It includes the Notch1 processing point mutant Val1744Gly (Notch1v!g/v!g) which has a phenotype very similar to, but slightly less penetrant than, a Notch1 null [15]. It also includes Notch1+/null heterozygotes that have mild Notch1 signaling defects uncovered in competition assays [19] or by close examination of specific cell types [18,20,21]. Haploinsufficiency of NOTCH1 is the basis of aortic valve disease in humans [51]. We predict that Notch1+/lbd and Notch1+/12f heterozygotes have slightly less Notch1 signaling than Notch1+/tm1Con and should display evidence of more extensive Notch1 signaling defects in particular cell types. The range of Notch1 mutant alleles available in the mouse should be helpful in identifying new in vivo functions for Notch1.
Figure 7.
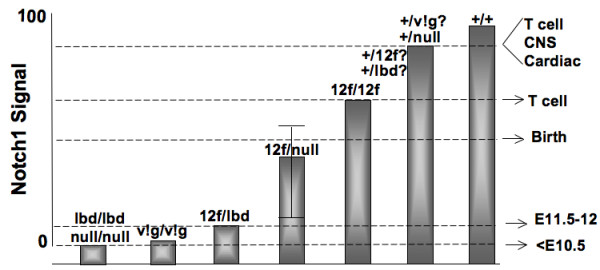
An allelic series of Notch1 mutants. Based on data reported herein and from the literature, the relative signaling strength of Notch1 mutant alleles in various combinations with wild type or other Notch1 mutant alleles is represented as discussed in the Discussion. The consequences with respect to time of death of embryos with severe Notch1 signaling defects, or more subtle defects in T cell, CNS or cardiac development are noted.
Methods
Mice
Mice carrying Notch1 lacking the O-fucose site in EGF12 (Notch112f) and mice carrying Notch1 lacking the ligand binding domain (Notch1lbd) were generated by gene targeting as previously described [8,12]. They were backcrossed 6-7 generations to C57/Bl6 mice before being used in these experiments. Notch1l2f/lbd embryos were obtained by crossing Notch112f/12f and Notch1+/lbd mice. Embryos were collected from E9.5 and yolk sac DNA was genotyped by PCR using primers 5F: GTATGTATATGGGACTTGTAGGCAG and 6R: CTATGAGGGGTCACAGGACCAT, that give a 466 bp product for the Notch112f allele and a 363 bp product for the wild type Notch1 allele; and primers 5F and 9R: CTTCATAACCTGTGGACGGGAG that give a 575 bp product for the Notch1lbd allele. The Notch1 null allele (Notch1tm1Con) encoding Notch1 lacking the transmembrane domain [14] backcrossed extensively to C57Bl/6 was kindly provided by Cynthia Guidos, University of Toronto. Notch112f/tm1Con embryos were obtained by crossing Notch112f/12f and Notch1+/tm1Con mice and genotyped by PCR using primers neo-F: CTTGGGTGGAGAGGCTATTC and neo-R: AGGTGAGATGACAGGAGATC for the Notch1tm1Con allele and primers loxF: GGCGAGCTCGAATTGATCC and 9R for Notch112f allele. Mice were housed under conventional barrier protection in accordance with Einstein and NIH guidelines. Protocols were approved by the Albert Einstein Animal Institute Committee.
Embryonic stem cell isolation
ES cells were isolated from E3.5 blastocysts as described [52], and genomic DNA was genotyped by PCR as described above. ES cells were routinely cultured on an SNL2 γ-irradiated feeder layer [53] in DMEM supplemented with 15% fetal bovine serum (Gemini, West Sacramento, CA), non-essential amino acids, L-glutamine, 1000 U ESGRO® (Chemicon, Temecula, CA), 1% β-mercaptoethanol, 25 mM HEPES, penicillin (50 U/ml) and streptomycin (50 μg/ml). All reagents were from SpecialtyMedia, Lavellette, NJ. Before use in experiments, ES cells were passaged on gelatinized plates for 2-3 generations to remove feeder cells.
