

Abstract
Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) is a multifaceted protein that is involved in numerous processes including glycolysis, translational silencing, transcriptional regulation of specific genes, and acting as a nitric oxide sensor. The precise mechanism on how GAPDH is targeted to these different roles is unclear but believed to involve specific posttranslational modification to the protein. Numerous studies have demonstrated that GAPDH is a target for tyrosine nitration. However, the site of modification and the molecular consequence have not been defined. Rabbit GAPDH with a reversibly protected catalytic cysteine was nitrated in vitro with tetranitromethane, resulting in complete loss of GAPDH catalytic activity. Nitration was estimated as 0.32 mol of nitrotyrosine residue per mole of GAPDH. Mass spectrometry analysis of nitrated GAPDH indicated that Tyr311 and Tyr317 were the sole sites of nitration. The X-ray crystal structure revealed that the distances between Tyr311 and Tyr317 and the cofactor nicotinamide adenine dinucleotide (NAD+) were less than 7.2 and 3.7 Å, respectively, implying that nitration of these two residues may affect NAD+ binding. This possibility was assessed using an NAD+ binding assay, which showed that nitrated GAPDH was incapable of binding NAD+. Thus, these results strongly suggest that Tyr311 and Tyr317 nitration prohibits NAD+ binding, leading to the loss of catalytic activity.
Keywords: nitration, glyceraldehyde-3-phosphate dehydrogenase, GAPDH, nicotinamide adenine dinucleotide, mechanism of inactivation
Introduction
The nitration of protein tyrosine residues is one of the several possible chemical modifications that can occur when a cell or organism is undergoing oxidative stress.1–4 A number of inflammatory and neurodegenerative disorders have been associated with tyrosine nitration, including ocular inflammation,5,6 retinal ischemia,7 lung infection,8 cancer,9 and neurodegenerative disorders.10,11 However, the consequences of nitration in the pathogenesis of these disorders remain unclear.
We recently found that alterations in protein nitration occur in the outer segments of photoreceptor cells upon intense light exposure.12 One of the major target proteins for nitration was glyceraldehyde-3-phosphate dehydrogenase (GAPDH), suggesting possible involvement of GAPDH in the mechanism of light-induced photoreceptor cell death. GAPDH has also been reported to be nitrated in the rat model of sepsis,1 diabetic retinopathy,13 Alzheimer's disease,14 and aging skeletal and cardiac muscle.15,16 However, the effects of this modification on the functions of GAPDH remain unanswered.
GAPDH has long been known as a metabolic enzyme involved in energy production. However, its presence in multiple organelles of the cell suggested that it serves other important functions. Recent investigations have begun to illuminate these roles. These include endocytosis and membrane fusion,17 translational control,18 nuclear tRNA transport,19 signaling,20 pathogen virulence,21 and cell death.22 It is not clear how GAPDH can be targeted for these specific functions in the cell, but is believed to involve posttranslational modification of the protein.23 One such modification that has been extensively studied involves nitric oxide (NO)-induced modification of cysteine. Increased NO generation in cells induces S-nitrosylation of the active site cysteine, which inhibits the enzymatic activity but confers the ability to bind Siah1, a E3 ubiquitin-ligase with a nuclear localization signal.23 This GAPDH-Siah1 complex then migrates to the nucleus and, then following other posttranslational modification of GAPDH, mediates cell death.
Several reports have documented that the catalytic cysteine residue of GAPDH is susceptible to oxidative modifications by nitrating agents, and that these modifications result in the loss of enzymatic activity.24,25 However, the same nitrating agents can also cause nitration of tyrosine residues in GAPDH resulting in as yet unknown effects on the enzyme's activity. The purpose of this study was to examine solely the effect of tyrosine nitration on GAPDH enzymatic activity to better understand the consequences of this modification that might be occurring in vivo. To achieve this goal, an in vitro system is required in which S-nitrosylation of the catalytic cysteine residue is blocked. Therefore, we used rabbit GAPDH whose catalytic cysteine residue had been reversibly protected with 5,5′-dithiobis-2-nitrobenzoic acid (DTNB) and modified the S-protected GAPDH with a nitrating agent, tetranitromethane (TNM). The extent of nitration and localization of nitrotyrosine residues were determined by mass spectrometry. The effect of nitration on GAPDH enzymatic activity was studied, and the mechanism behind the observed effect was uncovered.
Results
Effect of nitration on GAPDH enzymatic activity
The catalytic cysteine residue, Cys149, has been implicated in GAPDH inactivation following the exposure to nitrating agents such as peroxynitrite. Therefore, to study the effects of tyrosine nitration on the enzymatic activity of GAPDH, it was necessary to protect the sulfhydryl group of this residue. This was achieved by treating GAPDH with DTNB, which forms a disulfide bond with the catalytic sulfhydryl group.26
To determine the efficacy of protection and ensure that the use of the protecting agent would not affect the enzymatic assay for GAPDH activity, the enzyme was incubated with DTNB for 30 min. As shown in Figure 1(a), GAPDH activity was reduced more than 95% following incubation with DTNB [Fig. 1(a), bar 2], suggesting that the protection of the catalytic sulfhydryl group by DTNB was nearly complete. S-protected GAPDH regained greater than 95% of its activity following reduction with DTT [Fig. 1(a), bar 3], indicating that the loss of activity induced by DTNB was entirely reversible. No differences in activity were observed upon the addition of DTT [Fig. 1(a), bar 4], showing that the use of the protective agent and DTT did not interfere with the enzyme activity assay.
