

Abstract
Human tissue transglutaminase (TGM2) is implicated in the pathogenesis of several neurodegenerative disorders including Alzheimer's, Parkinson's and expanded polyglutamine (polyQ) diseases. TGM2 promotes formation of soluble and insoluble high molecular weight aggregates by catalyzing a covalent linkage between peptide-bound Q residues in polyQ proteins and a peptide-bound Lys residue. Therapeutic approaches to modulate the activity of TGM2 are needed to proceed with studies to test the efficacy of TGM2 inhibition in disease processes. We investigated whether acetylation of Lys-residues by sulfosuccinimidyl acetate (SNA) or aspirin (ASA) would alter the crosslinking activity of TGM2. Acetylation by either SNA and/or ASA resulted in a loss of >90% of crosslinking activity. The Lys residues that were critical for inhibition were identified by mass spectrometry as Lys444, Lys468, and Lys663. Hence, acetylation of Lys-residues may modulate the enzymatic function of TGM2 in vivo and offer a novel approach to treatment of TGM2 mediated disorders.
Keywords: transglutaminase, neurodegenerative diseases, acetylation, deacetylation, Huntington's disease, polyglutamine repeats, Parkinson's disease, Alzheimer's disease
Introduction
Tissue transglutaminase (TGM2) is a member of the transglutaminase (TG, E.C. 2.3.2.13, protein-glutamine γ-glutamyltransferase) gene family, known to catalyze both inter- and intra-molecular isopeptide bonds between specific glutamine γ-carboxamide groups and ɛ-amino groups in Lys and free primary amines (TGase).1–3 The crosslinking reaction functions to promote extracellular matrix stabilization and wound healing; however, abnormal crosslinking contributes to tissue fibrosis and several neurodegenerative diseases including Huntington's, Parkinson's, and Alzheimer's diseases.4 TGM2 was validated as a therapeutic target in expanded polyQ diseases based on evidence from TGM2 knock-out experiments5,6 and TGM2 inhibition studies.7–9 TGM2-mediated crosslinking alters the solubility of proteins that contain a poly-glutamine (polyQ) repeat,10 alpha-synuclein11,12 and Tau.12,13 TGM2 is a likely mediator of the apparently irreversible crosslinking of neuronal proteins, leading to the formation of soluble aggregates, as well as insoluble inclusions, which are distinctive pathologic features of neurodegenerative diseases.4 Given the fundamental significance of TGM2 in both human biology and pathology, the development of a practical approach to modulate TGM-2 activity could have widespread application.
TGM2 activity can be inhibited by developing specific inhibitors targeting the enzyme14,15 but this approach has not led to orally active TGM2 inhibitors that are safe and effective. An ideal strategy for drug development would be to preserve the essential TGM2 crosslinking reaction and limit excessive TGM2 activity that promotes pathology. We investigated whether the Lys residues on TGM2 could be modified by acetylation using either chemical reagent or a common pharmacologic agent. Lys residues are critical for protein function, solubility, protein–protein interactions and are common targets for post-translational modification.16,17 Ubiquitination, sumoylation, as well as acetylation, all modify Lys residues.18 Acetylation of free Lys-residues may modulate the crosslinking reaction in a manner that limits the contribution of the enzyme to pathologic processes. In this study, we investigated the effect of acetylation on the crosslinking activity of TGM2 and identified specific Lys residues through Matrix-Assisted Laser Desorption/Ionization Time-of-Flight (MALDI-TOF) mass spectrometry (MS).
Results
We initially tested whether a reactive acetylating agent, sulfo-NHS-acetate (SNA), a water soluble analog of N-hydroxysuccinimide (NHS) (Fig. 1), would react with the primary amines (ɛ-amino groups of Lys-residues) in TGM2 and alter enzymatic function. The acetylation of ɛ-NH3 groups produces an amide bond and releases sulfo-NHS as a by-product. This reagent was selected because of its specificity and reactivity with primary amines under nondenaturing conditions to ensure Lys residues were targeted rather than other potential targets such as histidine and cysteine residues. SNA was chosen previously to probe the surface Lys residue on the plasma transglutaminase, Factor XIIIA20 and several other enzymes.21 Another pharmacologic acetylating agent, aspirin (acetyl salicylic; ASA), which is a common anti-inflammatory agent, acetylated Lys residues in several molecules including antithrombin III,22 hemoglobin,23,24 fibrinogen25 and BSA.26 Since ASA can be administered orally to humans, it could serve as a practical method to modulate the activity of TGM2.
