

Abstract
We have prepared 23 new dehydroartemisinin (DART) trioxane derivatives (11 thiazoles, 2 oxadiazoles, and 10 carboxamides) and have screened them for in vitro activity in the Toxoplasma lytic cycle. Fifteen (65%) of the derivatives were non-cytotoxic to host cells (TD50 ≥ 320 μM). Eight thiazole derivatives and two carboxamide derivatives displayed effective inhibition of Toxoplasma growth (IC50 = 0.25-0.42 μM), comparable in potency to artemether (IC50 = 0.31 μM) and >100 times more inhibitory than the currently employed front-line drug trimethoprim (IC50 = 46 μM). The thiazoles as a group were more effective than the other derivatives at inhibiting growth of extracellular as well as intracellular parasites. Unexpectedly, two thiazole trioxanes (5 and 6) were parasiticidal; both inhibited parasite replication irreversibly after parasite exposure to 10 μM of drug for 24 hours, whereas the standard trioxane drugs artemisinin and artemether were not parasiticidal. Some of the new derivatives of artemisinin described here represent effective anti-Toxoplasma trioxanes as well as molecular probes for elucidating the mechanism of action of the DART class of artemisinin derivatives.
Introduction
Toxoplasma gondii is an obligate, intracellular, apicomplexan protozoan with worldwide distribution. The complete life cycle of T. gondii comprises two phases: sexual and asexual. These phases involve several distinct life forms: tachyzoites, bradyzoites, merozoites, and sporozoites. Tachyzoites and merozoites function largely to expand the parasite population within the host while bradyzoites and sporozoites are capable of spreading infection to new hosts via the environment.1 The complete life cycle takes place only within members of the Felidae family making them the definitive hosts. Only the asexual phase takes place inside intermediate hosts, any warm-blooded animal including humans. All hosts are infected by ingesting sporulated oocysts shed into the environment by infected cats, by consuming bradyzoites in the form of tissue cysts from infected animals, or by drinking water contaminated with either of these.2 Human fetuses also can become infected transplacentally from their mother.
Globally, estimates are that one person in three is infected with T. gondii.3 In the United States, nearly 25% of the adult population has been infected with this organism. The percentage of people infected correlates with demographic factors such as age, race, and socioeconomic status and environmental factors such as ambient temperature, humidity, and altitude. In some areas of the world the seropositivity in adults ranges from 80%4,5 to 95%.6
T. gondii is implicated in several maladies in humans including encephalitis, spontaneous abortions in pregnant women, and ocular disease. Recently, a report was published linking Toxoplasma infection with schizophrenia.7 Infected immunocompetent adults rarely experience acute symptoms beyond fever, malaise, and adenopathy. Individuals with HIV/AIDS, cancer chemotherapy patients, or those with otherwise compromised immune systems can experience neurologic, ocular, or systemic toxoplasmosis with widespread organ damage. The potential impact of the disease is sizeable considering the near 50 million people with HIV/AIDS or cancer accounting for almost one percent of the total world population.8,9 Toxoplasma infection can lead to seizures and life-threatening illnesses, such as encephalitis in immunocompromised individuals and, if left untreated, can be fatal.10
Current therapies for treating Toxoplasma infections show limited efficacy and are often associated with severe side effects. These therapies include inhibition of folate metabolism, of protein synthesis, and of electron transport. Antifolate combination therapies employing diaminopyrimidines, such as trimethoprim or pyrimethamine, combined with sulfonamides, such as sulfadiazine or sulfamethoxazole, act synergistically against various bacterial and parasitic microorganisms. Protein synthesis inhibitors, such as macrolide and lincosamide antibiotics, are a second class of medications with anti-Toxoplasma activity. Their mechanism of action in T. gondii is assumed to inhibit plastid or mitochondrial organellar protein synthesis. The third class of anti-Toxoplasma drugs comprises the electron-transport inhibitors such as atovaquone. Atovaquone is occasionally used with pyrimethamine to treat Toxoplasma, as a potential substitute for the sulfa combination therapy. Because of the close resemblance of such compounds to ubiquinone, a suggested mechanism of action for atovaquone and related medications involves disruption of the mitochondrial membrane potential.11
Recently developed Toxoplasma inhibitors have shown a broad range of efficacy, toxicity, and therapeutic index (TI).12-19 The TI is a measure of tolerability of a drug expressed as a ratio of the median cytotoxic dose (TD50) divided by the median inhibitory concentration (ID50). The limited efficacy of current therapies due to patient drug tolerance and the relatively large quantities of drug(s) required to treat the disease necessitates the development of non-toxic, well-tolerated alternatives. Ideally these alternatives will inhibit both the tachyzoite and bradyzoite form in all intermediate hosts and will have selectivity towards the targeted lytic stage of Toxoplasma with minimal effect on human host cell metabolism. Additionally, therapies which target specific stages in the parasitic life cycle provide additional possibilities for synergistic combinations as well as for clarification of the drug's mechanism of action.
Artemisinin is a sesquiterpene lactone that possesses a 1,2,4-trioxane moiety. Artemisinin was first identified as the active constituent in Qinghao (Artemisia annua) in 1972. The artemisinin class of drugs has shown a broad range of activity against many parasites in vitro including Plasmodium, Leishmania, Schistosoma, Trypanosoma, and Toxoplasma.20,21 While the 1,2,4-trioxane function has been shown to be the pharmacophore responsible for the potent activity against malaria parasites in vitro,22,23 its role in anti-Toxoplasma activity is not clear. Deoxyartemisinins lacking the 1,2,4-trioxane moiety are unable to effectively block intracellular Toxoplasma tachyzoite replication but can moderately inhibit extracellular tachyzoite invastion of host cells.24 Thus the illumination of the exact mechanism of action of artemisinins against in vitro Toxoplasma is still an area of intense interest and investigation.25-28 Additionally, thiazole-containing compounds have been shown, in vitro, to possess anti-microbial29 and anti-parasitic properties.30,31 We have previously shown, in vitro, that some C9-C10 dehydroartemsinin (DART) derivatives, including one possessing a thiazole moiety, inhibit multiple steps in the lytic cycle of T. gondii.24 This report describes results with novel DART-thiazole, DART-oxadiazole, and DART-carboxamide derivatives.
Results and Discussion
Scheme 1 outlines the preparation of the 23 DART derivatives starting from the natural trioxane artemisinin. The specific C-10 functional groups in this study were chosen based largely on their chemical accessibility. The syntheses of the DART-1,3-thiazole derivatives were accomplished in a three-step, one-pot procedure while the DART-1,2,4-oxadiazoles and DART-carboxamides were prepared in four or five chemical steps. The DART-1,3-thiazole series were prepared by the addition to artemisinin of the lithium thiazole species, from the respective 2-bromo-1,3-thiazole that was synthesized from the corresponding aryl or alkyl ketone using published procedures.32 The resulting thiazolyl alcohol was acetylated and then acetic acid was eliminated, in situ. The DART-1,2,4-oxadiazole series was prepared from the DART-acid33 via the amidoxime34 followed by cyclization with tetrabutylammonium fluoride (TBAF).35 The DART-carboxamide series was prepared from this same DART-acid and the requisite amine via N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide (EDC)/hydroxybenzotriazole (HOBt) amide coupling.
