

Abstract
Activation of Janus kinase/signal transducers and activators of transcription (JAK/STAT) is an important mechanism by which hyperglycemia contributes to renal damage, suggesting that modulation of this pathway may prevent renal and vascular complications of diabetes. Here, we investigated the involvement of suppressors of cytokine signaling (SOCS) as intracellular negative regulators of JAK/STAT activation in diabetic nephropathy. In a rat model, inducing diabetes resulted in JAK/STAT activation and increased expression of SOCS1 and SOCS3. In humans, we observed increased expression of glomerular and tubulointerstitial SOCS proteins in biopsies of patients with diabetic nephropathy. In vitro, high concentrations of glucose activated JAK/STAT/SOCS in human mesangial and tubular cells. Overexpression of SOCS reversed the glucose-induced activation of the JAK/STAT pathway, expression of STAT-dependent genes (chemokines, growth factors, and extracellular matrix proteins), and cell proliferation. In vivo, intrarenal delivery of adenovirus expressing SOCS1 and SOCS3 to diabetic rats significantly improved renal function and reduced renal lesions associated with diabetes, such as mesangial expansion, fibrosis, and influx of macrophages. SOCS gene delivery also decreased the activation of STAT1 and STAT3 and the expression of proinflammatory and profibrotic proteins in the diabetic kidney. In summary, these results provide direct evidence for a link between the JAK/STAT/SOCS axis and hyperglycemia-induced cell responses in the kidney. Suppression of the JAK/STAT pathway by increasing intracellular SOCS proteins may have therapeutic potential in diabetic nephropathy.
Diabetic nephropathy, the most common cause of ESRD, is thought to result from interaction between metabolic and hemodynamic factors. The pathologic changes in diabetic nephropathy include renal hypertrophy and extracellular matrix accumulation, which contribute to glomerular sclerosis, which leads to proteinuria and renal failure through the tubular interstitial fibrosis.1 The basic underlying mechanisms of diabetic nephropathy involve high-glucose (HG)-induced production of cytokines and growth factors, which promote leukocyte infiltration, renal cell proliferation, and matrix production.2–4 No treatment options are currently available to prevent the renal complications of diabetes except for therapies that may slow the progression through intensive control of glycemia and blood pressure.1 Therefore, it is of great clinical importance to identify new therapeutic targets that might lead to the prevention of diabetes-induced nephropathy.
Hyperglycemia triggers a series of intracellular events in glomerular and tubular cells, including reactive oxygen species generation, protein kinase C and mitogen-activated protein kinase activation, and transcription factor induction.2,5–8 Acting through these different intracellular pathways, HG enhances proinflammatory and profibrotic factors as well as cellular hypertrophy.1,2,9 Recent findings also suggest that exposure of renal cells to HG activates the Janus kinase (JAK)/signal transducers and activators of transcription (STAT) signaling cascade.6,10–13
The JAK/STAT pathway is an essential intracellular mechanism of cytokines and other stimuli that regulates gene expression and cellular activation, proliferation, and differentiation. Four JAK and seven STAT family members constitute the JAK/STAT system, and cell-specific JAK/STAT combinations have been paired with each receptor type.14,15 Receptor engagement activates the associated JAK, which phosphorylates the receptor cytoplasmic domain to allow recruitment and tyrosine phosphorylation of STAT. Activated STATs dimerize and translocate into the nucleus to activate specific gene expression. The JAK/STAT pathway is controlled through different mechanisms16: receptor internalization, protein tyrosine phosphatases, protein inhibitors of activated STAT, and suppressors of cytokine signaling (SOCS). SOCS comprise a family