

Abstract
Protein aggregation into insoluble fibrillar structures known as amyloid characterizes several neurodegenerative diseases, including Alzheimer's, Huntington's and Creutzfeldt-Jakob. Transthyretin (TTR), a homotetrameric plasma protein, is known to be the causative agent of amyloid pathologies such as FAP (familial amyloid polyneuropathy), FAC (familial amyloid cardiomiopathy) and SSA (senile systemic amyloidosis). It is generally accepted that TTR tetramer dissociation and monomer partial unfolding precedes amyloid fibril formation. To explore the TTR unfolding landscape and to identify potential intermediate conformations with high tendency for amyloid formation, we have performed molecular dynamics unfolding simulations of WT-TTR and L55P-TTR, a highly amyloidogenic TTR variant. Our simulations in explicit water allow the identification of events that clearly discriminate the unfolding behavior of WT and L55P-TTR. Analysis of the simulation trajectories show that (i) the L55P monomers unfold earlier and to a larger extent than the WT; (ii) the single α-helix in the TTR monomer completely unfolds in most of the L55P simulations while remain folded in WT simulations; (iii) L55P forms, early in the simulations, aggregation-prone conformations characterized by full displacement of strands C and D from the main β-sandwich core of the monomer; (iv) L55P shows, late in the simulations, severe loss of the H-bond network and consequent destabilization of the CBEF β-sheet of the β-sandwich; (v) WT forms aggregation-compatible conformations only late in the simulations and upon extensive unfolding of the monomer. These results clearly show that, in comparison with WT, L55P-TTR does present a much higher probability of forming transient conformations compatible with aggregation and amyloid formation.
Keywords: amyloid, amyloidogenic intermediates, molecular dynamics, protein misfolding, protein unfolding, transthyretin, L55P-TTR
Introduction
Protein aggregation into insoluble ordered structures known as amyloid has been shown to occur in more than 20 distinct proteins.1 Probably, for most of the amyloidoses, in particular extracellular amyloidoses, amyloid formation is not due to errors in the normal folding pathway of a protein but to decreased structural stability, unfolding events or even conformational fluctuations around the native structure ensemble, allowing for alternative stable conformations. Mutations, in particular, seem to play a critical role in the decrease of protein conformational stability and in triggering unfolding events.2
Amyloid fibril formation has been linked to several pathologies ranging from Huntington's disease and Creutzfeldt-Jakob, with variable age of onset, to Alzheimer's, Parkinson's and other neurodegenerative diseases affecting mostly the elderly.1 Familial amyloid polyneuropathy (FAP) and senile systemic amyloidosis (SSA) account for another group of such diseases. FAP is characterised by impairment of temperature and pain sensation in the feet, at early stages of the disease, which then evolves to autonomic dysfunction and, in many cases, to patient death.3 Costa and coworkers first identified transthyretin (TTR) as the principal protein constituent of amyloid fibrils in Portuguese patients with FAP.4
Human TTR is mainly produced by the liver and secreted as a 55 kDa homotetramer into the plasma, and its function has been linked to thyroxine and retinol transport. More than 80 mutations of TTR have been reported, most of them amyloidogenic.5 Amongst TTR variants, V30M is the most prevalent and L55P yields the most aggressive symptoms.6 The 3D structures of the wild-type form of TTR (WT-TTR) and several of its variants have been solved to high-resolution by X-ray crystallography (see reference7 and references therein). Remarkably, the crystal structures of the TTR variants are, in general, similar to that of the wild-type protein. TTR is composed by four subunits with 127 residues each, containing two four-strand β-sheets packed in a β-sandwich topology and mainly stabilized by hydrogen bonds and hydrophobic contacts (Fig. 1). Experimental evidences based on aging experiments of tetrameric TTR and chemically induced unfolding experiments, revealed that tetramer dissociation into compact monomers with low conformational stability precedes amyloid fibril formation.8 It has also become apparent that amyloidogenic single-point mutations influence not only TTR dissociation kinetics9 but yield monomers with lower thermodynamic stability,8 in spite of the high structural similarity between the mutants and the wild-type protein.7 Additionally, amyloidogenic TTR variants were shown to produce, in solution and even at neutral pH, higher amounts of partially unfolded monomeric species with enhanced propensity for ordered aggregation into amyloid fibrils.10,11 Therefore, the conformational stability of the TTR monomers undoubtedly plays a critical role in the process of amyloid formation.
Figure 1.
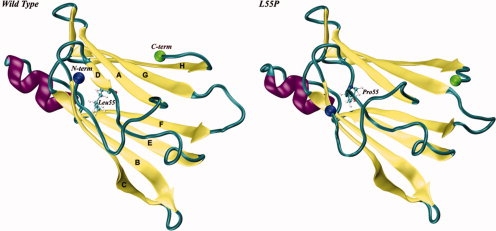
Schematic representation of the X-ray crystal structures of wild type (on the left) and L55P (on the right) subunits of human transthyretin (TTR; PDB entries 1TTA36 and 5TTR,52 respectively). Residue 55, located in β-strand D, is represented in balls-and-sticks. The N- and C-termini are represented by two spheres. [Color figure can be viewed in the online issue, which is available at www.interscience.wiley.com.]
Among the structural changes necessary for amyloid formation by TTR, the displacement of the D-strand-loop-C-strand segment (see Fig. 1) from its native position is believed to be essential in order to expose a new interface comprised by strands A and B, necessary for monomer assembly into aggregates.12–14 Site-directed spin labeling combined with electron paramagnetic resonance (EPR)15 and hydrogen/deuterium (H/D) exchange studies by nuclear magnetic resonance (NMR) spectroscopy have supported this hypothesis16 and, recently, a computational model of an amyloid protofilament of TTR was proposed17 where proper subunit-subunit docking was only achieved if the D-strand-loop-C-strand segment was fully displaced from the β-sandwich core of the TTR subunit.
Molecular dynamics (MD) simulations may be used to study in detail conformational changes in atomic models of proteins and to gain insight into time- and environment-dependent events, such as protein folding and unfolding processes. Despite the great increase of computational power seen in the past decade, it is still unpractical to simulate, in reasonable times, an all-atom model of a protein in explicit solvent for more than tens of nanoseconds, while both folding and unfolding events typically take milliseconds, seconds or more to occur.18–22 Although some groups have proposed promising strategies to track the protein folding pathways by computer simulations, computer limitations have made protein unfolding more tractable than protein folding.23–27 In fact, simulation protocols making use of high temperature do increase the rate of these processes and in addition provide unfolding conditions. As pointed out by Finkelstein,28 though, care must be taken while extrapolating MD results obtained at extreme unfolding conditions to the protein folding problem. Even so, unfolding simulations at high temperature allow for the exploration of the conformational space available to a protein at several points of the unfolding process, supplying atomic-scale details to experimental results of protein unfolding.29
Previously, based on a smaller set of TTR MD unfolding data, we have discussed the importance of collapsed denaturated states in sampling alternative stable conformations for the TTR polypeptide chain.30 Here, based on a larger set of multiple MD unfolding simulations at high temperature and in explicit solvent, we investigate the conformational stability and the unfolding behavior of WT-TTR and its highly amyloidogenic variant L55P-TTR. Employing classical and geometry-based methods as well as several property-fluctuation analyses and cluster analysis, we shed new light on the molecular mechanism of amyloidogenesis by TTR and extend the observations by other authors.31–34 Our simulations corroborate experimental observations correlating the conformational stability of monomeric TTR species and their amyloidogenic potential,8 and highlight some of the amino acid residues that may have a relevant role in the early stages of amyloid formation. Additionally, our simulations allow the identification of unfolding events leading to aggregation-prone intermediates, and thus allow the discrimination between amyloidogenic and nonamyloidogenic behavior.
Results
To better characterize the unfolding landscape of transthyretin monomeric species and its relation to amyloid formation, we have performed five independent 10-ns long MD simulations, at 500 K, of WT-TTR, the naturally occurring form of the protein, and L55P-TTR, a highly amyloidogenic variant. In the following paragraphs, a detailed analysis of these simulations is presented.
Analysis of global structural properties
The root mean square deviation (RMSD) provides an overall measure of the departure of the structures from the initial X-ray Cartesian coordinates, at each frame of the MD trajectory, which may be valuable for preliminary estimation of the effect of mutations over the conformational stability of one protein. However, differences in the RMSD values among multiple unfolding trajectories must be carefully interpreted. In many instances, high RMSDs between structures may suggest the exploration of different regions of the protein conformational landscape, however careful analysis of changes in experimentally observable properties for these structures may be considerably smaller, suggesting one single dominant pathway.35 The Cα-RMSD variation from the crystal structures of WT- and L55P-TTR along each simulation is reported in Figure 2. At physiological temperature (310 K), both TTR variants show small fluctuations throughout the 10 ns simulations, with values below 2 Å, and therefore agreeing with previous results on native-state simulations at room temperature.31 High temperature, on the other hand, clearly induces the unfolding of all replicas, despite the distinct Cα-RMSD profiles observed. Actually, while comparable patterns are observed along the first nanosecond of simulation among both TTR variants, in general L55P-TTR undergoes a faster increase in RMSD than WT-TTR. This difference becomes more obvious after the 2.5 ns time point. After 6.2 ns, two trajectories of WT-TTR – runs 1 and 4 – and other two of L55P-TTR – runs 1 and 2 – exhibit an analogous trend characterized by rapid-increasing and fluctuating RMSD until the end of the simulation, whereas the remaining three trajectories present lower RMSD variations. The mean Cα-RMSD over the 50,000 snapshots of the five unfolding simulations is 5.2 ± 2.7 Å for WT-TTR and 7.2 ± 3.9 Å for L55P-TTR, showing on average a larger departure from the initial structure in the case of L55P-TTR.
Figure 2.