Figure 1.
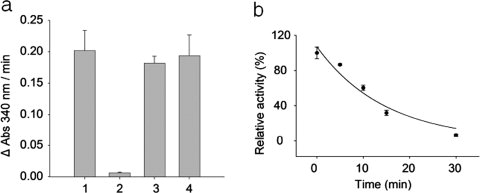
Protection of the catalytic cysteine residue of GAPDH by DTNB and inactivation of S-protected GAPDH by TNM. (a) Rabbit GAPDH was incubated with 5 mM DTNB for 30 min at 25°C. Excess DTNB was removed and the remaining enzymatic activity was measured. Alternatively, an aliquot of S-protected GAPDH was reduced with 10 mM DTT and enzyme activity measured. Bar 1: positive control GAPDH, bar 2: DTNB-treated (S-protected) GAPDH, bar 3: DTT-treated S-protected GAPDH, and bar 4: DTT-treated GAPDH. (b) S-protected GAPDH was nitrated with TNM and aliquots were taken at 0, 5, 10, 15, and 30 min. Excess TNM was removed and the desalted fraction was reduced with 10 mM DTT before the measurement of enzyme activity. The results represent the mean ± standard deviation of three independent experiments.
S-protected GAPDH was nitrated with TNM for different time intervals (0, 5, 10, 15, and 30 min), and residual enzymatic activity was measured after reductive cleavage of the S-protecting group. Figure 1(b) shows a plot of GAPDH activity following nitration relative to that of unmodified enzyme. After 5 min, enzyme activity decreased exponentially from 100 to 86%. The activity was further reduced to 60% after a 10-min treatment. Likewise, treatment for 15 and 30 min reduced GAPDH activity to 32 and 6%, respectively.
Identification of nitrated residues in GAPDH
To identify nitrated residues, GAPDH was S-protected and nitrated with TNM for 30 min followed by reduction with DTT and S-alkylation with iodoacetamide. The resulting product was then digested by trypsin before analysis by LC-MS/MS. A thorough manual analysis of the data revealed only one nitrated peptide, LISWYDNEFGYSNR, which contained Tyr311 and Tyr317, under the experimental conditions used. Supporting Information Figure S1 shows tandem mass spectra of the peptide with nitration at only Tyr311 (nitroTyr311-peptide), Tyr317 (nitroTyr317-peptide), and both sites (nitroTyr311/317-peptide). These spectra verify that Tyr311 and/or Tyr317 are nitration sites within the peptide. Manual analysis did not identify any other modifications that might be possible due to the use of TNM. Only a carboxamide methylated catalytic cysteine residue, Cys149, was found within a peptide (position: 143–159, amino acid sequence: IVSNASCTTNCLAPLAK) after a 30-min TNM treatment (data not shown), suggesting that the catalytic cysteine residue was successfully protected against modification during the treatment, and that the loss of catalytic activity was not caused by modifications to Cys149.
Quantitation of nitrated peptide by mass spectrometry
Although nitroTyr311-peptide and nitroTyr317-peptide exhibited the same m/z value, it was possible to separate them chromatographically and estimate the fractional amount of the individual nonnitrated and nitrated peptide species based on the peak intensity of each species (Supporting Information Fig. S2). The increase in fractional amount of nitration for each nitrated peptide is shown for the different time points in Supporting Information Figure S3(a), and the decrease in the amount of nonnitrated peptide is shown in Supporting Information Figure S3(b). The amount of nitroTyr311-peptide increased from ∼4% at 5 min to 10% at 10 min without any further significant increase at the subsequent time points assessed. Likewise, the amount of nitroTyr317-peptide increased from 2% at 5 min to 4.5% at 10 min and remained the same thereafter. The amount of nitroTyr311/317-peptide increased from 0% at 5 min to ∼17.5% at 30 min. Thus, although the total amount of nonnitrated peptide decreased from 100% at the start of the experiment to about 68%, the total amount of nitrated peptide increased from 0% to about 32%.
Biophysical characterization of the effects of nitration on GAPDH
GAPDH is an asymmetric homotetrameric molecule composed of four functionally nonidentical monomers, which consist of both catalytic and nicotinamide adenine dinucleotide (NAD+)-binding domains.27 The tetramer binds only two molecules of NAD+.27,28 Interactions through NAD+ and the catalytic domains exist only between adjacent monomers.29 These complex interactions form the structural basis for the functional interplay between the oligomers within the tetramer.30 The most important manifestations of such interplay are negative cooperativity in NAD+ binding and half-of-sites reactivity, a nonequivalence of active centers upon interaction with some substrate analogs or inhibitors.30 Thus, the monomers are not functionally similar even though their covalent structures are identical.
It is possible that nitration could disrupt oligomerization of the enzyme. Size exclusion chromatography was used to test this possibility. Unmodified and nitrated GAPDH eluted at the same time at 19.9 min [Supporting Information Fig. S4(b,c)], suggesting that nitration did not disturb the oligomeric organization of GAPDH. Circular dichroism spectroscopy was used to determine whether nitration affected the secondary structure of the enzyme. The spectrum of unmodified GAPDH showed the ellipticity to be higher at 208–210 nm than at 200–222 nm [Supporting Information Fig. S5(a)]. However, the ellipticity of nitrated GAPDH was approximately equal at both wavelengths, suggesting the loss of some secondary structure. Incubation with NAD+ did not alter the circular dichroism spectra [Supporting Information Fig. S5(b)], indicating that NAD+ binding also did not cause significant changes in secondary structure.
