

Abstract
ClpXP, an AAA+ protease, plays key roles in protein-quality control and many regulatory processes in bacteria. The N-terminal domain of the ClpX component of ClpXP is involved in recognition of many protein substrates, either directly or by binding the SspB adaptor protein, which delivers specific classes of substrates for degradation. Despite very limited sequence homology between the E. coli and C. crescentus SspB orthologs, each of these adaptors can deliver substrates to the ClpXP enzyme from the other bacterial species. We show that the ClpX N domain recognizes different sequence determinants in the ClpX-binding (XB) peptides of C. crescentus SspBα and E. coli SspB. The C. crescentus XB determinants span 10 residues and involve interactions with multiple side chains, whereas the E. coli XB determinants span half as many residues with only a few important side chain contacts. These results demonstrate that the N domain of ClpX functions as a highly versatile platform for peptide recognition, allowing the emergence during evolution of alternative adaptor-binding specificities. Our results also reveal highly conserved residues in the XB peptides of both E. coli SspB and C. crescentus SspBα that play no detectable role in ClpX-binding or substrate delivery.
Keywords: regulated proteolysis, AAA+, adaptor, SspB, ClpX N domain, ClpP
Introduction
Proteolysis of damaged or misfolded proteins by AAA+ proteases is essential for quality control and recycling of amino acids for new protein synthesis. Protein degradation also plays a regulatory role in numerous cellular processes, including responses to DNA damage and cell-cycle progression.1 Because proteolysis occurs in crowded cellular environments with thousands of potential substrates, it is important to understand how the proper proteins are chosen for destruction. For bacterial systems, peptide signals (called degradation tags or degrons) in substrates and adaptor proteins play central roles in determining the specificity of proteolytic recognition. How adaptor proteins and degrons are recognized by AAA+ proteases is an active area of study, but only a handful of these interactions have been characterized in detail.
ClpX and ClpP assemble to form ClpXP, one of the best understood AAA+ proteases. Most biochemical studies have focused on ClpXP from Escherichia coli, a member of the γ-proteobacteria, but orthologs from other bacteria and mitochondria appear to have similar structures and mechanisms.2–4 ClpP is a multisubunit serine peptidase, in which the proteolytic active sites reside within a barrel-shaped structure.5 ClpX is a hexameric AAA+ enzyme (ATPases associated with a variety of cellular activities), which recognizes substrates and uses cycles of ATP-powered conformational changes to unfold the native protein and to translocate the denatured polypeptide into the proteolytic chamber of ClpP for degradation.6–8
ClpX typically identifies substrates by binding to degrons located near the protein termini. For example, when ribosomes stall during translation, the ssrA tag is appended onto the C-terminus of incomplete polypeptides and subsequently targets these failed translation products to ClpXP and other proteases.6,7 The 11-residue ssrA tag can be recognized directly by ClpX but is also bound by an adaptor protein, SspB, which aids in delivery of substrates to ClpXP.8 Indeed, adaptor proteins facilitate ClpXP degradation of numerous substrates.9–11 Each ClpX subunit consists of an AAA+ domain and a ClpX-family-specific N-terminal domain, which binds zinc via a conserved set of cysteine residues and forms a stable dimer.12,13 ClpX lacking the N domain (ClpXΔN) can still bind ClpP and power degradation of some substrates,12,14 establishing that the N domain is not required for the basic enzymatic functions of ClpX. However, ClpXΔN fails to recognize certain substrates and does not support degradation mediated by many adaptors.10,12,14–18
SspB consists of a dimeric substrate-binding domain, followed by a flexible linker and a C-terminal peptide that binds to the ClpX N domain.8,15,19–23 By binding to ClpX and specific substrates, simultaneously, SspB increases the local concentration of substrate relative to the protease.8,11,15,21,24 As a consequence of this tethering-mediated avidity increase, SspB enhances the rate of ClpXP degradation at sub-KM substrate concentrations. SspB orthologs were first identified in the γ- and β-proteobacteria8 and were later discovered in α-proteobacteria, including Caulobacter crescentus.25,26 The domain organization and structure of all SspB proteins are similar, but those from α-proteobacteria comprise a distinct and more distant subfamily and are therefore called SspBα. For example, the orthologs from E. coli (EcSspB) and C. crescentus (CcSspBα) share only 16% sequence identity. Nevertheless, CcSspBα delivers substrates efficiently to E. coli ClpXP (EcClpXP).17 The C-terminal residues of EcSspB are known to bind the isolated N domain of EcClpX,23,27 and a co-crystal structure has been solved (PDB ID: 2DS8) [Fig. 1(A)].23 The N domain of CcClpXP and the five C-terminal amino acids of CcSspBα are also required for adaptor-mediated substrate delivery,25 suggesting a corresponding binding relationship.
Figure 1.
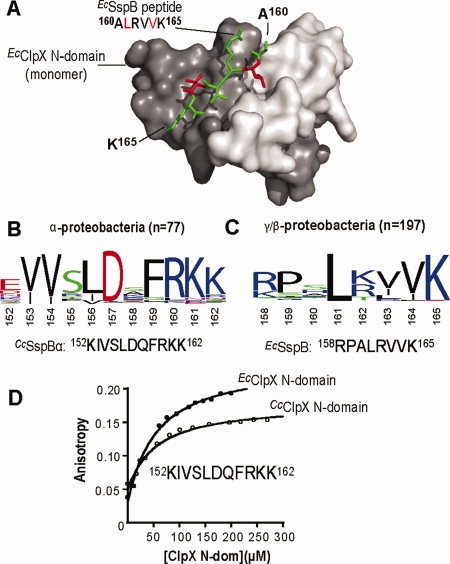
XB conservation and cross-species interaction of SspB and ClpX from α- and γ/β-proteobacteria. A: The structure of the E. coli ClpX N-domain dimer bound to the C-terminal peptide of E. coli SspB (PDB ID: 2DS8) shows the hydrophobic pockets of the N domain monomers occupied by the L161 and V164 residues of the peptide.23 The N-domain monomers are shown in dark and pale gray and the peptide in green with L161 and V164 highlighted in red. B: Weblogo28 depiction of sequence conservation within the C-terminal regions of 77 SspB proteins from α-proteobacteria. Alignments were performed using Jalview.29 The C-terminal region of the α-proteobacteria C. crescentus SspB is also depicted. C: Sequence conservation in the C-terminal regions of 197 SspB proteins from γ/β-proteobacteria reveals a very different pattern than observed in panel A. The C-terminal region of the γ-proteobacteria E. coli SspB is also shown. D: Binding of the N domains from C. crescentus (KD 25 μM) or E. coli (KD 25 μM) ClpX to a fluorescein-labeled peptide (60 nM) corresponding to the XB region of CcSspBα.
Here, we probe the fine specificity of the interaction of CcSspBα with CcClpX and EcClpX. In both cases, the 10 C-terminal residues of CcSspBα comprise the ClpX-binding (XB) region that tethers the adaptor to the N domain. Mutational analyses show that seven side chains in CcSspBα XB contribute to adaptor-enzyme recognition, and all SspBαs have homologous sequences that maintain the chemical character of these residues. Surprisingly, however, the corresponding XB peptide of EcSspB is shorter, shares little meaningful homology, and displays a radically different mutational profile with just a few residues playing major roles in recognition. Again, these features appear to be shared by SspB orthologs in other γ- and β-proteobacteria. Nevertheless, we find that EcSspB delivers substrates to CcClpXP for degradation. Thus, the N domains of both EcClpX and CcClpX have the ability to recognize two different XB peptides. Apparently, these domains possess distinct peptide-binding specificities and have adopted alternative but nonexclusive modes of adaptor-binding during the evolution of different bacterial lineages. We also find that some highly conserved amino acids in the XB peptides of CcSspBa and EcSspB play no obvious roles in substrate delivery or ClpX-binding and suggest that these amino acids may help protect the adaptors from degradation during substrate delivery.