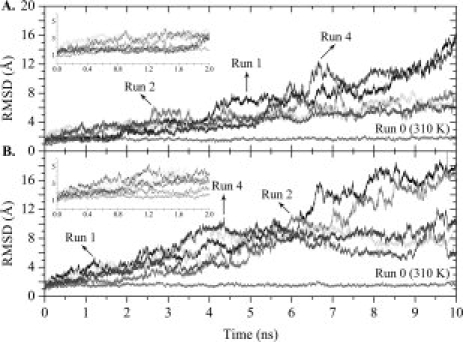
Variation along the simulation trajectories of the Cα atom root mean square deviation (RMSD) with respect to the crystal structure. RMSDs were calculated over all residues except the N- and C-terminal tails. Control trajectories performed at 310 K are labelled accordingly. Results for 2 × 5 unfolding simulations at 500 K for WT-TTR (A) and L55P-TTR (B) are presented. The insets represent expansions of the first 2 ns of simulation.
Figure 3 shows the Cα atoms root mean square fluctuation (RMSF) per residue, compared with the RMSF values derived from the crystallographic B-factors of WT-TTR,36 using the relationship RMSF = (3B/8π2)1/2.37,38 The largest values of RMSF correspond mainly to the N- and C-terminal tails of the peptide chain and to loop regions. It is clear that both WT-TTR and L55P-TTR present modest and nearly superimposable fluctuations for the equilibrium simulations, at 310 K. On the contrary, L55P-TTR shows significantly larger fluctuations than WT-TTR for all residues, during the unfolding process at 500 K. In L55P-TTR, it is observed a somewhat expected broadening of the peak between residues 47 and 58, centred on the β-bulge region initiated by residue 55. This may be because the hydrogen bonding network near the mutation site is extensively disrupted even in the native structure of L55P-TTR. Additionally, in L55P-TTR, larger fluctuations extend to the DE loop, namely from residue 56 to 59. Intriguingly, a sharp increase on RMSF is also observed in the region of residues 75 to 88, which is primarily constituted by the only α-helix in the structure.
Figure 3.
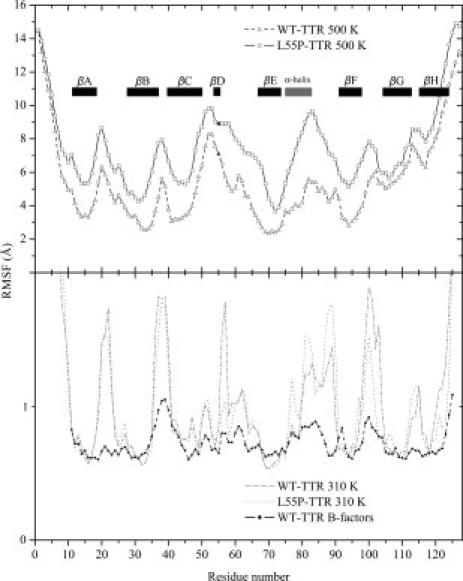
Cα atom root mean square fluctuation (RMSF) as a function of residue number and calculated over all trajectories at 500 K (top panel) and 310 K (bottom panel) for WT-TTR (hollow triangles) and L55P-TTR (hollow squares). Residue 55 is colored in black. All 127 residues were considered and the secondary structure motifs are identified by eight black rectangles corresponding to the β-strands, and a gray rectangle corresponding to the α-helix. The line with black circles (bottom panel) represents the fluctuations derived from the crystallographic B-factors of WT-TTR (PDB entry 2QGB). For clarity, RMSF values in the top and bottom panels were represented with different scales.
Structural compactness along the MD unfolding simulations may be assessed by parameters such as the radius of gyration (Rgyr) or the solvent accessible surface area (SASA). Despite the rather erratic variation of the Rgyr along the 10 ns simulations, several highly expanded and several collapsed protein conformations are sampled along the unfolding pathways of both WT-TTR and L55P-TTR (Supporting Information, Fig. S1). It is interesting to contrast these results with the variation in SASA of the TTR hydrophobic residues (Supporting Information, Fig. S2). The progressive increase in solvent exposure of all types of residues is an expected outcome of the denaturation process. Nonpolar residues, in particular, represent a key driving force for protein folding and also a pivotal basis for protein thermodynamic stability. The simulations show that on average the amyloidogenic mutant exposes its hydrophobic residues to a larger extent when compared to WT-TTR, starting right from the first nanosecond of simulation (Fig. S2 in Supporting Information).
To test the idea that strands C and D must be displaced from the TTR monomer β-sandwich for amyloid formation to occur,12–17 we have also inspected the variation of SASA for the side-chains belonging to residues in strands A and B, against that of the side-chains of all other residues in the TTR monomer (Fig. 4). These two selections present similar variation profiles (regardless of the different scaling), changing at comparable rates within each variant. As seen for RMSD and Rgyr variations, two simulations of WT-TTR — runs 1 and 4 — and other two of L55P-TTR — runs 1 and 2 — present significantly larger property variations than the remaining three runs. Interestingly, the differences are more evident for strands A and B in L55P-TTR where several peaks are observed in the SASA variation profiles (Fig. 4), suggesting that runs 1, 2, and 4 of L55P may be sampling the most amyloidogenic pathways. Moreover, the L55P variant is more prone to expose strands A and B, starting as early as the fourth nanosecond of simulation in run 4.
Figure 4.
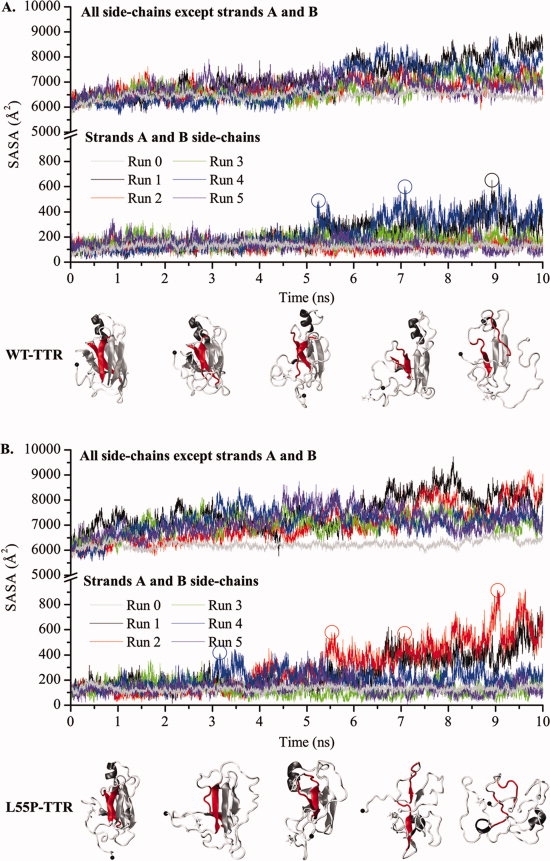
Variation of the solvent accessible surface area (SASA) of the monomers' side-chains along MD simulations of WT-TTR (A) and L55P-TTR (B). The SASA of residues located in β-strands A and B (lower panels) and of all other residues (upper panels) are represented separately. Runs 0 are control simulations performed at 310 K. All other simulations are unfolding simulations performed at 500 K. Protein conformations shown underneath the plots correspond to SASA increases at different stages of the simulations. Strands A and B are highlighted in red.
Whether the mutation in position 55 produces conformational instability exclusively at the β-sandwich edge-region encompassed by the D-strand-loop-C-strand segment or exerts global effects over the monomeric structure of TTR is a crucial question, for it may conceal the underlying mechanisms by which remote and apparently unrelated point mutations may yield amyloid aggregates sharing a common molecular architecture. To this end, we have also calculated the relative difference in SASA per residue between the L55P variant and WT-TTR, across all probed conformations (Fig. 5). The results suggest that the L55P mutation is capable of inducing an odd increase in solvent exposure of the majority of the TTR residues, as illustrated in Figure 5 by the positive displacements. Most of the observed negative and null differences correspond to residues located either in turns and coil regions or in strands G and H. Strands B, E, and F and residue 42 in strand C exhibit the most significant overall increases, followed by the α-helix and strand A, whose residues all present a positive increase. In particular, the mutated residue Pro55, located in strand D, appears ∼2.9 times more exposed than Leu55 in WT-TTR. Residues His31, Phe33, Arg34, Glu42, Tyr69, Lys70, Val93, and Phe95 show, on average, tens of times larger SASA in L55P-TTR than their counterparts in WT-TTR, thereby accounting for the most dramatic differences in solvent exposure patterns. The hypothetical role of these residues will be discussed in the following sections.
Figure 5.
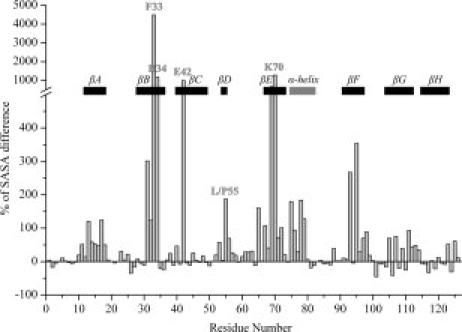
Relative difference in SASA per residue (% of SASA difference) between L55P-TTR and WT-TTR, across all sampled conformations. The secondary structure motifs in native TTR are identified by eight black rectangles corresponding to the beta-strands, and a gray rectangle corresponding to the alpha-helix. Additionally, residue 55 and all other residues displaying exceptionally high SASA increases are labeled.
Analysis of secondary structure, native contacts, and H-bonds
The changes in protein secondary structure along control and unfolding simulations are reported in Figure 6. In the crystal structure of WT-TTR, 55.1% of the amino acid residues are present in regular secondary structure, while in L55P-TTR this percentage equals 52.8%. As expected, both in the WT- and L55P-TTR equilibrium simulations [Fig. 6(A, G)] the secondary structure motifs are well maintained over the 10 ns of simulation. However, for the simulations carried out at 500 K, highly unfolded conformations are sampled in some of the runs of WT-TTR and L55P-TTR (Fig. 6). The loss of secondary structure upon thermal unfolding occurs earlier and to a larger extent in the simulations of the L55P variant [Fig. 6(H–L)] than in those of the wild–type form of TTR [Fig. 6(B–F)]. While in the control simulations the percentage of residues in regular secondary structure is only slightly inferior to that of the crystal structures, 49.7 ± 1.5% and 49.4 ± 1.1% in WT- and L55P-TTR, respectively, in the unfolding simulations a much larger decrease is found when analysing the average percentage of residues in regular secondary structure over all the simulations. The average regular secondary structure content for all simulations of WT-TTR is 34.5 ± 9.7%, but only 28.9 ± 10.8% for L55P-TTR.