Western blot analysis
ES cells cultured on gelatinized plates were lysed in RIPA buffer (Upstate, Lake Placid, NY) containing complete protease inhibitor cocktail (Roche, Basel, Switzerland) for 30 min on ice and debris was removed by low speed centrifugation. Lysates were resolved by SDS-PAGE, transferred to polyvinyldifluoride (PVDF) membrane and probed with 8G10 anti-Notch1 mAb (Upstate, 57-557, 1:500, Lake Placid, NY) for full-length Notch1 or Val1744 Notch1 antibody (Cell Signaling Technology, Val1744, 1:1000, Beverly, MA) for cleaved, activated Notch1, followed by horseradish peroxidase(HRP)-conjugated secondary antibodies. Reactive bands were visualized with Enhanced Chemiluminescence Reagent (Amersham Pharmacia Biotech, Piscataway, NJ). β-tubulin-III specific antibody T8660 (Sigma Chemical Co., St. Louis, MO) was used as a loading control.
Flow cytometry
For cell surface Notch1 expression, 70-80% confluent ES cells were dissociated from plates using phosphate-buffered saline (PBS)-based enzyme-free dissociation solution (SpecialtyMedia, Lavellette, NJ) for 10 min at 37°C. After washing, ES cells (5 × 105) were incubated with 0.5 μg 8G10 anti-Notch1 antibody in Hank's balanced salt solution containing 3% bovine serum albumin Fraction V (Sigma Chemical Co., St. Louis, MO), 1 mM CaCl2 and 0.05% Na azide (HBSS/BSA) for 1 h at 4°C, washed and incubated in Alexa-488 conjugated anti-Hamster IgG (1:100) in HBSS/BSA in the dark (Invitrogen, Carlsbad, CA) for 30 min at 4°C. Immunofluorescence was analyzed on a FACSCalibur flow cytometer (BD Biosciences, San Diego, CA), gating on live cells determined by 7-AAD staining. Data were analyzed using Flowjo software (Tree Star, San Carlos, CA).
Notch co-culture signaling assay
Notch signaling assays were performed in duplicate as previously described [54,55]. ES cells were plated at 2 × 105 cells per well of a six-well plate in ES medium, and co-transfected the next day with 0.2 μg of TP1-luciferase Notch reporter plasmid and 0.05 μg of Renilla luciferase reporter (pRL-TK; Promega, Madison, WI) along with 1.8 μg empty vector alone using FuGene 6 (Roche, Basel, Switzerland). At 16 h post-transfection, ES cells were overlaid with 1 × 106 rat Jagged1-expressing L cells (Jagged1/L), Delta1-expressing L cells (Delta1/L) or parental L cells [56]. At 48 h after transfection, firefly and Renilla luciferase activities were quantitated in cell lysates using a dual luciferase assay (Promega, Madison, WI). Ligand-dependent Notch activation was expressed as relative fold-activation of normalized luciferase activity stimulated by ligand/L cells compared to L cells.
Notch ligand binding assay
Soluble Notch ligand Delta1 with human Fc tag [57,58] was prepared form HEK-293T cells expressing Delta1-Fc [17] cultured in α-MEM containing 10% FBS until 70~80% confluence. The medium was changed to 293 SFM II serum-free medium (Invitrogen) and conditioned medium was collected after 3 days. Cellular debris was removed by low-speed centrifugation, the supernatant was filtered and stored at 4°C. Soluble ligand concentration was determined by western blotting using HRP-conjugated anti-human IgG antibody (Jackson Immunoresearch, West Grove, PA). For the binding assay, ES cells on plates were dissociated using PBS-based Enzyme-free dissociation medium for 10 min at 37°C, and the single cell suspension of ES was incubated with 2 μg/ml Delta1-Fc in HBSS/BSA for 1 h at 4°C, followed by incubation with 1:100 phycoerythrin (PE)-conjugated anti-human Fc antibody (Jackson Immunoresearch, West Grove, PA) for 30 min at 4°C. After washing, live cells determined by gating on the 7-AAD negative population were analyzed on a FACS Calibur flow cytometer (BD Biosciences, San Jose, CA). Ligand binding ability was measured as mean fluorescence intensity (MFI) using Flowjo software (Tree Star, San Carlos, CA).
