

Abstract
The polyomavirus JC (JCV) is the etiologic agent of progressive multifocal leukoencephalopathy(PML) and of JCV granule cell neuronopathy(JCV GCN). We present an HIV-negative patient who developed multiple cortical lesions, aphasia and progressive cognitive decline after chemotherapy for non-small-cell lung cancer. Brain biopsy and CSF PCR demonstrated JCV and she had a rapidly fatal outcome. Post-mortem analysis showed diffuse cortical lesions and areas of necrosis at the gray-white junction. Immunostaining revealed a productive JCV-infection of cortical pyramidal neurons, confirmed by electron microscopy, with limited demyelination. This novel gray matter syndrome expands the scope of JCV clinical presentation and pathogenesis.
Introduction
Progressive multifocal leukoencephalopathy (PML)1 is a demyelinating disease of the central nervous system (CNS) occurring in immunocompromised individuals. The etiologic of PML is the polyomavirus JC (JCV). This virus remains latent in kidneys and lymphoid organs and can be found in the urine of healthy individuals, where it harbors a stable, and non-pathogenic regulatory region (RR) called the “archetype”. JCV reactivation, associated with rearrangement of its RR2 leads to a lytic infection of oligodendrocytes, while infected astrocytes appear enlarged and bizarre. Although lesions of PML are always located in the white matter or at the gray-white junction, we and others1, 3 have recognized that demyelinating lesions of PML can also occur within the cortical gray matter of the cerebrum. However, productive JCV infection of cortical hemispheric neurons has never been demonstrated. We have recently described a second clinical entity caused by infection of cerebellar granule cell neurons with a JCV VP1 capsid protein deletion mutant, which we called JCV granule cell neuronopathy (JCV GCN)4–7. We present an HIV-negative patient who developed aphasia and cognitive dysfunction with brain lesions that were initially restricted to the hemispheric cortex. Histological analysis showed the novel finding of productive JCV infection in cortical pyramidal neurons.
Case Report
A 74 year old woman with non-small-cell lung cancer, hypertension, and chronic obstructive pulmonary disease had one month of progressive aphasia and cognitive decline. Her lung cancer was treated with lobectomy, radiation, carboplatin and taxol chemotherapy, which had been completed five months prior to her neurological symptoms. Two weeks after her neurological presentation she was treated with a 7 week Prednisone taper from 60mg to 5mg daily, but continued have worsening aphasia and cognition MRI showed multiple patchy hyperintense signals in the cortex of both cerebral hemispheres on FLAIR sequences (Fig 1 A) as well as periventricular white matter changes that were consistent with microvascular ischemic disease. CSF showed 1 WBC, 13 RBC and normal protein and glucose concentration. Flow cytometry on CSF, an echocardiogram, ANA, Hepatitis B and C, Bartonella, Lyme, and PANCA titers were normal. EEG showed asymmetric slowing with rare sharp components suggesting underlying cortical irritability.
Fig 1.
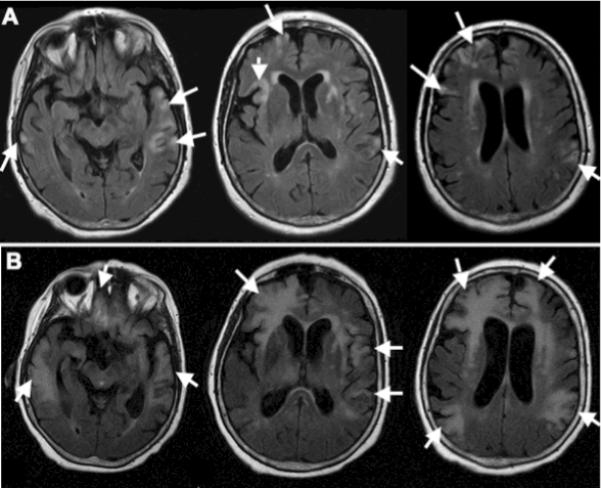
Multiple cortical lesions are present in both cerebral hemispheres.