Results
Phylogenetic comparisons suggest use of different adaptor tethering contacts
A multiple sequence alignment of the C-terminal regions of more than 100 SspBα orthologs revealed a conserved block of residues [Fig. 1(B)]. Within this region, CcSspBα residues 153, 154, 156, 157, 159, 160, and 161 were most highly conserved. Deletion of a portion of this C-terminal region prevents substrate delivery by CcSspBα to ClpXP.25 Sequence conservation near the C-terminus of SspB orthologs from γ- and β-proteobacteria revealed a very different pattern of homology [Fig. 1(C)], suggesting that SspBα interacts with ClpX in a fashion distinct from their γ and β counterparts.
Previous studies showed that the EcClpX N domain binds to the EcSspB XB peptide and is important for efficient substrate delivery by CcSspBα,17,23,27 suggesting that the CcSspBα XB region binds directly to the N domains of EcClpX and CcClpX. To test this idea, a peptide consisting of the C-terminal decapeptide of CcSspBα preceded by a fluorescent dye and tyrosine was synthesized for binding studies monitored by fluorescence anisotropy. As shown in Figure 1(D), this CcSspBα peptide was bound with similar affinity (KD ∼ 25 μM) by the purified CcClpX and EcClpX N domains. Thus, the N domains of both ClpX enzymes, which share ∼60% sequence identity (Supporting Information Figure 1), have the ability to recognize very different XB-peptide sequences.
Adaptor delivery of cognate substrates to CcClpXP and EcClpXP
For studies of adaptor stimulation of degradation, we used green fluorescent protein (GFP) bearing either a C. crescentus ssrA tag (AANDNFAEEFAVAA; GFP-CcssrA), which binds well to CcSspBα, or an E. coli ssrA tag (AANDENYALAA; GFP-EcssrA), which binds well to EcSspB.17,21 As anticipated,17,25 CcSspBα stimulated degradation of GFP-CcssrA by the CcClpXP protease and by the EcClpXP enzyme (Fig. 2). Importantly, EcSspB also stimulated degradation of GFP-EcssrA by both CcClpXP and EcClpXP [Fig. 2(B,C)]. Thus, despite minimal XB-sequence homology, the adaptors from E. coli and C. crescentus were both able to stimulate degradation of cognate substrates by the ClpXP enzyme from the other species.
Figure 2.
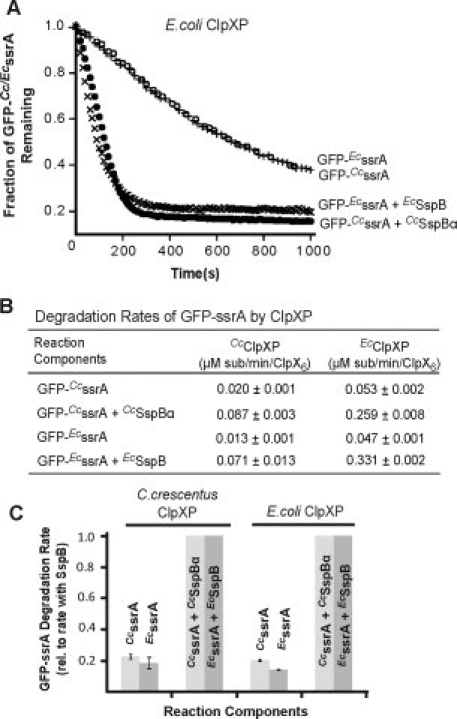
C. crescentus and E. coli ClpXP interact with both CcSspBα and EcSspB. A: CcSspBα and EcSspB (1.2 μM) both enhanced degradation of their cognate GFP-ssrA by EcClpXP as monitored by decreases in fluorescence. ClpXP concentration in each case was 0.1 μM and the substrate concentration was 0.1 μM. This low substrate concentration (sub-KM) was used to help ensure that degradation was adaptor-stimulated. B: Both CcClpX and EcClpXP were able to degrade GFP containing either the CcssrA or the EcssrA tags. Protein concentrations used were as in (A). The rate of substrate degradation was enhanced by the cognate adaptor SspB. Under the purification conditions used in this study, CcClpX was less active compared to EcClpX. C: Normalized degradation rates of GFP-CcssrA (pale gray bars) or GFP-EcssrA (dark gray bars) by CcClpXP and EcClpXP in the presence or absence of CcSspBα or EcSspB; in this case, the adaptor species (Cc vs. Ec) matched that of the ssrA tag sequence on the substrate. Protein concentrations were as described in (A).
We also constructed a chimera, consisting of the substrate-binding domain of EcSspB followed by the CcSspBα C-terminal linker and XB region. This chimeric adaptor enhanced EcClpXP degradation of GFP-EcssrA [Fig. 3(A)], establishing that tethering interactions mediated by CcSspBα XB can replace the interactions normally mediated by EcSspB XB.
Figure 3.
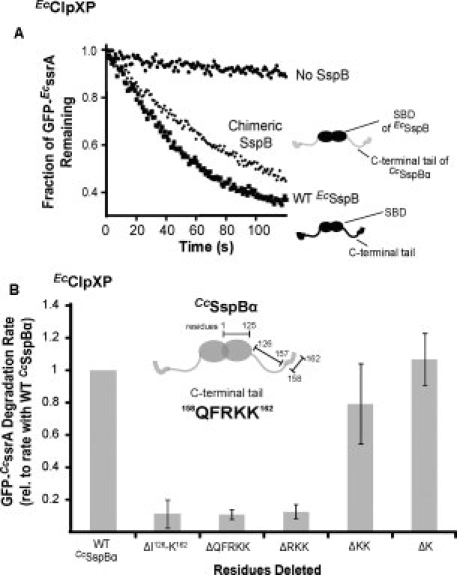
CcSspBα interacts with ClpXP via its C-terminal region. A: A chimeric SspB (0.3 μM), obtained by substituting the C-terminal region of EcSspB with that of CcSspBα, enhanced the degradation of GFP-EcssrA (0.1 μM) by EcClpXP (0.05 μM). B: Different segments of the C-terminal region of CcSspBα were removed and the variant adaptors tested for their ability to enhance degradation of GFP-EcssrA (0.1 μM) by EcClpXP (0.05 μM).
To address the importance of residues near the C-terminus of CcSspBα, we constructed truncated variants and assayed their adaptor activity. The last two lysine residues (K161K162) could be deleted without a major effect on delivery, whereas deletion of additional upstream residues eliminated activity [Fig. 3(B)]. However, the substrate-binding domain of EcSspB followed by the C-terminal residues 158QFRKK162 of CcSspBα was inactive as an adaptor (data not shown), establishing that the 158QFR sequence may be necessary but is not sufficient for adaptor function.
Residues involved in CcSspBα tethering to ClpX
To determine which residues in the XB region of CcSspBα are important for ClpX binding, we individually mutated the 10 C-terminal residues to alanine and purified these variants. In one set of assays, we tested stimulation of CcClpXP degradation of 0.1 μM GFP-CcssrA (Fig. 4). This substrate concentration is below KM (∼1 μM) for unassisted CcClpXP degradation, allowing adaptor-mediated stimulation to be observed. Alanine substitutions at I153, V154, L156, R160, and K162 caused the largest defects in substrate delivery [Fig. 4(A)]. Milder effects were observed for substitutions at the other positions, with mutations at S155 and D157 having essentially no effect on delivery activity. The alanine mutations in CcSspBα had generally similar effects on adaptor-mediated degradation by EcClpXP. However, V154A appeared to be more active with the EcClpXP protease than with CcClpXP [Fig. 4(A,B)], suggesting that this residue plays a somewhat different role in binding the two enzymes.