of eight intracellular, cytokine-inducible proteins (CIS; SOCS1 to SOCS7), each of which has a variable N-terminal domain, a central SH2 domain, and a conserved C-terminal SOCS box.14,15 SOCS family members, particularly SOCS1 and SOCS3, control the magnitude and duration of JAK/STAT signaling through several mechanisms, including direct JAK inhibition, STAT binding, and targeting receptor complex and other signaling proteins for proteasomal degradation.14,15,17 In addition to cytokines, SOCS1 and SOCS3 are induced by many pathologic stimuli (e.g., angiotensin II, chemokines, insulin, immunoglobulins, and lipoproteins), thus indicating their involvement in many biologic processes.15,18–21
Members of the JAK/STAT pathway have been claimed as new molecular targets of anti-inflammatory treatment in acute and chronic inflammatory diseases,22 and their activation is involved in the development of atherosclerosis, hypertension, and the renal and vascular complications of diabetes.12,23,24 In this study, we explored the negative regulation of the JAK/STAT pathway during diabetes-induced renal damage, focusing on the two main members of SOCS family (SOCS1 and SOCS3). We first evaluated the renal expression of SOCS1 and SOCS3 in human and experimental diabetic nephropathy. Next, we analyzed whether SOCS proteins modulate JAK/STAT-mediated gene expression and cell responses to hyperglycemia in cultured renal cells. Finally, we investigated the effects of SOCS renal delivery by recombinant adenovirus on the progression of diabetic nephropathy in rats.
Results
Renal Expression of SOCS in Diabetic Nephropathy
SOCS1 and SOCS3 were increased in the renal cortex of streptozotocin (STZ)-induced diabetic rats at the mRNA (Figure 1A) and protein level (n-fold versus controls: SOCS1, 4.5 ± 0.7; SOCS3, 2.1 ± 0.2; P < 0.05; Figure 1B). Immunohistochemistry revealed a broad distribution of SOCS1 and SOCS3 in glomerular mesangial cells (MCs), podocytes, and proximal tubular cells of diabetic rats (Figure 1C). Moreover, and consistent with previous reports,13,25 diabetes induction resulted in the activation of the JAK/STAT pathway, as assessed by JAK2, STAT1, and STAT3 tyrosine phosphorylation (n-fold versus controls: 5.2 ± 1.5, 8.5 ± 2.5, and 1.8 ± 0.1, respectively; P < 0.05; Figure 1B).
Figure 1.

Renal SOCS expression increases after diabetes induction in rats. (A) SOCS mRNA expression in renal samples from control and diabetic rats was determined by real-time PCR. (B) Representative Western blots for SOCS1, SOCS3, P-JAK2, P-STAT1, and P-STAT3 in control and diabetic rats. (C) Representative micrographs showing positive SOCS immunostaining in glomeruli (magnification: ×400) and tubulointerstitium (×200) of diabetic rats. Lower panel: Quantitative analysis data in control and diabetic groups. Data are mean ± SEM of three to five animals per group (*P < 0.05 versus control).
The histologic distribution of SOCS1 and SOCS3 proteins was also evaluated in renal samples from diabetic nephropathy patients. In glomeruli, SOCS were observed mainly in podocytes and, to a lesser extent, in MCs. In the interstitial area, SOCS was expressed by proximal and distal tubular cells and inflammatory cells (Figure 2A). Quantification of positive staining in glomerular and tubulointerstitial compartments showed significant differences in diabetic patients compared with control subjects (Figure 2B). Additional micrographs of diabetic patients and animals are shown in Supplemental Figure I.
Figure 2.

SOCS proteins are expressed in renal biopsies of diabetic patients. (A) Representative micrographs of SOCS1 and SOCS3 immunostaining in serial sections of kidney biopsies from a patient with progressive diabetic nephropathy and a control subject. SOCS expression was increased in glomerular (magnification: ×400) and tubulointerstitial (×200) areas of a diabetic patient compared with the control (×100). (B) Quantitative analysis data in control and diabetic groups (mean ± SEM, n = 5 per group, *P < 0.05 versus control).