Whole mount immunohistochemistry
Embryos were collected on E9.5 and DNA from yolk sac was genotyped by PCR. Embryos were fixed in 4% paraformaldehyde (PFA) in PBS overnight at 4°C, dehydrated through a methanol series, and bleached in 5% H2O2/methanol for 5 h. Embryos were rehydrated and placed in PBSMT (PBS containing 3% nonfat milk, 0.1% Triton X-100). After 2 h, embryos were incubated with anti-mouse Pecam1 (1:200; BD Biosciences, San Jose, CA) in PBSMT overnight at 4°C. After 5 washes with PBSMT embryos were incubated in a 1:200 dilution of HRP-conjugated secondary antibody (Zymed, South San Francisco, CA) overnight. Embryos were washed 5 times in PBSMT and rinsed in PBT (PBS containing containing 0.2% BSA, 0.1% Triton X-100), followed developing with DAB kit (Vector Laboratories, Burlingame, CA). Finally, embryos were washed in PBT and postfixed in 4% PFA, dehydrated through a methanol series and cleared in BABB (benzyl alcohol: benzyl benzoate - 1:2) in a glass Petri dish. Photos were taken in PBS or BABB using an inverted phase contrast microscope (Olympus IMT-2, Olympus America Inc., Center Valley, PA) and a Canon S40 camera with T-mount adaptor.
Real-Time PCR
Total RNA was extracted from yolk sac or embryo head using TRIZOL® reagent (Invitrogen, Carlsbad, CA) according to the manufacturer's instructions. Aliquots of 1 μg RNA were digested by DNase I and cDNA was prepared using RNA PCR Kit ver. 3.0 (Takara Mirus Bio, Madison, WI) with oligo dT priming. Real-time PCR reactions with SybrGreen quantification were established with 1/20 of each cDNA preparation in an Opticon2 DNA Engine (MJ Research, Cambridge, MA). Relative expression levels after normalization using β-actin were calculated using the 2-ΔΔCT method ()([59] and confirmed by the absolute quantification method using standard curves. Primer pairs for real-time PCR were Ang1 (CATTCTTCGCTGCCATTCTG, GCACATTGCCCATGTTGAATC)[60], Pecam1 (GAGCCCAATCACGTTTCAGTTT, TCCTTCCTGCTTCTTGCTAGCT) [60], Vegf (GGAGATCCTTCGAGGAGCACTT, GCGATTTAGCAGCAGATATAAGAA)[60], Tie2 (ATGTGGAAGTCGAGAGGCGAT, CGAATAGCCATCCACTATTGTCC)[60], Hey1 (TGAGCTGAGAAGGCTGGTAC, ACCCCAAACTCCGATAGTCC)[39], Hey2 (TGAGAAGACTAGTGCCAACAGC, TGGGCATCAAAGTAGCCTTTA)[39], Ephrin-B2 (GCGGGATCCAGGAGATCCCCACTTGGACT, GTGCGCAACCTTCTCCTAAG)[39], Hes1 (AAGGCGGACATTCTGGAAAT, GTCACCTCGTTCATGCACTC) [61]. Hes5 (TACCTGAAACACAGCAAAGC, GCTGGAGTGGTAAGCAG) [62] and β-actin (GTGGGCCGCTCTAGGCACCA, TGGCCTTAGGGTTCAGGGGG). All real-time PCR experiments were performed in duplicate from ≥ 4 independent samples.
Statistical analysis
Statistical significance was calculated using the unpaired Student's t-test (two-tailed) using Graphpad Prism (GraphPad Software, Inc., San Diego, CA) unless otherwise noted.
Authors' contributions
PS conceived the project, obtained funding, participated in the design of experiments and analysis of data, and co-wrote the manuscript; CG partipated in the design of the experiments, performed or participated in all experiments, analysed data and co-wrote the paper. All authors read and approved the final version of the manuscript.
Contributor Information
Changhui Ge, Email: changhuige@yahoo.com.cn.
Pamela Stanley, Email: pamela.stanley@einstein.yu.edu.
Acknowledgements
We thank Wen Dong for excellent technical assistance, Linchao Lu for helpful suggestions and Bin Zhou for helpful comments on the manuscript. This work was supported by NIH grant NCI RO1 95022 to PS and in part by NCI grant PO1 13330 to the Albert Einstein Cancer Center.