Scheme 1.

For all the prepared DART derivatives, the in vitro inhibition of growth, invasion, and replication were determined using published methods24 (see Experimental Section). In the five day growth inhibition assay wherein HFF (human foreskin fibroblast) cells are grown in the presence of test compounds and tachyzoites for five days (Tables 1-4), 21 of the 23 compounds displayed at least moderate inhibition (log TI > 1.4) of the parasite, greater than that of trimethoprim, with 12 displaying very strong inhibition (log TI ≥ 2.9). These 12 compounds inhibited the parasite at a therapeutic index level similar to that of the widely-used, antiparasitic trioxane drugs artemisinin and artemether (log TI = 2.9 and 3.1, respectively). Of the 23 prepared compounds, only two (8 and 16) were ineffective in this assay as anti-Toxoplasma agents owing to their cytotoxicity and thus were not investigated further in the secondary assays described below. Additionally, N-methyl amide 17 was not subjected to further assays because it was not more active than the corresponding N-ethyl amide 15.
Table 1. Standards.

Table 4. DART-carboxamides.
 | ||||||
|---|---|---|---|---|---|---|
| Compound | R4 | R5 | IC50 | TD50 | TI | Log TI |
| 1524 | H | Et | 6.8 | 259 | 38 | 1.6 |
| 16 | Et | Et | 11 | 23 | 2.1 | 0.3 |
| 17 | H | Me | 15 | ≥320 | 37 | 1.6 |
| 18 | H | Bn | 0.67 | ≥320 | 839 | 2.9 |
| 19 | H | 4-MeThiazole | 0.65 | ≥320 | 865 | 2.9 |
| 20 | H | p-FBn | 0.47 | 72 | 153 | 2.2 |
| 21 | H | p-CH3Bn | 0.51 | 169 | 331 | 2.5 |
| 22 | H | p-CF3Bn | 0.34 | ≥320 | 1654 | 3.2 |
| 23 | H | 3,4-F2Bn | 0.47 | 78 | 166 | 2.2 |
| 24 | H | 3,5-F2Bn | 0.36 | ≥320 | 1562 | 3.2 |
| 25 | H | 3,5-Br2Bn | 0.81 | ≥320 | 694 | 2.8 |
T. gondii establishes its lytic growth cycle in host cells through the active process of invasion. This process is initiated by attachment of the tachyzoite to the cell and ordinarily quickly progresses to penetration of that cell. The red/green invasion assay tests the ability of compounds to affect the attachment and/or penetration steps of host cell invasion. In this assay, cell-free tachyzoites are incubated with test compounds for 20 minutes before being added to host HFF cell monolayers. Sequential fluorescent staining of the monolayers results in extracellular/attached parasites labeled red and intracellular/penetrated parasites labeled green. (Figure 1) Thus this assay demarcates compounds that can act extracellularly, i.e., directly on the parasite, from those that require being in the intracellular environment, i.e., inside the host cell, in order to manifest anti-parasitic activity. Generally, the DART-thiazoles appear to have the ability to operate extracellularly, whereas the DART-oxadiazoles and DART-carboxamides do not (Figure 1). Specifically, compounds 1-3 and 7-12 caused significant reductions in the number of penetrated (green) parasites. In this assay an effect on attachment of parasites to host cells is defined as a decrease of numbers of both penetrated (green) and attached (red) parasites relative to same of the vehicle.36 As shown in Figure 1, only the DART-thiazoles 1-3 and 7-12 exerted significant inhibition of attachment. It is important to note that the inhibition of attachment and the inhibition of penetration are not necessarily interrelated. It is possible to inhibit penetration while not inhibiting attachment as illustrated by cytochalasin D, a powerful inhibitor of penetration (Figure 1).
Figure 1.
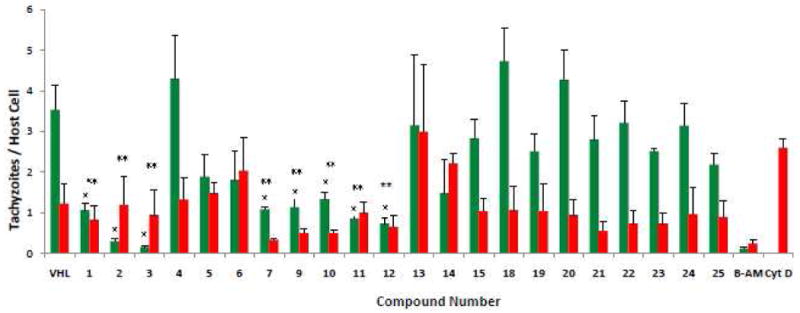
Quantification of invasion/attachment inhibition using red/green assay. The red/green invasion assay was performed using published methods.24 Tachyzoites (5 × 106) were mixed with DMSO [vehicle (VHL)] or compound (final 10 μM) and allowed to sit at room temperature for 20 min. before being added to HFF monolayers growing in 8-well chamber slides. After 1 hour at 37 °C, 5% CO2, the cells were rinsed and fixed. Attached/extracellular parasites were detected using Rb anti-p30 (SAG1) (AbD Serotec, UK) followed by Alexa Fluoro 594 (red) (Invitrogen, CA). After permeabilization, penetrated/intracellular parasites were stained with MAb 9e11 anti-SAG1 (Argene Inc., NY) followed by Gt antimouse Alexa Fluoro 488 (green). DAPI (Invitrogen) for staining nuclei was added to secondary antibody. BAPTA-AM (20 μM) and cytochalasin D (2 μM) are included as controls for defects in attachment and penetration, respectively.24 Stained cells were examined by phase contrast and reflected fluorescence using an Olympus BX41 microscope. Numbers of green and red tachyzoites per host cell were enumerated by visual counting. Data are mean values ±SEM of three independent experiments, counting 10 random fields per well at 600× magnification. A single asterisk indicates tachyzoite penetration significantly lower (P ≤ 0.05, two-tailed Students' t-test) than vehicle. A double asterisk indicates a significant effect (P ≤ 0.05, one-tailed Students' t-test) on parasite attachment relative to vehicle.
