

Abstract
Although sinus node bradycardia is a very common clinical condition, the cellular mechanisms contributing to abnormal sinus node function are not clearly delineated. In recent publications, mutations in the hyperpolarization-activated, cyclic nucleotide-gated (HCN) 4 channels have been associated with sinus bradycardia. These channels are thought to be crucial in generating the spontaneous sinus node action potential, in accelerating the heart rate during sympathetic drive, and decelerating heart rate during vagal stimulation. Humans carrying HCN4 mutations indeed display significant bradycardia. Recent studies generating HCN4 knock out mice suggested that although HCN4 is crucial in early development, other mechanisms may also play a role in the accelerated heat rate achieved during sympathetic drive. In this review, we focus on genotype–phenotype correlation of these mutations and discuss the relative contribution of various ion channels to sinus node function. We also discuss the importance of HCN in treating clinical conditions such as brady- and tachycardia.
Keywords: genetics, ion channels, bradycardia
Introduction
Sinus bradycardia is a common condition. However, in some individuals, such as athletes, bradycardia may be benign; in others it may have more serious consequences. In some, the bradycardia is associated with other myocardial conditions, such as congenital abnormalities, myocarditis, dystrophies, cardiomyopathies, and is often associated with structural remodeling and fibrosis of the sinoatrial (SA) node (SAN).1–8 Although there are many etiologies for slow heart rate, in terms of symptomatic bradycardia, the only current effective treatment is pacemaker implantation. The predominant ion currents, which contribute to pacemaker activity in the SA node, include currents flowing through hyperpolarization-activated, cyclic nucleotide-gated (HCN) channels9; L-type Ca, T-type Ca.,10 delayed rectifier K11,12; and acetylcholine-activated13,14 channels. However, their relative roles remain controversial and the cellular mechanisms contributing to abnormal sinus node function, such as bradycardia, remain unknown. In contrast to acquired bradycardia, familial diseases of the sinus node are rare. Sinus bradycardia syndromes are commonly associated with other cardiac and extracardiac abnormalities.1–8 In most of the reported cases, permanent pacing was required. Some families with sinus bradycardia (usually without any extracardiac abnormalities) have a more benign course and may even be asymptomatic.1,2,15–18
Mutations in the HCN4 gene have been shown to be responsible for inherited sinus bradycardia.19–22 HCN4 encodes the protein responsible for the channels that carry If, the hyperpolarizing-activated current or the “funny” current that participates in spontaneous diastolic membrane depolarization of SA node cells.23–26 The HCN channels have a structure of six transmembrane segments (Fig. 1). cAMP binds to the cyclic nucleotide-binding domain (CNBD) located in the COOH terminus.27 The modulation of these channels by cAMP is believed to be responsible for acceleration of the heart rate.24 Muscarinic agonists shift the activation curve to more negative voltages; thus, less inward current is available at diastolic potentials, resulting in deceleration of the heart rate.27 Four HCN gene family members have been cloned; all of them were found to be expressed in the heart at some level.9,28,29 HCN4 is the most prominent HCN transcript in SA node, whereas HCN2 is the dominant transcript in the ventricles of all the species investigated.9,28 In rabbits, HCN1 is also expressed in the atria, representing approximately 20% of the total HCN mRNA.9,30 SA cells from knockout mice lacking HCN4 have 75% less If31 and SA cells from mice lacking HCN2 have 25% less HCN current.32 Of note in humans, HCN2 and HCN4 were found to be the dominant mRNA transcripts.33 Characteristics of If in atrial myocytes closely resemble those of HCN4 + HCN130 or HCN4 + HCN234 expressed in heterologous systems, supporting the thesis that atrial myocytes may carry heteromeric complexes composed not only of HCN4 but also of HCN1 or HCN2. Moreover, an ion pore mutation in HCN2 has been shown not only to suppress the HCN2 current in a dominant negative manner, but also to reduce HCN4 current, further substantiating the fact that these channels may co-assemble.35 However, the exact roles of HCN2 and HCN1 in the human sinus node are still in debate. A recent paper36 demonstrated that HCN2 is actually more abundant in the right atrium than in sinus node area while HCN1 and HCN4 are more abundant in the sinus node itself.
Figure 1.
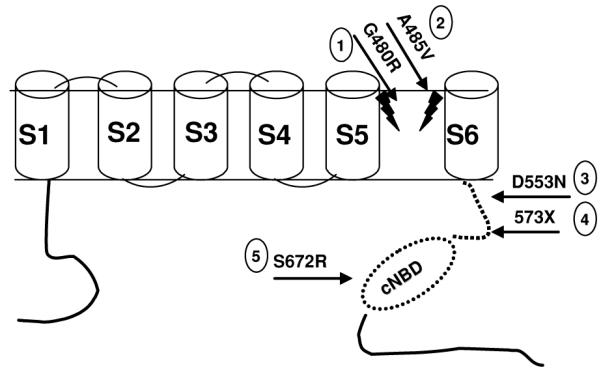
Schematic topology of HCN4 and related mutations in humans. Mutations 122 and 246 are in the ion channel pore. Mutations 321 and 420 are in the region between the core domain and cyclic nucleotide binding domain (CNBD; dashed line). Mutation 519 is in the CNBD itself. Reproduced from Nof et al.22 with permission.