References
- Lai EC. Notch signaling: control of cell communication and cell fate. Development. 2004;131:965–73. doi: 10.1242/dev.01074. [DOI] [PubMed] [Google Scholar]
- Schweisguth F. Regulation of notch signaling activity. Curr Biol. 2004;14:R129–38. [PubMed] [Google Scholar]
- Kopan R, Ilagan MX. The canonical Notch signaling pathway: unfolding the activation mechanism. Cell. 2009;137:216–33. doi: 10.1016/j.cell.2009.03.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moloney DJ, Shair LH, Lu FM, Xia J, Locke R, Matta KL, Haltiwanger RS. Mammalian Notch1 is modified with two unusual forms of O-linked glycosylation found on epidermal growth factor-like modules. J Biol Chem. 2000;275:9604–11. doi: 10.1074/jbc.275.13.9604. [DOI] [PubMed] [Google Scholar]
- Rebay I, Fleming RJ, Fehon RG, Cherbas L, Cherbas P, Artavanis-Tsakonas S. Specific EGF repeats of Notch mediate interactions with Delta and Serrate: implications for Notch as a multifunctional receptor. Cell. 1991;67:687–99. doi: 10.1016/0092-8674(91)90064-6. [DOI] [PubMed] [Google Scholar]
- Lieber T, Wesley CS, Alcamo E, Hassel B, Krane JF, Campos-Ortega JA, Young MW. Single amino acid substitutions in EGF-like elements of Notch and Delta modify Drosophila development and affect cell adhesion in vitro. Neuron. 1992;9:847–59. doi: 10.1016/0896-6273(92)90238-9. [DOI] [PubMed] [Google Scholar]
- Xu A, Lei L, Irvine KD. Regions of Drosophila Notch that contribute to ligand binding and the modulatory influence of Fringe. J Biol Chem. 2005;280:30158–65. doi: 10.1074/jbc.M505569200. [DOI] [PubMed] [Google Scholar]
- Ge C, Liu T, Hou X, Stanley P. In vivo consequences of deleting EGF repeats 8-12 including the ligand binding domain of mouse Notch1. BMC Dev Biol. 2008;8:48. doi: 10.1186/1471-213X-8-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei L, Xu A, Panin VM, Irvine KD. An O-fucose site in the ligand binding domain inhibits Notch activation. Development. 2003;130:6411–21. doi: 10.1242/dev.00883. [DOI] [PubMed] [Google Scholar]
- Rampal R, Arboleda-Velasquez JF, Nita-Lazar A, Kosik KS, Haltiwanger RS. Highly conserved O-fucose sites have distinct effects on Notch1 function. J Biol Chem. 2005;280:32133–40. doi: 10.1074/jbc.M506104200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi S, Ge C, Luo Y, Hou X, Haltiwanger RS, Stanley P. The threonine that carries fucose, but not fucose, is required for Cripto to facilitate Nodal signaling. J Biol Chem. 2007;282:20133–41. doi: 10.1074/jbc.M702593200. [DOI] [PubMed] [Google Scholar]
- Ge C, Stanley P. The O-fucose glycan in the ligand-binding domain of Notch1 regulates embryogenesis and T cell development. Proc Natl Acad Sci USA. 2008;105:1539–44. doi: 10.1073/pnas.0702846105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swiatek PJ, Lindsell CE, del Amo FF, Weinmaster G, Gridley T. Notch1 is essential for postimplantation development in mice. Genes Dev. 1994;8:707–19. doi: 10.1101/gad.8.6.707. [DOI] [PubMed] [Google Scholar]
- Conlon RA, Reaume AG, Rossant J. Notch1 is required for the coordinate segmentation of somites. Development. 1995;121:1533–45. doi: 10.1242/dev.121.5.1533. [DOI] [PubMed] [Google Scholar]
- Huppert SS, Le A, Schroeter EH, Mumm JS, Saxena MT, Milner LA, Kopan R. Embryonic lethality in mice homozygous for a processing-deficient allele of Notch1. Nature. 2000;405:966–70. doi: 10.1038/35016111. [DOI] [PubMed] [Google Scholar]