(A) Magnetic resonance imaging shows patchy areas of hyperintense signal in FLAIR images (TR: 8002.0 ms, TE: 128.4 ms, slice thickness 5 mm) in the temporal, parietal and frontal cortex bilaterally (arrows). (B) Three months later, additional cortical areas are affected, and the abnormal signal extend to the subcortical white matter (arrows).
Nine weeks after the onset of aphasia she was alert, with a normal affect but was only oriented to her name. Her speech was halting and nonfluent, containing many paraphasic errors and frequently displayed echolalia. She had great difficulty with repetition, naming, reading, and writing and would follow only one step commands. Her ability to produce salutations and sing simple songs was preserved. There was no cranial nerve dysfunction. Strength and tone were normal. Reflexes were mildly increased but symmetric and plantar responses were flexor. Sensation and coordination were preserved. Her gait was wide-based but steady. Serology for HIV was negative. Her absolute CD4+ T cell count was 461/μl with a CD4/CD8 ratio at 1.8.
A second MRI showed enlargement of the cortical lesions that now extended to the subcortical white matter, and there was no gadolinium enhancement. The CSF had 2 WBC, 0 RBC, protein of 47.8 mg/dL, glucose of 67 mg/dL and no organisms on gram stain and culture. Paraneoplastic antibodies (Hu, Yo, Tr, Ri, Ma1, Ma2, CV2/CRMP5, amphiphysin, GAD, VGKC and NMDA receptor) and 14-3-3 protein were not detected in the serum. A right frontal brain biopsy demonstrated prominent reactive gliosis and cells with viral cytopathic changes which stained positively for polyomavirus. CSF PCR confirmed the presence of JCV. Immunostaining for Toxoplasma, CMV, HSV 1 and 2, and VZV were negative. A third MRI performed 3 months after the initial study showed new areas of cortical involvement as well as further spread of the lesions in the white matter (Fig 1B). She had one episode of seizures, and her health rapidly declined. She passed away few days later, 4 ½ months after the onset of her neurological symptoms.
Quantified PCR for JCV DNA was performed as previously described8 using JCV-specific primers and DNA extracted from 5 fresh frozen cortical sites corresponding to lesions of both hemispheres seen on MRI, showing a very high mean viral load of 4.6 ×108 cps/μg brain DNA (range 1.2 108 −1.2 109). Negative controls included JCV-negative human genomic DNA as well as BK virus and simian virus 40 (SV40) DNA.
Post-mortem examination revealed diffuse cortical involvement of both cerebral hemispheres with areas of laminar necrosis in the subcortical white matter and deep cortical layers (Fig 2). Histological examination showed giant multinucleated astrocytes (Fig 2 A) and numerous cells with morphology consistent with pyramidal neurons, harboring enlarged nuclei with central clearing of the chromatin (Fig 2 B) throughout the cortex of both cerebral hemispheres and at the gray-white junction.
Fig 2.

Numerous astrocytes and neurons in the cortex of both cerebral hemispheres and at the gray-white junction and are infected by JCV. (A) Typical appearance of giant, bizarre astrocytes with multiple nuclei (arrrows) located in proximity to a neuron with an enlarged nucleus and marginated chromatin (arrowhead, bar= 25 μm). (B) Cortical pyramidal neurons harboring atypical nuclei 5 to 6 times their normal size with central clearing of the chromatin (arrows) are interspersed among unaffected cells. Bar= 25 μm. Hematoxylin and eosin stain. (C, D) Double immunohistochemistry staining for JCV T Ag (sc-20800, dark blue) and myelin and oligodendrocytes (C, CNPase, C5922, brown) or astrocytes (D, GFAP, M0761, light brown), displayed in 25,000,000 pixels pictures of a 1cm2 representative area of cerebrum, shows a linear area of tissue necrosis at the gray-white junction (C,D, arrowheads), and patchy areas of cell loss in the cortex (D, arrows). Most JCV-infected cells (C, dark blue, arrows) which are located at the gray-white junction and the cortex, are not oligodendrocytes (C, inset, absence of staining for myelin (brown) in JCV-infected cells (blue, arrow)). Conversely, numerous JCV-infected astrocytes (D, inset, light brown cell with blue nucleus, arrowhead) can be found within or in proximity to areas of tissue necrosis, interspersed among JCV-infected non-glial cells with neuronal morphology (D, inset, arrows). Bars = 1mm. (E, F) Double immunofluorescence (IF) staining for the neuronal marker MAP-2 (E, M4403, F, AB5622, AlexaFluor 488, green) and for JCV T Ag (sc-20800) or VP1 (PAB597, AlexaFluor 568, red) shows that numerous neurons are infected by JCV and express T Ag (E, arrows) and/or VP1 (F, arrows), interspersed among JCV-infected glial cells (E, F, arrowheads), some of them with clear astrocytic morphology (E, asterisk). Bars = 50μm. GM, gray matter; GWJ, gray-white junction; WM, white matter.