Figure 1.
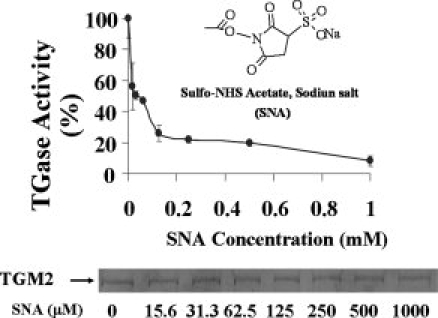
Sulfo-NHS acetate inhibits TGase activity in a concentration-dependent manner. Recombinant TGM2 (0.7 μM) was incubated with 0–1 mM of SNA in HBS (50 mM HEPES, pH 8.5, 100 mM NaCl) at room temperature for 30 min. Following reaction, the reaction mixtures were dialyzed against large excess of dialysis buffer containing 10 mM Tris-Cl, pH 7.5, 10% glycerol, and 1 mM EDTA. After dialysis, equal amounts (∼2 μg) of acetylated TGM2 were loaded on 8–15% SDS-PAGE gel and stained with Coomassie blue (bottom panel). TGase activity was measured using the established BP incorporation assay.19 The data represent the average (± standard deviation) of a total of three independent experiments.
Initial acetylating experiments were carried out using 0–1 mM of SNA and unreacted SNA and/or by products removed by extensive dialysis. We found a concentration-dependent inhibition of TGase activity with an IC50 of ∼35 μM (Fig. 1). At 1 mM SNA, more than 90% of TGase activity was inhibited. The inhibition of TGase activity was not due to a change in solubility because SDS-PAGE revealed the protein remained soluble and confirmed that an equal amount of TGM-2 was present in each of the TGase assays (Fig. 1, bottom panel).
Aspirin (ASA) inhibited TGase activity with an IC50 of ∼400 μM. The apparent higher concentration of ASA needed to inhibit TGM2 could be due in part to either ASA targeting different Lys residue(s), a short-half life and/or lower efficiency in the acetylation reaction. It is known that acetylating efficiency is dependent on the hydrolysis rate of ASA, which is time and temperature dependent.27 A time course study was performed to determine the optimal incubation time for ASA to inhibit TGase activity (Fig. 2). We found there was a two-phase time-dependent inhibition of TGase activity when recombinant TGM2 was incubated with ASA. At 500 μM of ASA, TGase was inhibited rapidly by 80% after 1 hour and then a much slower inhibition to 90% occurred after 16 hours of incubation. The inhibition of TGase activity was not due to a change in TGM2 solubility after acetylation since equal amounts of TGM2 were recovered after dialysis (Fig. 2, bottom panel).
Figure 2.
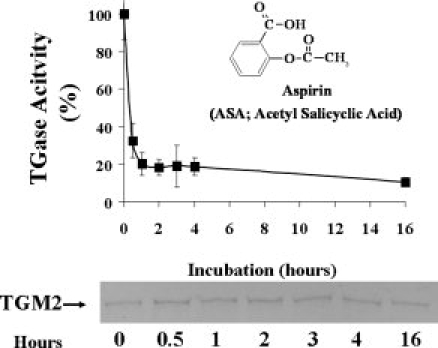
Inhibition of TGase activity by aspirin is time dependent. Recombinant TGM2 (0.7 μM) was incubated with 500 μM of ASA in HEPES-buffered saline (HBS; 50 mM HEPES, pH 8.5, 100 mM NaCl) at room temperature for 0.5, 1, 2, 3, 4, and 16 hours. Following reaction, the reaction mixtures were dialyzed against large excess of dialysis buffer containing 10 mM Tris-Cl, pH 7.5, 10% glycerol, and 1 mM EDTA. After dialysis, equal amounts (∼2 μg) of acetylated TGM2 were loaded on 8–15% SDS-PAGE gel and stained with Coomassie blue (bottom panel). TGase activity was measured using the established BP incorporation assay.19 The data represent the average (±standard deviation) of a total of three independent experiments.