Figure 4.
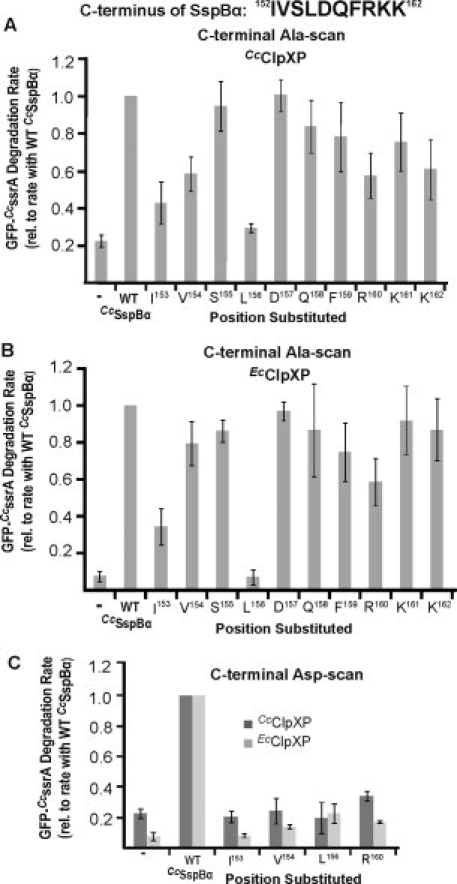
Substrate-delivery activity of CcSspBα variants with substitution mutations. A: Variants of CcSspBα with Ala substitutions in the C-terminal residues were assayed for their delivery activity by determining how well they enhanced degradation of GFP-CcssrA (0.1 μM) by CcClpXP (0.2 μM). All rates were normalized to the degradation rate of GFP-CcssrA in the presence of WT CcSspBα. Wild-type CcSspBα or variants were present at 0.3 μM. B: Analysis is the same as in (A), except that the protease was EcClpXP (at 0.05 μM). Similar results were observed, however the V154A variant appeared to be more active with EcClpXP than with CcClpXP. C: Aspartate-substitutions were made at four positions (I153, V154, L156, and R160) in CcSspBα and the variants (0.3 μM) tested for adaptor function with CcClpXP (0.2 μM) and EcClpXP (0.05 μM). The rates were normalized to the degradation rate of GFP-CcssrA in the presence of WT CcSspBα.
Most alanine substitutions did not reduce activity to the level of unassisted ClpXP degradation, suggesting that these mutations weaken but do not eliminate tethering. To test if more dramatic mutations had larger effects, we also constructed and purified variants in which I153, V154, L156, and R160 were changed to aspartic acid. Except for R160D, these substitutions decreased CcClpXP degradation to the level of the no-SspB control when assayed with CcClpXP [Fig. 4(C)]. The aspartate substitutions were also more severe than the alanine substitutions in EcClpXP degradation assays [Fig. 4(C)].
Assays of CcSspBα-mediated stimulation of GFP-CcssrA degradation have limited dynamic range because this substrate is degraded reasonably well (KM ∼ 1–2 μM) by ClpXP alone [Fig. 5(B)]. To address this concern, we changed the C-terminal residues of this substrate from VAA to DAS (GFP-CcDAS) [Fig. 5(A)]. This substitution weakens EcClpX recognition of the ssrA tag and increases the adaptor-dependence of degradation.30 When we assayed CcSspBα stimulation of CcClpXP degradation of GFP-CcDAS, the I153A, V154A, L156A, R160A, and K162A mutations caused substantial reductions in the stimulated degradation rate whereas the D157A, F159A, and K161A substitutions had only modest effects [Fig. 5(C)].
Figure 5.
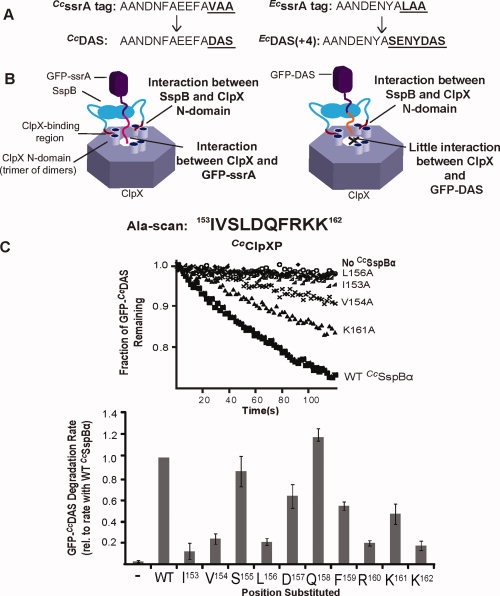
Degradation of GFP-CcDAS by CcClpXP. A: The ssrA tag in C. crescentus (CcssrA) consists of 14 amino acids, the last three (VAA) resembling the terminal “LAA” residues of the 11-residue E. coli ssrA tag (EcssrA). Substituting the last three residues of either tag with the sequence “DAS” made the tag a weaker degron, and thus proteolysis of tagged protein more adaptor-dependent. For EcssrA, the tag was also elongated (to remove an SspB-ClpX clash) to generate the DAS + 4 tag. B: Cartoon depicting ssrA-DAS recognition. The efficiency of substrate degradation depends on the protein-protein interactions occurring in the ternary complex formed between the substrate (GFP-ssrA), the adaptor (SspB), and ClpX (left panel). When the tag is modified such that there is only a weak substrate-ClpX interaction (e.g. the DAS tag), adaptor-ClpX interactions dictate the efficiency of degradation (right panel). C: The CcSspBα C-terminal residues were individually changed to alanine and assayed for their ability to enhance degradation of GFP-CcDAS (0.1 μM) by CcClpXP (0.2 μM). CcSspBα variants were present at 0.3 μM. (Top) Degradation traces for representative CcSspBα variants. (Bottom) Summary of the alanine-scan results of the C-terminal region of CcSspBα. The rates were normalized to the degradation rate of GFP-CcDAS (0.1 μM) in the presence of WT CcSspBα (0.3 μM).
Substitution of alanine for S155 or D157 in CcSspBα did not have a large effect on substrate delivery. It seemed possible, however, that proline substitutions at these positions might interfere with binding to ClpX by disrupting conformations (for example, an α-helix or β-strand) of the entire XB peptide and thus interfering with contacts made by residues flanking these positions. However, proline substitutions at either position caused only minor reductions in the ability of these CcSspBα variants to stimulate CcClpXP degradation (Fig. 6). It appears, therefore, that the XB peptide of CcSspBα binds in a conformation compatible with the restrictions of the backbone dihedral angle that would be enforced by proline at these positions.
Figure 6.
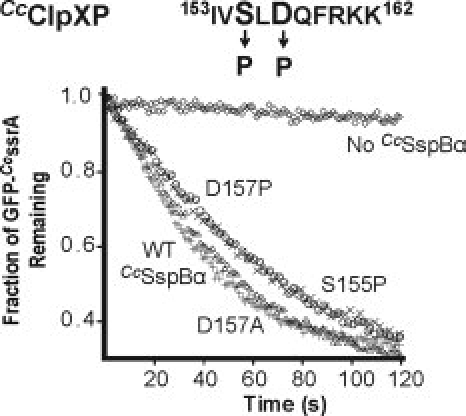
Secondary structure in the CcSspBα C-terminal region is not critical. S155 or D157 (shown in bold in sequence, Top) in the C-terminal region of CcSspBα were substituted with proline, which disrupts secondary structure, and the proteins were assayed for their ability to deliver GFP-CcssrA (0.1 μM) to CcClpXP (0.2 μM). Neither substitution inhibited adaptor function.