- Hadland BK, Huppert SS, Kanungo J, Xue Y, Jiang R, Gridley T, Conlon RA, Cheng AM, Kopan R, Longmore GD. A requirement for Notch1 distinguishes 2 phases of definitive hematopoiesis during development. Book A requirement for Notch1 distinguishes 2 phases of definitive hematopoiesis during development City. 2004;104:3097–3105. doi: 10.1182/blood-2004-03-1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stahl M, Uemura K, Ge C, Shi S, Tashima Y, Stanley P. Roles of Pofut1 and O-fucose in mammalian Notch signaling. J Biol Chem. 2008;283:13638–51. doi: 10.1074/jbc.M802027200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Givogri MI, Costa RM, Schonmann V, Silva AJ, Campagnoni AT, Bongarzone ER. Central nervous system myelination in mice with deficient expression of Notch1 receptor. J Neurosci Res. 2002;67:309–20. doi: 10.1002/jnr.10128. [DOI] [PubMed] [Google Scholar]
- Visan I, Tan JB, Yuan JS, Harper JA, Koch U, Guidos CJ. Regulation of T lymphopoiesis by Notch1 and Lunatic fringe-mediated competition for intrathymic niches. Nat Immunol. 2006;7:634–43. doi: 10.1038/ni1345. [DOI] [PubMed] [Google Scholar]
- Loomes KM, Stevens SA, O'Brien ML, Gonzalez DM, Ryan MJ, Segalov M, Dormans NJ, Mimoto MS, Gibson JD, Sewell W. Dll3 and Notch1 genetic interactions model axial segmental and craniofacial malformations of human birth defects. Dev Dyn. 2007;236:2943–51. doi: 10.1002/dvdy.21296. [DOI] [PubMed] [Google Scholar]
- Li Y, Takeshita K, Liu PY, Satoh M, Oyama N, Mukai Y, Chin MT, Krebs L, Kotlikoff MI, Radtke F. Smooth muscle Notch1 mediates neointimal formation after vascular injury. Circulation. 2009;119:2686–92. doi: 10.1161/CIRCULATIONAHA.108.790485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi S, Stanley P. Protein O-fucosyltransferase 1 is an essential component of Notch signaling pathways. Proc Natl Acad Sci USA. 2003;100:5234–9. doi: 10.1073/pnas.0831126100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oka C, Nakano T, Wakeham A, de la Pompa JL, Mori C, Sakai T, Okazaki S, Kawaichi M, Shiota K, Mak TW. Disruption of the mouse RBP-J kappa gene results in early embryonic death. Development. 1995;121:3291–301. doi: 10.1242/dev.121.10.3291. [DOI] [PubMed] [Google Scholar]
- Donoviel DB, Hadjantonakis AK, Ikeda M, Zheng H, Hyslop PS, Bernstein A. Mice lacking both presenilin genes exhibit early embryonic patterning defects. Genes Dev. 1999;13:2801–10. doi: 10.1101/gad.13.21.2801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swift MR, Weinstein BM. Arterial-venous specification during development. Circ Res. 2009;104:576–88. doi: 10.1161/CIRCRESAHA.108.188805. [DOI] [PubMed] [Google Scholar]
- Krebs LT. Notch signaling is essential for vascular morphogenesis in mice. Genes Dev. 2000;14:1343–1352. [PMC free article] [PubMed] [Google Scholar]
- Limbourg FP, Takeshita K, Radtke F, Bronson RT, Chin MT, Liao JK. Essential role of endothelial Notch1 in angiogenesis. Circulation. 2005;111:1826–32. doi: 10.1161/01.CIR.0000160870.93058.DD. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Risau W, Flamme I. Vasculogenesis. Annu Rev Cell Dev Biol. 1995;11:73–91. doi: 10.1146/annurev.cb.11.110195.000445. [DOI] [PubMed] [Google Scholar]
- Flamme I, Frolich T, Risau W. Molecular mechanisms of vasculogenesis and embryonic angiogenesis. J Cell Physiol. 1997;173:206–10. doi: 10.1002/(SICI)1097-4652(199711)173:2<206::AID-JCP22>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- Risau W. Mechanisms of angiogenesis. Nature. 1997;386:671–4. doi: 10.1038/386671a0. [DOI] [PubMed] [Google Scholar]
- Carmeliet P, Ferreira V, Breier G, Pollefeyt S, Kieckens L, Gertsenstein M, Fahrig M, Vandenhoeck A, Harpal K, Eberhardt C. Abnormal blood vessel development and lethality in embryos lacking a single VEGF allele. Nature. 1996;380:435–9. doi: 10.1038/380435a0. [DOI] [PubMed] [Google Scholar]