Identification of Lys-residues that are acetylated
To identify Lys residues that were acetylated and to confirm the specificity of the reaction, TGM2 was acetylated with either SNA or ASA and then dialyzed. The dialyzed samples were digested with trypsin and MALDI-TOF MS was performed to identify each acetylated peptide, as described in “Materials and Methods.” The mass of peptides containing an acetylated Lys residue was identified as m/z 42 greater than its unmodified counterpart. When TGM2 was acetylated with 150 μM SNA, Lys173, Lys379, Lys429, Lys468, Lys590, Lys598, Lys600, Lys663, Lys674, and Lys677 were found to be acetylated. In 50% (four determinations) of the samples, Lys74, Lys444 and Lys649 also were found also to be acetylated. When TGM2 was treated with 1 mM ASA, Lys444, Lys468, and Lys663 were found to be acetylated.
Discussion
TGM2 has been considered as a therapeutic target for the treatment of several neurodegenerative diseases.4,28 Current strategies to inhibit TGM2 include the use of peptides and small molecules inhibitors to directly inhibit crosslinking activity.14,29 In this study, we discovered there were critical Lys residues on TGM2 that are susceptible to modification by acetylation, resulting in the loss of TGase crosslinking activity. Our results have important implications in the treatment of neurodegenerative diseases.
The abundance of lysine residues in many proteins and their tendency to be located on surfaces rather than in the interior makes this amino acid an attractive target for modification.30 Furthermore, lysines are critical residues for protein function, protein–protein interactions, chemical conjugation and are targets for post-translational modification.16,17,31,32 Remarkably, several important post-translational reactions, including TGM2-mediated crosslinking, ubiquitination, sumoylation, and acetylation, all target Lys residues.18,32 In some cases, the same Lys residues are modified, but the biological consequences are distinct,18,32 whereas, in other cases, one modification may preclude the other and could have a significant impact on biology. Recent studies indicate that reversible-acetylation of the ɛ-NH3 group of Lys residues occurs not only in histones and transcription factors, but in other cellular proteins involved in cell motility, protein trafficking, immune function, synapse formation and apoptosis.32,33 In this study, we demonstrated that acetylation of critical Lys residue(s) not only will render specific Lys-containing substrates unavailable for TGM2 to perform crosslinking, but also will affect TGM2 crosslinking.
The reactivity of individual Lys residues is highly dependent upon the degree to which they are exposed to the solvent.32 Investigation of three-dimensional structure and surface probability analysis of TGM2 indicate that 30 Lys residues are solvent accessible. Lys265, Lys273, Lys364, and Lys672 appear to occupy surface positions within the catalytic core domain and other exposed Lys residues are located at β-barrel 1 and 2 domains. The results from acetylation demonstrated that SNA and ASA targeted only a few of the same residues, that is, Lys444, Lys468, and Lys663, indicating that they play a role in inhibiting enzyme function. As acetylation of these three Lys residues resulted in a loss of TGase activity after ASA treatment, they are likely to play an important role in regulating TGase activity. The inhibition by acetylation was not due to the presence of excess SNA, ASA or the hydrolysis product, salicylic acid, as they were removed by extensive dialysis.
To investigate how acetylation of the three common lysines (Lys444, Lys468, and Lys663) that reacted with both SNA and ASA might impact enzymatic activity, the positions of these residues were examined in the structure of TGM2 (Figs. 3 and 4). The crystal structure of TGM2 has been determined in two states: an “open” state in complex with a peptide inhibitor that is believed to represent the active form of the enzyme37 and a GDP-bound “closed” form in which the active site is inaccessible and the enzyme is inactive.34 In the closed form, the C-terminal β1 and β2 domains pack against the core (catalytic) domain, but in the open form these interactions are lost and the four domains are arranged in a linear fashion (Fig. 3).
Figure 3.