The replication assay is a relatively short duration assay that helps determine the time required for activity onset as well as the compound's ability to inhibit an established, intracellular infection. All of the newly synthesized derivatives subjected to the replication assay inhibited replication to varying degrees (Figure 2). Specifically, trioxanes 13, 15, 18, 21, 23, and 24 inhibited replication of the parasite by 50% while trioxanes 1-10, 12, 14, 19, 20, and 22 inhibited replication by 75%. Trioxanes 11 and 25 completely inhibited replication, prohibiting even one round of replication, i.e. one parasite doubling. Generally then, the DART-thiazoles appear to act quickly with all members of this thiazole series restraining replication to ≤1 parasite doubling. On the other hand, only five of the eleven DART-oxadiazole and carboxamide series (14, 19, 20, 22, and 25) effectively inhibited replication. By comparison, parent trioxane drug artemisinin showed no measurable activity and artemether showed only modest inhibition.
Figure 2.

Quantification of replication inhibition using replication assay. Compounds were tested for replication inhibition using established procedures.24 HFF monolayers were inoculated with tachyzoites and then incubated (37 °C, 5% CO2) for 2 h. Compounds (final 10 μM) or DMSO (vehicle) were then added to the medium. Parasite replication proceeded for 24-26 h after which time the monolayers were fixed, permeablilized, and immunolabeled with Rb anti-p30 followed by Gt antirabbit Alexa Fluoro 594 (red). DAPI was added to secondary antibody. Cells were examined by reflected fluorescence as in Figure 1. Data were compiled from three independent experiments, each from 10 random fields per sample. The number of vacuoles containing 1, 2, 4, or 8+ parasites/vacuole were enumerated by eye. Data are expressed as the modal number of parasites per vacuole.
In separate experiments, compounds 2-6 and 14 displayed effective replication inhibition when tested over a range of concentrations (1, 5, and 10 μM), allowing no more than 2 parasite doublings at any concentration. Thiazoles 2, 3, and 4 showed the same activity at both 5 and 10 μM with only 1 doubling/vacuole. However, at 1 μM, DART thiazoles 2 and 4 allowed 2 doublings while thiazole 3 continued to allow just 1 doubling. Thiazole 6 displayed a shallow dose-response curve never allowing more than 2 doublings. Further in these dose-response experiments, both compounds 5 and 14 limited replication to 0.5 doubling at 10 μM and 1 doubling at both 5 and 1 μM.37
Several structure-activity relationships (SAR) became apparent. Monomethyl thiazoles 2 and 4 had higher therapeutic indices than the corresponding dimethyl thiazole 3. DART thiazoles bearing monosubstitution in the 4-position encompassed the rest of the thiazoles tested. DART thiazole 8, a saturated cyclohexyl version of DART phenyl thiazole 5, only weakly inhibited replication. N,N-Diethyl carboxamide 16 demonstrated that the amide nitrogen needs to bear at least one hydrogen atom otherwise activity is lost. N-Monobenzyl amides 18-25 comprised the remainder of the NH-amide class. 3,5-Dibromobenzyl NH-amide 25 showed potent parasite inhibition, as did the corresponding 3,5-difluorobenzyl NH-amide 24. 4-Substitution of the benzyl ring with a fluorine atom was detrimental to inhibitory activity (see NH-amides 20 and 23). 4-Trifluoromethylbenzyl NH-amide 22, however, was considerably more potent than 4-methylbenzyl NH-amide 21.
Finally, when DART thiazoles 5 and 6 were tested for their ability to permanently (i.e. irreversibly) inhibit tachyzoite replication, we found that, after 24 hours of treatment of intracellular parasites followed by no treatment for 96 hours, the parasites were effectively killed.37,38 Thus, DART thiazoles 5 and 6 are parasiticidal; the standard trioxane drugs artemisinin and artemether are not parasiticidal.
In conclusion, we report synthesis and characterization of three new classes of DART derivatives. Nearly all of the new derivatives described here are non-cytotoxic, while many are also highly growth inhibitory against T. gondii tachyzoites, in vitro. Compounds 2-12 represent a new series of very potent, thiazole-containing,19 anti-Toxoplasma compounds. Two of these derivatives (5 and 6) stand out as selective, parasiticidal compounds, a first for the artemisinin-based family of anti-Toxoplasma drugs. This lytic cycle stage selectivity is facilitating use of these trioxanes also as molecular probes to elucidate the mechanism of action of these classes of DART derivatives in T. gondii.
Experimental Section
1H NMR and 13C NMR spectra were recorded on Bruker spectrometer at 400 and 100 MHz or 300 and 75 MHz, respectively, using the residual solvent peak as the internal standard. High Resolution Mass Spectrum-Fast Atom Bombardment (HRMS-FAB) mass spectra were obtained using a VG70SE double focusing magnetic sector mass spectrometer (VG Analytical, Manchester, UK now Micromass/Waters) equipped with a Cs+ ion gun (28 kV @ 2 μA), an off-axis multiplier and a MSS data system (MasCom, Bremen, Germany). The resolution of the instrument was set at 10,000 (100 ppm peak width). Samples were mixed with m-nitrobenzyl alcohol matrix deposited on the target of a direct insertion probe for introduction into the source. For accurate mass measurements, a mass scan range was employed with the matrix containing 10% polyethylene glycol (PEG) or polyethylene glycol, monomethyl ether (PEGMME) mass calibrant. Fourier Transform-Infrared (FT-IR) experiments were performed on a Bruker Vector 22 instrument. Optical rotation values were obtained using a 100 mm quartz cell on a JASCO P-1010 polarimeter with a 589 nm source. Thin-layer chromatography was performed with Silicycle glass-backed 20 cm × 20 cm extra-hard layer 250 μm thickness 60 Å plates with F254 indicator cut down to 20 mm × 67 mm for analytical purposes. The purity of all DART trioxanes was determined to be >95% by analytical thin-layer chromatography (TLC).
General Synthesis of Compounds 4-12
To an oven-dried vial were added the 4-substituted 2-bromothiazole (0.62 mmol) and tetrahydrofuran (500 μL). The resulting solution was cooled to -78 °C. n-Butyllithium (1.6 M in hexanes, 0.62 mmol) was added drop wise, via syringe, over five min. Once the addition was complete, the reaction was stirred at -78 °C for 30 min. Then a solution of artemisinin (0.45 mmol) in tetrahydrofuran (500 μL) was added drop wise, over five min. The resulting reaction mixture was stirred at -78 °C for 30 min and then at -65 °C for 30 min. Then acetic anhydride (3.54 mmol) was added to the reaction drop wise over five min, and the reaction was allowed to warm to 0 °C and to stir at 0 °C for 10 min. Boron trifluoride etherate (3.97 mmol) was added, drop wise, over five min and the reaction was allowed to warm to room temperature (RT) and stir at RT for 16 hours. The reaction was placed in a separatory funnel containing dichloromethane (20 mL) and saturated aqueous sodium bicarbonate solution (10 mL). The organic layer was washed with saturated aqueous sodium bicarbonate solution (2 × 10 mL) followed by saturated aqueous sodium chloride solution (10 mL). The organic layer was dried over magnesium sulfate and was concentrated, in vacuo, at 40 °C to afford crude DART-thiazole. The crude DART-thiazole was purified by flash silica gel chromatography (100 mg of silica gel/mg of crude product).