NAD+-binding studies
Computational analysis of the X-ray crystal structure of GAPDH revealed that the catalytic cysteine residue is located near the two tyrosine residues that undergo nitration [Fig. 2(a)]. Tyr317 can be seen at the base of an α-helix, whereas Tyr311 resides at the junction of the end of a strand and the beginning of a turn. The phenolic ring side chains of these two tyrosine residues are each ∼4.2 Å away from the sulfhydryl group of the catalytic cysteine residue. Although the phenolic ring of Tyr311 is ∼7.2 Å away from the NAD+ [Fig. 2(a)], only 3.7 Å separates the phenolic side chain of Tyr317 from the nicotinamide moiety of NAD+ in GAPDH [Fig. 2(b)]. This is approximately the same distance as that between the sulfhydryl groups of the catalytic cysteine residue to NAD+, suggesting that NAD+ binding may be affected by nitration. Molecular modeling demonstrated that adding a nitro group to Tyr317 would reduce this distance to 1.4 Å [Fig. 2(c)], which is so short that the modeling software predicts a bond between the nitro group and NAD+. Thus, nitration might affect the binding of NAD+ through steric effects or destabilization of the base of the α-helix on which the tyrosine resides.
Figure 2.
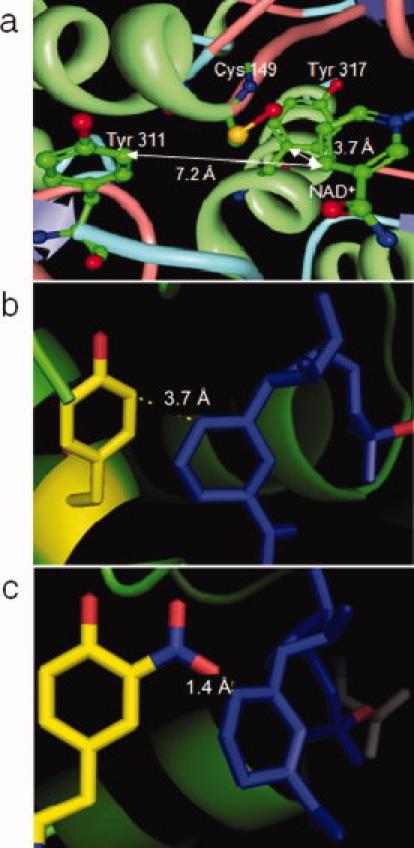
Enlarged view of the GAPDH active site. (a) The active site cysteine (Cys149), Tyr311, Tyr317, and NAD+ reside in close proximity to each other. Distance between Tyr317 and NAD+ in native (b) and nitrated (c) GAPDH. This image was derived from the GAPDH crystal structure stored in the Protein Data Bank.31 The distance measurements were performed using either the molecular biology toolkit viewer32 (a) or the PyMOL Molecular Graphics System software (b, c). The following color coding was used: blue = turn, pink = coil, green = helix, violet = strand, and unknown regions = gray.
Studies that monitor changes in tryptophan fluorescence upon incubation with NAD+ were performed on nitrated GAPDH to test whether nitration affects NAD+ binding. Unmodified and nitrated GAPDH were incubated with different concentrations of NAD+ for 30 min before determining fluorescence [Fig. 3(a)]. Unmodified GAPDH showed a significant decrease (P < 0.05 as per the Student's t-test) in fluorescence at all concentrations of NAD+ compared with nitrated GAPDH. These results suggest that nitrated GAPDH may not be able to bind NAD+ efficiently.
Figure 3.
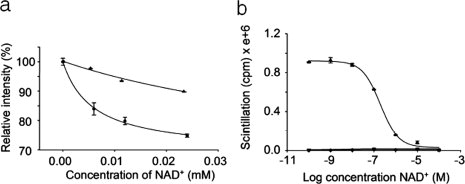
NAD+-binding studies. (a) Change in fluorescence of unmodified and nitrated GAPDH upon binding NAD+. GAPDH (•) and nitrated GAPDH (▴) were incubated with different concentrations of NAD+ for 30 min at room temperature. The fluorescence of tryptophan within the enzyme was measured by monitoring the emission at 340 nm following excitation at 277 nm in a fluorescence spectrophotometer at 25°C. Tryptophan fluorescence in the enzyme before NAD+ addition was considered to be 100%. (b) Competitive NAD+-binding assay of native and nitrated GAPDH. Native (•) or nitrated GAPDH (▴) was incubated with a constant amount of 32P-labeled NAD+ along with different concentrations of unlabeled NAD+ for 1 h at room temperature. Excess NAD+ was removed and bound radioactivity was determined by scintillation counting. The results represent the mean ± standard deviation of three independent experiments.
Radioactive ligand-binding studies were performed with 32P-labeled NAD+ to confirm the results of the fluorescence studies. Unmodified and nitrated GAPDH were incubated with a constant amount of 32P-labeled NAD+, and different concentrations of NAD+ were added to competitively inhibit the binding of labeled ligand to the enzyme. GAPDH was incubated with labeled and unlabeled NAD+ for 1 h before excess ligand was removed using gel filtration. The amount of radioactivity bound to GAPDH was determined by scintillation counting [Fig. 3(b)]. GAPDH exhibited ligand binding and the Kd was calculated to be ∼200 nM. In contrast, nitrated GAPDH essentially did not bind NAD+ as shown by the straight line of the binding curve, thereby confirming that nitration of GAPDH causes the loss of NAD+ binding.