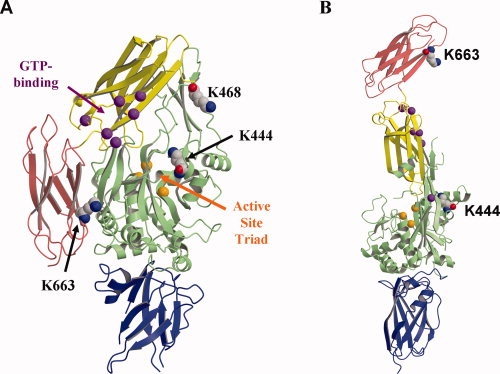
The open and closed forms of TGM2. (A) The “closed”, inactive form of TGM2. (PDB ID: 1KV3) (B) The “open,” active form (PDB ID: 2Q3Z). In both cases, the molecule is shown in ribbon format where each domain colored as follows: N-terminal domain, blue; core domain, green; β1 domain, yellow and β2 domain, red. The lysines acetylated by ASA are shown in space-filling format and labeled. The TGase active site triad (Cys277, His335, and Asp358) is marked by orange spheres corresponding to the alpha carbon residues and, similarly, the alpha carbons of the proposed GTP-binding residues (Lys174, Arg476, Arg478, Met483, Arg580, and Tyr583) are shown as purple spheres.34 These regions are indicated by arrows in (A). Figures 3 and 4 both produced using MOLSCRIPT35 and RASTER3D.36
Figure 4.
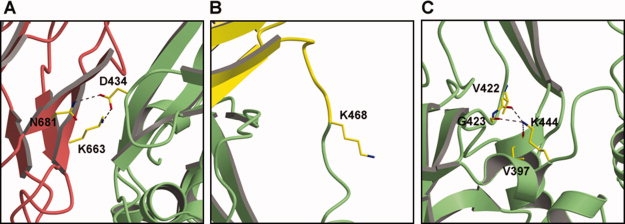
Structural context of the three Lys residues in TGM2 that are acetylated by ASA. (A) Lys663 hydrogen bonds with Asp434 across the β2-core domain interface in the closed form of TGM2. This interaction is absent in the open form [see Fig. 3(B)]. (B) Lys468 is located within a disordered loop of TGM2 that connects the core and β1 domains. Although Lys468 was included in the crystal structure of the closed form of TGM2, the electron density is weak or absent in all molecules of the asymmetric unit, indicating its disorder. (C) Lys444 is relatively buried within the core domain where it forms hydrogen bonds with three main chain carbonyls. Shown here is the closed form of TGM2, but the structure is essentially the same in the open form. For all three panels, the views used are approximately the same as in Figure 3 and the same colors are used for the domains.
In the closed form of TGM2, Lys663, which lies on domain β2, forms a domain–domain interaction through an electrostatic linkage with Asp434 of the core domain [Fig. 4(A)]. A nearby residue, Asn681, also hydrogen bonds with Asp434. When Lys663 is acetylated, the electrostatic linkage with Asp434 would be lost, which may weaken the domain–domain interaction and thus increase the likelihood of the enzyme existing in the open form. How this would lead to inhibition of the enzyme, rather than activation, however, is unclear.
Lys468 is also located in an interesting region of TGM2 [Fig. 4(B)]. This residue lies on a loop that connects the core domain and domain β1 and, in both the open and closed forms of the enzyme, this loop is disordered. This loop appears to act as the hinge between the two forms of the molecule and thus one explanation for enzyme inhibition is that acetylation of Lys468 hinders the hinge motion, thus preventing closed-to-open, that is inactive-to-active, transitions.
The third lysine, Lys444, is located within the core domain and forms hydrogen bonds with carbonyl groups of Val397, Val422, and Gly423. These interactions, which also occur in the open form of the enzyme, may stabilize this region of the molecule. Acetylation of Lys444 would disrupt the hydrogen bonding arrangement and might lead to structural changes. In particular, since the side chain of Val422 is only 6Å from Phe6 of the peptide inhibitor in the structure of the open form of TGM2 (not shown), these changes could alter the architecture of the active site.
Hence, modification of all three residues would likely cause structural perturbations consistent with the lowering of enzyme activity. Elucidating the precise mechanism of each, however, will require more detailed structural studies.