Compound 4
Crude 4 was purified by flash silica gel chromatography (100% hexanes then 10% ethyl acetate in hexanes) to afford 4 as a white solid (47.2%). TLC one spot Rf = 0.90 (40% ethyl acetate in hexanes); 1H NMR (400 MHz, CDCl3): δ 7.45 (s, 1H), 5.74 (s, 1H), 2.49-2.37 (m, 4H), 2.19 (s, 3H), 2.12-2.02 (m, 2H), 1.96-1.88 (m, 2H), 1.73-1.69 (m, 1H), 1.62-1.24 (m, 7H), 1.18-1.09 (m, 1H), 1.03-0.96 (m, 3H); 13C NMR (100 MHz, CDCl3): δ 140.4, 138.0, 110.0, 104.5, 90.2, 78.4, 50.7, 48.0, 37.6, 36.1, 34.2, 29.0, 25.6, 24.5, 20.1, 17.1, 11.8; HRMS-FAB (m/z): [M + H]+ calcd for C19H26NO4S, 364.1577; found, 364.1581; [α]D25 + 53.6 (c 0.640, CHCl3); FT-IR (NaCl, thin film) (cm-1) 2925, 2891, 1653, 1520, 1445, 1408, 1367, 1314, 1267, 1234, 1215, 1191, 1168, 1143, 1117, 1091, 1062, 1025, 1004.
Compound 5
Crude 5 was purified by flash silica gel chromatography (100% hexanes then 2% ethyl acetate in hexanes) to afford 5 as an amorphous white solid (52.2%). TLC one spot Rf = 0.72 (40% ethyl acetate in hexanes); 1H NMR (400 MHz, CDCl3): δ 7.96-7.94 (d, 2H, J = 7.6Hz), 7.51 (s, 1H), 7.45-7.41 (t, 2H, J = 14.0Hz), 7.35-7.31 (t, 1H, J = 14.8Hz), 5.80 (s, 1H), 2.43-2.34 (m, 4H), 2.16-2.04 (m, 2H), 2.00-1.93 (m, 2H), 1.76-1.70 (m, 1H), 1.63-1.56 (m, 1H), 1.50-1.41 (m, 5H), 1.35-1.30 (m, 1H), 1.22-1.13 (m, 1H), 1.02-0.98 (m, 3H); 13C NMR (100 MHz, CDCl3): δ 165.0, 155.7, 138.3, 135.2, 129.0, 128.3, 126.6, 113.5, 112.6, 111.8, 105.0, 90.9, 78.8, 51.1, 48.4, 38.0, 36.5, 34.5, 29.4, 25.8, 24.8, 20.2, 17.4; HRMS-FAB (m/z): [M + H]+ calcd for C24H28NO4S, 426.1739; found, 426.1729; [α]D25 + 55.5 (c 38.0, CHCl3); FT-IR (NaCl, thin film) (cm-1) 2927, 2863, 1653, 1606, 1496, 1449, 1379, 1346, 1281, 1235, 1211, 1141, 1105, 1066, 1001.
Compound 6
Crude 6 was purified by flash silica gel chromatography (100% hexanes then 2% ethyl acetate in hexanes) to afford 6 as an amorphous white solid (54.8%) TLC one spot Rf = 0.81 (20% ethyl acetate in hexanes); 1H NMR (400 MHz, CDCl3): δ 6.82 (s, 1H), 5.75 (s, 1H), 2.46-2.39 (m, 1H), 2.28 (s, 3H), 2.12-2.02 (m, 2H), 1.98-1.90 (m, 2H), 1.74-1.70 (m, 1H), 1.63-1.57 (m, 1H), 1.44 (s, 4H), 1.34 (s, 10H), 1.33-1.09 (m, 1H), 0.99-0.98 (m, 3H); 13C NMR (100 MHz, CDCl3): δ 166.7, 163.6, 138.2, 110.1, 109.6, 104.5, 90.2, 78.5, 50.7, 48.1, 37.6, 36.1, 34.8, 34.2, 30.1, 29.1, 25.7, 24.5, 20.2, 17.2; HRMS-FAB (m/z): [M + H]+ calcd for C22H32NO4S, 406.2052; found, 406.2048; [α]D25 + 72.2 (c 28.5, CHCl3); FT-IR (NaCl, thin film) (cm-1) 2962, 2925, 2865, 1745, 1655, 1506, 1451, 1374, 1361, 1350, 1281, 1237, 1212, 1142, 1104, 1042, 1001.
Compound 7
Crude 7 was purified by flash silica gel chromatography (100% hexanes then 15% ethyl ether in petroleum ether) to afford 7 as a white foam (71.8%). TLC one spot Rf = 0.57 (20% ethyl acetate in hexanes); 1H NMR (400 MHz, CDCl3): δ 6.86 (s, 1H), 5.75 (s, 1H), 2.82-2.75 (q, 2H, J = 7.5Hz), 2.42-2.32 (m, 1H), 2.21 (s, 3H), 2.12-2.02 (m, 2H), 2.00-1.88 (m, 2H), 1.73-1.67 (m, 1H), 1.63-1.44 (m, 3H), 1.40 (s, 3H), 1.32-1.27 (t, 3H, J = 7.5Hz), 1.18-1.11 (m, 1H), 1.00-0.98 (m, 3H); 13C NMR (100 MHz, CDCl3): δ 164.2, 159.6, 138.4, 112.0, 110.8, 104.8, 90.7, 78.8, 51.0, 48.2, 37.9, 36.4, 34.4, 29.4, 25.8, 25.4, 24.8, 20.2, 17.1, 13.7; HRMS-FAB (m/z): [M + H]+ calcd for C20H28NO4S, 378.1739; found, 378.1732; [α]D25 + 61.3 (c 51.0, CHCl3); FT-IR (NaCl, thin film) (cm-1) 2967, 2929, 2872, 1656, 1515, 1450, 1382, 1349, 1280, 1233, 1213, 1135, 1106, 1042, 1004.