In Vitro SOCS Induction by Hyperglycemia
Incubation of human glomerular MCs and human renal proximal tubular cells (HK2) under hyperglycemic conditions (medium containing 30 mM d-glucose) time-dependently induced the gene and protein expression of SOCS compared with control conditions (medium with 10 mM d-glucose) (Figure 3, A and B). SOCS3 expression was maximal at 4 to 8 hours and then decreased to baseline, whereas SOCS1 was expressed later and remained elevated after longer incubation times (48 to 56 hours). In agreement with previous findings,26,27 hyperglycemia induced tyrosine phosphorylation of JAK2 and STAT1, peaking at 24 hours (Figure 3B). In all of the experiments presented in this study, no significant effects of hyperosmolarity were seen in cells incubated in medium with 20 mM d-mannitol (Figures 3A, 5A, 5C, and Supplemental Figure IIB).
Figure 3.

HG induces SOCS expression in cultured renal cells. (A) HK2 cells were incubated for different time intervals in culture medium containing HG (30 mM d-glucose, black bars) or mannitol (10 mM d-glucose + 20 mM d-mannitol; gray bars). Gene expression of SOCS1 (upper panel) and SOCS3 (lower panel) was analyzed by real-time PCR and values were normalized by 18S expression. (B) Representative immunoblots and densitometric analysis showing time course of SOCS1, SOCS3, P-JAK2, and P-STAT1 induction by HG in human MCs. Fold increases versus basal conditions (10 mM d-glucose) are mean ± SEM of three to five experiments in duplicate (*P < 0.05 versus basal).
Figure 5.
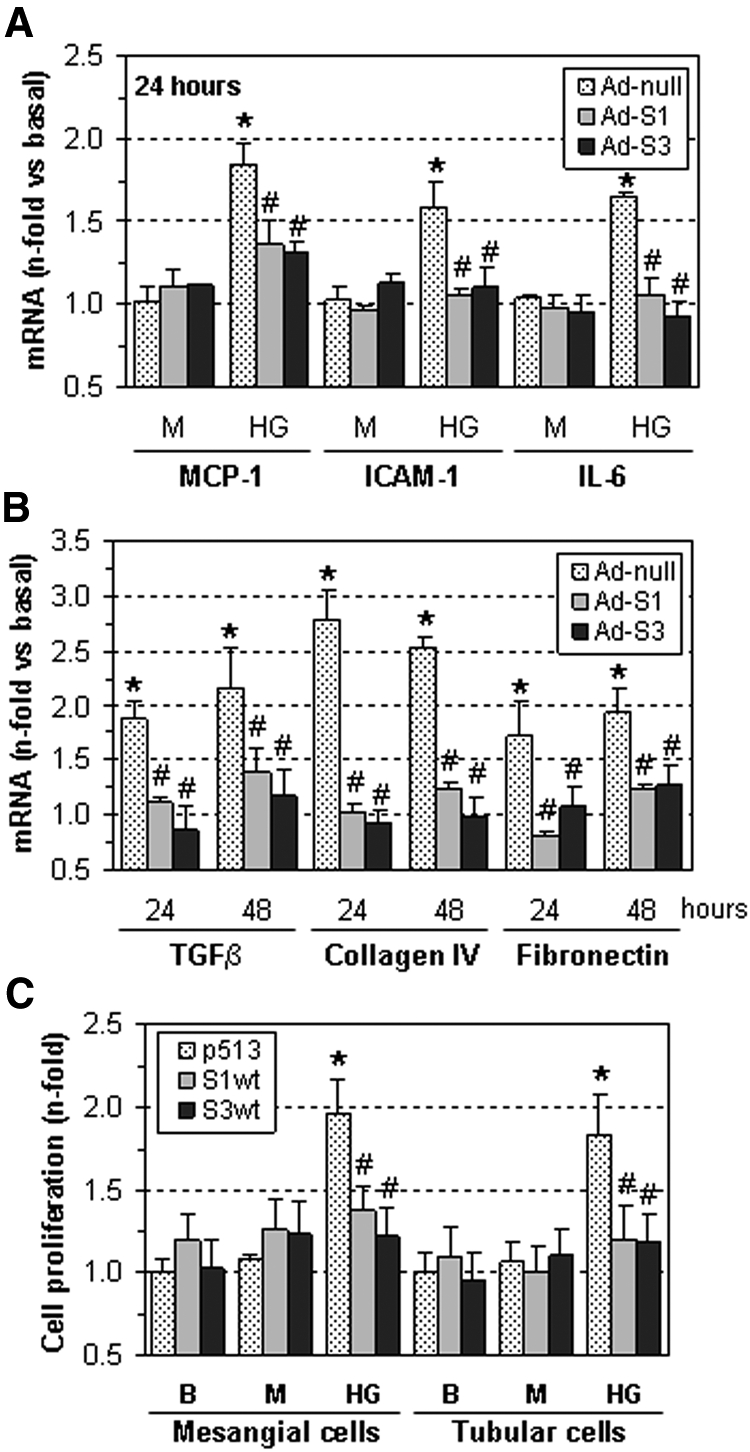
SOCS overexpression prevents HG-induced gene expression and cell proliferation. (A) Human MC and (B) tubular HK2 cells treated with SOCS-expressing adenovirus (Ad-S1, Ad-S3) or control adenovirus (Ad-null) were then incubated for 24 to 48 hours in medium containing HG or mannitol (M). The mRNA expression of