CcSspBα XB mutations decrease N-domain affinity
We anticipated that the alanine substitutions in the XB region of CcSspBα would reduce affinity for the CcClpXP N domain. To test this ideal directly, we synthesized fluorescent XB-peptide variants and assayed binding. Alanine substitutions for I153, V154, L156, F159, R160, and K162 decreased affinity to varying extents (Fig. 7, Table I). Substitutions at the remaining positions had very small effects. These results largely mirror the defects in substrate delivery for the corresponding substitutions in full-length CcSspBα.
Figure 7.
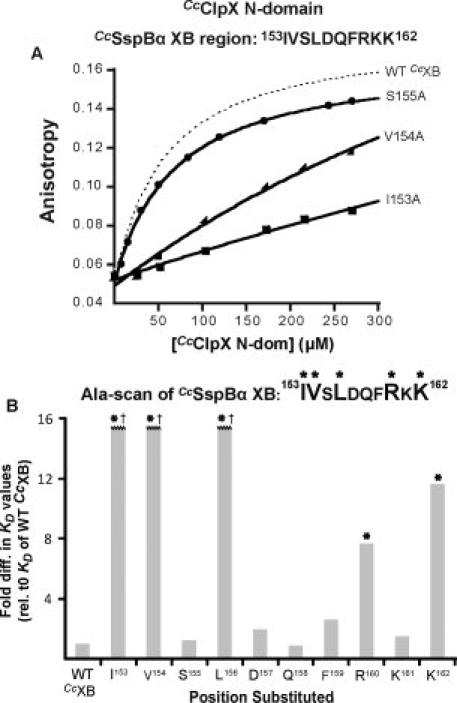
Binding interaction between CcSspBα ClpX N domain and CcSspBα. A: An alanine-scan of a peptide consisting of the C-terminal residues of CcSspBa was done and the peptides were tested for their ability to bind the CcClpX N domain. Binding of the wild-type sequence is shown in dashed gray [see Fig. 1(C)]. Peptide concentration was 60 nM. KD values of all the CcSspBα C-terminal peptide variants are shown in Table I. B: Comparison of the change in KD value caused by the Ala-substitution (relative to the value of WT CcSspBα XB). Residues I153, V154, L156, R160, and K162 (marked with asterisk) were most important for binding CcClpX N domain. The KD values of Ala-substitutions at I153, V154, and L156 (marked with †) were >16-fold higher than that of WT adaptor and could not be depicted within the scale of the y-axis.
Table I.
KD Values of CcSspBα C-Terminal Peptides Binding to C. crescentus ClpX N-Domain (Dimer)
| Peptide | Sequence | KD (μM) |
|---|---|---|
| WT | YKIVSLDQFRKK | 26.0 ± 4.8 |
| I153A | YKAVSLDQFRKK | >400 |
| V154A | YKIASLDQFRKK | >400 |
| S155A | YKIVALDQFRKK | 32.1 ± 2.3 |
| L156A | YKIVSADQFRKK | >400 |
| D157A | YKIVSLAQFRKK | 50.9 ± 12 |
| Q158A | YKIVSLDAFRKK | 23.1 ± 3.9 |
| F159A | YKIVSLDQARKK | 66.9 ± 13 |
| R160A | YKIVSLDQFAKK | 200 ± 82 |
| K161A | YKIVSLDQFRAK | 39.3 ± 5.4 |
| K162A | YKIVSLDQFRKA | 303 ± 100 |
Effects of EcSspB XB mutations on substrate delivery and N-domain affinity
As shown in Figure 1(B,C), phylogenetic comparisons reveal very different patterns of sequence conservation for the XB peptides of SspB orthologs from the γ- and β-proteobacteria as opposed to those from α-proteobacteria. To probe the functional importance of residues in the EcSspB XB peptide, we purified alanine-substituted variants and assayed their ability to deliver GFP-EcDAS to EcClpXP. The largest defects were observed for the L161A and V164A variants [Fig. 8(A)]. For comparison, this figure also shows the relative abilities of the same variants to deliver a normal ssrA-tagged substrate, a similar but less sensitive assay.21
Figure 8.
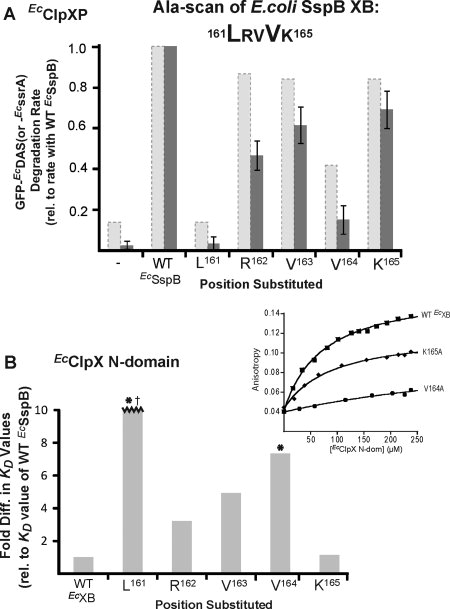
Functional and binding interactions between EcClpX N domain and EcSspB. A: The five C-terminal residues of EcSspB were changed individually to Ala and the variants tested for function by monitoring degradation of GFP- EcDAS (dark gray bars). Reactions contained 0.05 μM EcClpXP, 0.3 μM EcSspB variant and 0.1 μM substrate. Rates were normalized to the rate of degradation of GFP-EcDAS in the presence of WT EcSspB. The L161A and V164A variants were the most defective. A similar result was observed by Wah et al.21 who used GFP-EcssrA as a substrate to test the function of these Ala-variants (shown in pale gray/dashed bars). As expected, the activities of all variants were higher for degradation of WT GFP-ssrA than the DAS variant. B: Peptides (60 nM) corresponding to an alanine-scan of the EcSspB C-terminal region were tested for binding to the EcClpX N domain. The KD values were determined by fluorescence anisotropy (inset). Residues L161 and V164 (marked with asterisk) were the most important for the interaction. The KD value of L161A (marked with †) was >10-fold higher than that of WT adaptor and could not be depicted within the scale of the y-axis.
To determine the effects of the alanine substitutions on the affinity of the EcSspB XB peptide for the EcClpX N domain, we carried out peptide-binding experiments [Fig. 8(B)]. The L161A substitution made N-domain binding too weak to measure, the V164A substitution decreased binding approximately seven-fold, whereas smaller effects were detected for the remaining substitutions. These results agree well with the functional studies. Importantly, they establish that the N domain of ClpX recognizes CcSspBa XB peptides in a substantially different manner than the XB peptides from c- and b-proteobacterial SspB adaptors.
Different XB sequences compete for binding to the N domain
Because CcXB and EcXB sequences have such distinct features, we sought to determine if they bound to distinct sites in the N domains of CcClpX and EcClpX. Therefore, degradation of GFP-EcDAS by either CcClpXP or EcClpXP was performed in the presence of EcSspB with or without high concentrations of CcXB peptide. As shown in Figure 9(A), this peptide inhibited degradation by both proteases, indicating that it competes with EcSspB for interaction with these enzymes. Furthermore, GFP-EcssrA degradation, which is less adaptor-dependent, was also inhibited by the CcXB peptide [Fig. 9(B)]. Importantly, CcXB only inhibited reactions in which substrate delivery was promoted by an adaptor (EcSspB). Thus, these results indicate that the C. crescentus and E. coli XB peptides bind to the same or overlapping sites on the N domain of ClpX.