Figure 6.

Secondary structure variation along MD simulations of WT- and L55P-TTR. (A): WT-TTR, control run (at 310 K); (B–F): WT-TTR, runs 1 to 5; (G): L55P-TTR, control run (at 310 K); (H–L): L55P-TTR, runs 1 to 5. All runs, except the control runs, were performed at 500 K. Secondary structure was assigned with the program STRIDE.65 Residues in β-sheet are shown in gray, helical motifs are represented in dark gray, and residues in nonregular or other secondary structure are shown in white.
A closer look at the plots in Figure 6 points out interesting details on the structural stability of the individual elements of secondary structure. As mentioned earlier, each TTR monomer has a β-sandwich fold composed of two four-stranded β-sheets labeled DAGH and CBEF (Fig. 1). β-strands A and B present comparable overall stability in WT-TTR and L55P-TTR multiple trajectories, though strand B is more stable than strand A. Strand C shows good persistence in the simulations of WT-TTR. However, in L55P-TTR, strand C completely unfolds in 2 of the 5 simulations. Although the short strand D — formed by residues 54 and 55 — is clearly defined during the unfolding trajectories of WT-TTR, and only absent at the end of runs 1 and 4, it remains unstructured over all simulations of L55P-TTR, just like in the crystal structure. Strand E is completely lost in the second run of L55P-TTR and in no other simulation, thereby standing as the most stable β-sheet-forming sequence. By contrast, strand F and mainly strand H appear to be the most sensitive to thermal unfolding conditions, in both variants of TTR. Concerning the stability of the α-helical structure formed by residues 75–83, in the trajectories of WT-TTR the helix is normally maintained until the end of the simulations. However, as anticipated in the RMSF plots (Fig. 3), in L55P-TTR the α-helix experiences important conformational conversions and is only maintained in runs 2 and 5, being replaced by coils and turns around the first nanosecond in the other three runs.
Sets of 241 and 228 interresidue native contacts were first defined from the crystal structures of WT- and L55P-TTR, respectively, as described in the “Methods” section. The difference in the number of contacts between the two proteins is mostly due to the first few residues, as the N-terminal unstructured tail of WT-TTR participates in a number of contacts. For the rest of the sequence, and focusing on the contacts involving the β-strands, the number of native contacts defined for the initial structures of WT-TTR and L55P-TTR is similar. These two sets of contacts were followed along the 10 ns of each simulation to identify meaningful trends among the simulation replicas and/or to recognize consistent differences between the two TTR variants. Contact matrices were rendered with this purpose (Fig. 7). For the control runs, the number of native contacts remains roughly constant throughout the simulations. At 500 K however, we observe a gradual loss of native contacts over time, generally more severe for L55P-TTR than for wild type. This global tendency is even clearer for WT-TTR runs 1 and 4, and for runs 1 and 2 in L55P-TTR, which is in agreement with what was observed for the global structural properties reported in the previous section. Perhaps the most intriguing observations out of the native contacts analysis come out of the identification of those contacts which are more preserved throughout the 10 ns of each simulation. For example, contacts between residues Phe33-Lys70 (contact number 133) and Arg34-Glu42 (contact number 137) are kept across all simulations of WT-TTR at 500 K. This behavior does not apply to the L55P-TTR simulations, where these contacts (here numbered 119 and 122, correspondingly) are much less persistent, and almost unseen in runs 1 and 2. An even more conspicuous divergence can be found for contacts Val32-Phe44 (number 124) and Phe33-Glu42 (number 128), connecting strands B and C; contact Phe33-Tyr69 (number 132), linking strands B and E; contact Arg34-Trp41 (number 136) between strands B and C; and finally contact Tyr69-Phe95 (number 174), between strands E and F. All these contacts are well maintained in all the simulations of WT-TTR, yet significantly disrupted in runs 1 and 2 of L55P-TTR. Most of these residues correspond to those presenting the largest differences between their average solvent exposure in all simulations of the wild type and L55P-TTR (Fig. 5). Possible similarities among contacts with low prevalence levels have also been checked, excluding those relating to residues located in the terminal tails of the monomer (and thus, undertaking constant fluctuations). Although good consistency is seen among the replicas of each variant, no common drifts are detected between WT and L55P-TTR, and most of the contacts with low prevalence involve residues located in strand A, loops and coils, and the helical motif.
Figure 7.
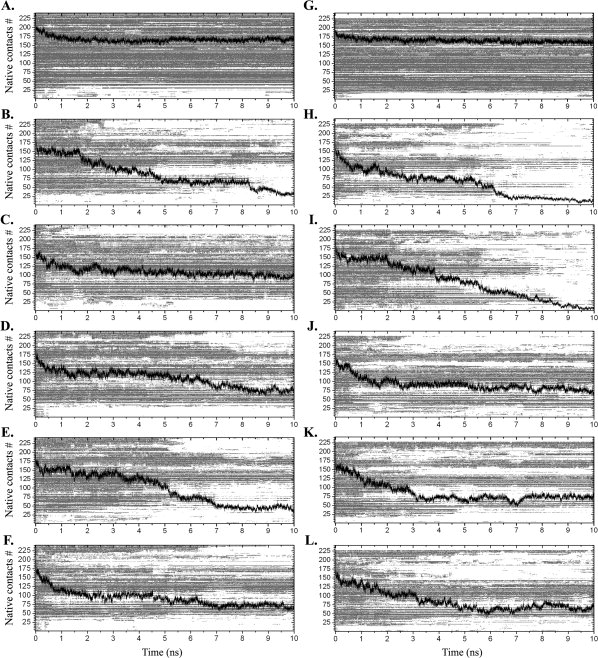
Persistence of native contacts along MD simulations of WT-TTR (on the left) and L55P-TTR (on the right), for the control runs at 310 K (panels A and G) and for unfolding runs 1 to 5, at 500 K (panels B–F and H–L). Black traces represent the total number of enduring native contacts along the simulations. The light gray matrices on the background illustrate the persistence of each contact along the simulation.
Because of the importance of hydrogen bonds between backbone amide groups in the β-sandwich structural motif, we have studied the changes in the hydrogen bond network and its behaviour along the unfolding trajectories (Table I). Hydrogen bonds were defined according to the geometrical criteria presented in the “Methods” section, resulting in 37 interstrand H-bonds involved in the two β-sheets in the crystal structure of WT-TTR. The mutated monomer presents a markedly disrupted H-bond network with only 26 H-bonds defined between β-strands. The absent bonds in L55P-TTR are identified as dotted-line arrows in the schematic drawing provided in Figure S3 of the Supporting Information. Most of these H-bonds lie between β-strands C and B and between strands G and H. Moreover, the H-bonds between strand D (holding the mutation site) and strand A are also absent, as Pro55 is not capable of establishing H-bonds with Val14. The variation of the total number of hydrogen bonds along the trajectories at 500 K may be directly related with changes in interresidue contacts and secondary structure. During the unfolding process, native contacts are broken and helices and strands are converted into nonregular secondary structure elements containing none or few hydrogen bonds. The levels of relative occupancy of each hydrogen bond defined in the β-sandwich are, in general, lower in L55P-TTR (Table I). In particular, all hydrogen bonds between strands C and B have reduced percentages of occupancy. These calculations were also repeated considering only the most disruptive unfolding simulations: runs 1 and 4 of WT-TTR and runs 1 and 2 of L55P-TTR. Considerable decreases from the overall mean occupancies were obtained in these runs (Table I). Oddly, while in WT-TTR most of these differences are centred in the hydrogen bonds linking strands D and A, and strands A and G (hydrogen bond number 21 being the exception), in L55P-TTR the difference is more global and centred on the H-bond network of the CBEF β-sheet.
Table I.