Discussion
GAPDH was one of the proteins found to be tyrosine nitrated in many tissues and cells, including the photoreceptor outer segments following light exposure. Studies on the effect of nitrating agents on the catalytic activity of GAPDH have generally focused on S-nitrosylation of the catalytic cysteine residue. In this article, we attempted to examine the effects of tyrosine nitration alone on GAPDH activity. As in vitro treatment of GAPDH by peroxynitrite causes rapid S-nitrosylation of the catalytic cysteine residue before appreciable amount of tyrosine nitration is detected,33 we used GAPDH whose catalytic sulfhydryl group had been protected. The protecting agent, DTNB, was identified as an agent capable of protecting the catalytic cysteine residue in a reversible manner without affecting enzymatic activity [Fig. 1(a)]. Other agents, such as dimethyldisulfide and 2,2′-dithiodiethanol, were also tested; however, these were ineffective as the extent of protection was too low or the agents caused precipitation of the protein (data not shown). Peroxynitrite is believed to be one of the major precursor nitrating agents in vivo.34 However, peroxynitrite can react with multiple targets resulting in numerous reaction products.35 Because the focus of this study was to understand the mechanism behind the effects observed upon tyrosine nitration of GAPDH, TNM was chosen as a nitrating agent.
The effect of nitration by TNM was studied by following the loss of enzymatic activity with increased nitration of S-protected GAPDH in vitro. The enzyme showed an exponential loss of activity over time [Fig. 1(b)], indicating that tryrosine nitration was adequate for the loss of activity. This loss ranged from 14% at 5 min to 94% at 30 min, whereas the quantity of nitrated tyrosine residues increased from 6.0% at 5 min to 32% at 30 min [Supporting Information Fig. S3(a)]. These results indicate that ∼0.32 mol of tyrosine nitration per mole of GAPDH can lead to a near complete loss of activity. This may suggest that nitration of only one tyrosine residue within a tetramer is sufficient for the loss of activity. Nitration also appeared to affect the secondary structure of the enzyme (see Supporting Information Fig. S5). This loss of secondary structure may affect the intersubunit interactions formed between the catalytic domains of adjacent monomers. This, along with the inability of the enzyme to bind NAD+ could be a contributing factor for the loss of activity with only 32% of the enzyme being affected. Tyr311 and Tyr317 were found to be nitrated and located close to the NAD+-binding site. Unfortunately, no conclusion can be made regarding whether only one or both of the nitrated tyrosine residues was responsible for this loss of activity. Only one nitrated peptide could be confirmed by analyzing tryptic digests of nitrated GAPDH by LC-MS/MS. Other possible modifications that were investigated included nitrotryptophane,36 nitrophenylalanine,37 hydroxyphenylalanine,37 nitrohistidine,35 and conversion of sulfhydryl to sulfenic and sulfonic acid in cysteine.38 The mass spectral data did not show evidence for any of these modifications. This suggests that these other modifications either did not occur or, if they did, were present in such a low yield that they were not likely to be significant.
The major physicochemical difference between tyrosine and nitrotyrosine is the decrease in pKa of the phenolic −OH group from 10.1 in tyrosine to 6.8 in nitrotyrosine.39 This may result in different ionization efficiencies between nitrated and unmodified peptides. However, like phenolic −OH group of tyrosine, the majority of phenolic −OH group of nitrotyrosine should be protonated under the acidic conditions used (pH < 2.5) for LC-MS/MS analysis; therefore, the lower pKa value of nitrotyrosine is unlikely to affect significantly to the ionization efficiencies of nitrated peptides. Thus, the estimated fractions of nitrated peptide species are unlikely to be far different from the true values.
Studies assessing changes in tryptophan fluorescence were performed to determine the binding of NAD+ to GAPDH. Our data showed that the change in tryptophan fluorescence following incubation with NAD+ was significantly different for native GAPDH, whereas the nitrated enzyme showed only a minimal change [Fig. 3(a)], implying that nitrated GAPDH may not bind NAD+ efficiently. These results are consistent with the concept that when small ligands like NAD+ bind larger proteins like GAPDH, the rotational and translational diffusion coefficients will change leading to alterations in fluorescence intensity.40 Competitive ligand-binding studies using radioactively labeled ligands clearly showed that nitrated GAPDH lost its capability to bind NAD+, whereas native GAPDH was able to bind NAD+ with a Kd of 200 nM [Fig. 3(b)]. We also investigated whether incubation of GAPDH with NAD+ could protect the enzyme from losing activity upon exposure to TNM. However, the enzyme still showed the loss of activity (data not shown) that could be attributed to the fact that the GAPDH tetramer binds only two molecules of NAD+.27,28 Therefore, even in the presence of NAD+, the other two vacant subunits are subject to nitration. This hypothesis is reasonable because only 0.32 mol of tyrosine nitration per mole of GAPDH is sufficient to cause a near complete loss of activity.