Compound 9
Crude 9 was purified by flash silica gel chromatography (100% hexanes then 2.5% ethyl acetate in hexanes) to afford 9 as an amorphous white solid (68%). TLC one spot Rf = 0.51 (20% ethyl acetate in hexanes); 1H NMR (400 MHz, CDCl3): δ 7.83-7.81 (d, 2H, J = 7.6Hz), 7.39 (s, 1H), 7.23-7.21 (d, 2H, J = 7.6Hz), 5.78 (s, 1H), 2.43-2.37 (m, 7H), 2.14-2.05 (m, 2H), 2.00-1.95 (m, 2H), 1.75-1.72 (m, 1H), 1.56-1.41 (m, 5H), 1.36-1.26 (m, 1H), 1.21-1.10 (m, 1H), 1.01 (m, 3H); 13C NMR (100 MHz, CDCl3): δ 164.5, 155.4, 138.1, 137.6, 132.2, 129.3, 126.8, 126.2, 111.4, 110.9, 104.6, 90.3, 84.7, 78.4, 50.7, 48.1, 37.6, 36.1, 34.2, 29.1, 25.7, 24.5, 21.3, 20.2, 17.3; HRMS-FAB (m/z): [M + H]+ calcd for C25H30NO4S, 440.1890; found, 440.1888; [α]D25 + 56.6 (c 0.800, CHCl3); FT-IR (NaCl, thin film) (cm-1) 3107, 2925, 2870, 1653, 1493, 1455, 1376, 1347, 1322, 1277, 1240, 1222, 1181, 1157, 1134, 1108, 1084, 1059, 1035, 1003.
Compound 11
Crude 11 was purified by flash silica gel chromatography (100% hexanes then 4-6% ethyl acetate in hexanes) to afford 11 as a white foam (64%). TLC one spot Rf = 0.53 (20% ethyl acetate in hexanes); 1H NMR (400 MHz, CDCl3): δ 7.87-7.85 (d, 2H, J = 8.34Hz), 7.40 (s, 1 H); 7.31-7.28 (d, 2 H, J = 8.34Hz), 5.78 (s, 1 H), 2.51 (s, 3 H), 2.46-2.39 (m, 1 H), 2.36 (s, 3 H), 2.14-2.04 (m, 2 H), 1.98-1.94 (m, 2 H), 1.75-1.71 (m, 1 H), 1.64-1.59 (m, 1 H), 1.58-1.53 (m, 1H), 1.50-1.46 (m, 3 H, including singlet at 1.46), 1.39-1.26 (m, 2H, including singlet at 1.26), 1.21-1.12 (m, 1 H), 1.02-1.00 (d, 3 H, J = 5.81Hz); 13C NMR (100 MHz, CDCl3): δ 164.7, 162.4, 138.0, 126.7, 126.6, 111.7, 111.1, 104.6, 90.3, 78.4, 50.7, 48.1, 37.6, 36.1, 34.2, 33.0, 29.7, 29.1, 25.7, 25.6, 24.5, 24.5, 20.1, 17.4, 15.8; HRMS-FAB (m/z): [M + H]+ calcd for C25H30NO4S2, 472.1616; found, 472.1610; [a]D25 + 16.2 (c 0.32, CHCl3); FT-IR (NaCl, thin film) (cm-1) 2922, 2849, 1761, 1652, 1558, 1539, 1489, 1456, 1221, 1108, 1058, 1002, 919, 879, 828, 751, 665.
General Synthesis of Compound 13 and 14
A solution of DART-acid (0.15 mmol) in dichloromethane (1 mL) was added over 20 min to a solution of N,N′-diisopropylcarbodiimide (0.18 mmol), HOBt (0.18 mmol) and the substituted N-hydroxyalkylimidamide (0.26 mmol) in dichloromethane (3 mL) at -10 °C. The reaction was stirred for 20 min at this temperature and then over night at room temperature. The solvent was removed under reduced pressure and the residue was dissolved in ethyl acetate. The organic layer was subsequently washed with a saturated sodium bicarbonate solution, water, a 0.5 M potassium bisulfate solution (2 × mL) and saturated sodium chloride solution. The organics were dried over magnesium sulfate. A 1M TBAF solution in tetrahydrofuran (218 μL, 0.21 mmol) was added drop wise to a solution of acetimidamide (62 mg, 0.15 mmol) in tetrahydrofuran (2 mL). The reaction mixture was stirred for 5 h. The mixture was poured in ethyl acetate (10 mL), washed with water and saturated sodium chloride solution, and dried over magnesium sulfate.
Compound 14
The crude product was purified by flash silica gel chromatography (11% ethyl acetate in hexanes) to afford 14 as a amorphous white solid (82% overall). TLC one spot Rf = 0.30 (20% ethyl acetate in hexanes); 1H NMR (400 MHz, CD2Cl2): δ 8.11-8.13 (m, 2H), 7.48 (m, 3H), 5.81 (s, 1H), 2.35-2.40 (m, 1H), 2.25 (s, 3H), 1.95-2.15 (m, 4H), 1.75 (dq, J = 12.0 Hz, 4.0 Hz, 1H), 1.46-1.61 (m, 3H), 1.43 (s, 3H), 1.32 (dq, J = 12.0 Hz, 4.0 Hz, 1H), 1.15-1.25 (m, 1H), 1.01 (d, J = 8.00 Hz, 3H); 13C NMR (100 MHz, CD2Cl2): δ 172.4, 168.7, 133.0, 131.7, 129.4 (2C), 128.0 (2C), 127.6, 120.9, 105.4, 91.0, 78.8, 51.3, 48.3, 38.1, 36.6, 35.4, 29.6, 26.0, 24.9, 20.4, 17.9; HRMS-FAB (m/z): [M + H]+ calcd for C23H27N2O9, 411.1920; found, 411.1931; [α]D20 + 32.3 (c 0.98, CH2Cl2); FT-IR (KBr) (cm-1) 3067, 2928, 2872, 1658, 1562, 1446, 1364, 1348, 1268, 1252, 1221, 1207, 1181, 1158, 1127, 1114, 1184, 1037, 1003, 968, 880, 830.
General Procedure for the Synthesis of Compounds 16-25
Into a flame dried 5 mL RBF was charged DART-acid (0.10 mmol), EDC, (0.16 mmol), and HOBt, (0.12 mmol). Dichloromethane (2.5 mL) was then added and the mixture was stirred for an hour at which time, the substituted amine (0.38 mmol) was added. The reaction was allowed to stir at room temperature for 3 hours. It was then quenched with 1N hydrochloric acid, extracted with dichloromethane (3 × 5 mL), washed with saturated sodium chloride solution, dried over magnesium sulfate, and evaporated. The crude product was purified by preparative thin layer chromatography.