Inter-β-Strand Hydrogen Bond Occupancy During MD Unfolding Simulations of WT- and L55P-TTR
| WT-TTR |
L55P-TTR |
||||||
|---|---|---|---|---|---|---|---|
| β-strands | # | Donor-acceptor | % of occupancy in all runs | % of occupancy in runs 1 and 4 | Donor-acceptor | % of occupancy in all runs | % of occupancy in runs 1 and 2 |
| D and A | 1 | L55-V14 | 32.5 | 13.4 | P55-V14 | 0.0 | 0.0 |
| 2 | V14-L55 | 16.1 | 4.7 | V14-P55 | 0.0 | 0.0 | |
| 3 | L111-L17 | 53.8 | 44.7 | L111-L17 | 42.9 | 34.7 | |
| 4 | L17-A109 | 60.4 | 52.9 | L17-A109 | 54.7 | 37.6 | |
| A and G | 5 | A109-K15 | 58.5 | 48.9 | A109-K15 | 53.3 | 38.9 |
| 6 | K15-I107 | 60.3 | 48.0 | K15-I107 | 48.9 | 41.3 | |
| 7 | I107-M13 | 48.9 | 37.8 | I107-M13 | 40.7 | 42.8 | |
| 8 | M13-Y105 | 31.9 | 23.6 | M13-Y105 | 27.5 | 33.6 | |
| 9 | S112-S115 | 4.8 | 2.0 | S112-S115 | 0.0 | 0.0 | |
| 10 | L110-S117 | 25.2 | 21.5 | L110-S117 | 0.0 | 0.0 | |
| 11 | S117-L110 | 12.0 | 9.4 | S117-L110 | 0.0 | 0.0 | |
| 12 | A108-T119 | 27.4 | 24.1 | A108-T119 | 36.4 | 34.6 | |
| G and H | 13 | T119-A108 | 21.3 | 16.9 | T119-A108 | 0.0 | 0.0 |
| 14 | T106-V121 | 20.6 | 22.9 | T106-V121 | 21.0 | 11.4 | |
| 15 | V121-T106 | 24.5 | 25.3 | V121-T106 | 29.0 | 23.4 | |
| 16 | R104-T123 | 6.6 | 7.5 | R104-T123 | 4.3 | 0.5 | |
| 17 | T123-R104 | 14.5 | 16.5 | T123-R104 | 10.6 | 3.5 | |
| 18 | T49-V28 | 13.3 | 16.3 | T49-V28 | 0.0 | 0.0 | |
| 19 | G47-V30 | 22.8 | 20.9 | G47-V30 | 0.0 | 0.0 | |
| 20 | V30-G47 | 32.4 | 32.0 | V30-G47 | 0.0 | 0.0 | |
| C and B | 21 | V32-A45 | 49.6 | 37.6 | V32-A45 | 37.7 | 25.2 |
| 22 | F44-V32 | 40.7 | 35.1 | F44-V32 | 39.3 | 27.9 | |
| 23 | E42-R34 | 72.6 | 71.0 | E42-R34 | 60.8 | 48.2 | |
| 24 | R34-E42 | 74.6 | 74.5 | R34-E42 | 0.0 | 0.0 | |
| 25 | A36-T40 | 35.0 | 37.8 | A36-T40 | 33.3 | 28.7 | |
| 26 | H31-E72 | 55.0 | 58.0 | H31-E72 | 53.4 | 46.0 | |
| 27 | E72-H31 | 65.9 | 67.3 | E72-H31 | 60.7 | 54.0 | |
| B and E | 28 | F33-K70 | 80.7 | 80.9 | F33-K70 | 73.9 | 65.5 |
| 29 | K70-F33 | 68.8 | 69.6 | K70-F33 | 0.0 | 0.0 | |
| 30 | K35-I68 | 42.4 | 40.0 | K35-I68 | 36.0 | 28.2 | |
| 31 | I73-A91 | 27.4 | 31.0 | I73-A91 | 27.5 | 20.9 | |
| 32 | A91-I73 | 17.6 | 19.7 | A91-173 | 11.0 | 12.7 | |
| 33 | V71-V93 | 54.4 | 60.3 | V71-V93 | 43.0 | 29.2 | |
| E and F | 34 | V93-V71 | 33.0 | 36.0 | V93-V71 | 33.4 | 22.7 |
| 35 | Y69-F95 | 65.4 | 61.9 | Y69-F95 | 34.7 | 25.9 | |
| 36 | F95-Y69 | 67.2 | 68.0 | F95-Y69 | 41.6 | 29.9 | |
| 37 | A97-G67 | 37.5 | 36.5 | A97-G67 | 14.7 | 13.3 | |
For each protein, the occupancy was calculated over all runs performed at 500 K, and also over those trajectories exploring potentially amyloidogenic routes.
Hierarchical agglomerative clustering analysis
Clustering is the arrangement of similar objects into different groups, or more precisely, the partitioning of a data set into subsets (clusters) so that the data in each subset ideally share some common trait, often the proximity according to some defined distance measure.39 For the protein unfolding problem, insightful analyses of multiple trajectories can be attained by gathering snapshots from each simulation and clustering those with similar properties.
The clustering analysis of the structures sampled from the unfolding simulations of WT- and L55P-TTR was based on the similarity matrices constructed from the pairwise RMSD between all structures for each TTR variant. The analysis of the sampled structures from both WT- and L55P-TTR original data sets led to 10 clusters for each variant. Representative structures of each cluster were chosen based on four molecular properties: number of H-bonds, number of native contacts, secondary structure and nonpolar SASA. Figure 8, and Tables SI and SII in the Supporting Information, summarize information on cluster populations, average molecular properties of the centroids, the contribution of each run to the clusters and the contribution of ten nonoverlapping time intervals of 1 ns. The clusters are ranked by the number of structures. In the case of WT-TTR, the most populated cluster contains 30.9% of the total population, followed by two clusters holding ∼27 and 24%. Cluster 2 contains the X-ray structure and its centroid presents average molecular properties that illustrate the native ensemble. These first three clusters sum up ∼82% of the whole structure population. The fourth cluster represents 12.7% and the remaining clusters have populations ranging from 0.2 to 2%. For L55P-TTR, the distribution of the structures in the clusters is different. The first three clusters sum up ∼64% of the total population. The first one contains about 29%, and includes the X-ray structure, and two clusters of ∼19% and one of 15% follow. Consequentially, the remaining clusters represent a larger number of structures than their counterparts in WT-TTR, except for the smallest cluster, representing 0.1% of the population. For WT- and L55P-TTR, the centroids of the most populated clusters mainly represent native-like conformations sampled within the first nanoseconds of the simulations. For example, in both variants the average nonpolar SASA of the first five centroids is below 6000 Å2. On the contrary, the centroids of the five less populated clusters tend to represent unfolded structures sampled on later stages of the simulations, with average nonpolar SASA ranging up to 7000 Å2. Exceptions are the seventh and the ninth clusters of L55P-TTR, holding average molecular properties that bring them closer to the native-like monomer conformations but presenting significant displacement of strands C and D from the β-sandwich core.
Figure 8.
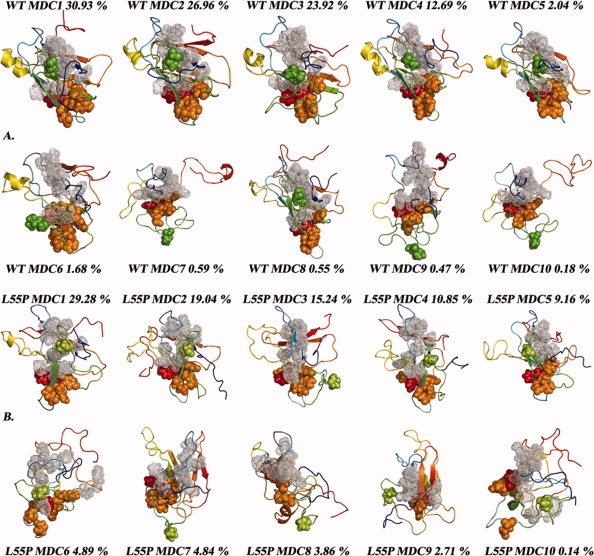
Minimum distance conformations (MDC) for each of the 10 clusters obtained by hierarchical agglomerative clustering for WT-TTR (A) and L55P-TTR (B). The residue at the mutation site is represented with spheres: Leu55 is colored in green and Pro55 in light green. Residues undergoing larger increases in SASA throughout the L55P-TTR simulations are represented as orange spheres; Phe33—showing the highest increase — is represented in red. Residues belonging to β-strands A and B and displaying significant increases in SASA are represented with gray dots. The orientation of all models was obtained by least-squares fitting to the initial crystal model of WT-TTR shown in Figure 1, except MDCs 7 and 9 which are slightly rotated from the other conformations.
Discussion
General considerations
Molecular dynamics simulations are the archetypal computational technique for conducting atomic-resolution studies of biological systems, providing links between structure and dynamics through the exploration of the conformational landscape accessible to biomolecules. The use of high temperature simulations to overcome the conformation sampling problem is well documented.23–27 The additional kinetic energy introduced in the simulation allows crossing of high energy barriers, ensuring a broad sampling of the conformational space, and simultaneously promoting the thermal unfolding of the proteins under study. Moreover, if reasonable comparisons with experimental denaturation studies are to be made, performing multiple simulations becomes critical to probe the multitude of conformations accessible to the protein between the native and the unfolded states. Daggett and coworkers have shown that a small number of simulations, five to ten runs, can be adequate to extract the average properties and the essence of the unfolding pathway.40
Our simulations aimed at an atomic-scale understanding of the effect of a point mutation on the structural stability and conformational unfolding landscape of TTR monomers. High temperature was employed as a mean to accelerate the unfolding process and thus to evidence potential dissimilarities between the wild–type and the mutant TTR, rather than to study the pathways of thermal unfolding of the protein in depth. Furthermore, explicit water molecules were used in all our simulations because solvent molecules have been shown to play an active part in the denaturation process of some proteins.23
Significant efforts have been made by several authors to identify the key events in amyloid fibril formation by TTR. Tetramer dissociation into monomers has been pointed out as the rate-limiting step for TTR amyloid formation.41 However, tetramer dissociation thermodynamics and kinetics do not fully correlate with the amyloidogenic potential of TTR variants.2 Thus, the detailed understanding of the unfolding process of TTR monomers may therefore provide important clues to define new strategies towards the development of efficient treatments for FAP and SSA.
High-temperature MD simulations provided us with countless details on the structural and dynamic properties of the TTR monomer unfolding. Multiple runs allowed the exploration of the conformational space accessible to the subunits through distinct unfolding pathways. Correlating the data extracted from secondary structure, interresidue contacts, hydrogen-bonding and clustering analyses, we were able to: (i) assess differences in the conformational stability of WT- and L55P-TTR – identifying, for each variant, trajectories that are more likely to yield amyloidogenic intermediates; and (ii) define critical protein regions displaying higher lability, particularly in the mutated TTR.
Comparison with other computer simulations
Yang and coworkers first addressed the stability and flexibility of TTR monomers using MD simulations in explicit water, at room temperature.31 It was shown that, on average, the nonbonded interaction energy (Coulomb and van der Waals interactions) between the two β-sheets of the TTR β-sandwich was more negative for WT-TTR than for the monomers of L55P-TTR or V30M-TTR, suggesting a stronger packing of the β-sandwich in the former. More recently, the same authors reported on the use of MD simulations to characterize the unfolding pathways of WT-, V30M-, and L55P-TTR and their role in amyloidogenicity.34 In this study, the generalized Born (GB) implicit solvent model was employed to simulate monomers at different temperatures. The potential energy of multiple conformers was used to render energy landscapes for each protein variant as a function of conformational similarities simplified by principal coordinate analysis.34 Multiple and dissimilar routes of unfolding were reported for each of the three variants, again suggesting that single point mutations strongly affect the unfolding mechanisms of TTR. Moreover, initial disruption of strand D was seen for the three variants under study, with subsequent unfolding of strand H and ultimate disruption and dislocation of strand F from the AG strands.