Figure 9.
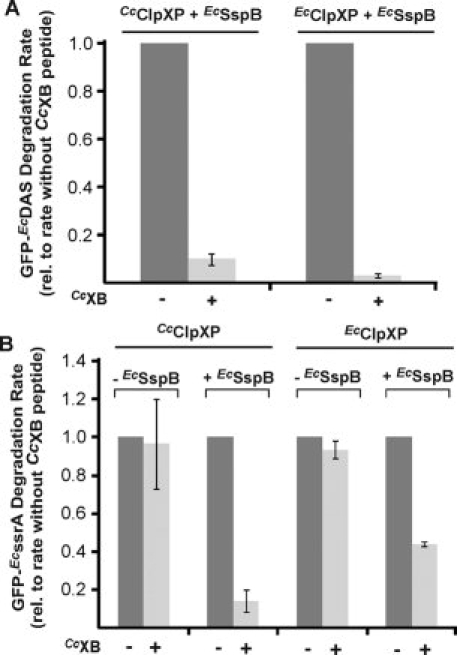
Competition between CcSspB XB and EcSspB XB motifs for binding CcClpX and EcClpX. A: GFP-EcDAS (0.1lM) degradation reactions were set up using either EcClpXP (0.05 lM) or CcClpXP (0.2 lM) as the protease. When a high concentration of CcXB peptide (200 μM) was added, GFP-EcDAS (0.1 μM) degradation was inhibited. Degradation rates were normalized to that of GFP-EcDAS in the presence of WT EcSspB without any competitor peptide. B: ClpXP-mediated degradation of GFP- EcssrA (0.1 μM) in the presence of EcSspB (0.3 μM) was inhibited when 200 μM CcXB (YKIVSLDQFRKK) peptide was added to the reaction. This result was observed with ClpXP from both C. crescentus and E. coli. In the absence of EcSspB, inhibition was not observed, indicating that peptide-inhibition was due to disruption of the EcSspB-ClpX interaction rather than a direct effect on the enzyme.
Discussion
As previously shown for E. coli SspB, the C-terminal residues of C. crescentus SspBα mediate its binding to ClpX. Surprisingly, however, phylogenetic conservation and studies of mutant adaptors and XB peptides indicate that ClpX recognizes CcSspBα and EcSspB in very different ways. For instance, the XB regions of orthologs from α-proteobacteria are longer and appear to make many more side chain contacts with ClpX than the XB regions of orthologs from γ- and β-proteobacteria. Nevertheless, the N-terminal domain of ClpX from either E. coli or C. crescentus is able to bind both XB peptides. As a consequence, both CcSspBα and EcSspB can deliver cognate substrates to the ClpXP proteases from E. coli or C. crescentus. These results establish that the N domains of ClpX from both species have at least two distinct peptide-binding specificities. One of these modes of binding appears to be exclusively used for SspBα recognition in the α-proteobacteria, whereas the other seems to be employed for SspB tethering in all γ- and β-proteobacteria.
Mutant studies presented here and previously21 indicate that just two residues in the XB peptide of EcSspB, L161 and V164 (LRVVK165), play major roles in ClpX recognition, with the leucine side chain being most important. Because the XB peptide of CcSspBα (IVSLDQFRKK162) contains a leucine separated by two residues from a phenylalanine, it might be argued that these peptides bind the N domain of ClpX in generally similar ways. However, several results are difficult to reconcile with this model. First, the LxxF sequence in the CcSspBα peptide is still present in the ΔRKK chimeric variant, which fails to deliver substrates [see Fig. 3(B)], and in the ΔRKK CcSspBα XB peptide, which fails to bind the N domain of CcClpX (data not shown). Second, the crystal structure of a complex of EcClpX N domain with EcXB peptide shows that the residue immediately following L161 (LRVVK) adopts dihedral angles that would be inaccessible to proline.23 By contrast, our results show that the CcSspBα XB variant IVSLPQFRKK is active in delivering substrates to ClpXP (see Fig. 6). Third, the side chain of V164 in the EcSspB XB peptide packs into a hydrophobic pocket in the EcClpX N domain that is too small to accommodate a phenylalanine side chain.23 It seems most likely, therefore, that these peptides bind the N domain in fundamentally different fashions. However, competition experiments suggest that both XB peptides bind the same or overlapping sites in the ClpX N domain (see Fig. 9). Thus, the same general peptide-binding pocket may have an unusual amount of flexibility in potential modes of binding specificity.
One minor anomaly in analysis of the CcSspBα XB region concerns differences between experiments using deletions and alanine substitutions. Specifically, we found that one (K162) or two (K161K162) C-terminal residues could be deleted from the CcSspBα XB peptide without causing significant defects in substrate delivery (see Fig. 3). We also synthesized ΔKK and ΔRKK XB peptides and assayed binding to the N domain of CcClpX. The ΔKK peptide bound the N domain with reduced but substantial affinity, whereas almost no binding was detected for the ΔRKK peptide (data not shown). By contrast, alanine substitutions at positions 161 and 162 reduced substrate-delivery activity in some assays [see Fig. 5(C)]. These results could be reconciled if contacts between these lysine side chains and ClpX stabilize the complex, whereas contacts mediated by the peptide backbone of these residues destabilize binding to a roughly comparable extent. Prior studies have also shown that deletion of the C-terminal lysine (K165) of EcSspB does not affect substrate delivery.21 However, the co-crystal structure shows that this lysine side chain makes numerous intimate contacts with the EcClpX N domain.23 Indeed, based on the structure alone, it would be reasonable to suggest that this lysine plays an important role in ClpX-binding, and yet we detected only a marginal decrease in ClpX N domain affinity when this residue was changed to alanine, indicating that the side chain contacts are not critical.
If the C-terminal lysine residues of EcSspB and CcSspBα are not needed for binding ClpX or for delivery of substrates for degradation, then why have these residues been conserved in adaptors and most of their orthologs? We propose that these terminal amino acids might function to help protect SspB from degradation. SspB is a dimer, and both C-terminal tails normally bind N domains in the ClpX hexamer.27 However, EcSspB tethered via a single XB tail also functions as an adaptor,27,31 which would potentially allow the second XB peptide of a dimer to be engaged by the translocation pore of ClpX, leading to degradation of that subunit. However, replacing the C-terminal residue of the ssrA degron with lysine makes it an exceptionally poor degradation tag for ClpXP.32 Thus, having lysine at the C-terminus of the XB peptide should minimize inadvertent ClpXP-mediated degradation. Similar considerations may explain the strong phylogenetic conservation of D157 in SspBα orthologs [see Fig. 1(A)]. This acidic amino acid is present in more than 95% of all SspBα XB sequences, but we detected no effects of an alanine substitution either in substrate delivery or in ClpX-binding [see Figs. 4(A,B), 5(C), and 7(B); Table I]. Numerous experiments have shown that mutation of residues in degrons to aspartic acid also weakens binding to the translocation pore of ClpX.7,33 Testing these ideas will require further analysis as the CcSspB variants used in this study are not ideally suited for degradation experiments because they carry N-terminal affinity tags.