ASA is among the most widely used drugs worldwide.38 ASA is being prescribed as nonsteroid anti-inflammatory medication and the recommended dosage ranges from 75 to 325 mg per day up to 5 g/day. These doses of ASA correspond to plasma concentration of ∼ 0.017 to 15 mM.39 Low and high therapeutic doses of ASA apparently have different targets and applications. The concentrations of ASA used in this study to inhibit TGM2 are within the range of therapeutic dosage and therefore relevant for consideration as therapy to modulate pathologic TGM2 activity. As inflammation plays an important role in the pathogenesis of neurodegenerative diseases, ASA has been used in several in vivo studies for potential benefits in Alzheimer's, Parkinson's, and Huntington's diseases.38,40,41 Although the results are mixed, it remains to be determined whether the optimization of the therapeutic dose of ASA will increase the therapeutic potential in these disorders.40
In summary, we identified critical Lys residues in TGM2 that are important for modulating its crosslinking activity. As TGM2 is a validated target for inhibition in HD and other neurodegenerative diseases, our study offers an alternative therapeutic approach to inhibit TGM2 activity. In addition to the effects on TGM2, the acetylation of the available Lys-residue(s) on polyQ containing proteins such as truncated huntingtin protein in HD disease would prevent them from serving as the Lys-substrates in the TGM2-mediated crosslinking reaction. Two acetylating reagents were used in this study and common Lys residues were targeted by both agents indicating that these Lys residues are a sensitive site for post-translational modification. As post-translation modification events are cell- and location-specific, the manipulation of these events could offer as alternative approaches to specifically inhibit TGM2. Further studies are needed to investigate whether TGM2 and/polyQ proteins are indeed modified by post-translational modification. Our results have potential implication in treating HD and other neurodegenerative diseases.
Materials and Methods
Materials
Streptavidin-alkaline phosphatase, LB medium, and yeast extract were obtained from Invitrogen. 5-biotin-amido-pentylamine (BP) and sulfosuccinimidyl acetate (sulfo-NHS-acetate, SNA) were obtained from Pierce (Rockford, IL). The TG100 monoclonal antibody against TGM2 was purchased from NeoMarker (Labvision, CA). Acetylsalicylic acid (Aspirin, ASA) and all other reagents used in this investigation were purchased from Sigma (St. Louis, MO), unless stated otherwise.
Expression and purification of recombinant TGM2
Recombinant TGM2 was expressed and purified in E. coli as a glutathione S-transferase (GST) fusion protein as described.42 Protein concentrations were quantified using the Bradford method (Bio-Rad). The purified protein was stored in 50 mM Tris-acetate buffer, pH 7.5, at −80°C.
Acetylation of Lys residues using Sulfo-NHS-acetate (SNA) or aspirin (ASA)
SNA stock solution (in water) was freshly prepared and diluted immediately before each reaction. Aspirin was freshly diluted from concentrated stock solution (in 100% ethanol) before each assay. For the acetylating reaction, TGM2 was incubated with different concentrations of SNA or ASA in HBS buffer (50 mM HEPES, pH 8.5, 100 mM NaCl) at room temperature for 1 hour. After reaction, the mixture was dialyzed against 2 × 2 liters of HBS buffer and stored in ice before determining transglutaminase activity.
Matrix-assisted laser desorption/ionization time-of-flight (MALDI-TOF) mass spectrometry (MS)
MALDI/TOF MS was performed at Michael Hooker proteomic core facility, University of North Carolina, Chapel Hill, using the tryptic peptides derived from acetylated TGM2 and Reflex I (Bruker Instruments Co., Bremen, Germany) and Reflex III (Billerica, MA) instruments. Mascot™ was utilized for searching and interpreting the product ion spectra.43 When searching product ion spectra, a mass accuracy of ±0.1 Da in the masses of both precursor and product ions was selected.
Transglutaminase (TGase) BP-incorporation assay
TGase activity was determined by quantifying the incorporation of 5-biotinamidopentylamine (BP) into N, N′-dimethylcasein in the presence of recombinant TGM2 and 10 mM Ca+2 in a microtiter plate as described previously.19 The amount of BP incorporated into the N, N′-dimethylcasein was determined after a 45-min incubation at 37°C and color developed using streptavidin-conjugated alkaline phosphatase and PNPP.19 For IC50 determination, all assays were determined in the presence of 1 mM BP.
SDS-PAGE and immunoblotting
After SDS-polyacrylamide gel electrophoresis (PAGE) and transfer to a nitrocellulose membrane, the bands were detected using a monoclonal antibody against TGM2 and visualized using Super Signal West Pico Luminol (Pierce). For experiments involving biotinylated pentylamine (BP), the bands were also detected using streptavidin-peroxidase and visualized by Super Signal West Pico Luminol (Pierce).
Acknowledgments
The authors thank Dr. David C. Sane at Wake Forest Medical Center for critical reading of this manuscript. The technical assistance of Kevin Peng and Brett Rollins was greatly appreciated.