Despite the alternative approach followed by Yang and coworkers, our results are in agreement with theirs in several aspects, which evidences the sucesses being made in the optimization of implicit solvent models.42 A somewhat comparable sequence of events is seen in our simulations of L55P-TTR. However, in our WT-TTR simulations, ordered secondary structure is generally maintained in strand D, and strand H undergoes progressive disruption and displacement from the DAG strands, even before significant modifications occur in strand D. In addition, the secondary structure and positioning of strand F are well conserved in our simulations of WT-TTR. Moreover, in our simulations of L55P-TTR, both strands C and D undergo marked unfolding and displacement from the core of the TTR subunit, exposing specific hydrophobic residues at the surface of strands A and B. In addition, strong disruption of the α-helical motif is another key event seen in 4 out 5 of our simulations of L55P-TTR, but not in WT-TTR. These observations consistently discriminate the behaviour of WT-TTR from that of L55P-TTR, suggesting a possible explanation for the augmented amyloidogenicity of L55P-TTR. Thus, our simulations in explicit solvent seem to be able to probe in greater detail the differences in unfolding patterns between WT- and L55P-TTR.
Comparison with chemical denaturation experiments
Although the accuracy of computer simulation predictions may be estimated by considering the approximations and simplifications of the models and the computational procedures used, the ultimate test lies in the comparison between the predicted and the experimentally measured properties. The conformational stability of monomeric forms of WT-, L55P-, V30M-, and T119M-TTR was previously measured in our group through chemically-induced unfolding experiments followed by intrinsinc tryptophan fluorescence.8 The analysis of structural properties extracted from the simulations, reported in the previous sections, show consistency with this previous experimental work. In fact, the variation of all analysed properties, but in particular Cα-RMSD (Fig. 2), solvent accessible surface area (Fig. 5), secondary structure (Fig. 6), and native contacts (Fig. 7) clearly indicate a higher tendency for unfolding of the L55P-TTR monomer when compared with the wild type, agreeing with the lower experimentally determined conformational stability of the L55P-TTR monomer.8 Interestingly, while the side-chain SASA of residue Trp41 changes erratically across all simulations, Trp79, belonging to the helical motif and hidden from the solvent in the native state, appears more exposed in all simulations of L55P-TTR, with an overall average SASA of 26.9 ± 24.8 Å2, than in those of WT-TTR, with an average SASA of 11.8 ± 17.6 Å2. These results are in agreement with the hypothesis that residue Trp79 is the main responsible for the fluorescence increase observed in the chemically-induced unfolding experiments.8 Thus, the reduced propensity of L55P-TTR to retain the initial native structure, as seen in the simulations, correlates well with the experimental evidence showing its lower monomer conformational stability8 and higher tendency to form a larger amount of partially unfolded monomeric species.11
Comparison with H/D exchange studies by NMR
Wemmer and coworkers, using H/D exchange studies followed by NMR, determined solvent protection factors for WT- and L55P-TTR.43,44 These authors reported that at low pH the amide protons within the CBEF β-sheet of WT-TTR β-sandwich are more labile, showing selective destabilization of one half of the beta-sandwich structure, which could favor the formation of an amyloidogenic intermediate.44 Additionally, a similar behaviour was observed for L55P-TTR at neutral pH.43 Our simulations, at high temperature and TTR protonation state corresponding to neutral pH, show this type of behaviour late in the simulations for L55P-TTR but not for WT-TTR. As pointed out in the “Results” section, and focusing in the most disruptive trajectories, the L55P mutant undergoes a severe loss of the hydrogen bond network connecting the CBEF β-sheet. In WT-TTR, however, a much smaller number of hydrogen bonds is lost and β-strands D, A, and G are the ones mostly affected. As the hydrogen bonds between β-strands D and A remained partially intact in the experiments at low pH, Wemmer and co-workers have also proposed that the concomitant disruption of H-bonds connecting strands C, B, E, and F – resulting in an increase in mobility and ultimate displacement of this β-sheet from the core – could represent the hypothetical monomeric amyloidogenic intermediate.44 Conformations sharing analogous topology with these species were sampled at the later stages of the L55P-TTR simulations, namely in runs 1 and 2. In opposition, in WT-TTR simulations the hydrogen bond occupancies between backbone atoms of β-strands B, E and F are reasonably high, particularly in runs 1 and 4 (Table I). Thus, the conformational intermediates detected by NMR for WT-TTR at low pH,41 could be structurally similar to those detected late in some of our simulations for L55P-TTR, suggesting that the CBEF β-sheet of the TTR subunit is destabilized either by low pH or by amyloidogenic mutations.
Insights on the exposure of β-strands A and B
The elucidation of the structural details of amyloid fibrils and the identification of the molecular species at the source of fibril formation are essential for the understanding of the mechanisms of pathogenesis in amyloid diseases. We have previously published a molecular model of a TTR protofilament17 based on experimental constraints obtained by EPR and NMR.15,16 To build a model having the continuous extended cross-β structure characteristic of amyloid,12 full displacement of strands C and D from the TTR β-sandwich was required to expose a new interface formed by strands A and B, and allow monomer to monomer docking.17
The positioning of strands C and D and exposure of strands A and B were followed by analysis of the variation of SASA along the MD simulation trajectories. Full displacement of the D-strand-loop-C-strand segment from the core of the monomer, with consequent increase in SASA of strands A and B, are recognizable events on L55P-TTR trajectories, starting at around 3.5 ns of simulation and well before the destabilization of the whole CBEF β-sheet occurs (Fig. 4). Albeit transiently in WT-TTR, around the fifth nanosecond of simulation, a partial displacement of strand C at its C-terminus is seen, shortly after the displacement of strand D takes place. Additionally, in WT-TTR, strands F and H are notably unstable, loosing regular secondary structure in most trajectories. Strand H, in particular, shows strong propensity to move away from strand G in WT-TTR, generally before strand D starts separating from the monomer's core. These observations are consistent with the H/D exchange rates in the native tetrameric state which show a slowly exchanging core (comprised of strands A, B, E, and G) and faster exchange associated with the edge regions of WT-TTR.45 These observations clearly suggest that L55P-TTR has a higher propensity than WT-TTR for exposing strands A and B, while the global β-sandwich fold is still retained.
Differences in solvent exposure for WT-TTR and L55P-TTR were analysed exhaustively on an individual residue basis (Fig. 5). This comparison unequivocally reveals high increase in solvent exposure for a set of residues whose backbone amides had also shown increased H/D exchange rates at low pH, namely His31, Phe33, Arg34, Glu42, Tyr69, Val93, and Phe95. In the WT-TTR unfolding simulations, several native contacts involving this set of residues are well conserved, whereas permanent loss occurs in L55P-TTR. As pointed out in the “Results” section, these residues are located in strands C, B, E, and F, forming interstrand hydrogen bonds and acting as guardians of those strands' relative position. Disruption of the H-bond network in the CBEF sheet can indeed be one underlying cause for the high solvent exposure and deprotection of the afore mentioned residues, disclosing the global effects of the L55P mutation on the structural stability of the TTR monomer. However, it should be kept in mind that this phenomenon occurs late in the simulations. On the contrary, the extensive H-bond disruption between strands D and A, and between strands C and B, and consequent partial (WT-TTR) or total (L55P-TTR) displacement of strands C and D from the β-sandwich core, occurs earlier in the simulations, and especially in putative amyloidogenic unfolding pathways (Table I). Both in WT- and L55P-TTR, the displacement of strand D from strand A (and resulting increase in SASA) seems to trigger unfolding by opening routes to subsequent steps leading to a conformation that may correspond to an amyloidogenic intermediate.
Exposure of a hydrophobic patch might favor amyloid assembly
Results on the relative difference in SASA per residue between the L55P-TTR and WT-TTR simulations (Fig. 5), highlight the behavior of Phe33, located in β-strand B, which presents the largest overall difference. The average SASA for Phe33 across all WT-TTR simulations is ∼0.079 Å2, the lowest value among all residues. Lys70 is positioned in strand E, aligned with Phe33 in strand B, and also presents a low average SASA (∼0.204 Å2). These two residues are highly protected from solvent interactions, mainly by Trp41 and also by Pro43 (in hydrophobic contact with Phe33), located at the N-terminus of β-strand C. Interestingly, it was suggested that β-sandwich proteins naturally place charged side-chains and combinations of prolines and β-bulges close to hydrophobic patches in edge β-strands, in order to prevent edge-to-edge aggregation.46 Pro43 and the β-bulge comprising Phe44 and Ala45 in TTR somehow resemble this natural design. In L55P-TTR simulations, both Phe33 and Lys70 are on average considerably more exposed (∼3.609 Å2 and 2.830 Å2, respectively). These increases in SASA may be related to the larger conformational fluctuations and displacement of strand C in the amyloidogenic variant. Furthermore, changes in strand C end up being reflected in the higher exposure of Arg34 and Glu42, located in strand B and strand C, respectively. In this sense, one may speculate on the role of residues, such as Phe33, in the aggregation of TTR. These residues may actually be involved in key hydrophobic interactions between monomers in the aggregation process, and alongside other interactions may account for the formation of a non-native interface between strands B and B′, and A and A′, in consecutive monomers of the TTR amyloid aggregates and fibrils. In fact, aromatic interactions have been previously reported to be of importance in amyloid formation by the islet amyloid polypeptide, Alzheimer's Aβ peptide, gelsolin and β2-microglobulin.47–50
Insights on the stability of the helical motif
Transitions from α-helix to β-sheet (α/β transitions) are known to be linked with some amyloid diseases, namely BSE and Creutzfeldt-Jakob.51 We analyzed the behaviour of the only α-helix in the TTR monomer, formed by residues 75–83, throughout the MD unfolding simulations. The mutation in position 55, located in β-strand D, seems to induce a perturbation in the helical motif, despite their distance of more than 12 Å. L55P-TTR simulations show evident increases in residue fluctuations and SASA in the α-helix region compared to WT-TTR. In addition, the analysis of secondary structure variation along the trajectories reveals that the α-helix is well conserved in all runs of WT-TTR, whereas it is converted into coils and turns in 3 of the 5 trajectories of L55P-TTR (Fig. 6). Though no α/β transitions were detected in our simulations, one cannot discard this type of events in TTR monomers. High temperature may not provide constructive conditions for the formation of ordered secondary structure motifs. Re-equilibration of the system at room-temperature for a given conformation and at a given stage of the trajectory, followed by equilibrium production runs, would probably be required to sample such phenomenon.