- Suri C, Jones PF, Patan S, Bartunkova S, Maisonpierre PC, Davis S, Sato TN, Yancopoulos GD. Requisite role of angiopoietin-1, a ligand for the TIE2 receptor, during embryonic angiogenesis. Cell. 1996;87:1171–80. doi: 10.1016/S0092-8674(00)81813-9. [DOI] [PubMed] [Google Scholar]
- Wang HU, Chen ZF, Anderson DJ. Molecular distinction and angiogenic interaction between embryonic arteries and veins revealed by ephrin-B2 and its receptor Eph-B4. Cell. 1998;93:741–53. doi: 10.1016/S0092-8674(00)81436-1. [DOI] [PubMed] [Google Scholar]
- Oshima M, Oshima H, Taketo MM. TGF-beta receptor type II deficiency results in defects of yolk sac hematopoiesis and vasculogenesis. Dev Biol. 1996;179:297–302. doi: 10.1006/dbio.1996.0259. [DOI] [PubMed] [Google Scholar]
- Shawber CJ, Kitajewski J. Notch function in the vasculature: Insights from zebrafish, mouse and man. Bioessays. 2004;26:225–234. doi: 10.1002/bies.20004. [DOI] [PubMed] [Google Scholar]
- Krebs LT. Haploinsufficienct lethality and formation of arteriovenous malformations in Notch pathway mutants. Genes Dev. 2004;18:2469–2473. doi: 10.1101/gad.1239204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue Y, Gao X, Lindsell CE, Norton CR, Chang B, Hicks C, Gendron-Maguire M, Rand EB, Weinmaster G, Gridley T. Embryonic lethality and vascular defects in mice lacking the Notch ligand Jagged1. Hum Mol Genet. 1999;8:723–30. doi: 10.1093/hmg/8.5.723. [DOI] [PubMed] [Google Scholar]
- Duarte A. Dosage-sensitive requirement for mouse Dll4 in artery development. Genes Dev. 2004;18:2474–2478. doi: 10.1101/gad.1239004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer A, Schumacher N, Maier M, Sendtner M, Gessler M. The Notch target genes Hey1 and Hey2 are required for embryonic vascular development. Genes Dev. 2004;18:901–11. doi: 10.1101/gad.291004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dou GR, Wang YC, Hu XB, Hou LH, Wang CM, Xu JF, Wang YS, Liang YM, Yao LB, Yang AG. RBP-J, the transcription factor downstream of Notch receptors, is essential for the maintenance of vascular homeostasis in adult mice. FASEB J. 2008;22:1606–17. doi: 10.1096/fj.07-9998com. [DOI] [PubMed] [Google Scholar]
- Taylor KL, Henderson AM, Hughes CC. Notch activation during endothelial cell network formation in vitro targets the basic HLH transcription factor HESR-1 and downregulates VEGFR-2/KDR expression. Microvasc Res. 2002;64:372–83. doi: 10.1006/mvre.2002.2443. [DOI] [PubMed] [Google Scholar]
- Lawson ND. Notch signaling is required for arterial-venous differentiation during embryonic vascular development. Development. 2001;128:3675–3683. doi: 10.1242/dev.128.19.3675. [DOI] [PubMed] [Google Scholar]
- Shawber CJ, Das I, Francisco E, Kitajewski J. Notch signaling in primary endothelial cells. Ann N Y Acad Sci. 2003;995:162–70. doi: 10.1111/j.1749-6632.2003.tb03219.x. [DOI] [PubMed] [Google Scholar]
- Morrow D, Cullen JP, Cahill PA, Redmond EM. Cyclic strain regulates the Notch/CBF-1 signaling pathway in endothelial cells: role in angiogenic activity. Arterioscler Thromb Vasc Biol. 2007;27:1289–96. doi: 10.1161/ATVBAHA.107.142778. [DOI] [PubMed] [Google Scholar]