the indicated genes was measured by real-time PCR, and values were normalized by 18S endogenous control. (C) Cell proliferation assay in cells transfected with SOCS expression vectors (S1wt and S3wt) or empty plasmid (p513) after 48 hours of incubation in basal conditions (B), HG or mannitol (M). Fold increases versus basal are mean ± SEM of three to six experiments. *P < 0.05 versus basal, #P < 0.05 versus control (Ad-null or empty plasmid).
SOCS Proteins Prevent HG-Induced JAK/STAT Activation
To investigate the modulation of the JAK/STAT pathway by SOCS in renal cells, overexpression of SOCS1 and SOCS3 proteins was induced by plasmid transfection (S1wt, S3wt, and control p513) or adenovirus infection (Ad-S1, Ad-S3, and control Ad-null). In human MCs, SOCS overexpression significantly inhibited HG-induced JAK2 and STAT1 tyrosine phosphorylation (Figure 4A). Similarly, SOCS-expressing adenovirus prevented the STAT1 phosphorylation and STAT-transcriptional activity by HG in HK2 cells (Figure 4, B and C).
Figure 4.

SOCS inhibit JAK/STAT activation by HG in renal cells. (A) Human MCs were transfected with SOCS expression vectors (S1wt and S3wt) or empty plasmid (p513). (B) Tubular HK2 cells were infected with SOCS-expressing adenovirus (Ad-S1, Ad-S3) or control adenovirus (Ad-null). After 24 hours, cells were stimulated for an additional 24 hours with HG. Representative immunoblots and densitometric analysis of the indicated proteins are shown. Fold increases versus basal conditions (10 mM d-glucose) are mean ± SEM of three independent experiments. (C) Adenovirus-treated HK2 cells were transfected with the STAT-responsive luciferase vectors and then stimulated for 24 hours with HG. Data of relative luciferase units (Firefly/Renilla/mg protein) are mean ± SEM of three to five experiments. *P < 0.05 versus basal, #P < 0.05 versus control (empty plasmid or Ad-null).