Debate continues over the precise ionic mechanisms responsible for diastolic depolarization in the human SAN. Ion channel currents thought to participate include the HCN current or “funny” current, If; deactivation of the delayed rectifier current, IK11,12; sustained inward current (Ist)37,38; and the L-type (ICa-L) and T-type (ICa-T) calcium channel currents,10 which participate in the final portion of phase 410 (Fig. 2). In addition to these ion channel currents, an intracellular “calcium clock” has been proposed as contributing to spontaneous SA nodal activity.39 Spontaneous rhythmic submembrane local calcium release from the sarcoplasmic reticulum is thought to activate the sodium–calcium exchanger. This inward current contributes to depolarization of the SAN cell. The frequency of this release is modulated by cAMP and can thus respond to sympathetic stimulation-mediated increase in heart rate.
Figure 2.
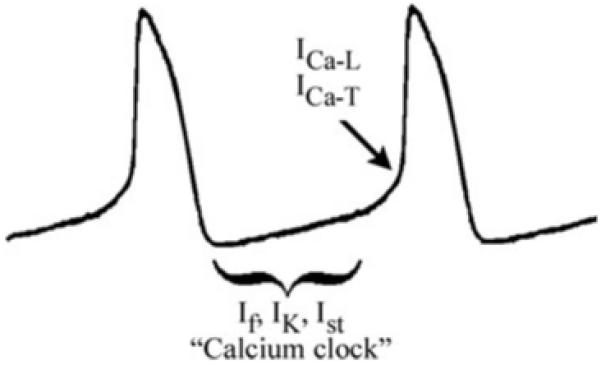
Schematic of the sinus node action potential and key players thought to contribute to spontaneous diastolic depolarization: If = hyperpolarization-activated non-selective cation current or “funny” current; IK = deactivation of delayed rectifier current; Ist = sustained inward current; ICa-L = L-type calcium channel current and ICa-T = T-type calcium channel current; “Calcium Clock” (see text for further details).
In this review, we will focus on the contribution of HCN4 to sinus node function in murine models and human cases.
The Role of HCN4 in Animal Models
There is clear evidence from several studies that mouse embryos lacking functional HCN4 fail to develop during the embryonic period.23,35,40,41 Homozygous embryo mice, lacking exon 4 encoding the HCN4 ion channel pore and the transmembrane segment 6, died in utero.23 Likewise, overexpression of a pore mutation of HCN2 causing a dominant negative suppression or “knockout” of If, eliminated the spontaneous beating activity of neonatal rat myocytes.35 A recent study40 demonstrated that even mice lacking the ability of cAMP to bind to the CNBD, died in utero. In contrast, the role of If in adult mice is controversial. Because HCN4 knockout mice fail to develop, it has not been clear until recently whether If has the same prominent role in adult as in embryonic mice. Recently, Herrmann and colleagues41 succeeded in generating an adult mouse lacking HCN4 channels. They used the cre/loxP system. Adding tamoxifen by week 8 to the floxed mice resulted in the deletion of exon 4 of HCN4. Surprisingly, in contrast to embryonic mice lacking HCN4, adult mice developed normally. If in SAN cells was 75% less intense as expected and the adult mice displayed sinus pauses only at rest, which diminished when the mice were active. Mice lacking HCN4 did not show any differences in maximum heart rate achieved after isoproterenol infusion when compared to normal controls. When the heart rate of the knockout mice returned to basal rate, the sinus pauses reappeared. Even after the addition of an If blocker to abolish the remaining current, heart rate could still be accelerated. This study clearly demonstrated that in adult mice, HCN4 did not appear to be the only mechanism required for adrenergic stimulation of sinus activity. In another adult mouse model, Harzheim and colleagues40 disrupted the binding of cAMP to the CNBD with a single nucleotide missense mutation. As in previous reports, homozygous mice died in utero, but heterozygous mice developed normally. Similar to the previous report, the adult knockout mice had a basal heart rate comparable to the wild type, although sinus pauses were evident at rest. However, these mice exhibited more sinus pauses during exercise compared to rest.
These studies suggest that although If is crucial in early development, it may not be the sole mechanism responsible for determining heart rate at rest or during sympathetic drive in adult mice. Other ionic mechanisms contributing to the spontaneous diastolic depolarization, such as described above, may take over when HCN4 is knocked out or may even be the main mechanism responsible for the increase in heart rate in the murine adult heart.
The Role of HCN4 in Humans
To date, only five HCN4 mutations have been described in humans. The first report20 described a single patient with malignant syncope, sinus bradycardia, and bouts of atrial fibrillation. During exercise, the patient did not reach the predicted maximal heart rate for her age and gender. There was no family history of bradycardia. Genetic analysis revealed a heterozygous stop codon at position 573, creating a protein lacking the CNBD unit (Fig. 1, No. 4). A functional expression revealed that the mutant channels were insensitive to cAMP and had slower activation and faster deactivation kinetics. These findings correlate with the patient’s inability to accelerate her heart rate during exercise. The altered kinetics of the mutant channel explains the sinus bradycardia at rest.