Identification of potentially amyloidogenic conformational ensembles
As pointed out before, the use of multiple MD simulations is of major importance for tracking different regions of the conformational space accessible to a polypetide chain. However, the computation of a few trajectories is sufficient to generate considerable amounts of data. In this work, 50,000 structural snapshots were collected for each of the two TTR variants under study. This scenario raises obvious questions: Which trajectories are similar? Are some trajectories and conformations more meaningful than others? Can we identify a narrow set of molecular species able to represent relevant trends in our simulations and, hence, to describe the essence of the biological ensemble?
The 50,000 conformations sampled from the five trajectories of each TTR variant were clustered using a hierarchical agglomerative method, as described in the “Methods” section. The minimum distance conformations (MDCs) representing the 10 resulting clusters for each TTR variant are shown in Figure 8, and a detailed description of the clusters can be found in Tables SI and SII (Supporting Information).
The first five clusters of WT-TTR are more populated than those of L55P-TTR and mostly by conformations retaining a high proportion of native structure (i.e., more hydrogen bonds, more native contacts and/or more regular secondary structure content). In fact, the first five MDCs of the WT clusters all resemble the native topology of the TTR monomer (Fig. 8). On the contrary, the very first MDC of L55P-TTR, along with all MDCs of the most populated clusters, shows important deviations from the native TTR topology. For example, the helical motif is barely present in the L55P MDCs. Moreover, the second and third most populated clusters of L55P-TTR are represented by markedly unfolded conformations, with high average nonpolar SASA and a reduced amount of secondary structure elements.
Mostly for WT-TTR, sparsely populated clusters reflect the extensive conformational diversity of protein unfolded states. The major contributions for clusters 6–10 come from the second half of the trajectories, essentially from the sixth to tenth ns. Interestingly, WT MDCs 7, 9 and 10 feature fully displaced strands C and D (shown as green coils in Fig. 8), and capture the unstable nature of strand H (red segment) in WT-TTR. Despite showing highly exposed hydrophobic residues, these structures are more likely to represent unfolded states and would hardly correspond to aggregation-competent species capable of building ordered oligomers because they have low amounts of β-structure. Contributions for clusters 7, 9, and 10 of WT-TTR come exclusively from runs 1 and 4.
In the case of L55P-TTR, interesting partially unfolded conformations were found. MDCs 7 and 9 illustrate some of these conformations. Cluster number 7 holds 4.8% of the pool of L55P structures and the corresponding representative conformation (MDC 7) is a partially unfolded monomer with fully disrupted α-helix and fully displaced strands C and D, and exposing a large hydrophobic interface formed by residues in strands A and B. Considering these structural characteristics and the weight of this cluster over the entire population of conformations, MDC 7 of L55P-TTR might represent an ensemble of aggregation-prone intermediate structures, i.e., an amyloidogenic conformation. MDC 9, in addition, featuring an ordered β-sandwich topology, disrupted α-helix and halfway displaced strands C and D, may represent an earlier intermediate in the formation of the amyloidogenic conformation. Major contributions for cluster number 9 come from structures taken between the second and the fifth ns of simulation.
Methods
Simulation details
The initial coordinates of the native monomeric forms of WT-TTR and L55P-TTR were obtained from X-ray structures deposited in the Protein Data Bank (PDB entries 1TTA 36 and 5TTR52), after preliminary structure quality assessment and subunit selection with the program WHAT_CHECK.53 The missing N- and C-termini in L55P-TTR were added and subjected to simulated annealing with the rest of the protein held fixed. All equilibration and MD procedures were carried out with NAMD,54 using the all-atom CHARMM 27 forcefield55 and running on a cluster of 100 Intel Pentium-4 processors (Centopeia at University of Coimbra).
A strict and unique protocol was employed to set up both TTR variant model systems. Water molecules were first placed in hydrophilic internal cavities of the monomer with the program Dowser56 and hydrogen atoms were added to the protein structures using the psfgen plug-in within VMD.57 Protein and internal waters were submitted to energy minimization in vacuum for three cycles of 1000 steps each: a first cycle for the hydrogen atoms, a second cycle for the side-chains and internal waters, and a final cycle for all atoms. Each monomer was then embedded in a large shell of explicit TIP3P water molecules,58 and the spherical volume was trimmed into a large cubic box afterwards. A solvent layer of at least 18 Å was maintained between protein atoms and box boundaries. Sodium and chloride ions were included to bring the net electric charge of the system to zero and an average ionic concentration of 145 mM was reached in order to better mimic the physiological environment. Both solvation and charge neutralization were accomplished with the program SOLVATE.59
After one last energy minimization cycle applied on solvent and protein side-chain atoms, the system was equilibrated up to the target temperature under Langevin dynamics. For the initial 20 ps, the protein backbone was maintained fixed. Then, heating proceeded with harmonic restraints placed on the protein Cα carbon atoms for another 20 ps. Finally, a 10 ps step of unrestrained Langevin dynamics was executed. Pressure was maintained at 1 atm using the Langevin piston method.60 From this point on, the volume was kept constant and the system was tested for 20 ps of classical MD. To avoid hypothetical biases taking place in the equilibration stage, each production run was independently equilibrated.
Five independent 10-ns long MD simulations at 500 K and one control simulation at 310 K were conducted for each of the two systems under study. The simulations were performed in the microcanonical ensemble (NVE) and periodic boundary conditions (PBC) were employed. The particle mesh Ewald summation method61 was elected for the quantification of long-range electrostatic interactions at every step, in spite of its high computational demand.62 A 12 Å cut-off with a switching function at 10 Å was used for the treatment of the van der Waals interactions, with the pair list distances being evaluated every 10 steps. All hydrogen-heavy atom bond lengths were constrained with the SHAKE algorithm.63 An integration time step of 2 fs was used and the coordinates for the whole system were recorded every 500 steps (i.e., at each ps of simulation).
Analysis details
The considerable amount of information emerging from each MD simulation renders the comparison of multiple unfolding trajectories a nontrivial task. Several parameters were analyzed to characterize the behavior of the two TTR variants along the unfolding trajectories. Variation of the protein secondary structure, number of hydrogen bonds and native contacts, as well as fluctuation of geometric measures along each simulation, were calculated using the program VMD.57 In addition, the variation of the solvent-accessible surface area (SASA) of the protein along the unfolding simulations was computed with NACCESS.64 Secondary structure was assigned with the program STRIDE.65 Hydrogen bonds were defined for donor (D) ··· acceptor (A) distances shorter than 3.2 Å and D···H···A angles deviating less than 35° from linearity. Pairwise distances between all heavy non-neighbor atoms were calculated for each conformation in the trajectories; two residues were considered to be in contact when at least one heavy atom of one residue was within 4.2 Å of a heavy atom of another residue. Contacts present in the crystallographic structures of the protein were taken as native.
Hierarchical agglomerative clustering analysis was applied to sort protein conformations sharing similar structural properties. Overall, 50,001 structures are available for each TTR variant. Because of computational constraints, systematic sampling was applied to the original data sets: every Nth record was selected from a set of conformations. The new data sets were constructed using N = 5, thereby containing 10,001 structures each. The similarity matrix used in the clustering was constructed based on the pairwise root mean square deviation (RMSD) between the 10,001 structures. Agglomerative clustering starts by identifying each object with a cluster. Next, it merges clusters, successively, into larger clusters. The procedure stops when all objects are covered by a cluster or when a certain pre-defined number of clusters is produced. The hierarchical agglomerative clustering was computed using the scluster program within the CLUTO clustering toolkit.66 Different parameter settings were tested to find the best clustering solution. This corresponds to a solution with ten clusters, using the complete linkage function, i.e., the distance between clusters is determined by the most distant pair of objects of each cluster. Moreover, four molecular properties were used to characterize all TTR conformations sampled in the MD unfolding simulations: the number of hydrogen bonds, the number of native contacts, the nonpolar SASA and the number of residues in regular secondary structure (α-helix and β-sheet). The centroid of the clusters is a vector characterizing the average values for these properties in the cluster. These data were used to characterize the clusters and to select their representative structures. Each cluster is described in terms of (a) the number of structures, and if it contains the X-ray structure, (b) average values of the four molecular properties defined earlier, (c) the contribution of each simulation to the cluster, (d) the contribution of each of 10 nonoverlapping time intervals of 1 ns. Based on the molecular properties defined, representative structures were identified as the ones having the highest number of closer neighbors (minimum distance conformations, or MDCs).