Immunohistochemistry (IHC) and immunofluorescence (IF) staining were done with the following antibodies (Abs): For JCV-infected cells, we used Abs against the simian virus 40 (SV40) that cross-react with JCV: mouse monoclonal anti-SV40 VP1 (PAB597.9) and rabbit polyclonal anti-SV40 T Ag (v-300; sc-20800, Santa Cruz Biotechnology, Santa Cruz, CA); For neurons: Mouse monoclonal anti-MAP-2 (HM-2; M-4403; Sigma, St-Louis, MO), rabbit polyclonal anti-MAP-2 (AB5622; Chemicon, Temecula, CA) and mouse monoclonal anti-NeuN (A60; MAB377; Chemicon). For astrocytes: Mouse monoclonal and rabbit polyclonal anti-GFAP (6F2; M0761 and Z0334; Dako, Carpinteria, CA). For oligodendrocytes and myelin: Mouse monoclonal anti-CNPase (11-5B; C5922; Sigma). IHC staining assays were performed as described in10. After incubation with primary Abs, the Envision G/2 double stain kit (Dako) was used according to the manufacturer's instructions.
IF staining was done as previously described11. In double IF staining, primary antibodies from different species (mouse monoclonals and rabbit polyclonals) were used with directly conjugated Goat anti Mouse (IgG and/or IgM) and/or Goat anti-Rabbit (IgG) Alexa Fluor 488 & 568 secondary Abs (Invitrogen), according to the manufacturer's instructions. Negative controls, for IHC and IF, included omission of the primary Abs and use of sections from healthy individuals.
Double IHC staining with cellular and JCV markers showed that JCV-infected oligodendrocytes were present in few area only and that demyelination was limited (Fig 2 C). On the contrary, numerous JCV-infected astrocytes were located at the gray-white junction and in the cortex (Fig 2 D). For the first time, using double immunostaining with neuronal marker MAP-2 and JCV markers, we found areas of cortex with extensive infection of cortical pyramidal neurons by JCV containing either JCV T Ag, a regulatory protein expressed early in the viral life cycle (Fig 2 E), or JCV VP1 protein, the major component of the viral capsid, expressed at the time of viral assembly (Fig 2 F).
To confirm the nature of JCV-infected cells in the cortex, double immunostaining was repeated with the neuronal marker NeuN and JCV markers, which revealed that cortical pyramidal neurons not only contained JCV proteins in their nucleus and perinuclear cytoplasm (Fig 3A), but also in their axons and dendritic processes (Fig 3 B–C). Typical aggregates of inflammatory cells were present around dying neurons, consistent with neuronophagia (Supplementary Figure 1 A and B).
Fig 3.