- Morrow D, Cullen JP, Cahill PA, Redmond EM. Ethanol stimulates endothelial cell angiogenic activity via a Notch- and angiopoietin-1-dependent pathway. Cardiovasc Res. 2008;79:313–21. doi: 10.1093/cvr/cvn108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suchting S, Freitas C, le Noble F, Benedito R, Breant C, Duarte A, Eichmann A. The Notch ligand Delta-like 4 negatively regulates endothelial tip cell formation and vessel branching. Proc Natl Acad Sci USA. 2007;104:3225–30. doi: 10.1073/pnas.0611177104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster GG. Negative complementation at the notch locus of Drosophila melanogaster. Genetics. 1975;81:99–120. doi: 10.1093/genetics/81.1.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Portin P. Allelic negative complementation at the Abruptex locus of Drosophila melanogaster. Genetics. 1975;81:121–33. doi: 10.1093/genetics/81.1.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakamoto K, Chao WS, Katsube K, Yamaguchi A. Distinct roles of EGF repeats for the Notch signaling system. Exp Cell Res. 2005;302:281–91. doi: 10.1016/j.yexcr.2004.09.016. [DOI] [PubMed] [Google Scholar]
- Vooijs M, Schroeter EH, Pan Y, Blandford M, Kopan R. Ectodomain shedding and intramembrane cleavage of mammalian Notch proteins is not regulated through oligomerization. J Biol Chem. 2004;279:50864–73. doi: 10.1074/jbc.M409430200. [DOI] [PubMed] [Google Scholar]
- Garg V. Molecular genetics of aortic valve disease. Curr Opin Cardiol. 2006;21:180–4. doi: 10.1097/01.hco.0000221578.18254.70. [DOI] [PubMed] [Google Scholar]
- Roach ML, McNeish JD. Methods for the isolation and maintenance of murine embryonic stem cells. Methods Mol Biol. 2002;185:1–16. doi: 10.1385/1-59259-241-4:1. [DOI] [PubMed] [Google Scholar]
- Ioffe E, Stanley P. Mice lacking N-acetylglucosaminyltransferase I activity die at mid-gestation, revealing an essential role for complex or hybrid N-linked carbohydrates. Proc Natl Acad Sci USA. 1994;91:728–32. doi: 10.1073/pnas.91.2.728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Moloney DJ, Stanley P. Fringe modulation of Jagged1-induced Notch signaling requires the action of beta 4galactosyltransferase-1. Proc Natl Acad Sci USA. 2001;98:13716–21. doi: 10.1073/pnas.241398098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi S, Stahl M, Lu L, Stanley P. Canonical Notch signaling is dispensable for early cell fate specifications in mammals. Mol Cell Biol. 2005;25:9503–8. doi: 10.1128/MCB.25.21.9503-9508.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hicks C, Johnston SH, diSibio G, Collazo A, Vogt TF, Weinmaster G. Fringe differentially modulates Jagged1 and Delta1 signalling through Notch1 and Notch2. Nat Cell Biol. 2000;2:515–20. doi: 10.1038/35019553. [DOI] [PubMed] [Google Scholar]
- Wang S, Sdrulla AD, diSibio G, Bush G, Nofziger D, Hicks C, Weinmaster G, Barres BA. Notch receptor activation inhibits oligodendrocyte differentiation. Neuron. 1998;21:63–75. doi: 10.1016/S0896-6273(00)80515-2. [DOI] [PubMed] [Google Scholar]
- Hicks C, Ladi E, Lindsell C, Hsieh JJ, Hayward SD, Collazo A, Weinmaster G. A secreted Delta1-Fc fusion protein functions both as an activator and inhibitor of Notch1 signaling. J Neurosci Res. 2002;68:655–67. doi: 10.1002/jnr.10263. [DOI] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–8. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- Shih SC, Robinson GS, Perruzzi CA, Calvo A, Desai K, Green JE, Ali IU, Smith LE, Senger DR. Molecular profiling of angiogenesis markers. Am J Pathol. 2002;161:35–41. doi: 10.1016/S0002-9440(10)64154-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin D, Xie D, Sakajiri S, Miller CW, Zhu H, Popoviciu ML, Said JW, Black KL, Koeffler HP. DLK1: increased expression in gliomas and associated with oncogenic activities. Oncogene. 2006;25:1852–61. doi: 10.1038/sj.onc.1209219. [DOI] [PubMed] [Google Scholar]
- Ikawa T, Kawamoto H, Goldrath AW, Murre C. E proteins and Notch signaling cooperate to promote T cell lineage specification and commitment. J Exp Med. 2006;203:1329–42. doi: 10.1084/jem.20060268. [DOI] [PMC free article] [PubMed] [Google Scholar]