Conclusions
Multiple molecular dynamics simulations of WT- and L55P-TTR monomers were performed at high temperature and in explicit water to assess the structural stability of transthyretin, explore the conformational space available to the polypeptide chain upon protein unfolding, and identify potential structural changes leading to amyloid assembly. As a general observation, the analysis of molecular properties such as secondary structure, hydrogen bonds, and solvent accessible surface area along the MD unfolding trajectories clearly demonstrate that L55P-TTR has a much higher tendency to unfold than WT-TTR, agreeing well with previously published experimental data on the conformational stability of monomeric forms of several TTR variants.8
Additionally, according to our simulations, two main events appear to be of critical importance in distinguishing the unfolding behavior of L55P-TTR monomers from that of WT-TTR: (i) Premature disruption and displacement of β-strand D, followed by separation of β-strand C from the core of the monomer β-sandwich, exposing key residues and a hydrophobic interface, embodied by β-strands A and B, that may promote monomer-monomer interactions; (ii) Pronounced unfolding of the α-helical motif into coils and turns. These events characterize early intermediate structures in the unfolding pathways of L55P-TTR. On the contrary, in WT-TTR extensive unfolding of the entire monomeric subunit must occur in order to observe full displacement of strands C and D from the rest of the structure. Moreover, the single α-helix in WT-TTR remains folded through out most of the simulations. Thus, L55P-TTR does show a significantly increased tendency to form aggregation-prone monomeric conformations with all the structural requirements for monomer-monomer docking and formation of an extended cross-β structure characteristic of amyloid and previously reported for a model of an amyloid protofilament of TTR.17
Molecular property variation analysis and hierarchical clustering of multiple unfolding simulations helped us to identify and characterize the structure of potentially amyloidogenic intermediates in the unfolding landscape of TTR. Whether these species are on- or off-pathway intermediates of fibril formation is still a matter of debate, but our simulations lead us to conclude that these intermediates can be formed along multiple unfolding routes, and some of these may actually yield these structures in relatively high amounts. Clearly, specific mutations, such as L55P, have an important role in facilitating the unfolding reaction, but the link between this and the type or location of different mutations needs further clarification. Careful comparison of unfolding simulations of multiple TTR variants, such as Y78F-TTR, a highly amyloidogenic variant with a mutation located in the α-helix, or V122I-TTR, a variant producing strong cardiomyopathic effects, may help in the future to clarify the relation between protein unfolding and amyloid formation and, ultimately, help dissect the mechanisms of tissue specificity for different TTR variants.
Acknowledgments
The authors thank the Center for Computational Physics, Departamento de Física, Universidade de Coimbra, Coimbra, Portugal, and the Computer Science and Technology Center, Departamento de Informática, Universidade do Minho, Braga, Portugal, for the computer resources provided.
Glossary
Abbreviations:
- FAC
Familial Amyloid Cardiomiopathy
- FAP
Familial Amyloid Polyneuropathy
- H/D exchange
Hydrogen/Deuterium exchange
- L55P-TTR
Transthyretin with a proline replacing a leucine in position 55
- MD
Molecular Dynamics
- MDC
Minimum Distance Conformation
- NMR
Nuclear Magnetic Resonance
- RMSD
Root Mean Square Deviation
- RMSF
Root Mean Square Fluctuation
- Rgyr
Radius of Gyration
- SASA
Solvent Accessible Surface Area
- SSA
Senile Systemic Amyloidosis
- TTR
Transthyretin
- WT-TTR
Wild-type transthyretin
References
- 1.Chiti F, Dobson CM. Protein misfolding, functional amyloid, and human disease. Annu Rev Biochem. 2006;75:333–366. doi: 10.1146/annurev.biochem.75.101304.123901. [DOI] [PubMed] [Google Scholar]
- 2.Brito RMM, Damas AM, Saraiva MJ. Amyloid formation by transthyretfrom protein stability to protein aggregation. Curr Med Chem Immunol Endocr Metab Agents. 2003;3:349–360. [Google Scholar]
- 3.Andrade C. A peculiar form of peripheral neuropathy. Familial atypical generalized amyloidosis with special involvement of peripheral nerves. Brain. 1952;75:408–427. doi: 10.1093/brain/75.3.408. [DOI] [PubMed] [Google Scholar]
- 4.Costa PP, Figueira AS, Bravo SR. Amyloid fibril protein related to prealbumin in familial amyloidotic polyneuropathy. Proc Natl Acad Sci USA. 1978;75:4499–4503. doi: 10.1073/pnas.75.9.4499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Connors LH, Richardson AM, Theberge R, Costello CE. Tabulation of transthyretin (TTR) variants as of 1/1/2000. Proc Natl Acad Sci USA. 2000;7:54–69. doi: 10.3109/13506120009146826. [DOI] [PubMed] [Google Scholar]
- 6.Jacobson DR, McFarlin DE, Kane I, Buxbaum JN. Transthyretin Pro55, a variant associated with early-onset, aggressive, diffuse amyloidosis with cardiac and neurologic involvement. Hum Genet. 1992;89:353–356. doi: 10.1007/BF00220559. [DOI] [PubMed] [Google Scholar]
- 7.Hörnberg A, Eneqvist T, Olofsson A, Lundgren E, Sauer-Eriksson AE. A comparative analysis of 23 structures of the amyloidogenic protein transthyretin. J Mol Biol. 2000;302:649–669. doi: 10.1006/jmbi.2000.4078. [DOI] [PubMed] [Google Scholar]
- 8.Quintas A, Vaz DC, Cardoso I, Saraiva MJ, Brito RMM. Tetramer dissociation and monomer partial unfolding precedes protofibril formation in amyloidogenic transthyretin variants. J Biol Chem. 2001;276:27207–27213. doi: 10.1074/jbc.M101024200. [DOI] [PubMed] [Google Scholar]
- 9.Jian X, Buxbaum JN, Kelly JW. The V122I cardiomyopathy variant of transthyretin increases the velocity of rate-limiting tetramer dissociation, resulting in accelerated amyloidosis. Proc Natl Acad Sci USA. 2001;98:14943–14948. doi: 10.1073/pnas.261419998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lai Z, Colón W, Kelly JW. The acid-mediated denaturation pathway of transthyretin yields a conformational intermediate that can self-assemble into amyloid. Biochemistry. 1996;35:6470–6482. doi: 10.1021/bi952501g. [DOI] [PubMed] [Google Scholar]
- 11.Quintas A, Saraiva MJ, Brito RMM. The amyloidogenic potential of transthyretin variants correlates with their tendency to aggregate in solution. FEBS Lett. 1997;418:297–300. doi: 10.1016/s0014-5793(97)01398-7. [DOI] [PubMed] [Google Scholar]
- 12.Blake C, Serpell L. Synchrotron X-ray studies suggest that the core of the transthyretin amyloid fibril is a continuous β-sheet helix. Structure. 1996;4:989–998. doi: 10.1016/s0969-2126(96)00104-9. [DOI] [PubMed] [Google Scholar]
- 13.Sunde M, Blake CC. From the globular to the fibrous state: protein structure and structural conversion in amyloid formation. Q Rev Biophys. 1998;31:1–39. doi: 10.1017/s0033583598003400. [DOI] [PubMed] [Google Scholar]
- 14.Goldsteins G, Persson H, Andersson K, Olofsson A, Dacklin I, Edvinsson A, Saraiva MJ, Lundgren E. Exposure of cryptic epitopes on transthyretin only in amyloid and in amyloidogenic mutants. Proc Natl Acad Sci USA. 1999;96:3108–3113. doi: 10.1073/pnas.96.6.3108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Serag AA, Altenbach C, Gingery M, Hubbell WL, Yeates TO. Arrangement of subunits and ordering of β-strands in an amyloid sheet. Nat Struct Biol. 2002;9:734–739. doi: 10.1038/nsb838. [DOI] [PubMed] [Google Scholar]
- 16.Olofsson A, Ippel JH, Wijmenga SS, Lundgren E, Ohman A. Probing solvent accessibility of transthyretin amyloid by solution NMR spectroscopy. J Biol Chem. 2004;279:5699–5707. doi: 10.1074/jbc.M310605200. [DOI] [PubMed] [Google Scholar]
- 17.Correia BE, Loureiro-Ferreira N, Rodrigues JR, Brito RMM. A structural model of an amyloid protofilament of transthyretin. Protein Sci. 2006;15:28–32. doi: 10.1110/ps.051787106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Segawa S, Sugihara M. Characterization of the transition state of lysozyme unfolding. I. Effect of protein-solvent interactions on the transition state. Biopolymers. 1984;23:2473–2488. doi: 10.1002/bip.360231122. [DOI] [PubMed] [Google Scholar]
- 19.Radford SE, Dobson CM, Evans PA. The folding of hen lysozyme involves partially structured intermediates and multiple pathways. Nature. 1992;358:302–307. doi: 10.1038/358302a0. [DOI] [PubMed] [Google Scholar]
- 20.Itzhaki LS, Evans PA, Dobson CM, Radford SE. Tertiary interactions in the folding pathway of hen lysozyme: kinetic studies using fluorescent probes. Biochemistry. 1994;33:5212–5220. doi: 10.1021/bi00183a026. [DOI] [PubMed] [Google Scholar]
- 21.Eaton WA, Munoz V, Thompson PA, Chan CK, Hofrichter J. Submillisecond kinetics of protein folding. Curr Opin Struct Biol. 1997;7:10–14. doi: 10.1016/s0959-440x(97)80003-6. [DOI] [PubMed] [Google Scholar]
- 22.Roder H, Colon W. Kinetic role of early intermediates in protein folding. Curr Opin Struct Biol. 1997;7:15–28. doi: 10.1016/s0959-440x(97)80004-8. [DOI] [PubMed] [Google Scholar]
- 23.Caflisch A, Karplus M. Molecular dynamics simulation of protein denaturation: solvation of the hydrophobic cores and secondary structure of barnase. Proc Natl Acad Sci USA. 1994;91:1746–1750. doi: 10.1073/pnas.91.5.1746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Caflisch A, Karplus M. Acid and thermal denaturation of barnase investigated by molecular dynamics simulations. J Mol Biol. 1995;252:672–708. doi: 10.1006/jmbi.1995.0528. [DOI] [PubMed] [Google Scholar]