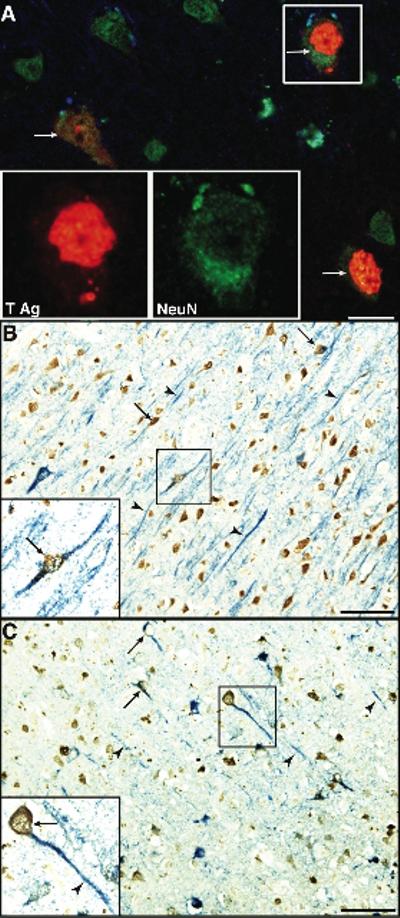
Expression of NeuN confirms the neuronal phenotype of JCV-infected cells. (A) Double immunofluorescence (IF) staining for the neuronal marker NeuN (MAB377, AlexaFluor 488, green) and for JCV T Ag (AlexaFluor 568, red) confirm the presence of JCV-infected neurons expressing T Ag (arrows), interspersed among uninfected neurons. Bar = 25μm. (B–C) Double immunohistochemistry (IHC) staining for NeuN (MAB377, brown) and JCV T Ag (B, sc-20800, blue) or the VP1 capsid protein (C, PAB597, blue). Numerous JCV-infected neuron processes (B, C, arrows) contain JCV T-Ag and VP1 in their axons and dendritic processes (B, C, arrowheads). Bars = 50μm.
Quantitative analyses done on the cortical gray matter of 7 separate areas showed that the number of JCV-infected neurons expressing T Ag/mm2 (83–1183, median: 427, Standard error of the mean (SE): 60,) was significantly higher than JCV-infected astrocytes expressing T Ag/mm2 (0–688, median: 103, SE:24, Wilcoxon p<0.0001). Similarly, VP1-expressing neurons/mm2 (28–183, median: 84, SE: 11) were more numerous than VP1-expressing astrocytes/mm2 (0–69, median 0, SE: 4, Wilcoxon p<0.0001). Similar Wilcoxon tests performed on the same values showed that the number of cells expressing T Ag/mm2 were significantly higher than those expressing VP1/mm2 for both neurons (p<0.0001) and astrocytes (p<0.0001).
To determine whether mature viral particles were present in cortical neurons, areas encompassing neurons expressing JCV VP1 capsid protein were first selected by light microscopy, and were then embedded in resin and processed for electron microscopy (EM), as previously described5. Polyoma virions of 40 nanometers could be visualized in the nuclei of cells with a morphology consistent with neurons (Fig 4).
Fig 4.
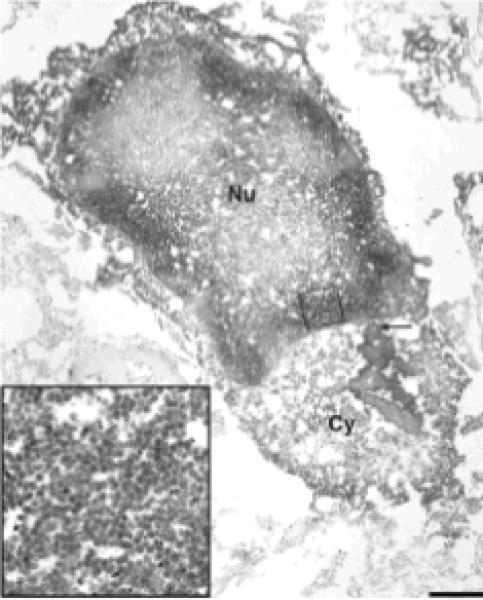
Electron microscopy of a cortical area containing JCV infected neurons.
JCV VP1 immunoreactivity is demonstrated by electron dense DAB bound to anti VP1 ab clumping in the cytoplasm (Cy, arrow). Large amounts of 40 nm viral particles, consistent with the size of polyomaviruses, are present in the nucleus (Nu), close to the nuclear membrane (inset). Bar 2 μm.
To ascertain whether JCV tropism for cortical pyramidal neurons was caused by the same VP1 capsid deletion found in cerebellar granule cell neurons, PCR amplification of the C terminus of the VP1 gene was performed as previously described followed by sequencing4, which demonstrated an intact VP1 gene (data not shown). However, JCV RR amplification using JCV-specific primers as described in12, followed by sequencing revealed an archetype-like RR, without the tandem repeat and duplications usually seen in JCV isolated from the CNS of PML patients2 (data not shown).