Another report21 described a patient with syncope, prolonged QTc (670 ms), and torsade de pointes (TdP). This patient was found to have a heterozygous missense mutation (Fig. 1, No. 3) affecting trafficking of the mutant channel to the membrane. A functional expression showed decreased current amplitudes for both homozygous and heterozygous states. This mutation segregated with long QTc among family members. No other mutations were found in KCNQ1, KCNH2, SCN5A, KCNE1, KCNE2, and RYR2. The reason for a prolonged QTc association with a decrease in HCN4 current amplitude is unclear, since a loss of If function is not expected to prolong the QT interval, other than through a reduction in heart rate. Studies in both humans42,43 and mice44 have not observed prolongation of the QTc in response to If blockers. It is also noteworthy that HCN2 rather than HCN49 is the predominant HCN in the ventricle. Of note, bradycardia by itself has been shown to contribute to QT and prolongation and the subsequent development of TdP.45
Two large families with mutations in HCN4 causing asymptomatic bradycardia have been reported by us and others.19,22 A missense mutation (S672R; Fig. 1, No. 5) found by Milanesi et al.19 was associated with an asymptomatic bradycardia. Despite its location in the CNBD, this mutation did not affect the binding properties of cAMP, but changed the biophysical properties of the channel. Mutant channels deactivated more slowly and showed a voltage-dependent shift of activation toward hyperpolarization, leading to a decrease in If, which was in turn responsible for slowing down the heart rate.19 We described22 a family with asymptomatic sinus bradycardia with no extracardiac abnormalities, managed conservatively during long-term follow-up (14 ± 11 years). Eight family members were classified as affected. All the affected family members were asymptomatic with a normal exercise capacity during long-term follow-up. A genetic analysis revealed a missense mutation (G480R) in the HCN4 channel pore (Fig. 1, No. 1). The reduction in If in this family was due to both functional changes in channels that reach the plasma membrane, as well as reduced membrane expression of the HCN4 channels.22
We recently46 identified three additional families with symptomatic bradycardia. The affected members presented with a history of presyncope, except for one subject who had a poorly documented event of loss of consciousness with apparent cardiopulmonary arrest, which resolved following basic cardiopulmonary resuscitation; he recovered without defibrillation. There were no documented events of syncope and all had a normal exercise test. Sequencing of the HCN4 gene in the probands of these families revealed a new heterozygous C to T transition in exon 4. This change predicted a substitution of alanine to valine at position 485 within the pore-forming region of the membrane (Fig. 1, No. 2). A485 is a conserved residue and the mutation was not found in unaffected relatives as well as in 50 controls. The reason why the affected family members in our first family reported and some of the affected family members in our previous report carry such a benign prognosis without any chronotropic incompetence despite a mutation in such a conserved pore region is not clear. Among possible explanations are the following: (1) the WT HCN4 protein is up-regulated resulting in only mild reduction of If; (2) expression of other currents may increase to compensate for the loss of HCN4 current; or (3) If may not be the only current responsible for spontaneous diastolic depolarization of the sinus node or responsible for sympathetic stimulation in humans.
Clinical Correlation between Animal Models and Human Studies
The families described19,22,46 and the knockout mice lacking HCN440,41 both display abnormalities of the sinus node function. Moreover, as in the adult HCN4 knockout mouse model reported by Herrmann et al.,41 the affected family members and the knockout mice had no impairment in a heart rate response during exercise. It was not clear whether or which channel/current compensated for the loss of HCN4 in this model. Herrmann et al.41 did not observe upregulation of HCN2, L-type and T-type calcium channels, potassium channels, or Na/Ca exchanger. In another adult mouse model,40 neither chronotropic incompetence nor bradycardia was observed at rest, but sinus pauses were evident mainly during exercise. The main difference between the two mouse models and the families with bradycardia described above was the resting heart rate. While normal basal heart rates were observed in the knockout mice, basal heart rate was reduced in the case of the humans,19,22 a finding that may be explained by the difference in basal heart rate between humans and mice. Mouse heart rate frequency is 10 times faster than that of the human heart, and If may play a more significant role at slower heart rates.40,41 Another explanation may be that a current, not expressed in the human heart, but expressed in the knockout mouse model, provides a compensatory force. Taken together, the available animal and human studies suggest that If is a major contributor to spontaneous diastolic depolarization at rest, evident by the significant bradycardia that all humans carrying mutations in HCN4 display, some of them symptomatic. The role in response to sympathetic stimulation is less clear. It seems that If makes a critical contribution during embryonic development. In adult life, there seems to be some discrepancy between animal models and humans. In most cases, humans with heterozygous mutations in HCN4 did not have chronotropic incompetence with the exception of one case with a stop codon mutation creating an HCN4 channel lacking the CNBD. In adult mice, one study40 in which adult mice had a heterozygous mutation reported that sinus pauses were accentuated during exercise, whereas in another report40 in which the adult mice lacked HCN4, all had a normal increase in heart rate during sympathetic drive. The reason for this different observation between the two models is not clear. One possibility is that mice completely lacking HCN4 have some kind of compensatory mechanism that mice only deficient in HCN4 do not have. This hypothesis remains to be tested.
The Role of HCN4 in Other Cardiac Diseases
HCN4 may also contribute to other cardiac pathological conditions. A decrease in HCN4 in the SAN of tachy-paced dogs that develop congestive heart failure (CHF) was found to be associated with prolonged sinus node recovery time.47 This may contribute, at least in part, to sinus node dysfunction in CHF. If was also found to be increased in ventricular myocytes of rats suffering from ventricular hypertrophy due to hypertension.48 HCN4 is present in other regions of the rat’s right atrium in addition to the SAN,49 which under certain conditions may lead to atrial ectopy and tachyarrhythmias.