- 25.Hunenberger PH, Mark AE, van Gunsteren WF. Computational approaches to study protein unfolding: hen egg white lysozyme as a case study. Proteins. 1995;21:196–213. doi: 10.1002/prot.340210303. [DOI] [PubMed] [Google Scholar]
- 26.Li A, Daggett V. Identification and characterization of the unfolding transition state of chymotrypsin inhibitor 2 by molecular dynamics simulations. J Mol Biol. 1996;257:412–429. doi: 10.1006/jmbi.1996.0172. [DOI] [PubMed] [Google Scholar]
- 27.Williams MA, Thornton JM, Goodfellow JM. Modelling protein unfolding: hen egg-white lysozyme. Protein Eng. 1997;10:895–903. doi: 10.1093/protein/10.8.895. [DOI] [PubMed] [Google Scholar]
- 28.Finkelstein AV. Can protein unfolding simulate protein folding? Protein Eng. 1997;10:843–845. doi: 10.1093/protein/10.8.843. [DOI] [PubMed] [Google Scholar]
- 29.Alonso DO, Alm E, Daggett V. Characterization of the unfolding pathway of the cell-cycle protein p13suc1 by molecular dynamics simulations: implications for domain swapping. Structure. 2000;8:101–110. doi: 10.1016/s0969-2126(00)00083-6. [DOI] [PubMed] [Google Scholar]
- 30.Rodrigues JR, Brito RMM. How important is the role of compact denatured states on amyloid formation by transthyretin? In: Grateau G, Kyle RA, Skinner M, editors. Amyloid and Amyloidosis. CRC Press; 2004. pp. 323–325. [Google Scholar]
- 31.Yang M, Lei M, Huo S. Why is Leu55→Pro55 transthyretin variant the most amyloidogenic: insights from molecular dynamics simulations of transthyretin monomers. Protein Sci. 2003;12:1222–1231. doi: 10.1110/ps.0239703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Armen RS, DeMarco ML, Alonso DO, Daggett V. Pauling and Corey's alpha-pleated sheet structure may define the prefibrillar amyloidogenic intermediate in amyloid disease. Proc Natl Acad Sci USA. 2004;101:11622–11627. doi: 10.1073/pnas.0401781101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Armen RS, Alonso DO, Daggett V. Anatomy of an amyloidogenic intermediate: conversion of beta-sheet to alpha-sheet structure in transthyretin at acidic pH. Structure. 2004;12:1847–1863. doi: 10.1016/j.str.2004.08.005. [DOI] [PubMed] [Google Scholar]
- 34.Yang M, Yordanov B, Levy Y, Brüschweiler R, Huo S. The sequence-dependent unfolding pathway plays a critical role in the amyloidogenicity of transthyretin. Biochemistry. 2006;45:11992–12002. doi: 10.1021/bi0609927. [DOI] [PubMed] [Google Scholar]
- 35.Kazmirski SL, Li A, Daggett V. Analysis methods for comparison of multiple molecular dynamics trajectories: applications to protein unfolding pathways and denatured ensembles. J Mol Biol. 1999;290:283–304. doi: 10.1006/jmbi.1999.2843. [DOI] [PubMed] [Google Scholar]
- 36.Hamilton JA, Steinrauf LK, Braden BC, Liepnieks J, Benson MD, Holmgren G, Sandgren O, Steen L. The x-ray crystal structure refinements of normal human transthyretin and the amyloidogenic Val-30→Met variant to 1.7-Å resolution. J Biol Chem. 1993;268:2416–2424. [PubMed] [Google Scholar]
- 37.Hunenberger PH, Mark AE, van Gunsteren WF. Fluctuation and cross-correlation analysis of protein motions observed in nanosecond molecular dynamics simulations. J Mol Biol. 1995;252:492–503. doi: 10.1006/jmbi.1995.0514. [DOI] [PubMed] [Google Scholar]
- 38.van Gunsteren WF, Mark AE. Validation of molecular dynamics. J Chem Phys. 1998;108:6109–6116. [Google Scholar]
- 39.Zhao Y, Karypis G. Data clustering in life sciences. Mol Biotechnol. 2005;31:55–80. doi: 10.1385/MB:31:1:055. [DOI] [PubMed] [Google Scholar]
- 40.Day R, Daggett V. Ensemble versus single-molecule protein unfolding. Proc Natl Acad Sci USA. 2005;102:13445–13450. doi: 10.1073/pnas.0501773102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hammarström P, Jiang X, Hurshman AR, Powers ET, Kelly JW. Sequence-dependent denaturation energetics: a major determinant in amyloid disease diversity. Proc Natl Acad Sci USA. 2002;99:16427–16432. doi: 10.1073/pnas.202495199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Roux B, Simonson T. Implicit solvent models. Biophys Chem. 1999;78:1–20. doi: 10.1016/s0301-4622(98)00226-9. [DOI] [PubMed] [Google Scholar]
- 43.Liu K, Kelly JW, Wemmer DE. Native state hydrogen exchange study of suppressor and pathogenic variants of transthyretin. J Mol Biol. 2002;320:821–832. doi: 10.1016/s0022-2836(02)00471-0. [DOI] [PubMed] [Google Scholar]
- 44.Liu K, Cho HS, Lashuel HA, Kelly JW, Wemmer DE. A glimpse of a possible amyloidogenic intermediate of transthyretin. Nat Struct Biol. 2000;7:754–757. doi: 10.1038/78980. [DOI] [PubMed] [Google Scholar]
- 45.Liu K, Cho HS, Hoyt DW, Nguyen TN, Olds P, Kelly JW, Wemmer DE. Deuterium-proton exchange on the native wild-type transthyretin tetramer identifies the stable core of the individual subunits and indicates mobility at the subunit interface. J Mol Biol. 2000;303:555–565. doi: 10.1006/jmbi.2000.4164. [DOI] [PubMed] [Google Scholar]
- 46.Richardson JS, Richardson DC. Natural β-sheet proteins use negative design to avoid edge-to-edge aggregation. Proc Natl Acad Sci USA. 2002;99:2754–2759. doi: 10.1073/pnas.052706099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Aggeli A, Bell M, Boden N, Keen JN, Knowles PF, McLeish TC, Pitkeathly M, Radford SE. Responsive gels formed by the spontaneous self-assembly of peptides into polymeric β-sheet tapes. Nature. 1997;386:259–262. doi: 10.1038/386259a0. [DOI] [PubMed] [Google Scholar]
- 48.Gazit E. A possible role for π-stacking in the self-assembly of amyloid fibrils. FASEB J. 2002;16:77–83. doi: 10.1096/fj.01-0442hyp. [DOI] [PubMed] [Google Scholar]
- 49.Colombo G, Daidone I, Gazit E, Amadei A, Di Nola A. Molecular dynamics simulation of the aggregation of the core-recognition motif of the islet amyloid polypeptide in explicit water. Proteins. 2005;59:519–527. doi: 10.1002/prot.20426. [DOI] [PubMed] [Google Scholar]
- 50.Gazit E. Mechanisms of amyloid fibril self-assembly and inhibition: Model short peptides as a key research tool. FEBS J. 2005;272:5971–5978. doi: 10.1111/j.1742-4658.2005.05022.x. [DOI] [PubMed] [Google Scholar]
- 51.Pan KM, Baldwin M, Nguyen J, Gasset M, Serban A, Groth D, Mehlhorn I, Huang Z, Fletterick RJ, Cohen FE, Prusiner SB. Conversion of α-helices into β-sheets features in the formation of the scrapie prion proteins. Proc Natl Acad Sci USA. 1993;90:10962–10966. doi: 10.1073/pnas.90.23.10962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sebastião MP, Saraiva MJ, Damas AM. The crystal structure of amyloidogenic Leu55→Pro transthyretin variant reveals a possible pathway for trans thyretin polymerization into amyloid fibrils. J Biol Chem. 1998;273:24715–24722. doi: 10.1074/jbc.273.38.24715. [DOI] [PubMed] [Google Scholar]
- 53.Hooft RWW, Vriend G, Sander C, Abola EE. Errors in protein structures. Nature. 1996;381:272. doi: 10.1038/381272a0. [DOI] [PubMed] [Google Scholar]
- 54.Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, Chipot C, Skeel RD, Kale L, Schulten K. Scalable molecular dynamics with NAMD. J Comput Chem. 2005;26:1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.MacKerell AD, Jr, Banavali N, Foloppe N. Development and current status of the CHARMM force field for nucleic acids. Biopolymers. 2001;56:257–265. doi: 10.1002/1097-0282(2000)56:4<257::AID-BIP10029>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- 56.Zhang L, Hermans J. Hydrophilicity of cavities in proteins. Proteins. 1996;24:433–438. doi: 10.1002/(SICI)1097-0134(199604)24:4<433::AID-PROT3>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 57.Humphrey W, Dalke A, Schulten K. VMD: visual molecular dynamics. J Mol Graph. 1996;14:33–38. 27–81. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 58.Jorgensen WL. Revised TIPS for simulations of liquid water and aqueous solutions. J Chem Phys. 1982;77:4156–4163. [Google Scholar]
- 59.Grubmuller H, Heymann B, Tavan P. Ligand binding: molecular mechanics calculation of the streptavidin-biotin rupture force. Science. 1996;271:997–999. doi: 10.1126/science.271.5251.997. [DOI] [PubMed] [Google Scholar]
- 60.Feller SE, Zhang Y, Pastor RW, Brooks BR. Constant pressure molecular dynamics simulation: the Langevin piston method. J Chem Phys. 1995;103:4613–4621. [Google Scholar]
- 61.Darden T, York D, Pedersen L. Particle mesh Ewald. An N. log (N) method for Ewald sums in large systems. J Chem Phys. 1993;98:10089–10092. [Google Scholar]
- 62.Monticelli L, Simões C, Belvisi L, Colombo G. Assessing the influence of electrostatic schemes on molecular dynamics simulations of secondary structure forming peptides. J Phys: Condens Matter. 2006;18:S329–S345. [Google Scholar]
- 63.Ryckaert JP, Ciccotti G, Berendsen HJC. Numerical integration of the cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. J Comput Phys. 1977;23:327–341. [Google Scholar]
- 64.Hubbard SJ, Thornton JM. ‘NACCESS’, computer program. Department Biochemistry and Molecular Biology. London: University College; 1993. [Google Scholar]
- 65.Frishman D, Argos P. Knowledge-based protein secondary structure assignment. Proteins. 1995;23:566–579. doi: 10.1002/prot.340230412. [DOI] [PubMed] [Google Scholar]
- 66.Karypis G. CLUTO — A clustering toolkit. Department of Computer Science and Engineering. Minneapolis: University of Minnesota; 2003. [Google Scholar]