Why has recognition of the two peptide-binding motifs, exemplified by the XB peptides of EcSspB and CcSspBα, been retained by ClpX orthologs that no longer need to interact with the other class of adaptor? The obvious possibility is that these binding sites in the ClpX N domain are maintained because they are also used in recognition of other substrates. For example, an LREI12 sequence in E. coli UmuD helps mediate ClpXP degradation10,34 and is a good match to the β/γ- XB consensus motif. Similarly, a peptide from the λO substrate, which binds to the N domain of ClpX, contains an LLAI56 sequence.16 Moreover, the C-terminal residues of the phage MuA protein (LDILEQNRRKKAI662) target it for ClpXP degradation in a partially N-domain-dependent manner and share homology with the CcSspBα XB peptide.12,18,35
Peptide-binding domains (PDZ, WW, SH2, SH3, PTB, FHA, 14-3-3, EVH1, etc.) are used in modular fashions in an enormous number of biological processes to ensure specificity. In virtually all of these cases, each type of domain has a single binding specificity. Thus, it is somewhat unusual that the N domain of ClpX has at least two peptide-binding specificities. Similarly, the SspB adaptor has more than one binding specificity.11,17,19,20,33,36 For example, crystal structures show that the peptide-binding groove of EcSspB binds to a peptide sequence in the ssrA tag in one way and binds to a nonhomologous recognition sequence in the RseA protein in a completely different fashion. Nevertheless, ssrA and RseA peptides compete for SspB because the binding sites for these peptides overlap.36 Our competition experiments suggest that the binding sites for the β/γ-XB peptides and α-XB peptides in the ClpX N domain also overlap, although structural experiments will be needed to confirm this surmise.
ClpXP has hundreds of natural substrates, which are recognized via five classes of degrons.37 Moreover, other AAA+ proteases interact with multiple types of peptide signals to identify the correct substrates.7,38–45 The AAA+ p97 protein also employs its N domain to interact with disparate sequences in a wide variety of adaptors.46 The peptide-binding versatility exhibited by the ClpX N domain and SspB ensures that ClpXP can recognize many different substrates and adaptors in different ways but with high specificity. This feature allows ClpXP to carry out quality-control surveillance of a large fraction of the proteome and to participate in numerous regulatory circuits without the need for a single type of degron. Moreover, the ability of ClpXP and other AAA+ proteases to recognize multiple classes of degrons permits the recognition of several weak sequence signals to be coupled via avidity effects. These properties of the system free protein substrates to evolve sequence signals that are both compatible with function and only result in degradation under specific circumstances, such as unfolding, complex dissociation, complex assembly, chemical, or proteolytic modification.47 Competition of different substrates and/or adaptors for distinct but overlapping binding sites provides an additional level of potential regulation of intracellular proteolysis.
Materials and Methods
Buffers
PD buffer contained 25 mM HEPES-KOH (pH 7.6), 5 mM KCl, 15 mM NaCl, 5 mM MgCl2, 0.032% NP-40, and 10% glycerol. The ATP-regeneration system contained 5 mM ATP, 50 μg/mL creatine kinase, and 5 mM creatine phosphate. Buffers S1, W20, and W500 contained 50 mM sodium phosphate (pH 8.0), 300 mM NaCl, and imidazole at concentrations of 10 mM, 20 mM, or 500 mM, respectively. For purification of the CcClpX N domain, buffer S1 contained 5 mM imidazole and buffers S1 and W20 were supplemented with 10 mM β-mercaptoethanol (BME) and 10% glycerol. Buffer A contained 20 mM HEPES-KOH (pH 8.0), 150 mM KCl, 10% glycerol, and 10 mM BME. Buffer S contained 20 mM HEPES-KOH (pH 8.0) and 100 mM KCl.
Protein and peptide purification
EcClpX, EcClpP, EcSspB, CcClpX, CcClpP, CcSspBα, and GFP proteins bearing the E. coli or C. crescentus ssrA tags were purified as described.8,25,35,48,49 GFP-EcDAS(+4) protein was a gift from J.S. Butler (MIT).
The CcSspBα variants with an N-terminal His6 tag were cloned into a pET28b vector under T7-promotor control and transformed into E. coli strain BL21(DE3)/pLysS. The N-terminal His6-tagged EcSspB variants, cloned in pET14b vector, were expressed in BL21(DE3) strains (strains provided by laboratory of RT Sauer). Cells were grown at 37°C to OD600 ≈ 0.5 in Luria-Bertani broth containing 50 μg/mL kanamycin. Protein expression was induced for 2 h by addition of 0.5 mM isopropyl β-d-thiogalactoside. The culture was harvested by centrifugation, resuspended in 10 mL of buffer S1 per liter of initial cell culture, and 1 μL/mL protease inhibitor cocktail set III (Novagen, Madison, WI) was added. Cells were frozen in liquid nitrogen, stored, thawed, and lysed by incubating with lysozyme. The lysate was treated with benzonase nuclease (Novagen), cleared by centrifugation for 20 min at 30,000g at 4°C, and incubated with Ni-NTA agarose beads (Qiagen, Valencia, CA) equilibrated in S1 buffer for 1 h at 4°C. The beads were collected by centrifugation, resuspended, and washed sequentially with buffer S1 and buffer W20. Bound protein was eluted in five fractions using buffer W500. Fractions containing SspB variants were identified by SDS-PAGE, buffer-exchanged into buffer S using PD-10 desalting columns (GE Healthcare, Piscataway, NJ), pooled, and the concentration determined by UV absorption at 280 nm (ɛ = 13,000 M−1 cm−1).
GFP-CcDAS was constructed using a Gateway cloning system as previously published50 and the protocol described earlier used to purify the protein (ɛ = 55,000 M−1 cm−1).
CcClpX (residues 1–61) and EcClpX (residues 1–64) N domains with cleavable N-terminal His6 tags were expressed in E. coli strains BLR(DE3) (provided by S. Glynn, MIT) and BL21(DE3)/pLysS, respectively, using the protocol described for expression of CcSspBα variants. Harvested cells were resuspended in 10 mL S1 buffer plus 10 mM BME and 10% glycerol per liter of initial culture and lysed using a French Press (25,000 psi) at 4°C. The protocol for purification of CcSspBα variants was then followed up to the wash step. After washing with buffer W20 plus 10 mM BME and 10% glycerol, the Ni-NTA beads were resuspended in wash buffer, recombinant thrombin (Novagen) was added, and the mixture was incubated overnight at 4°C to cleave the His6 tag. The Ni-NTA resin was removed by centrifugation, and the supernatant was chromatographed on a Superdex-75 gel filtration column (GE Healthcare) equilibrated in buffer A. Fractions containing the ClpX N domain were identified by SDS-PAGE, pooled, concentrated using Amicon (MWCO 5k) (Millipore, Billerica, MA) tubes, and the protein concentration was determined by UV absorption at 280 nm.
Fluorescein-labeled peptides corresponding to the XB regions of CcSspBα (YKIVSLDQFRKK), EcSspB (RGGRPALRVVK), and variants containing single alanine substitutions were synthesized by using FMOC techniques on an Apex 396 solid-phase synthesizer.
Protein degradation assays
GFP substrates (100 nM) were incubated with EcClpXP (50 nM EcClpX6; 100 nM EcClpP14) or CcClpXP (200 nM CcClpX6; 400 nM CcClpP14) in the presence or absence of adaptor (300 nM monomer) at 30°C in PD buffer plus an ATP-regeneration system.33 Degradation was monitored by decreased fluorescence (excitation 488 nm; emission 511 nm) using a Photon Technology International fluorimeter (Birmingham, NJ). The rates of reaction were determined by the slopes of linear fits to the decrease in fluorescence within the first 10–30 s of reaction. The error bars indicate the standard deviation of three or more independent measurements.