Compound 22
The crude product was purified by preparative thin layer chromatography (silica gel, 40% ethyl acetate in hexanes) to afford 22 as an amorphous white solid (69%): 1H NMR (400 MHz, CDCl3): δ 7.56 (m, 2H), 7.40 (m, 2H), 7.14 (m, 1H), 5.64 (s, 1H), 4.55 (m, 2H), 2.38 (m, 1H), 2.16 (s, 3H), 2.03 (m, 2H), 1.94 (m, 1H), 1.84 (m, 1H), 1.69 (m, 1H), 1.54-1.37 (m, 6H, including singlet at 1.37), 1.29-1.07 (m, 2H), 0.97 (d, 3H, J=6.0 Hz); 13C NMR (100 MHz, CDCl3): δ 163.30, 142.80, 136.28, 127.60, 125.42, 118.21, 104.65, 90.39, 78.17, 50.43, 48.29, 42.26, 37.56, 35.97, 34.05, 28.75, 25.68, 24.28, 20.00, 16.89; [α]D28 + 93 (c 1.4, CHCl3); HRMS-FAB (m/z): [M + Na]+ calcd for C24H28F3NO5Na, 490.1812; found, 490.1814; IR (NaCl, thin film) 3441, 2930, 2873, 1717, 1653, 1619, 1558, 1540, 1508, 1456, 1418, 1377, 1326, 1219, 1161, 1128, 1067, 1036, 1018, 1001, 964, 924, 881, 831 cm-1.
Compound 24
The crude product was purified by preparative thin layer chromatography (silica gel, 40% ethyl acetate in hexanes) to afford 24 as an amorphous, white solid (61%);1H NMR (400 MHz, CDCl3): δ 7.12 (m, 1H), 6.81 (m, 2H), 6.65 (m, 1H), 5.65 (s, 1H), 4.47 (m, 2H), 2.39 (m, 1H), 2.15 (s, 3H), 2.02 (m, 2H), 1.94 (m, 1H), 1.82 (m, 1H), 1.70 (m, 1H), 1.57-1.38 (m, 6H, including singlet at 1.38), 1.27-1.06 (m, 2H), 0.98 (d, 3H, J=5.2 Hz); 13C NMR (100 MHz, CDCl3): δ 163.27, 142.89, 136.24, 118.39, 110.07, 109.88, 102.47, 90.43, 78.16, 53.35, 50.45, 48.32, 41.96, 37.57, 36.00, 34.07, 28.73, 25.66, 24.30, 20.01, 16.88; [α]D24 + 99 (c 0.25, CHCl3); HRMS-FAB (m/z): [M + H]+ calcd for C23H28F2NO5, 436.1936; found, 436.1921; IR (NaCl, thin film) 3442, 2926, 2876, 1653, 1625, 1597, 1558, 1508, 1458, 1376, 1316, 1279, 1218, 1118, 1100, 1036, 1004, 924, 880, 832 cm-1.
Five Day Growth Inhibition Assay
The 5-Day growth inhibition assay was used to screen a test compound's ability to inhibit parasite growth, both intracellularly and extracellularly. Tests were performed using human foreskin fibroblast (HFF; ATCC, VA) host cells plated in 96-well plates and tachyzoites of T.gondii strain 2F (ATCC) which constitutively express β-galactosidase (β-gal). Test and control compounds were added to the first column of cells (final 320 μM) and then serially diluted across the plate by dilutions of 0.5 log10, leaving the final column drug-free (0 μM) (parasite control). Following this, tachyzoites were added to the top 6 rows of wells thus leaving the bottom 2 rows uninfected for cytotoxicity determinations. After 4 days incubation at 37°C/5% CO2 the substrate for β-gal, chlorophenol red-β-D-galactopyranoside (CPRG), was added to the Toxoplasma wells and the plates incubated for 1 additional day. The cell viability reagent, CellTiter 96®Aqueous One Solution Reagent (Promega Corp., WI) was then added to the bottom two rows of the plate. After three hours incubation color reactions in the wells were read in a Vmax microplate reader (Molecular Devices, CA). The amount of absorbance (570-650 nm) in wells containing drug, tachyzoites and CPRG was compared to that in parasite control wells. The amount of absorbance in these wells is directly proportional to the amount of β-gal activity and thus to the amount of viable tachyzoites. Thus, a decrease in the amount of absorbance indicates an inhibition of enzyme activity and, by extension, parasite growth. In the cytotoxicity wells, the bioreduction of the cell viability reagent by viable cells into a soluble, colored formazan product was captured by reading the plates at 490-650 nm. The median inhibitory concentration (IC50) and median cytotoxic dose (TD50) were calculated using CalcuSyn software (Biosoft, Cambridge, U.K.). For each compound, a therapeutic index (TI) was calculated with the formula TI = TD50/IC50. Data shown (Tables 1-4) are compiled from results from at least three independent experiments.
Supplementary Material
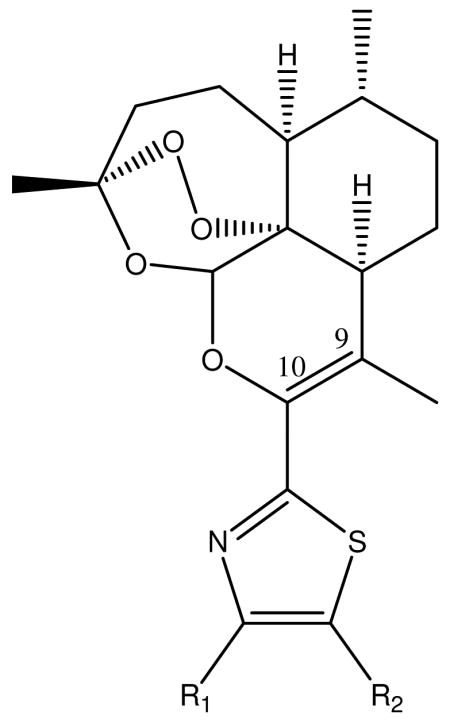
Table 2. DART-1,3-thiazoles.
 | ||||||
|---|---|---|---|---|---|---|
| Compound | R1 | R2 | IC50 (μM) | TD50 (μM) | TI | Log TI |
| 124 | H | H | 1.1 | ≥320 | 511 | 2.7 |
| 2 | Me | H | 0.40 | ≥320 | 1406 | 3.1 |
| 3 | Me | Me | 0.16 | 38 | 240 | 2.4 |
| 4 | H | Me | 0.37 | ≥320 | 1520 | 3.2 |
| 5 | Ph | H | 0.34 | ≥320 | 1654 | 3.2 |
| 6 | t-Bu | H | 0.25 | ≥320 | 2249 | 3.4 |
| 7 | Et | H | 0.40 | ≥320 | 1406 | 3.1 |
| 8 | C6H11 | H | 0.59 | 1.8 | 3.0 | 0.5 |
| 9 | p- CH3Ph | H | 0.35 | ≥320 | 1607 | 3.2 |
| 10 | p-MeOPh | H | 0.75 | 19 | 25 | 1.4 |
| 11 | p-MeSPh | H | 0.40 | ≥320 | 1406 | 3.1 |
| 12 | p-MeS(O2)Ph | H | 0.42 | ≥320 | 1339 | 3.1 |
Table 3. DART-1,2,4-oxadiazoles.
 | |||||
|---|---|---|---|---|---|
| Compound | R3 | IC50 | TD50 | TI | Log TI |
| 13 | Me | 0.84 | ≥320 | 669 | 2.8 |
| 14 | Ph | 0.44 | 141 | 320 | 2.5 |
Acknowledgments
We thank the Stanley Medical Research Foundation and the National Institutes of Health (Grant AI 34885 to G.H.P.) for their generous funding.