References
- 1.Folk JE. Mechanism and basis for specificity of transglutaminase-catalyzed epsilon-(gamma-glutamyl) lysine bond formation. Advam Enzymol Rel Areas Mol Biol. 1983;54:1–56. doi: 10.1002/9780470122990.ch1. [DOI] [PubMed] [Google Scholar]
- 2.Lorand L, Graham RM. Transglutaminases: crosslinking enzymes with pleiotropic functions. Nature Rev. 2003;4:140–156. doi: 10.1038/nrm1014. [DOI] [PubMed] [Google Scholar]
- 3.Greenberg CS, Birckbichler PJ, Rice RH. Transglutaminases: multifunctional cross-linking enzymes that stabilize tissues. FASEB J. 1991;5:3071–3077. doi: 10.1096/fasebj.5.15.1683845. [DOI] [PubMed] [Google Scholar]
- 4.Wilhelmus MM, van Dam AM, Drukarch B. Tissue transglutaminase: a novel pharmacological target in preventing toxic protein aggregation in neurodegenerative diseases. Eur J Pharmacol. 2008;585:464–472. doi: 10.1016/j.ejphar.2008.01.059. [DOI] [PubMed] [Google Scholar]
- 5.Mastroberardino PG, Iannicola C, Nardacci R, Bernassola F, De Laurenzi V, Melino G, Moreno S, Pavone F, Oliverio S, Fesus L, Piacentini M. ‘Tissue’ transglutaminase ablation reduces neuronal death and prolongs survival in a mouse model of Huntington's disease. Cell Death Differ. 2002;9:873–880. doi: 10.1038/sj.cdd.4401093. [DOI] [PubMed] [Google Scholar]
- 6.Bailey CD, Johnson GV. Tissue transglutaminase contributes to disease progression in the R6/2 Huntington's disease mouse model via aggregate-independent mechanisms. J Neurochem. 2005;92:83–92. doi: 10.1111/j.1471-4159.2004.02839.x. [DOI] [PubMed] [Google Scholar]
- 7.Dedeoglu A, Kubilus JK, Jeitner TM, Matson SA, Bogdanov M, Kowall NW, Matson WR, Cooper AJ, Ratan RR, Beal MF, Hersch SM, Ferrante RJ. Therapeutic effects of cystamine in a murine model of Huntington's disease. J Neurosci. 2002;22:8942–8950. doi: 10.1523/JNEUROSCI.22-20-08942.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jeitner TM, Delikatny EJ, Ahlqvist J, Capper H, Cooper AJ. Mechanism for the inhibition of transglutaminase 2 by cystamine. Biochem Pharmacol. 2005;69:961–970. doi: 10.1016/j.bcp.2004.12.011. [DOI] [PubMed] [Google Scholar]
- 9.Karpuj MV, Becher MW, Springer JE, Chabas D, Youssef S, Pedotti R, Mitchell D, Steinman L. Prolonged survival and decreased abnormal movements in transgenic model of Huntington disease, with administration of the transglutaminase inhibitor cystamine. Nat Med. 2002;8:143–149. doi: 10.1038/nm0202-143. [DOI] [PubMed] [Google Scholar]
- 10.Lai TS, Tucker T, Burke JR, Strittmatter WJ, Greenberg CS. Effect of tissue transglutaminase on the solubility of proteins containing expanded polyglutamine repeats. J Neurochem. 2004;88:1253–1260. doi: 10.1046/j.1471-4159.2003.02249.x. [DOI] [PubMed] [Google Scholar]
- 11.Segers-Nolten IM, Wilhelmus MM, Veldhuis G, van Rooijen BD, Drukarch B, Subramaniam V. Tissue transglutaminase modulates alpha-synuclein oligomerization. Protein Sci. 2008;17:1395–1402. doi: 10.1110/ps.036103.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Konno T, Morii T, Hirata A, Sato S, Oiki S, Ikura K. Covalent blocking of fibril formation and aggregation of intracellular amyloidgenic proteins by transglutaminase-catalyzed intramolecular cross-linking. Biochemistry. 2005;44:2072–2079. doi: 10.1021/bi047722d. [DOI] [PubMed] [Google Scholar]
- 13.Tucholski J, Kuret J, Johnson GV. Tau is modified by tissue transglutaminase in situ: possible functional and metabolic effects of polyamination. J Neurochem. 1999;73:1871–1880. [PubMed] [Google Scholar]