Peptide-binding assay
Fluorescein-labeled CcSspBα XB peptides (60 nM) were incubated with increasing amounts of CcClpX N domain in buffer A at 30°C, and fluorescence anisotropy was measured using a Photon Technology International fluorimeter (excitation 490 nm; emission 515 nm). The binding of fluorescent EcSspB XB peptides to the EcClpX N domain was assayed in the same way. The KD values from individual experiments were determined by fitting binding data to the quadratic equation determined from the following equilibrium:
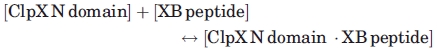 |
The binding equation used was y = a + ((b − a) ((d + x + c) − (SQRT((d + x + c)2 − 4dx)/2d))), where y (y-axis) = anisotropy, x (x-axis) = [ClpX N domain], a = anisotropy of free peptide, b = anisotropy when all peptide is bound to the N domain, c = KD, and d = [total peptide].
Acknowledgments
The authors thank Igor Levchenko, Jim Butler, and Steve Glynn and the Sauer laboratory for sharing reagents. They also thank Jennifer Hou for comments on the manuscript and the Baker laboratory. T.A.B. is an employee of the Howard Hughes Medical Institute.
References
- 1.Gottesman S. Proteolysis in bacterial regulatory circuits. Annu Rev Cell Dev Biol. 2003;19:565–587. doi: 10.1146/annurev.cellbio.19.110701.153228. [DOI] [PubMed] [Google Scholar]
- 2.Kang SG, Ortega J, Singh SK, Wang N, Huang NN, Steven AC, Maurizi MR. Functional proteolytic complexes of the human mitochondrial ATP-dependent protease, hClpXP. J Biol Chem. 2002;277:21095–21102. doi: 10.1074/jbc.M201642200. [DOI] [PubMed] [Google Scholar]
- 3.Jenal U, Fuchs T. An essential protease involved in bacterial cell-cycle control. Embo J. 1998;17:5658–5669. doi: 10.1093/emboj/17.19.5658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.van Dyck L, Dembowski M, Neupert W, Langer T. Mcx1p, a ClpX homologue in mitochondria of Saccharomyces cerevisiae. FEBS Lett. 1998;438:250–254. doi: 10.1016/s0014-5793(98)01310-6. [DOI] [PubMed] [Google Scholar]
- 5.Wang J, Hartling JA, Flanagan JM. The structure of ClpP at 2.3 A resolution suggests a model for ATP-dependent proteolysis. Cell. 1997;91:447–456. doi: 10.1016/s0092-8674(00)80431-6. [DOI] [PubMed] [Google Scholar]
- 6.Keiler KC, Waller PR, Sauer RT. Role of a peptide tagging system in degradation of proteins synthesized from damaged messenger RNA. Science. 1996;271:990–993. doi: 10.1126/science.271.5251.990. [DOI] [PubMed] [Google Scholar]
- 7.Gottesman S, Roche E, Zhou Y, Sauer RT. The ClpXP and ClpAP proteases degrade proteins with carboxy-terminal peptide tails added by the SsrA-tagging system. Genes Dev. 1998;12:1338–1347. doi: 10.1101/gad.12.9.1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Levchenko I, Seidel M, Sauer RT, Baker TA. A specificity-enhancing factor for the ClpXP degradation machine. Science. 2000;289:2354–2356. doi: 10.1126/science.289.5488.2354. [DOI] [PubMed] [Google Scholar]
- 9.Zhou Y, Gottesman S, Hoskins JR, Maurizi MR, Wickner S. The RssB response regulator directly targets sigma(S) for degradation by ClpXP. Genes Dev. 2001;15:627–637. doi: 10.1101/gad.864401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Neher SB, Sauer RT, Baker TA. Distinct peptide signals in the UmuD and UmuD' subunits of UmuD/D' mediate tethering and substrate processing by the ClpXP protease. Proc Natl Acad Sci USA. 2003;100:13219–13224. doi: 10.1073/pnas.2235804100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Flynn JM, Levchenko I, Sauer RT, Baker TA. Modulating substrate choice: the SspB adaptor delivers a regulator of the extracytoplasmic-stress response to the AAA+ protease ClpXP for degradation. Genes Dev. 2004;18:2292–2301. doi: 10.1101/gad.1240104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wojtyra UA, Thibault G, Tuite A, Houry WA. The N-terminal zinc binding domain of ClpX is a dimerization domain that modulates the chaperone function. J Biol Chem. 2003;278:48981–48990. doi: 10.1074/jbc.M307825200. [DOI] [PubMed] [Google Scholar]
- 13.Donaldson LW, Wojtyra U, Houry WA. Solution structure of the dimeric zinc binding domain of the chaperone ClpX. J Biol Chem. 2003;278:48991–48996. doi: 10.1074/jbc.M307826200. [DOI] [PubMed] [Google Scholar]
- 14.Singh SK, Rozycki J, Ortega J, Ishikawa T, Lo J, Steven AC, Maurizi MR. Functional domains of the ClpA and ClpX molecular chaperones identified by limited proteolysis and deletion analysis. J Biol Chem. 2001;276:29420–29429. doi: 10.1074/jbc.M103489200. [DOI] [PubMed] [Google Scholar]
- 15.Dougan DA, Weber-Ban E, Bukau B. Targeted delivery of an ssrA-tagged substrate by the adaptor protein SspB to its cognate AAA+ protein ClpX. Mol Cell. 2003;12:373–380. doi: 10.1016/j.molcel.2003.08.012. [DOI] [PubMed] [Google Scholar]
- 16.Thibault G, Yudin J, Wong P, Tsitrin V, Sprangers R, Zhao R, Houry WA. Specificity in substrate and cofactor recognition by the N-terminal domain of the chaperone ClpX. Proc Natl Acad Sci USA. 2006;103:17724–17729. doi: 10.1073/pnas.0601505103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chien P, Grant RA, Sauer RT, Baker TA. Structure and substrate specificity of an SspB ortholog: design implications for AAA+ adaptors. Structure. 2007;15:1296–1305. doi: 10.1016/j.str.2007.08.008. [DOI] [PubMed] [Google Scholar]
- 18.Abdelhakim AH, Oakes EC, Sauer RT, Baker TA. Unique contacts direct high-priority recognition of the tetrameric Mu transposase-DNA complex by the AAA+ unfoldase ClpX. Mol Cell. 2008;30:39–50. doi: 10.1016/j.molcel.2008.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Levchenko I, Grant RA, Wah DA, Sauer RT, Baker TA. Structure of a delivery protein for an AAA+ protease in complex with a peptide degradation tag. Mol Cell. 2003;12:365–372. doi: 10.1016/j.molcel.2003.08.014. [DOI] [PubMed] [Google Scholar]
- 20.Song HK, Eck MJ. Structural basis of degradation signal recognition by SspB, a specificity-enhancing factor for the ClpXP proteolytic machine. Mol Cell. 2003;12:75–86. doi: 10.1016/s1097-2765(03)00271-5. [DOI] [PubMed] [Google Scholar]
- 21.Wah DA, Levchenko I, Rieckhof GE, Bolon DN, Baker TA, Sauer RT. Flexible linkers leash the substrate binding domain of SspB to a peptide module that stabilizes delivery complexes with the AAA+ ClpXP protease. Mol Cell. 2003;12:355–363. doi: 10.1016/s1097-2765(03)00272-7. [DOI] [PubMed] [Google Scholar]
- 22.Bolon DN, Wah DA, Hersch GL, Baker TA, Sauer RT. Bivalent tethering of SspB to ClpXP is required for efficient substrate delivery: a protein-design study. Mol Cell. 2004;13:443–449. doi: 10.1016/s1097-2765(04)00027-9. [DOI] [PubMed] [Google Scholar]