The Role of HCN4 in Treatment of Brady- and Tachycardia
Blockade of If has the potential to reduce heart rate without reducing contractility. This is particularly important in patients with heart failure, ischemia, or both. Although several If blockers exist, ivabradine is the prototype and the most studied. Of note, these drugs, particularly at higher concentrations, have the ability to block other channels, such as ICa–L and IK,50,51 and therefore, some of their effects may not be attributed solely to the blockade of the “funny” current. Two large clinical studies evaluated the antianginal and antiischemic effects of ivabradine.52,43 The main conclusion was that ivabradine improves the time required to develop exercise-induced ischemia. Patients in the ivabradine group had lower resting and lower maximal heart rates during exercise. These effects were dose dependent. The highest dose used reduced maximal heart rate by an average of <20 beats/min. In studies on patients with CHF, ivabradine reduced heart rates without reducing left ventricular ejection fraction.53 The most serious and common adverse reaction in this study and others54 was visual symptoms thought to be related to blockade of ion channels in the retina. Currently, the effect of this drug is under investigation by researchers in two clinical trials, BEAUTIFUL55 and SHIFT.56 BEAUTIFUL is a multicenter, double-blind, placebo-controlled trial to evaluate the superiority of ivabradine over placebo as a lowering heart rate agent in reducing cardiovascular events in patients with stable coronary artery disease and left ventricular ejection fraction <40%. The primary end point is the composite of cardiovascular mortality and hospital admission for acute myocardial infarction or new onset or worsening of heart failure. The main results from this trial were recently published.57,58 Ivabradine significantly reduced heart rate over time. The difference in the mean heart rate (placebo minus ivabradine) at 12 months was 6.4 beats/min and 5.6 beats/min at 24 months. In patients with a baseline heart rate over 70 beats/min, the difference in heart rate was even greater. In this patient group (a baseline heart rate ≥70 beats/min), ivabradine reduced the incidence of hospitalization for myocardial infarction and subsequent coronary revascularization procedures. Interestingly, in this group ivabradine did not affect cardiovascular death or hospitalization for worsening heart failure. There were no major side effects reported. SHIFT will investigate patients with reduced left ventricular function because of various causes in addition to myocardial ischemia.56
If blockers may also serve as a tool to study the effect of “funny” current inhibition on the sinus node. Mice treated with ivabradine exhibited a decrease in heart rate and at higher doses displayed fluctuations in the R–R interval.44 Treated mice did not achieve the same maximal heart rate during exercise as before the drug. These results are somewhat at odds with the studies discussed above in which mice lacking HCN4 showed normal chronotropic competence even after adding Cilobradine (another If blocker).41 One explanation may be that the mice genetically lacking HCN4 may have another channel/current compensating for the loss of the “funny” current. Theoretically, HCN blockade may also play a role in the treatment of inappropriate sinus tachycardia although, with the exception of one case, clinical data are lacking.59
Recently another drug with the capability to block If was discovered.60 Clonidine decreases heart rate without lowering blood pressure when administered in low doses to mice. In isolated atrial cells, clonidine inhibits If in the low micromolar range (approximately 3 μmol/ L).
At present there is no effective alternative to pacing for patients with SN dysfunction. THEOPACE, a trial designed to assess the effect of theophylline,61 showed that although oral theophylline increases resting and exercise heart rate, it did not reduce the occurrence of syncope. Thus, there is a need for alternatives that more completely replicate the normal function of SN, other than electrical pacing. Overexpression of If may also serve as a therapeutic modality. Recent studies62,63 have implicated HCN channels in gene-based therapy. Modification of HCN1 or HCN2 DNA resulted in overexpression of If and an increase in heart rate, thus offering a potential alternative to artificial pacing in SN and conduction system disease. Cai and colleagues64 transfected an adenoviral vector containing HCN4 into the free left ventricular wall of pigs that underwent AV-nodal ablation. These pigs maintained a rapid idioventricular rhythm compared to pigs that were not transfected with HCN4. Whole cell recordings from the transduced myocytes demonstrated significant differences in If between transfected and nontransfected cells.
Summary and Conclusion
The role of If in maintaining slow diastolic depolarization in SAN cells and its contribution to adrenergic stimulation are still not completely understood. From recent animal and human studies, it appears that the role of HCN channels in autonomic regulation of heart rate in adults is important especially in maintaining normal heart rate at rest. Loss of their function has been shown to cause varying degrees of sinus bradycardia. The role during sympathetic drive is still under debate. Although most of the reported HCN4 mutation carriers had asymptomatic bradycardia, at present, there is no reason why clinical evaluation of these patients should differ from any other patient presenting with bradycardia. Understanding the pathophysiology of these channels provides an opportunity for new treatment options for a wide range of patients. Blocking these channels may help in reducing heart rate without causing undesirable systemic cardiovascular reactions in patients with CHF and coronary artery disease and in patients suffering from conditions such as inappropriate sinus tachycardia. Overexpression of these channels may provide exciting opportunities in the development of biological pacemakers.
Table 1.
Basic Clinical and Expressional Study Characteristics of Described Human Mutant HCN4 Channels
| Mutation | Patients (pt) |
Bradycardia at Rest |
Symptoms | Chronotropic Incompetence |
Need for Permanent Pacemaker |
Loss of Channel Function Due to |
|
|---|---|---|---|---|---|---|---|
| Trafficking Defect |
Change in Biophysical Properties |
||||||
| 573× 20 | Single pt | Yes | Syncope | Yes | Yes | No | Yes |
| D553N21 | Single pt | Yes | Syncope | No | Yes | Yes | No |
| S672R19 | One family | Yes | No | No | No | No | Yes |
| G480R22 | One family | Yes | No | No | No | Yes | Yes |
| A585V46 | Three families | Yes | Presyncope | No | No | Yes | Yes |
Acknowledgments
Supported by NIH grant HL47678 from NHLBI (CA) and by New York State and Florida Grand Lodges F. & A.M.