This article has focused on elucidating the mechanism of inactivation observed in GAPDH upon nitration. The situation in vivo is likely to be different. Peroxynitrite, one of the biological precursor nitrating agents, reacts with not only tyrosine residues but also with other amino acid residues including cysteine. Peroxynitrite reacts with greater efficiency at the cysteine residue when compared with the tyrosine residue, especially in GAPDH.33 The apparent second-order rate constant of peroxynitrite reacting with the catalytic Cys149 in GAPDH (6 × 104 M−1 s−1) (leading to loss of activity) is one order of magnitude faster than that with cysteine, glutathione, and the single thiol group of bovine serum albumin (1.3, 1.5, and 4.5 × 103 M−1 s−1, respectively).41,42 Thus, the modifications on GAPDH in vivo would probably include the combination of S-nitrosylation of cysteine, nitration of tyrosine, oxidation of methionine, and other minor modifications (e.g., oxidation of tryptophan), all of which play a role in determining the effects of nitration. Further studies are planned to determine the precise in vivo role of this specific modification in cells.
Materials and Methods
Materials
Rabbit GAPDH, diethylenetriaminepentaacetic acid (DTPA), 5,5′-dithiobis-2-nitrobenzoic acid (DTNB), tetranitromethane (TNM), and other chemical reagents used in this article were purchased from Sigma-Aldrich (St. Louis, MO). Sephadex-G25 columns and 32P-labeled nicotinamide dinucleotide (NAD+) were purchased from GE Healthcare (Piscataway, NJ). Trypsin for proteolytic digestion was purchased from Promega Corporation (Madison, WI).
Nitration of glyceraldehyde-3-phosphate dehydrogenase
Rabbit GAPDH was dissolved in degassed 50 mM Tris-HCl/0.1 mM DTPA, pH 8.0 buffer to a final concentration of 50 μM. GAPDH was then mixed with activated carbon (1:2 w/w) for 1 h at 4°C to remove any bound NAD+.28,43 The solution was centrifuged at 3000g for 1 min in a tabletop centrifuge and the supernatant removed. The sulfhydryl group of the catalytic cysteine residue (Cys149) was protected via a reaction with 10 mM DTNB in 50 mM Tris-HCl/0.1 mM DTPA, pH 8.0 buffer for 30 min at 25°C. Excess reagent and reaction byproducts were removed by desalting on a Sephadex-G25 column equilibrated with 50 mM Tris-HCl/0.1 mM DTPA, pH 8.0. The S-protected GAPDH (2.5 μM) was nitrated through reaction with 1 mM TNM for different time intervals (i.e., 0, 5, 10, 15, and 30 min) at 25°C. The reaction was stopped by immediately desalting the solution on a Sephadex-G25 column equilibrated with 50 mM sodium phosphate/0.1 mM DTPA, pH 8.0 buffer. The nitrated S-protected GAPDH was treated with 10 mM dithiothreitol (DTT) for 1 h at 25°C to reduce the disulfide bond between the catalytic cysteine residue and 2-nitro-5-thiobenzoic acid and then desalted on an equilibrated Sephadex-G25 column again.
Measurement of GAPDH enzymatic activity
In a 1-cm path length cuvette, 0.04 μM GAPDH or nitrated GAPDH was preincubated with 0.4 mM NAD+ in 50 mM sodium phosphate/0.1 mM DTPA for 15 min at 25°C. The enzyme reaction was initiated by adding 0.4 mM dl-glyceraldehye-3-phosphate to the solution, which was subsequently mixed. Reaction progress was tracked by monitoring the increase in the absorbance of NADH at 340 nm for 2 min using a diode array UV–vis spectrophotometer (Agilent Technologies, Santa Clara, CA).
Identification of the nitrated peptide
Nitrated GAPDH in 50 mM Tris-HCl/0.1 mM DTPA was prepared as described earlier except that sodium bicarbonate was used to elute the protein in the last desalting step. The nitrated protein was denatured by boiling at 80°C for 5 min and then reduced with 10 mM DTT and alkylated with 25 mM iodoacetamide before overnight trypsin digestion. The tryptic digests were concentrated using a Speed-vac and reconstituted in 0.1% formic acid before analysis by liquid chromatography tandem mass spectrometry. Data acquisition was performed on a LTQ Orbitrap hybrid mass spectrometer (Thermo Fisher Scientific, Waltham, MA) with a nanospray ion source connected to an Eksigent nanoLC-2D pump (Dublin, CA). After injection, the peptides were trapped using an Optipak trap cartridge packed with Magic C8 (Michrom Bioresources, Auburn, CA) plumbed into a Valco 10-port valve. Chromatographic separation was performed using a 15 cm × 200 μm C18 reverse phase column (Michrom Bioresources) with a 40-min linear gradient from 10 to 45% of acetonitrile in 0.15% formic acid/0.05% trifluoroacetic acid at a flow rate of 400 nL/min. The column effluent was passed to a capillary sprayer. The parameters used to obtain the total ion current in the positive ion mode with a mass range of 375–1600 m/z were as follows: resolution equals 60,000, isolation width of 1.0 for the precursor and 2.0 for the data dependent scans, normalized collision energy of 35 V, and an activation Q of 0.25. The mass spectrometer was operated in the data-dependent MS to MS/MS switching mode with the three most abundant ions in each precursor scan subjected to tandem mass spectrometry analysis. All acquisition and method development was performed using Xcalibur version 2.0 (Thermo Fisher Scientific). The data were interpreted manually to determine nitration sites.
Measurement of nitrated peptide amount by mass spectrometry
The amount of nitrotyrosine was estimated in a relative manner as follows. The intensity of the chromatographic peaks for individual peptides undergoing nitration and the corresponding nonnitrated peptide was determined. A relative estimate of the amount of nitrated and nonnitrated peptides was calculated by normalizing the individual intensities relative to the sum total of both the nitrated and corresponding non-nitrated peptides.