To explore whether the patient had sustained primary JCV infection or JCV reactivation, serum collected 4.5 months after disease onset was tested for anti-JCV Abs using viral-like particles based ELISA as previously described13. Serological analyses revealed the presence of a strong response with anti-JCV IgG (0.854 optical density (OD); cut off point 0.136) and IgA (0.167 OD; cut off point 0.100 ) and low level anti-JCV IgM (0.084 OD; cut off point 0.072).
Discussion
This patient's presentation markedly differs from two known clinical syndromes caused by JCV, namely PML and JCV GCN. First, she presented clinically with global cognitive decline and aphasia, consistent with an encephalopathy, rather than with focal alteration of mental function as well as with sensory or motor deficit common in PML, or with the cerebellar syndrome seen in JCV GCN. In addition, while PML lesions on MRI are primarily localized to white matter and the gray-white junction14, they have been reported occasionally in the intracortical myelinated fibers. Our patient's lesions were initially restricted to the hemispheric gray matter and only later extended to the subcortical regions. She had no evidence of cerebellar atrophy. Histologically, she predominantly had JCV infection of cerebral pyramidal neurons and of astrocytes located in the cortical gray matter and the gray-white junction, leading to areas of necrosis. Despite the apparent white matter involvement seen on the final MRI, she only had limited JCV infection of oligodendrocytes and did not display typical areas of demyelination found in PML. Finally, while there were few JCV infected cells in the cerebellum, these were not localized to the granule cell layer. Therefore, this is to our knowledge the first description of a clinical entity caused by JCV distinct from PML and JCV GCN, which we propose to call JCV encephalopathy (JCVE).
The finding that JCV can infect cortical pyramidal neurons expands the host cell range of this virus. Although cortical neurons are heterogeneous functionally and morphologically, we did not observe a preferential JCV tropism for a particular location, with frontal, parietal and temporal lobes being equally affected. While the morphology of infected neurons was mostly consistent with pyramidal cells, we cannot exclude that other neuronal populations were also involved, since infected neurons were found in all cortical layers in the brainstem. However, the presence of JCV within the axons and dendritic processes of pyramidal neurons is striking and suggests that the virus may have spread in the brain of this patient by migrating through the axons of infected neurons. This finding sheds a new light on JCV pathogenesis in the CNS.
Interestingly, more neurons (and astrocytes) contained JCV T Ag, a regulatory protein expressed early in the viral life cycle, than JCV VP1 capsid protein, produced at the time of viral assembly. This finding suggests that JCV infection of cortical pyramidal neurons (and astrocytes) may be latent or abortive in some cells. In fact, the presence of JCV T Ag, but not VP1 protein, has been previously reported in cortical dysplastic ganglion-like neurons of an HIV− patient with common variable immunodeficiency and PML15, and in cells with neuronal morphology of an immunocompetent patient with multiple sclerosis who died of a glioblastoma multiforme16. It is also possible that in our patient, neurons had been only recently infected by JCV, and that all would eventually sustain JCV VP1 expression and assembly of mature viral particles as seen on EM. Furthermore, there may only be a short window of time between production of VP1 protein, assembly of the viral capsid and neuronal cell lysis, which would be another explanation for the lower number of visible JCV-infected cells expressing VP1.
The restricted host cell range of JCV has been attributed mainly to the cellular requirements for transcription of the viral genome17. The RR, which is necessary for transcription and DNA replication, contains various binding sites for cellular proteins. Some of these, such as NF1-class X, are predominantly expressed in glial cells, and, to a lesser degree, in multipotent progenitor cells, when compared to neurons from fetus abortus, which were non permissive to JCV in vitro. Indeed, transfection of an NF1-X expression vector into progenitor-derived neuronal cells before infection resulted in viral protein production18. Furthermore, duplications in tandem repeat fashion of JCV RR, as seen in the CNS-derived JCV prototype Mad-12, were felt to be necessary for its neurotropism, by providing additional binding sites for cellular proteins. However, in our present case, JCV clearly infected numerous post-mitotic mature neurons in this elderly patient, and the RR had an archetype-like form, usually found in urine and kidney and not in CNS isolates of the virus.