Inhibition of STAT-Dependent Genes and Cell Proliferation by SOCS
Among the genes upregulated by HG in renal cells, we examined the effect of SOCS on the expression of classical STAT-responsive inflammatory genes, including chemokines, adhesion molecules, and cytokines.12,22 Adenovirus-mediated SOCS overexpression efficiently inhibited the induction of monocyte chemoattractant protein-1 (MCP-1), intercellular adhesion molecule-1 and IL-6 by hyperglycemia in human MCs (Figure 5A) and HK2 cells (Supplemental Figure IIA) as compared with control adenovirus. Moreover, treatment with Ad-S1 and Ad-S3 prevented the expression of transforming growth factor-β (TGFβ), collagen IV, and fibronectin, which are profibrotic genes known to be activated by HG via JAK/STAT during diabetic nephropathy6,8–10,13,27,28 (Figure 5B and Supplemental Figure IIB). SOCS also significantly reduced the accumulation of fibronectin protein in HK2 cells (% inhibition at 48 hours in HK2: Ad-S1, 71 ± 5; Ad-S3, 67 ± 7, P < 0.05, n = 3; not shown). Furthermore, the proliferative effect of HG on human MCs and HK2 cells was prevented by SOCS overexpression (Figure 5C).
In Vivo Gene Transfer of SOCS
To test the efficiency of the in vivo gene transfer, adenoviruses expressing green fluorescence protein (Ad-GFP) or SOCS proteins (Ad-S1 and Ad-S3) were injected into normal rats. As shown in Figure 6, A and B, intense green fluorescence was seen in kidney (glomerular and tubular cells) and liver of Ad-GFP transfected rats as compared with control adenovirus. Moreover, real-time PCR and Western blot analyses confirmed that the expression of SOCS1 and SOCS3 in renal tissue remained increased even at 7 days of gene transfer (Figure 6, C and D).
Figure 6.

Adenovirus-mediated gene delivery increases SOCS expression in control rats. Representative fluorescence photomicrographs of (A) kidney and (B) liver of rats 5 days after Ad-GFP injection showing intense fluorescence compared with mild autofluorescence in the control group (Ad-null). Arrows and arrowheads in panel A indicate positive cells in glomerular and tubulointerstitial areas, respectively. (C) Real-time PCR analysis of SOCS gene expression in renal tissues 3 and 7 days after adenovirus injection (Ad-S1 and Ad-S3). Fold increases versus control conditions are mean ± SEM of six experiments (*P < 0.05 versus control). (D) Representative immunoblots of SOCS1 and SOCS3 protein expression in kidney of rats treated with Ad-S1 and Ad-S3.
Effects of SOCS Gene Delivery in Diabetic Rats
Adenovirus expressing SOCS (Ad-S1 and Ad-S3) or the control virus (Ad-null) was injected into STZ-induced diabetic rats 3 weeks after the onset of diabetes, and animals were studied after 4 weeks of gene delivery. At this time point, SOCS transgene expression did not vary considerably among the diabetic groups (not shown). Interestingly, treatment with Ad-S1 and Ad-S3 tended to normalize creatinine clearance, urinary albumin excretion, and weight loss in diabetic rats (Table 1).
Table 1.
Functional and structural data from control and diabetic rats
| Control | Diabetes |
||||
|---|---|---|---|---|---|
| Sham | Ad-Null | Ad-S1 | Ad-S3 | ||
| Blood glucose (mg/dl) | 95 ± 8 | 581 ± 8a | 567 ± 20a | 576 ± 18a | 587 ± 9a |
| Body weight (g) | 283 ± 13 | 209 ± 14a | 196 ± 7a | 235 ± 15a,b | 230 ± 14a,b |
| Creatinine clearance (ml/min/kg) | 1.42 ± 0.28 | 3.34 ± 0.26a | 3.23 ± 0.17a | 2.01 ± 0.14b | 2.12 ± 0.22b |
| Urinary albumin (mg albumin/mg creatinin) | 0.19 ± 0.04 | 9.72 ± 3.16a | 9.47 ± 1.48a | 2.76 ± 0.64a,b | 4.75 ± 1.07a,b |
| Kidney-to-body-weight ratio (g/kg) | 8.87 ± 0.43 | 12.90 ± 0.57a | 13.41 ± 0.77a | 9.71 ± 1.07b | 9.42 ± 0.85b |
| Glomerular volume (μm3 × 106) | 0.45 ± 0.03 | 1.04 ± 0.06a | 1.00 ± 0.03a | 0.69 ± 0.04a,b | 0.61 ± 0.05a,b |
aP < 0.05 versus control nondiabetic rats.
bP < 0.05 versus sham-operated diabetic rats.