Circular dichroism
GAPDH and nitrated GAPDH were eluted with 10 mM Tris-HCl/0.2 mM DTPA from Sephadex-G25 columns after the reduction step. Equal amount of GAPDH and nitrated GAPDH (2.5 μM) was used to record circular dichroism spectra using a spectropolarimeter (Aviv Biomedical, Lakewood, NJ) from 190 to 250 nm with a 1-mm bandwidth and 1.356-mm slit. GAPDH and nitrated GAPDH (2.5 μM) were incubated with 0.2 mM NAD+ for 30 min at 25°C, and then circular dichroism spectra were recorded. The data were collected every 2 nm with an averaging time of 10 s per scan using quartz cells with a light path of 0.1 cm.
Size exclusion chromatography
Size exclusion chromatography was carried out using a Shodex KW-800 series column (Shoko America, La Jolla, CA) using 50 mM Tris–HCl, pH 8.0 as the buffer at a flow rate of 0.5 mL/min at 25°C on a Surveyor HPLC system (Thermo Fisher Scientific), and absorbance was monitored at 215 nm with a diode array UV–vis spectrophotometer for detection (Agilent Technologies, Santa Clara, CA).
Spectrofluorometry
GAPDH and nitrated GAPDH (2 μM) prepared were incubated for 30 min at 25°C with different concentrations of NAD+ (e.g., final concentrations were 0, 6, 12, and 24 μM). The fluorescence emission of tryptophan from the enzyme was measured by monitoring the emission spectra at 340 nm following excitation at 277 nm in a fluorescence spectrophotometer (HORIBA Jobin Yvon, Edison, NJ) at 25°C.
Binding assay
GAPDH and nitrated GAPDH (2.5 μM) were incubated with a constant amount of 32P-labeled NAD+ (0.5 pM) along with various concentrations of unlabeled NAD+ for 1 h at 25°C. Excess NAD+ was removed by desalting on a Sephadex G25 column equilibrated with 50 mM Tris-HCl/0.1 mM DTPA, pH 8. Radioactivity of bound protein was measured using a liquid scintillation detection system (Beckman Coulter, Fullerton, CA).
Modeling of nitrated GAPDH
The crystal structure of nitrated GAPDH was modeled based on that of rabbit GAPDH27 using either the PyMOL Molecular Graphics System software (DeLano Scientific, Palo Alto, CA) or the molecular biology toolkit.32
Acknowledgments
The authors thank Dr. Vernon Anderson, Dr. Krysztof Palczewski, Dr. Mark Chance, Dr. Michael Weiss, Dr. David A. Ahlquist, and Dr. Robert H. Bergen for providing access to their analytical equipment. They also thank Dr. Chandrasekhar Rao Kadiyala, Dr. David Lodowski, Dr. Benlian Wang, and Dr. Philip Kiser for their helpful discussions.
Glossary
Abbreviations:
- DTNB
5,5′-dithiobis-2-nitrobenzoic acid
- DTPA
diethylenetriaminepentaacetic acid
- DTT
dithiothreitol
- GAPDH
glyceraldehyde-3-phosphate dehydrogenase
- LC-MS/MS
liquid chromatography tandem mass spectrometry
- NAD+
nicotinamide adenine dinucleotide
- NO
nitric oxide
- NOS
nitric oxide synthase
- TNM
tetranitromethane.
References
- 1.Aulak KS, Miyagi M, Yan L, West KA, Massillon D, Crabb JW, Stuehr DJ. Proteomic method identifies proteins nitrated in vivo during inflammatory challenge. Proc Natl Acad Sci USA. 2001;98:12056–12061. doi: 10.1073/pnas.221269198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Miyagi M, Sakaguchi H, Darrow RM, Yan L, West KA, Aulak KS, Stuehr DJ, Hollyfield JG, Organisciak DT, Crabb JW. Evidence that light modulates protein nitration in rat retina. Mol Cell Proteomics. 2002;1:293–303. doi: 10.1074/mcp.m100034-mcp200. [DOI] [PubMed] [Google Scholar]
- 3.Zhan X, Desiderio DM. Nitroproteins from a human pituitary adenoma tissue discovered with a nitrotyrosine affinity column and tandem mass spectrometry. Anal Biochem. 2006;354:279–289. doi: 10.1016/j.ab.2006.05.024. [DOI] [PubMed] [Google Scholar]
- 4.Rubbo H, Radi R. Protein and lipid nitration: role in redox signaling and injury. Biochim Biophys Acta. 2008;1780:1318–1324. doi: 10.1016/j.bbagen.2008.03.007. [DOI] [PubMed] [Google Scholar]
- 5.Aslan M, Yucel I, Akar Y, Yucel G, Ciftcioglu MA, Sanlioglu S. Nitrotyrosine formation and apoptosis in rat models of ocular injury. Free Radic Res. 2006;40:147–153. doi: 10.1080/10715760500456219. [DOI] [PubMed] [Google Scholar]
- 6.Wu GS, Lee TD, Moore RE, Rao NA. Photoreceptor mitochondrial tyrosine nitration in experimental uveitis. Invest Ophthalmol Vis Sci. 2005;46:2271–2281. doi: 10.1167/iovs.04-1525. [DOI] [PubMed] [Google Scholar]