Dr. Eyal Nof is a recipient of fellowship grants from BIOTRONIK® and the “American Physicians Fellowship for Medicine in Israel.”
References
- 1.Albin G, Hayes DL, Holmes DR., Jr. Sinus node dysfunction in pediatric and young adult patients: Treatment by implantation of a permanent pacemaker in 39 cases. Mayo Clin Proc. 1985;60:667–672. doi: 10.1016/s0025-6196(12)60742-3. [DOI] [PubMed] [Google Scholar]
- 2.Yabek SM, Jarmakani JM. Sinus node dysfunction in children, adolescents, and young adults. Pediatrics. 1978;61:593–598. [PubMed] [Google Scholar]
- 3.Beder SD, Gillette PC, Garson A, Jr, Porter CB, McNamara DG. Symptomatic sick sinus syndrome in children and adolescents as the only manifestation of cardiac abnormality or associated with unoperated congenital heart disease. Am J Cardiol. 1983;51:1133–1136. doi: 10.1016/0002-9149(83)90358-2. [DOI] [PubMed] [Google Scholar]
- 4.Onat A. Familial sinus node disease and degenerative myopia–a new hereditary syndrome? Hum Genet. 1986;72:182–184. doi: 10.1007/BF00283944. [DOI] [PubMed] [Google Scholar]
- 5.von Zur MF, Klass C, Kreuzer H, Mall G, Giese A, Reimers CD. Cardiac involvement in proximal myotonic myopathy. Heart. 1998;79:619–621. doi: 10.1136/hrt.79.6.619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gulotta SJ, Gupta RD, Padmanabhan VT, Morrison J. Familial occurrence of sinus bradycardia, short PR interval, intraventricular conduction defects, recurrent supraventricular tachycardia, and cardiomegaly. Am Heart J. 1977;93:19–29. doi: 10.1016/s0002-8703(77)80167-1. [DOI] [PubMed] [Google Scholar]
- 7.Schneider MD, Roller DH, Morganroth J, Josephson ME. The syndromes of familial atrioventricular block with sinus bradycardia: Prognostic indices, electrophysiologic and histopathologic correlates. Eur J Cardiol. 1978;7:337–351. [PubMed] [Google Scholar]
- 8.Isobe M, Oka T, Takenaka H, Imamura H, Kinoshita O, Kasanuki H, Sekiguchi M. Familial sick sinus syndrome with atrioventricular conduction disturbance. Jpn Circ J. 1998;62:788–790. doi: 10.1253/jcj.62.788. [DOI] [PubMed] [Google Scholar]
- 9.Shi W, Wymore R, Yu H, Wu J, Wymore RT, Pan Z, Robinson RB, et al. Distribution and prevalence of hyperpolarization-activated cation channel (HCN) mRNA expression in cardiac tissues. Circ Res. 1999;85:e1–e6. doi: 10.1161/01.res.85.1.e1. [DOI] [PubMed] [Google Scholar]
- 10.Hagiwara N, Irisawa H, Kameyama M. Contribution of two types of calcium currents to the pacemaker potentials of rabbit sino-atrial node cells. J Physiol. 1988;395:233–253. doi: 10.1113/jphysiol.1988.sp016916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Brahmajothi MV, Morales MJ, Reimer KA, Strauss HC. Regional localization of HERG, the channel protein responsible for the rapid component of the delayed rectifier, K+ current in the ferret heart. Circ Res. 1997;81:128–135. doi: 10.1161/01.res.81.1.128. [DOI] [PubMed] [Google Scholar]
- 12.Brahmajothi MV, Morales MJ, Liu R, Rasmusson RL, Campbell DL, Strauss HC. In situ hybridization reveals extensive diversity of K+ channel mRNA in isolated ferret cardiac myocytes. Circ Res. 1996;78:1083–1089. doi: 10.1161/01.res.78.6.1083. [DOI] [PubMed] [Google Scholar]
- 13.Dobrzynski H, Marples DD, Musa H, Yamanushi TT, Henderson Z, Takagishi Y, Honjo H, et al. Distribution of the muscarinic K+ channel proteins Kir3.1 and Kir3.4 in the ventricle, atrium, and sinoatrial node of heart. J Histochem Cytochem. 2001;49:1221–1234. doi: 10.1177/002215540104901004. [DOI] [PubMed] [Google Scholar]
- 14.Wickman K, Nemec J, Gendler SJ, Clapham DE. Abnormal heart rate regulation in GIRK4 knockout mice. Neuron. 1998;20:103–114. doi: 10.1016/s0896-6273(00)80438-9. [DOI] [PubMed] [Google Scholar]
- 15.Sarachek NS, Leonard JL. Familial heart block and sinus bradycardia. Classification and natural history. Am J Cardiol. 1972;29:451–458. doi: 10.1016/0002-9149(72)90432-8. [DOI] [PubMed] [Google Scholar]
- 16.Lorber A, Maisuls E, Palant A. Autosomal dominant inheritance of sinus node disease. Int J Cardiol. 1987;15:252–256. doi: 10.1016/0167-5273(87)90324-x. [DOI] [PubMed] [Google Scholar]
- 17.Mehta AV, Chidambaram B, Garrett A. Familial symptomatic sinus bradycardia: Autosomal dominant inheritance. Pediatr Cardiol. 1995;16:231–234. doi: 10.1007/BF00795713. [DOI] [PubMed] [Google Scholar]