These data shed new light on the pathophysiology of JCV in the CNS, and should alert clinicians to look specifically for JCV in immunosuppressed patients presenting with unexplained cortical lesions and encephalopathy. In addition, JCV infection of cortical pyramidal neurons could possibly increase their excitability, leading to this patient's seizure. We had previously shown that 18% of PML patients develop seizures, especially when their lesions are immediately adjacent to the hemispheric cortex19. Whether some of these patients also had JCV infection of cortical pyramidal neurons in addition to PML remains to be determined. Of note, we have demonstrated a fulminant and productive infection of cortical pyramidal neurons by the JCV-related simian virus 40 (SV40) in immunosuppressed rhesus monkeys, after primary infection of the animals12.
Although most individuals get exposed to JCV in late childhood, this patient had a weak anti-JCV IgM response, in addition to a strong IgG and IgA response 4.5 months after disease onset. This is consistent with the late phase of a primary humoral immune response. Alternatively, reactivation of a latent infection could not be entirely ruled out, since such a pattern can be observed during the early anamnestic response. Unfortunately, the absence of premorbid sera or of serial samples precluded a more precise determination of the patient's humoral immune response against JCV.
We have previously reported that JCV tropism for GCN was associated with a 10 nt deletion in the C terminus of the VP1 gene, causing a frameshift and a total change of the last 13 aa of the VP1 protein4. Interestingly this deletion was not present in our patient. It is therefore possible that other mutations of JCV coding region may be associated with JCV tropism for cortical pyramidal neurons. Experiments are now in progress in our laboratory to characterize the entire sequence of this virus and study its phenotype in vitro.
Supplementary Material
Supplementary Fig 1. Occurrence of neuronophagia in the hemispheric cortex. Dying pyramidal neurons (A,B, arrows) are surrounded by inflammatory cells, interspersed among infected neurons with enlarged nuclei (A,B, arrowheads) . Bars = 50μm.
{kind=link}
Acknowledgements
We are grateful to Kristen Toohey for technical assistance with Fig 3, and Ali Ghazi Saad for interpretation of the brain biopsy of this patient. This study was supported in part by public health service grants R01 NS 047029 and 041198 and K24 NS 060950 to IJK, and grant R01 NS 057444 to MPA.
References
- 1.Koralnik IJ. Progressive multifocal leukoencephalopathy revisited: Has the disease outgrown its name? Ann Neurol. 2006;60:162–173. doi: 10.1002/ana.20933. [DOI] [PubMed] [Google Scholar]
- 2.Jensen PN, Major EO. A classification scheme for human polyomavirus JCV variants based on the nucleotide sequence of the noncoding regulatory region. J Neurovirol. 2001;7:280–287. doi: 10.1080/13550280152537102. [DOI] [PubMed] [Google Scholar]
- 3.Moll NM, Rietsch AM, Ransohoff AJ, et al. Cortical demyelination in PML and MS: Similarities and differences. Neurology. 2008;70:336–343. doi: 10.1212/01.WNL.0000284601.54436.e4. [DOI] [PubMed] [Google Scholar]
- 4.Dang X, Koralnik IJ. A granule cell neuron-associated JC virus variant has a unique deletion in the VP1 gene. J Gen Virol. 2006;87:2533–2537. doi: 10.1099/vir.0.81945-0. [DOI] [PubMed] [Google Scholar]
- 5.Du Pasquier RA, Corey S, Margolin DH, et al. Productive infection of cerebellar granule cell neurons by JC virus in an HIV+ individual. Neurology. 2003;61:775–782. doi: 10.1212/01.wnl.0000081306.86961.33. [DOI] [PubMed] [Google Scholar]