- 23.Park EY, Lee BG, Hong SB, Kim HW, Jeon H, Song HK. Structural basis of SspB-tail recognition by the zinc binding domain of ClpX. J Mol Biol. 2007;367:514–526. doi: 10.1016/j.jmb.2007.01.003. [DOI] [PubMed] [Google Scholar]
- 24.Wah DA, Levchenko I, Baker TA, Sauer RT. Characterization of a specificity factor for an AAA+ ATPase: assembly of SspB dimers with ssrA-tagged proteins and the ClpX hexamer. Chem Biol. 2002;9:1237–1245. doi: 10.1016/s1074-5521(02)00268-5. [DOI] [PubMed] [Google Scholar]
- 25.Chien P, Perchuk BS, Laub MT, Sauer RT, Baker TA. Direct and adaptor-mediated substrate recognition by an essential AAA+ protease. Proc Natl Acad Sci USA. 2007;104:6590–6595. doi: 10.1073/pnas.0701776104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lessner FH, Venters BJ, Keiler KC. Proteolytic adaptor for transfer-messenger RNA-tagged proteins from alpha-proteobacteria. J Bacteriol. 2007;189:272–275. doi: 10.1128/JB.01387-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bolon DN, Grant RA, Baker TA, Sauer RT. Nucleotide-dependent substrate handoff from the SspB adaptor to the AAA+ ClpXP protease. Mol Cell. 2004;16:343–350. doi: 10.1016/j.molcel.2004.10.001. [DOI] [PubMed] [Google Scholar]
- 28.Crooks GE, Hon G, Chandonia JM, Brenner SE. WebLogo: a sequence logo generator. Genome Res. 2004;14:1188–1190. doi: 10.1101/gr.849004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Clamp M, Cuff J, Searle SM, Barton GJ. The Jalview Java alignment editor. Bioinformatics. 2004;20:426–427. doi: 10.1093/bioinformatics/btg430. [DOI] [PubMed] [Google Scholar]
- 30.McGinness KE, Baker TA, Sauer RT. Engineering controllable protein degradation. Mol Cell. 2006;22:701–707. doi: 10.1016/j.molcel.2006.04.027. [DOI] [PubMed] [Google Scholar]
- 31.McGinness KE, Bolon DN, Kaganovich M, Baker TA, Sauer RT. Altered tethering of the SspB adaptor to the ClpXP protease causes changes in substrate delivery. J Biol Chem. 2007;282:11465–11473. doi: 10.1074/jbc.M610671200. [DOI] [PubMed] [Google Scholar]
- 32.Barkow SR. 2009. Mechanistic Studies of the AAA+ Motor Protein ClpXP. Ph.D Thesis, Department of Chemistry, Massachusetts Institute of Technology, Cambridge, Massachusetts. [Google Scholar]
- 33.Flynn JM, Levchenko I, Seidel M, Wickner SH, Sauer RT, Baker TA. Overlapping recognition determinants within the ssrA degradation tag allow modulation of proteolysis. Proc Natl Acad Sci USA. 2001;98:10584–10589. doi: 10.1073/pnas.191375298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gonzalez M, Rasulova F, Maurizi MR, Woodgate R. Subunit-specific degradation of the UmuD/D' heterodimer by the ClpXP protease: the role of trans recognition in UmuD' stability. Embo J. 2000;19:5251–5258. doi: 10.1093/emboj/19.19.5251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Levchenko I, Smith CK, Walsh NP, Sauer RT, Baker TA. PDZ-like domains mediate binding specificity in the Clp/Hsp100 family of chaperones and protease regulatory subunits. Cell. 1997;91:939–947. doi: 10.1016/s0092-8674(00)80485-7. [DOI] [PubMed] [Google Scholar]
- 36.Levchenko I, Grant RA, Flynn JM, Sauer RT, Baker TA. Versatile modes of peptide recognition by the AAA+ adaptor protein SspB. Nat Struct Mol Biol. 2005;12:520–525. doi: 10.1038/nsmb934. [DOI] [PubMed] [Google Scholar]
- 37.Flynn JM, Neher SB, Kim YI, Sauer RT, Baker TA. Proteomic discovery of cellular substrates of the ClpXP protease reveals five classes of ClpX-recognition signals. Mol Cell. 2003;11:671–683. doi: 10.1016/s1097-2765(03)00060-1. [DOI] [PubMed] [Google Scholar]
- 38.Gonzalez M, Frank EG, Levine AS, Woodgate R. Lon-mediated proteolysis of the Escherichia coli UmuD mutagenesis protein vitro degradation and identification of residues required for proteolysis. Genes Dev. 1998;12:3889–3899. doi: 10.1101/gad.12.24.3889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hoskins JR, Kim SY, Wickner S. Substrate recognition by the ClpA chaperone component of ClpAP protease. J Biol Chem. 2000;275:35361–35367. doi: 10.1074/jbc.M006288200. [DOI] [PubMed] [Google Scholar]
- 40.Ishii Y, Amano F. Regulation of SulA cleavage by Lon protease by the C-terminal amino acid of SulA, histidine. Biochem J. 2001;358:473–480. doi: 10.1042/0264-6021:3580473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Griffith KL, Shah IM, Wolf RE., Jr Proteolytic degradation of Escherichia coli transcription activators SoxS and MarA as the mechanism for reversing the induction of the superoxide (SoxRS) and multiple antibiotic resistance (Mar) regulons. Mol Microbiol. 2004;51:1801–1816. doi: 10.1046/j.1365-2958.2003.03952.x. [DOI] [PubMed] [Google Scholar]
- 42.Hoskins JR, Wickner S. Two peptide sequences can function cooperatively to facilitate binding and unfolding by ClpA and degradation by ClpAP. Proc Natl Acad Sci USA. 2006;103:909–914. doi: 10.1073/pnas.0509154103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Erbse A, Schmidt R, Bornemann T, Schneider-Mergener J, Mogk A, Zahn R, Dougan DA, Bukau B. ClpS is an essential component of the N-end rule pathway in Escherichia coli. Nature. 2006;439:753–756. doi: 10.1038/nature04412. [DOI] [PubMed] [Google Scholar]
- 44.Wang KH, Sauer RT, Baker TA. ClpS modulates but is not essential for bacterial N-end rule degradation. Genes Dev. 2007;21:403–408. doi: 10.1101/gad.1511907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gur E, Sauer RT. Recognition of misfolded proteins by Lon, a AAA(+) protease. Genes Dev. 2008;22:2267–2277. doi: 10.1101/gad.1670908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yeung HO, Kloppsteck P, Niwa H, Isaacson RL, Matthews S, Zhang X, Freemont PS. Insights into adaptor binding to the AAA protein p97. Biochem Soc Trans. 2008;36:62–67. doi: 10.1042/BST0360062. [DOI] [PubMed] [Google Scholar]
- 47.Baker TA, Sauer RT. ATP-dependent proteases of bacteria: recognition logic and operating principles. Trends Biochem Sci. 2006;31:647–653. doi: 10.1016/j.tibs.2006.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yakhnin AV, Vinokurov LM, Surin AK, Alakhov YB. Green fluorescent protein purification by organic extraction. Protein Expr Purif. 1998;14:382–386. doi: 10.1006/prep.1998.0981. [DOI] [PubMed] [Google Scholar]
- 49.Kim YI, Burton RE, Burton BM, Sauer RT, Baker TA. Dynamics of substrate denaturation and translocation by the ClpXP degradation machine. Mol Cell. 2000;5:639–648. doi: 10.1016/s1097-2765(00)80243-9. [DOI] [PubMed] [Google Scholar]
- 50.Skerker JM, Prasol MS, Perchuk BS, Biondi EG, Laub MT. Two-component signal transduction pathways regulating growth and cell cycle progression in a bacterium: a system-level analysis. PLoS Biol. 2005;3:1770–1788. doi: 10.1371/journal.pbio.0030334. [DOI] [PMC free article] [PubMed] [Google Scholar]