- 18.Caralis DG, Varghese PJ. Familial sinoatrial node dysfunction. Increased vagal tone a possible aetiology. Br Heart J. 1976;38:951–956. doi: 10.1136/hrt.38.9.951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Milanesi R, Baruscotti M, Gnecchi-Ruscone T, DiFrancesco D. Familial sinus bradycardia associated with a mutation in the cardiac pacemaker channel. N Engl J Med. 2006;354:151–157. doi: 10.1056/NEJMoa052475. [DOI] [PubMed] [Google Scholar]
- 20.Schulze-Bahr E, Neu A, Friederich P, Kaupp UB, Breithardt G, Pongs O, Isbrandt D. Pacemaker channel dysfunction in a patient with sinus node disease. J Clin Invest. 2003;111:1537–1545. doi: 10.1172/JCI16387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ueda K, Nakamura K, Hayashi T, Inagaki N, Takahashi M, Arimura T, Morita H, et al. Functional characterization of a trafficking-defective HCN4 mutation, D553N, associated with cardiac arrhythmia. J Biol Chem. 2004:1–26. doi: 10.1074/jbc.M311953200. [DOI] [PubMed] [Google Scholar]
- 22.Nof E, Luria D, Brass D, Marek D, Lahat H, Reznik-Wolf H, Pras E, et al. Point mutation in the HCN4 cardiac ion channel pore affecting synthesis, trafficking, and functional expression is associated with familial asymptomatic sinus bradycardia. Circulation. 2007;116:463–470. doi: 10.1161/CIRCULATIONAHA.107.706887. [DOI] [PubMed] [Google Scholar]
- 23.Stieber J, Herrmann S, Feil S, Loster J, Feil R, Biel M, Hofmann F, et al. The hyperpolarization-activated channel HCN4 is required for the generation of pacemaker action potentials in the embryonic heart. Proc Natl Acad Sci USA. 2003;100:15235–15240. doi: 10.1073/pnas.2434235100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.DiFrancesco D, Tortora P. Direct activation of cardiac pacemaker channels by intracellular cyclic AMP. Nature. 1991;351:145–147. doi: 10.1038/351145a0. [DOI] [PubMed] [Google Scholar]
- 25.DiFrancesco D. Pacemaker mechanisms in cardiac tissue. Annu Rev Physiol. 1993;55:455–472. doi: 10.1146/annurev.ph.55.030193.002323. [DOI] [PubMed] [Google Scholar]
- 26.Baruscotti M, Bucchi A, DiFrancesco D. Physiology and pharmacology of the cardiac pacemaker (“funny”) current. Pharmacol Ther. 2005;107:59–79. doi: 10.1016/j.pharmthera.2005.01.005. [DOI] [PubMed] [Google Scholar]
- 27.Accili EA, Proenza C, Baruscotti M, DiFrancesco D. From funny current to HCN channels: 20 years of excitation. News Physiol Sci. 2002;17:32–37. doi: 10.1152/physiologyonline.2002.17.1.32. [DOI] [PubMed] [Google Scholar]
- 28.Moosmang S, Stieber J, Zong X, Biel M, Hofmann F, Ludwig A. Cellular expression and functional characterization of four hyperpolarization-activated pacemaker channels in cardiac and neuronal tissues. Eur J Biochem. 2001;268:1646–1652. doi: 10.1046/j.1432-1327.2001.02036.x. [DOI] [PubMed] [Google Scholar]
- 29.Mistrik P, Mader R, Michalakis S, Weidinger M, Pfeifer A, Biel M. The murine HCN3 gene encodes a hyperpolarization-activated cation channel with slow kinetics and unique response to cyclic nucleotides. J Biol Chem. 2005;280:27056–27061. doi: 10.1074/jbc.M502696200. [DOI] [PubMed] [Google Scholar]
- 30.Moroni A, Gorza L, Beltrame M, Gravante B, Vaccari T, Bianchi ME, Altomare C, et al. Hyperpolarization-activated cyclic nucleotide-gated channel 1 is a molecular determinant of the cardiac pacemaker current I(f) J Biol Chem. 2001;276:29233–29241. doi: 10.1074/jbc.M100830200. [DOI] [PubMed] [Google Scholar]
- 31.Herrmann S, Stieber J, Ludwig A. Pathophysiology of HCN channels. Pflugers Arch. 2007;454:517–522. doi: 10.1007/s00424-007-0224-4. [DOI] [PubMed] [Google Scholar]
- 32.Ludwig A, Budde T, Stieber J, Moosmang S, Wahl C, Holthoff K, Langebartels A, et al. Absence epilepsy and sinus dysrhythmia in mice lacking the pacemaker channel HCN2. EMBO J. 2003;22:216–224. doi: 10.1093/emboj/cdg032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ludwig A, Zong X, Stieber J, Hullin R, Hofmann F, Biel M. Two pacemaker channels from human heart with profoundly different activation kinetics. EMBO J. 1999;18:2323–2329. doi: 10.1093/emboj/18.9.2323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Michels G, Er F, Khan I, Sudkamp M, Herzig S, Hoppe UC. Single-channel properties support a potential contribution of hyperpolarization-activated cyclic nucleotide-gated channels and If to cardiac arrhythmias. Circulation. 2005;111:399–404. doi: 10.1161/01.CIR.0000153799.65783.3A. [DOI] [PubMed] [Google Scholar]