- 6.Koralnik IJ, Wuthrich C, Dang X, et al. JC virus granule cell neuronopathy: A novel clinical syndrome distinct from progressive multifocal leukoencephalopathy. Ann Neurol. 2005;57:576–580. doi: 10.1002/ana.20431. [DOI] [PubMed] [Google Scholar]
- 7.Tyler KL. The uninvited guest: JC virus infection of neurons in PML. Neurology. 2003;61:734–735. doi: 10.1212/wnl.61.6.734. [DOI] [PubMed] [Google Scholar]
- 8.Lima MA, Marzocchetti A, Autissier P, et al. Frequency and phenotype of JC virus-specific CD8+ T lymphocytes in the peripheral blood of patients with progressive multifocal leukoencephalopathy. J Virol. 2007;81:3361–3368. doi: 10.1128/JVI.01809-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ashok A, Atwood WJ. Contrasting roles of endosomal pH and the cytoskeleton in infection of human glial cells by JC virus and simian virus 40. J Virol. 2003;77:1347–1356. doi: 10.1128/JVI.77.2.1347-1356.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wuthrich C, Kesari S, Kim WK, et al. Characterization of lymphocytic infiltrates in progressive multifocal leukoencephalopathy: co-localization of CD8(+) T cells with JCV-infected glial cells. J Neurovirol. 2006;12:116–128. doi: 10.1080/13550280600716604. [DOI] [PubMed] [Google Scholar]
- 11.Williams KC, Corey S, Westmoreland SV, et al. Perivascular macrophages are the primary cell type productively infected by simian immunodeficiency virus in the brains of macaques: implications for the neuropathogenesis of AIDS. J Exp Med. 2001;193:905–915. doi: 10.1084/jem.193.8.905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dang X, Wuthrich C, Axthelm MK, Koralnik IJ. Productive simian virus 40 infection of neurons in immunosuppressed Rhesus monkeys. J Neuropathol Exp Neurol. 2008;67:784–792. doi: 10.1097/NEN.0b013e318180f0d5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Randhawa P, Bohl D, Brennan D, et al. Longitudinal Analysis of Levels of Immunoglobulins against BK Virus Capsid Proteins in Kidney Transplant Recipients. Clin Vaccine Immunol. 2008;15:1564–1571. doi: 10.1128/CVI.00206-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Post MJ, Yiannoutsos C, Simpson D, et al. Progressive multifocal leukoencephalopathy in AIDS: are there any MR findings useful to patient management and predictive of patient survival? AIDS Clinical Trials Group, 243 Team. AJNR Am J Neuroradiol. 1999;20:1896–1906. [PMC free article] [PubMed] [Google Scholar]
- 15.Shintaku M, Matsumoto R, Sawa H, Nagashima K. Infection with JC virus and possible dysplastic ganglion-like transformation of the cerebral cortical neurons in a case of progressive multifocal leukoencephalopathy. J Neuropathol Exp Neurol. 2000;59:921–929. doi: 10.1093/jnen/59.10.921. [DOI] [PubMed] [Google Scholar]
- 16.Del Valle L, Delbue S, Gordon J, et al. Expression of JC virus T-antigen in a patient with MS and glioblastoma multiforme. Neurology. 2002;58:895–900. doi: 10.1212/wnl.58.6.895. [DOI] [PubMed] [Google Scholar]
- 17.Raj GV, Khalili K. Transcriptional regulation: lessons from the human neurotropic polyomavirus, JCV. Virology. 1995;213:283–291. doi: 10.1006/viro.1995.0001. [DOI] [PubMed] [Google Scholar]
- 18.Messam CA, Hou J, Gronostajski RM, Major EO. Lineage pathway of human brain progenitor cells identified by JC virus susceptibility. Ann Neurol. 2003;53:636–646. doi: 10.1002/ana.10523. [DOI] [PubMed] [Google Scholar]
- 19.Lima MA, Drislane FW, Koralnik IJ. Seizures and their outcome in progressive multifocal leukoencephalopathy. Neurology. 2006;66:262–264. doi: 10.1212/01.wnl.0000194227.16696.11. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Fig 1. Occurrence of neuronophagia in the hemispheric cortex. Dying pyramidal neurons (A,B, arrows) are surrounded by inflammatory cells, interspersed among infected neurons with enlarged nuclei (A,B, arrowheads) . Bars = 50μm.