- 7.Sennlaub F, Courtois Y, Goureau O. Inducible nitric oxide synthase mediates retinal apoptosis in ischemic proliferative retinopathy. J Neurosci. 2002;22:3987–3993. doi: 10.1523/JNEUROSCI.22-10-03987.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hopkins N, Cadogan E, Giles S, Bannigan J, McLoughlin P. Type 2 nitric oxide synthase and protein nitration in chronic lung infection. J Pathol. 2003;199:122–129. doi: 10.1002/path.1256. [DOI] [PubMed] [Google Scholar]
- 9.Maeda H, Akaike T. Nitric oxide and oxygen radicals in infection, inflammation, and cancer. Biochemistry (Mosc) 1998;63:854–865. [PubMed] [Google Scholar]
- 10.Ischiropoulos H, Beckman JS. Oxidative stress and nitration in neurodegeneration: cause, effect, or association? J Clin Invest. 2003;111:163–169. doi: 10.1172/JCI17638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Reynolds MR, Berry RW, Binder LI. Nitration in neurodegeneration: deciphering the “Hows” “nYs”. Biochemistry. 2007;46:7325–7336. doi: 10.1021/bi700430y. [DOI] [PubMed] [Google Scholar]
- 12.Palamalai V, Darrow RM, Organisciak DT, Miyagi M. Light-induced changes in protein nitration in photoreceptor rod outer segments. Mol Vis. 2006;12:1543–1551. [PubMed] [Google Scholar]
- 13.Kanwar M, Kowluru RA. Role of glyceraldehyde 3-phosphate dehydrogenase in the development and progression of diabetic retinopathy. Diabetes. 2009;58:227–234. doi: 10.2337/db08-1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sultana R, Poon HF, Cai J, Pierce WM, Merchant M, Klein JB, Markesbery WR, Butterfield DA. Identification of nitrated proteins in Alzheimer's disease brain using a redox proteomics approach. Neurobiol Dis. 2006;22:76–87. doi: 10.1016/j.nbd.2005.10.004. [DOI] [PubMed] [Google Scholar]
- 15.Kanski J, Alterman MA, Schoneich C. Proteomic identification of age-dependent protein nitration in rat skeletal muscle. Free Radic Biol Med. 2003;35:1229–1239. doi: 10.1016/s0891-5849(03)00500-8. [DOI] [PubMed] [Google Scholar]
- 16.Kanski J, Hong SJ, Schoneich C. Proteomic analysis of protein nitration in aging skeletal muscle and identification of nitrotyrosine-containing sequences in vivo by nanoelectrospray ionization tandem mass spectrometry. J Biol Chem. 2005;280:24261–24266. doi: 10.1074/jbc.M501773200. [DOI] [PubMed] [Google Scholar]
- 17.Bryksin AV, Laktionov PP. Role of glyceraldehyde-3-phosphate dehydrogenase in vesicular transport from golgi apparatus to endoplasmic reticulum. Biochemistry (Mosc) 2008;73:619–625. doi: 10.1134/s0006297908060011. [DOI] [PubMed] [Google Scholar]
- 18.Mukhopadhyay R, Jia J, Arif A, Ray PS, Fox PL. The GAIT system: a gatekeeper of inflammatory gene expression. Trends Biochem Sci. 2009;34:324–331. doi: 10.1016/j.tibs.2009.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jia J, Arif A, Ray PS, Fox PL. WHEP domains direct noncanonical function of glutamyl-Prolyl tRNA synthetase in translational control of gene expression. Mol Cell. 2008;29:679–690. doi: 10.1016/j.molcel.2008.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pumain R, Laschet J. A key glycolytic enzyme plays a dual role in GABAergic neurotransmission and in human epilepsy. Crit Rev Neurobiol. 2006;18:197–203. doi: 10.1615/critrevneurobiol.v18.i1-2.200. [DOI] [PubMed] [Google Scholar]
- 21.Fugier E, Salcedo SP, de Chastellier C, Pophillat M, Muller A, Arce-Gorvel V, Fourquet P, Gorvel JP. The glyceraldehyde-3-phosphate dehydrogenase and the small GTPase Rab 2 are crucial for Brucella replication. PLoS Pathog. 2009;5:e1000487. doi: 10.1371/journal.ppat.1000487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hara MR, Snyder SH. Nitric oxide-GAPDH-Siah: a novel cell death cascade. Cell Mol Neurobiol. 2006;26:527–538. doi: 10.1007/s10571-006-9011-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hara MR, Cascio MB, Sawa A. GAPDH as a sensor of NO stress. Biochim Biophys Acta. 2006;1762:502–509. doi: 10.1016/j.bbadis.2006.01.012. [DOI] [PubMed] [Google Scholar]
- 24.Mohr S, Stamler JS, Brune B. Posttranslational modification of glyceraldehyde-3-phosphate dehydrogenase by S-nitrosylation and subsequent NADH attachment. J Biol Chem. 1996;271:4209–4214. doi: 10.1074/jbc.271.8.4209. [DOI] [PubMed] [Google Scholar]
- 25.Souza JM, Radi R. Glyceraldehyde-3-phosphate dehydrogenase inactivation by peroxynitrite. Arch Biochem Biophys. 1998;360:187–194. doi: 10.1006/abbi.1998.0932. [DOI] [PubMed] [Google Scholar]