- 35.Er F, Larbig R, Ludwig A, Biel M, Hofmann F, Beuckelmann DJ, Hoppe UC. Dominant-negative suppression of HCN channels markedly reduces the native pacemaker current If and undermines spontaneous beating of neonatal cardiomyocytes. Circulation. 2003;107:485–489. doi: 10.1161/01.cir.0000045672.32920.cb. [DOI] [PubMed] [Google Scholar]
- 36.Chandler NJ, Greener ID, Tellez JO, Inada S, Musa H, Molenaar P, DiFrancesco D, et al. Molecular architecture of the human sinus node: Insights into the function of the cardiac pacemaker. Circulation. 2009;119:1562–1575. doi: 10.1161/CIRCULATIONAHA.108.804369. [DOI] [PubMed] [Google Scholar]
- 37.Guo J, Ono K, Noma A. A sustained inward current activated at the diastolic potential range in rabbit sino-atrial node cells. J Physiol. 1995;483:1–13. doi: 10.1113/jphysiol.1995.sp020563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhang Z, Xu Y, Song H, Rodriguez J, Tuteja D, Namkung Y, Shin HS, et al. Functional roles of Cav1.3 (a1D) calcium channel in sinoatrial nodes: Insight gained using gene-targeted null mutant mice. Circ Res. 2002;90:981–987. doi: 10.1161/01.res.0000018003.14304.e2. [DOI] [PubMed] [Google Scholar]
- 39.Maltsev VA, Vinogradova TM, Lakatta EG. The emergence of a general theory of the initiation and strength of the heartbeat. J Pharmacol Sci. 2006;100:338–369. doi: 10.1254/jphs.cr0060018. [DOI] [PubMed] [Google Scholar]
- 40.Harzheim D, Pfeiffer KH, Fabritz L, Kremmer E, Buch T, Waisman A, Kirchhof P, et al. Cardiac pacemaker function of HCN4 channels in mice is confined to embryonic development and requires cyclic AMP. EMBO J. 2008;27:692–703. doi: 10.1038/emboj.2008.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Herrmann S, Stieber J, Stockl G, Hofmann F, Ludwig A. HCN4 provides a ‘depolarization reserve’ and is not required for heart rate acceleration in mice. EMBO J. 2007;26:4423–4432. doi: 10.1038/sj.emboj.7601868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Redfern WS, Carlsson L, Davis AS, Lynch WG, MacKenzie I, Palethorpe S, Siegl PK, et al. Relationships between preclinical cardiac electrophysiology, clinical QT interval prolongation and torsade de pointes for a broad range of drugs: Evidence for a provisional safety margin in drug development. Cardiovasc Res. 2003;58:32–45. doi: 10.1016/s0008-6363(02)00846-5. [DOI] [PubMed] [Google Scholar]
- 43.Tardif JC, Ford I, Tendera M, Bourassa MG, Fox K. Efficacy of ivabradine, a new selective I(f) inhibitor, compared with atenolol in patients with chronic stable angina. Eur Heart J. 2005;26:2529–2536. doi: 10.1093/eurheartj/ehi586. [DOI] [PubMed] [Google Scholar]
- 44.Stieber J, Wieland K, Stockl G, Ludwig A, Hofmann F. Bradycardic and proarrhythmic properties of sinus node inhibitors. Mol Pharmacol. 2006;69:1328–1337. doi: 10.1124/mol.105.020701. [DOI] [PubMed] [Google Scholar]
- 45.Topilski I, Rogowski O, Rosso R, Justo D, Copperman Y, Glikson M, Belhassen B, et al. The morphology of the QT interval predicts torsade de pointes during acquired bradyarrhythmias. J Am Coll Cardiol. 2007;49:320–328. doi: 10.1016/j.jacc.2006.08.058. [DOI] [PubMed] [Google Scholar]
- 46.Laish-Farkash A, Marek D, Brass D, Pras E, Dascal N, Arad M, Nof E, et al. A novel mutation in the HCN4 gene causes familial sinus bradycardia in two unrelated Moroccan families. (abstract) Heart Rhythm. 2008;5S:S275. [Google Scholar]
- 47.Zicha S, Fernandez-Velasco M, Lonardo G, L’Heureux N, Nattel S. Sinus node dysfunction and hyperpolarization-activated (HCN) channel subunit remodeling in a canine heart failure model. Cardiovasc Res. 2005;66:472–481. doi: 10.1016/j.cardiores.2005.02.011. [DOI] [PubMed] [Google Scholar]
- 48.Cerbai E, Barbieri M, Mugelli A. Occurrence and properties of the hyperpolarization-activated current if in ventricular myocytes from normotensive and hypertensive rats during aging. Circulation. 1996;94:1674–1681. doi: 10.1161/01.cir.94.7.1674. [DOI] [PubMed] [Google Scholar]
- 49.Yamamoto M, Dobrzynski H, Tellez J, Niwa R, Billeter R, Honjo H, Kodama I, et al. Extended atrial conduction system characterised by the expression of the HCN4 channel and connexin45. Cardiovasc Res. 2006;72:271–281. doi: 10.1016/j.cardiores.2006.07.026. [DOI] [PubMed] [Google Scholar]