- 14.Lai TS, Liu Y, Tucker T, Daniel KR, Sane DC, Toone E, Burke JR, Strittmatter WJ, Greenberg CS. Identification of chemical inhibitors to human tissue transglutaminase by screening existing drug libraries. Chem Biol. 2008;15:969–978. doi: 10.1016/j.chembiol.2008.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hausch F, Halttunen T, Maki M, Khosla C. Design, synthesis, and evaluation of gluten peptide analogs as selective inhibitors of human tissue transglutaminase. Chem Biol. 2003;10:225–231. doi: 10.1016/s1074-5521(03)00045-0. [DOI] [PubMed] [Google Scholar]
- 16.Han KK, Martinage A. Post-translational chemical modification(s) of proteins. Int J Biochem. 1992;24:19–28. doi: 10.1016/0020-711x(92)90225-p. [DOI] [PubMed] [Google Scholar]
- 17.Herr FM, Matarese V, Bernlohr DA, Storch J. Surface lysine residues modulate the collisional transfer of fatty acid from adipocyte fatty acid binding protein to membranes. Biochemistry. 1995;34:11840–11845. doi: 10.1021/bi00037a023. [DOI] [PubMed] [Google Scholar]
- 18.Freiman RN, Tjian R. Regulating the regulators: Lysine modifications make their mark. Cell. 2003;112:11–17. doi: 10.1016/s0092-8674(02)01278-3. [DOI] [PubMed] [Google Scholar]
- 19.Slaughter TF, Achyuthan KE, Lai TS, Greenberg CS. A microtiter plate transglutaminase assay utilizing 5-(biotinamido)pentylamine as substrate. Analyt Biochem. 1992;205:166–171. doi: 10.1016/0003-2697(92)90594-w. [DOI] [PubMed] [Google Scholar]
- 20.Turner BT, Jr, Sabo TM, Wilding D, Maurer MC. Mapping of factor XIII solvent accessibility as a function of activation state using chemical modification methods. Biochemistry. 2004;43:9755–9765. doi: 10.1021/bi049260+. [DOI] [PubMed] [Google Scholar]
- 21.Wang X, Kim SH, Ablonczy Z, Crouch RK, Knapp DR. Probing rhodopsin-transducin interactions by surface modification and mass spectrometry. Biochemistry. 2004;43:11153–11162. doi: 10.1021/bi049642f. [DOI] [PubMed] [Google Scholar]
- 22.Villanueva GB, Allen N. Acetylation of antithrombin III by aspirin. Semin Thromb Hemost. 1986;12:213–215. doi: 10.1055/s-2007-1003553. [DOI] [PubMed] [Google Scholar]
- 23.Xu AS, Labotka RJ, London RE. Acetylation of human hemoglobin by methyl acetylphosphate. Evidence of broad regio-selectivity revealed by NMR studies. J Biol Chem. 1999;274:26629–26632. doi: 10.1074/jbc.274.38.26629. [DOI] [PubMed] [Google Scholar]
- 24.Xu AS, Macdonald JM, Labotka RJ, London RE. NMR study of the sites of human hemoglobin acetylated by aspirin. Biochim Biophys Acta. 1999;1432:333–349. doi: 10.1016/s0167-4838(99)00094-1. [DOI] [PubMed] [Google Scholar]
- 25.He S, Blomback M, Yoo G, Sinha R, Henschen-Edman AH. Modified clotting properties of fibrinogen in the presence of acetylsalicylic acid in a purified system. Ann NY Acad Sci. 2001;936:531–535. doi: 10.1111/j.1749-6632.2001.tb03540.x. [DOI] [PubMed] [Google Scholar]
- 26.Walker JE. Lysine residue 199 of human serum albumin is modified by acetylsalicyclic acid. FEBS Lett. 1976;66:173–175. doi: 10.1016/0014-5793(76)80496-6. [DOI] [PubMed] [Google Scholar]
- 27.Habib MJ, Rogers JA. Hydrolysis and stability of acetylsalicylic acid in stearylamine-containing liposomes. J Pharm Pharmacol. 1993;45:496–499. doi: 10.1111/j.2042-7158.1993.tb05586.x. [DOI] [PubMed] [Google Scholar]
- 28.De Vivo G, Gentile V. Transglutaminase-catalyzed post-translational modifications of proteins in the nervous system and their possible involvement in neurodegenerative diseases. CNS Neurol Disord Drug Targets. 2008;7:370–375. doi: 10.2174/187152708786441821. [DOI] [PubMed] [Google Scholar]