- 26.Riddles PW, Blakeley RL, Zerner B. Ellman's reagent: 5,5′-dithiobis(2-nitrobenzoic acid)—a reexamination. Anal Biochem. 1979;94:75–81. doi: 10.1016/0003-2697(79)90792-9. [DOI] [PubMed] [Google Scholar]
- 27.Cowan-Jacob SW, Kaufmann M, Anselmo AN, Stark W, Grutter MG. Structure of rabbit-muscle glyceraldehyde-3-phosphate dehydrogenase. Acta Crystallogr D. 2003;59:2218–2227. doi: 10.1107/s0907444903020493. [DOI] [PubMed] [Google Scholar]
- 28.Taylor JF, Velick SF, Cori GT, Cori CF, Slein MW. The prosthetic group of crystalline d-glyceraldehyde-3-phosphate dehydrogenase. J Biol Chem. 1948;173:619–626. [PubMed] [Google Scholar]
- 29.Duee E, Olivier-Deyris L, Fanchon E, Corbier C, Branlant G, Dideberg O. Comparison of the structures of wild-type and a N313T mutant of Escherichia coli glyceraldehyde 3-phosphate dehydrogenases: implication for NAD binding and cooperativity. J Mol Biol. 1996;257:814–838. doi: 10.1006/jmbi.1996.0204. [DOI] [PubMed] [Google Scholar]
- 30.Nagradova NK. Study of the properties of phosphorylating D-glyceraldehyde-3-phosphate dehydrogenase. Biochemistry (Mosc) 2001;66:1067–1076. doi: 10.1023/a:1012472627801. [DOI] [PubMed] [Google Scholar]
- 31.Cowan-Jacobs SW, Kaufmann M, Anselmo AN, Stark W, Grutter MG. Structure of rabbit-muscle glyceraldehyde-3-phosphate dehydrogenase. Acta Crystallogr D. 2007;59:2218–2227. doi: 10.1107/s0907444903020493. [DOI] [PubMed] [Google Scholar]
- 32.Moreland JL, Gramada A, Buzko OV, Zhang Q, Bourne PE. The molecular biology toolkit (MBT): a modular platform for developing molecular visualization applications. BMC Bioinformatics. 2005;6:21. doi: 10.1186/1471-2105-6-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Buchczyk DP, Grune T, Sies H, Klotz LO. Modifications of glyceraldehyde-3-phosphate dehydrogenase induced by increasing concentrations of peroxynitrite: early recognition by 20S proteasome. Biol Chem. 2003;384:237–241. doi: 10.1515/BC.2003.026. [DOI] [PubMed] [Google Scholar]
- 34.Szabo C, Ischiropoulos H, Radi R. Peroxynitrite: biochemistry, pathophysiology, and development of therapeutics. Nat Rev Drug Discov. 2007;6:662–680. doi: 10.1038/nrd2222. [DOI] [PubMed] [Google Scholar]
- 35.Alvarez B, Radi R. Peroxynitrite reactivity with amino acids and proteins. Amino Acids. 2003;25:295–311. doi: 10.1007/s00726-003-0018-8. [DOI] [PubMed] [Google Scholar]
- 36.Prieels JP, Dolmans M, Leonis J, Brew K. Nitration of tyrosyl residues in human alpha-lactalbumin. Effect on lactose synthase specifier activity. Eur J Biochem. 1975;60:533–539. doi: 10.1111/j.1432-1033.1975.tb21032.x. [DOI] [PubMed] [Google Scholar]
- 37.van der Vliet A, O'Neill CA, Halliwell B, Cross CE, Kaur H. Aromatic hydroxylation and nitration of phenylalanine and tyrosine by peroxynitrite. Evidence for hydroxyl radical production from peroxynitrite. FEBS Lett. 1994;339:89–92. doi: 10.1016/0014-5793(94)80391-9. [DOI] [PubMed] [Google Scholar]
- 38.Carballal S, Radi R, Kirk MC, Barnes S, Freeman BA, Alvarez B. Sulfenic acid formation in human serum albumin by hydrogen peroxide and peroxynitrite. Biochemistry. 2003;42:9906–9914. doi: 10.1021/bi027434m. [DOI] [PubMed] [Google Scholar]
- 39.De Filippis V, Frasson R, Fontana A. 3-Nitrotyrosine as a spectroscopic probe for investigating protein protein interactions. Protein Sci. 2006;15:976–986. doi: 10.1110/ps.051957006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hovius R, Vallotton P, Wohland T, Vogel H. Fluorescence techniques: shedding light on ligand-receptor interactions. Trends Pharmacol Sci. 2000;21:266–273. doi: 10.1016/s0165-6147(00)01503-0. [DOI] [PubMed] [Google Scholar]
- 41.Souza JM, Radi R. Glyceraldehyde-3-phosphate dehydrogenase inactivation by peroxynitrite. Arch Biochem Biophys. 1998;360:187–194. doi: 10.1006/abbi.1998.0932. [DOI] [PubMed] [Google Scholar]
- 42.Ducrocq C, Blanchard B, Pignatelli B, Ohshima H. Peroxynitrite: an endogenous oxidizing and nitrating agent. Cell Mol Life Sci. 1999;55:1068–1077. doi: 10.1007/s000180050357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dalziel K, McFerran NV, Wonacott AJ. Glyceraldehyde-3-phosphate dehydrogenase. Philos Trans R Soc Lond B Biol Sci. 1981;293:105–118. doi: 10.1098/rstb.1981.0064. [DOI] [PubMed] [Google Scholar]