- 50.Goethals M, Raes A, Van Bogaert PP. Use-dependent block of the pacemaker current I(f) in rabbit sinoatrial node cells by zatebradine (UL-FS 49). On the mode of action of sinus node inhibitors. Circulation. 1993;88:2389–2401. doi: 10.1161/01.cir.88.5.2389. [DOI] [PubMed] [Google Scholar]
- 51.DiFrancesco D, Camm JA. Heart rate lowering by specific and selective If current inhibition with ivabradine: A new therapeutic perspective in cardiovascular disease. Drugs. 2004;64:1757–1765. doi: 10.2165/00003495-200464160-00003. [DOI] [PubMed] [Google Scholar]
- 52.Borer JS, Fox K, Jaillon P, Lerebours G. Antianginal and antiischemic effects of ivabradine, an If inhibitor, in stable angina: A randomized, double-blind, multicentered, placebo-controlled trial. Circulation. 2003;107:817–823. doi: 10.1161/01.cir.0000048143.25023.87. [DOI] [PubMed] [Google Scholar]
- 53.Manz M, Reuter M, Lauck G, Omran H, Jung W. A single intravenous dose of ivabradine, a novel If inhibitor, lowers heart rate but does not depress left ventricular function in patients with left ventricular dysfunction. Cardiology. 2003;100:149–155. doi: 10.1159/000073933. [DOI] [PubMed] [Google Scholar]
- 54.Tardif JC. Clinical results of If current inhibition by ivabradine. Drugs. 2007;67:35–41. doi: 10.2165/00003495-200767002-00005. [DOI] [PubMed] [Google Scholar]
- 55.Fox K, Ferrari R, Tendera M, Steg PG, Ford I. Rationale and design of a randomized, double-blind, placebo-controlled trial of ivabradine in patients with stable coronary artery disease and left ventricular systolic dysfunction: The morBidity-mortality EvAlUaTion of the I(f) inhibitor ivabradine in patients with coronary disease and left ventricULar dysfunction (BEAUTIFUL) study. Am Heart J. 2006;152:860–866. doi: 10.1016/j.ahj.2006.01.013. [DOI] [PubMed] [Google Scholar]
- 56.Bohm M, Reil JC. Perspectives of If inhibition by ivabradine in cardiology. Drugs. 2007;67:43–49. doi: 10.2165/00003495-200767002-00006. [DOI] [PubMed] [Google Scholar]
- 57.Fox K, Ford I, Steg PG, Tendera M, Ferrari R. Ivabradine for patients with stable coronary artery disease and left-ventricular systolic dysfunction (BEAUTIFUL): A randomised, double-blind, placebo-controlled trial. Lancet. 2008;372:807–816. doi: 10.1016/S0140-6736(08)61170-8. [DOI] [PubMed] [Google Scholar]
- 58.Fox K, Ford I, Steg PG, Tendera M, Robertson M, Ferrari R. Heart rate as a prognostic risk factor in patients with coronary artery disease and left-ventricular systolic dysfunction (BEAUTIFUL): A subgroup analysis of a randomised controlled trial. Lancet. 2008;372:817–821. doi: 10.1016/S0140-6736(08)61171-X. [DOI] [PubMed] [Google Scholar]
- 59.Schulze V, Steiner S, Hennersdorf M, Strauer BE. Ivabradine as an alternative therapeutic trial in the therapy of inappropriate sinus tachycardia. A case report. Cardiology. 2007;110:206–208. doi: 10.1159/000111931. [DOI] [PubMed] [Google Scholar]
- 60.Knaus A, Zong X, Beetz N, Jahns R, Lohse MJ, Biel M, Hein L. Direct inhibition of cardiac hyperpolarization-activated cyclic nucleotide-gated pacemaker channels by clonidine. Circulation. 2007;115:872–880. doi: 10.1161/CIRCULATIONAHA.106.667675. [DOI] [PubMed] [Google Scholar]
- 61.Alboni P, Menozzi C, Brignole M, Paparella N, Gaggioli G, Lolli G, Cappato R. Effects of permanent pacemaker and oral theophylline in sick sinus syndrome the THEOPACE study: A randomized controlled trial. Circulation. 1997;96:260–266. doi: 10.1161/01.cir.96.1.260. [DOI] [PubMed] [Google Scholar]
- 62.Bucchi A, Plotnikov AN, Shlapakova I, Danilo P, Jr, Kryukova Y, Qu J, Lu Z, et al. Wild-type and mutant HCN channels in a tandem biological-electronic cardiac pacemaker. Circulation. 2006;114:992–999. doi: 10.1161/CIRCULATIONAHA.106.617613. [DOI] [PubMed] [Google Scholar]
- 63.Tse HF, Xue T, Lau CP, Siu CW, Wang K, Zhang QY, Tomaselli GF, et al. Bioartificial sinus node constructed via in vivo gene transfer of an engineered pacemaker HCN Channel reduces the dependence on electronic pacemaker in a sick-sinus syndrome model. Circulation. 2006;114:1000–1011. doi: 10.1161/CIRCULATIONAHA.106.615385. [DOI] [PubMed] [Google Scholar]
- 64.Cai J, Yi FF, Li YH, Yang XC, Song J, Jiang XJ, Jiang H, et al. Adenoviral gene transfer of HCN4 creates a genetic pacemaker in pigs with complete atrioventricular block. Life Sci. 2007;80:1746–1753. doi: 10.1016/j.lfs.2007.02.006. [DOI] [PubMed] [Google Scholar]