- 29.Siegel M, Khosla C. Transglutaminase 2 inhibitors and their therapeutic role in disease states. Pharm Therap. 2007;115:232–245. doi: 10.1016/j.pharmthera.2007.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rose GD, Geselowitz AR, Lesser GJ, Lee RH, Zehfus MH. Hydrophobicity of amino acid residues in globular proteins. Science. 1985;229:834–838. doi: 10.1126/science.4023714. [DOI] [PubMed] [Google Scholar]
- 31.Hanai R, Wang JC. Protein footprinting by the combined use of reversible and irreversible lysine modifications. Proc Natl Acad Sci USA. 1994;91:11904–11908. doi: 10.1073/pnas.91.25.11904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yang XJ, Seto E. Lysine acetylation: codified crosstalk with other posttranslational modifications. Mol Cell. 2008;31:449–461. doi: 10.1016/j.molcel.2008.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cohen T, Yao TP. AcK-knowledge reversible acetylation. Sci STKE. 2004:pe42. doi: 10.1126/stke.2452004pe42. 2004. [DOI] [PubMed] [Google Scholar]
- 34.Liu S, Cerione RA, Clardy J. Structural basis for the guanine nucleotide-binding activity of tissue transglutaminase and its regulation of transamidation activity. Proc Natl Acad Sci USA. 2002;99:2743–2747. doi: 10.1073/pnas.042454899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kraulis PJ. MOLSCRIPT: a program to produce both detailed and schematic plots of protein structures. J Appl Cryst. 1991;24:946–950. [Google Scholar]
- 36.Merritt EA, Murphy ME. Raster3D Version 2.0. A program for photorealistic molecular graphics. Acta Cryst D. 1994;50:869–873. doi: 10.1107/S0907444994006396. [DOI] [PubMed] [Google Scholar]
- 37.Pinkas DM, Strop P, Brunger AT, Khosla C. Transglutaminase 2 undergoes a large conformational change upon activation. PLoS Biol. 2007;5:e327. doi: 10.1371/journal.pbio.0050327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Asanuma M, Miyazaki I. Nonsteroidal anti-inflammatory drugs in experimental parkinsonian models and Parkinson's disease. Curr Pharm Des. 2008;14:1428–1434. doi: 10.2174/138161208784480153. [DOI] [PubMed] [Google Scholar]
- 39.Rowland M, Riegelman S, Harris PA, Sholkoff SD. Absorption kinetics of aspirin in man following oral administration of an aqueous solution. J Pharm Sci. 1972;61:379–385. doi: 10.1002/jps.2600610312. [DOI] [PubMed] [Google Scholar]
- 40.Aisen PS. An aspirin a day for Alzheimer's disease? Lancet Neurol. 2008;7:20–21. doi: 10.1016/S1474-4422(07)70296-X. [DOI] [PubMed] [Google Scholar]
- 41.Norflus F, Nanje A, Gutekunst CA, Shi G, Cohen J, Bejarano M, Fox J, Ferrante RJ, Hersch SM. Anti-inflammatory treatment with acetylsalicylate or rofecoxib is not neuroprotective in Huntington's disease transgenic mice. Neurobiol Dis. 2004;17:319–325. doi: 10.1016/j.nbd.2004.07.011. [DOI] [PubMed] [Google Scholar]
- 42.Lai TS, Slaughter TF, Peoples KA, Hettasch JM, Greenberg CS. Regulation of human tissue transglutaminase function by magnesium-nucleotide complexes. Identification of distinct binding sites for Mg- GTP and Mg-ATP. J Biol Chem. 1998;273:1776–1781. doi: 10.1074/jbc.273.3.1776. [DOI] [PubMed] [Google Scholar]
- 43.Perkins DN, Pappin DJ, Creasy DM, Cottrell JS. Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis. 1999;20:3551–3567. doi: 10.1002/(SICI)1522-2683(19991201)20:18<3551::AID-ELPS3551>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]