Abbreviations
- DART
Dehydroartemisinin
- IC50
Median inhibitory concentration
- TD50
Median cytotoxic dose
- TI
Therapeutic Index (Calculated as TD50/IC50)
- TBAF
Tetrabutylammonium fluoride
- EDC
N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide
- HOBt
Hydroxybenzotriazole
- HFF
Human foreskin fibroblast
- NMR
Nuclear magnetic resonance
- HRMS-FAB
High resolution mass spectrometry-fast atom bombardment
- PEG
Polyethylene glycol
- PEGMME
Polyethylene glycol monomethyl ether
- FT-IR
Fourier transform-infrared
- TLC
Thin layer chromatography
- RT
Room temperature
- CPRG
chlorophenol red-P-D-galactopyranoside
- VHL
Vehicle
- DAPI
4′,6-Diamidino-2-phenylindole
- DMSO
Dimethylsulfoxide
- BAPTA-AM
1,2-Bis(2-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid tetrakis(acetoxymethyl ester)
Footnotes
Supporting Information Available Spectral data for compounds 1-3, 8, 10, 12, 13, 15-21, 23, and 25. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Ferguson DJP, Dubremetz JF. The Ultrastructure of Toxoplasma Gondii. In: Weiss L, Kim K, editors. Toxoplasma gondii. First. Academic Press; London, U.K.: 2007. pp. 19–48. [Google Scholar]
- 2.Bowie WR, King AS, Werker DH, Isaac-Renton JL, Bell A, Eng SB, Marion SA. Outbreak of Toxoplasmosis Associated with Municipal Drinking Water. Lancet. 1997;350:173–177. doi: 10.1016/s0140-6736(96)11105-3. [DOI] [PubMed] [Google Scholar]
- 3.Jackson MH, Hutchison WM. The Prevalence and Source of Toxoplasma Infection in the Environment. Adv Parasitol. 1989;28:55–105. doi: 10.1016/s0065-308x(08)60331-0. [DOI] [PubMed] [Google Scholar]
- 4.Montoya JG, Liesenfeld O. Toxoplasmosis. Lancet. 2004;363:1965–1976. doi: 10.1016/S0140-6736(04)16412-X. [DOI] [PubMed] [Google Scholar]
- 5.Khan A, Jordan C, Muccioli C, Vallochi A, Rizzo L, Belfort R, Jr, Vitor RWA, Silveira C, Sibley LD. Genetic Divergence of Toxoplasma gondii Strains Associated with Ocular Toxoplasmosis, Brazil. Emer Infect Dis. 2006;12:942–949. doi: 10.3201/eid1206.060025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Toxoplasmosis: Epidemiology and Risk Factors. [Feb 12, 2010]; http://www.cdc.gov/toxoplasmosis/epi.html.
- 7.Yolken RH, Bachmann S, Rouslanova I, Lillehoj E, Ford G, Torrey EF, Schroeder J. Antibodies to Toxoplasma gondii in Individuals with First-episode Schizophrenia. Clin Infect Dis. 2001;32:842–844. doi: 10.1086/319221. [DOI] [PubMed] [Google Scholar]
- 8.World Health Organization Fact Sheet No. 297: Cancer. [Feb 12, 2010]; http://www.who.int/mediacentre/factsheets/fs297/en/index.html.
- 9.World Health Organization Global Summary of AIDS Epidemic. [Feb 12, 2010]; http://www.who.int/hiv/data/2009_global_summary.gif.
- 10.Mayo Foundation for Medical Education and Research: Toxoplasmosis. [Feb 12, 2010]; http://www.mayoclinic.com/print/toxoplasmosis/DS00510/DSECTION=all&METHOD=print.
- 11.Vercesi AE, Rodrigues CO, Uyemura SA, Li Z, Moreno SN. J. Repiration and Oxidative Phosphorylation in the Apicomplexan Parasite Toxoplasma gondii. J Biol Chem. 1998;273:31,040–31,047. doi: 10.1074/jbc.273.47.31040. [DOI] [PubMed] [Google Scholar]
- 12.Saudi MNS, Gaafar MR, El-Azzouni MZ, Ibrahim MA, Eissa MM. Synthesis and Evaluation of Some Pyrimidine Analogs Against Toxoplasmosis. Med Chem Res. 2008;17:541–563. [Google Scholar]
- 13.Mui EJ, Schiehser GA, Milhous WK, Hsu H, Roberts CW, Kirisits M, Muench S, Rice D, Dubey JP, Fowble JW, Rathod PK, Queener SF, Liu SR, Jacobus DP, McLeod R. Novel Triazine JPC-2067-B Inhibits Toxoplasma gondii In Vitro and In Vivo. PLoS Neglected Tropical Diseases. 2008;2:1–13. doi: 10.1371/journal.pntd.0000190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gangjee A, Adair OO, Pagley M, Queener SF. N9-Substituted 2,4-Diaminoquinazolines: Synthesis and Biological Evaluation of Lipophilic Inhibitors of Pneumocystis carinii and Toxoplasma gondii Dihydrofolate Reductase. J Med Chem. 2008;51:6195–6200. doi: 10.1021/jm800694g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Peuchmaur M, Saїdani N, Botté C, Maréchal E, Vial H, Wong Y. Enhanced Antimalarial Activity of Novel Synthetic Aculeatin Derivatives. J Med Chem. 2008;51:4870–4873. doi: 10.1021/jm8007322. [DOI] [PubMed] [Google Scholar]
- 16.Krivogorsky B, Grundt P, Yolken R, Jones-Brando L. Inhibition of Toxoplasma gondii by Indirubin and Tryptanthrin Analogs. Antimicrob Agents Chemother. 2008;52:4466–4469. doi: 10.1128/AAC.00903-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Strobl JS, Seibert CW, Li Y, Nagarkatti R, Mitchell SM, Rosypal AC, Rathore D, Lindsay DS. Inhibiton of Toxoplasma gondii and Plasmodium falciparum Infections In Vitro by NSC3852, A Redox Active Antiproliferative and Tumor Cell Differentiation Agent. J Parasitol. 2009;95:215–223. doi: 10.1645/GE-1608.1. [DOI] [PubMed] [Google Scholar]
- 18.Walton JGA, Patterson S, Liu G, Haraldsen JD, Hollick JJ, Slawin AMZ, Ward GE, Westwood NJ. Synthesis and Biological Evaluation of Functionalised Tetrahydro-β-carboline Analogues as Inhibitors of Toxoplasma gondii Invasion. Org Biomol Chem. 2009;7:3049–3060. [Google Scholar]
- 19.Chimenti F, Bizzarri B, Bolasco A, Secci D, Chimenti P, Carradori S, Granese A, Rivanera D, Frishberg N, Bordón C, Jones-Brando L. Synthesis and Evaluation of 4-Acyl-2-thiazolylhydrazone Derivatives for Anti-Toxoplasma Efficacy In Vitro. J Med Chem. 2009;52:4574–4577. doi: 10.1021/jm9005862. [DOI] [PubMed] [Google Scholar]
- 20.Hencken CP, Kalinda AS, D'Angelo JG. The Anti-infective and Anti-cancer Properties of Artemisinin and its Derivatives. Ann Rev Med Chem. 2009;44:359–378. [Google Scholar]
- 21.Krishna S, Bustamante L, Haynes RK, Staines HM. Artemisinins: Their Growing Importance in Medicine. Trends Pharmacol Sci. 2008;29:520–527. doi: 10.1016/j.tips.2008.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.O'Neill PM, Rawe SL, Borstnik K, Miller A, Ward SA, Bray PG, Davies J, Oh CH, Posner GH. Enantiomeric 1,2,4-Trioxanes Display Equivalent in vitro Antimalarial Activity Versus Plasmodium falciparum Malaria Parasites: Implications for the Molecular Mechanism of Action of the Artemisinins. ChemBioChem. 2005;6:2048–2054. doi: 10.1002/cbic.200500048. [DOI] [PubMed] [Google Scholar]
- 23.O'Neill PM, Posner GH. A Medicinal Chemistry Perspective on Artemisinin and Related Endoperoxides. J Med Chem. 2004;47:2945–2964. doi: 10.1021/jm030571c. [DOI] [PubMed] [Google Scholar]
- 24.D'Angelo JG, Bordón C, Posner GH, Yolken RH, Jones-Brando L. Artemisinin Derivatives Inhibit Toxoplasma gondii in vitro at Multiple Steps in the Lytic Cycle. J Antimicrob Chem. 2009;63:146–150. doi: 10.1093/jac/dkn451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nagamune K, Beatty WL, Sibley DL. Artemisinin Induces Calcium-dependent Protein Secretion in the Protozoan Parasite Toxoplasma gondii. Eukaryotic Cel. 2007;6:2147–2156. doi: 10.1128/EC.00262-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kawase O, Nishikawa Y, Bannai H, Zhang H, Zhang G, Jin S, Lee E, Xuan X. Proteomic Analysis of Calcium-dependent Secretion in Toxoplasma gondii. Proteomics. 2007;7:3718–3725. doi: 10.1002/pmic.200700362. [DOI] [PubMed] [Google Scholar]
- 27.Nagamune K, Moreno SNJ, Sibley DL. Artemisinin-resistant Mutants of Toxoplasma gondii have Altered Calcium Homeostasis. Antimicrob Agents Chemother. 2007;51:3816–3823. doi: 10.1128/AAC.00582-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bouchot A, Jaillet J, Bonhomme A, Pezzella-D'Alessandro N, Laquerriere P, Kilian L, Burlet H, Gomez-Marin J, Pluot M, Bonhomme P, Pinon J. Detection and Localization of a Ca2+-dependent ATPase Activity in Toxoplasma gondii. Cell Structure and Function. 2001;26:49–60. doi: 10.1247/csf.26.49. [DOI] [PubMed] [Google Scholar]
- 29.Karegoudar P, Karthikeyan MS, Prasad DJ, Mahalinga M, Holla BS, Kumari NS. Synthesis of Some Novel 2,4-Disubstituted Thiazoles as Possible Antimicrobial Agents. Eur J Med Chem. 2008;43:261–267. doi: 10.1016/j.ejmech.2007.03.014. [DOI] [PubMed] [Google Scholar]
- 30.Hemphill A, Mueller J, Esposito M. Nitazoxanide, A Broad Spectrum Thiazolide Anti-infective Agent for the Treatment of Gastrointestinal Infections. Expert Opin Pharmacother. 2006;7:953–964. doi: 10.1517/14656566.7.7.953. [DOI] [PubMed] [Google Scholar]
- 31.Karade Hitendra N, Acharya BN, Sathe Manisha, Kaushik MP. Design, Synthesis and Antimalarial Evaluation of Thiazole-derived Amino Acids. Med Chem Res. 2008;17:19–29. [Google Scholar]
- 32.Delbecq F, Cordonnier G, Pommery N, Barbry D, Henichart J. New Heteroarylbenzenesulphonamides as Matrix Metalloproteinase Inhibitors. Bioorg Med Chem Lett. 2004;14:1119–1121. doi: 10.1016/j.bmcl.2003.12.076. [DOI] [PubMed] [Google Scholar]
- 33.Brando LV, Posner GH, D'Angelo JG, Yolken RH, Hencken CP, Woodard L. PCT Int Appl. 2008 WO 2008127381. [Google Scholar]
- 34.Hamzé A, Hernandez J, Fulcrand P, Martinez J. Synthesis of Various 3-Substituted 1,2,4-Oxadiazole-containing Chiral β3 and α-Amino Acids from Fmoc-protected Aspartic Acid. J Org Chem. 2003;68:7316–7321. doi: 10.1021/jo0345953. [DOI] [PubMed] [Google Scholar]
- 35.Gangloff AR, Litvak J, Shelton EJ, Sperandio D, Wang VR, Rice KD. Synthesis of 3,5-disubstituted-1,2,4-oxadiazoles using tetrabutylammonium fluoride as a mild and efficient catalyst. Tetrahedron Lett. 2001;42:1441–1443. [Google Scholar]
- 36.Huynh MH, Rabenau KE, Harper JM, Beatty WL, Sibley LD, Carruthers VB. Rapid invasion of host cells by Toxoplasma requires secretion of the MIC2-M2AP adhesive protein complex. EMBO J. 2003;22:2082–2090. doi: 10.1093/emboj/cdg217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bordón, C.; Hencken, C. P.; Posner, G. H.; Yolken, R. H.; Jones-Brando, L. Unpublished results
- 38.Silverman JA, Hayes ML, Luft BJ, Joiner KA. Characterization of anti-Toxoplasma activity of SDZ 215-918, a cyclosporin derivative lacking immunosuppressive and peptidyl-prolyl-isomerase-inhibiting activity: possible role of a P glycoprotein in Toxoplasma physiology. Antimicrob Agents Chemother. 1997;41:1859–1866. doi: 10.1128/aac.41.9.1859. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.