Histologic assessment in renal samples revealed that SOCS gene delivery ameliorated the pathologic changes associated with diabetes, including glomerular hypertrophy, mesangial expansion, and tubular atrophy/dilation (Figure 7, A and B). Accordingly, fractional kidney weight and glomerular volume (parameters of kidney hypertrophy) were significantly lower in diabetic rats treated with Ad-S1 and Ad-S3 versus Ad-null or sham-operated rats (Table 1). Furthermore, analysis of collagen distribution by picrosirius red staining indicated that SOCS administration normalized the intraglomerular and tubulointerstitial fibrosis in diabetic rats (Figure 7, A and C). SOCS gene delivery also decreased the number of glomerular and interstitial macrophages in the diabetic kidneys (CD68 immunostaining; Figure 7, A and D).
Figure 7.

SOCS gene delivery ameliorates diabetes-induced renal damage. Renal histopathology in control nondiabetic rats, diabetic rats without treatment (sham-operation), treated with control adenovirus (Ad-null), and with SOCS-expressing adenovirus (Ad-S1 and Ad-S3). (A) Representative micrographs of renal sections stained with periodic acid–Schiff (glomerular capillary dilation, mesangial expansion, and tubular glycogen deposits are observed; magnification: ×400), picrosirius red (collagen deposition; ×200), and CD68 antibody (macrophage marker; ×400). (B) Semiquantitative assessment of renal lesions (periodic acid–Schiff score: 0 to 3). (C) Quantification of collagen-positive areas. (D) Number of CD68+ macrophages in all of the study groups. Upper and lower panels represent measurements in glomerular and tubulointerstitial areas, respectively. Data are mean ± SEM of n = 12 rats analyzed per group (*P < 0.05 versus control nondiabetic rats, #P < 0.05 versus sham-operated diabetic rats).
SOCS Reduces STAT-Mediated Gene Expression in Diabetes
Diabetic rats treated with Ad-S1 and Ad-S3 showed a decrease in the tyrosine phosphorylation of STAT1 and STAT3 (Figure 8, A and B). Furthermore, the renal expression of STAT-dependent genes associated with inflammation (MCP-1; regulated upon activation, normal T cell expressed and secreted; and intercellular adhesion molecule-1) and fibrosis (TGFβ and collagen IV) during diabetic nephropathy (Figure 8C) were significantly reduced by SOCS treatment. SOCS gene transfer also diminished the protein levels of MCP-1 and fibronectin (Figure 8B) and the positive staining for TGFβ in diabetic kidneys (% of total area: sham, 16.0 ± 1.8; Ad-null, 14.8 ± 2.7; Ad-S1, 2.8 ± 1.8; Ad-S3, 1.8 ± 0.9; P < 0.05 versus sham/null; Figure 8A). In these studies, no significant differences were observed between sham-operated and Ad-null groups.
Figure 8.

SOCS gene delivery reduces STAT activation and expression of inflammatory and fibrotic genes in the diabetic kidney. (A) Representative micrographs of P-STAT1, P-STAT3, and TGFβ immunodetection in diabetic rat groups as specified. (B) Western blot analyses of P-STAT1, P-STAT3, MCP-1, and fibronectin (FN) levels in the renal cortex of control nondiabetic rats and STZ-induced diabetic rats (sham-operated, Ad-null, Ad-S1, and Ad-S3). Representative immunoblots and quantitative analyses are shown. (C) Real-time PCR analysis of the indicated genes in renal samples was assessed in duplicate and normalized by 18S endogenous control. Fold increases versus control rats are mean ± SEM (*P < 0.05 versus controls, #P < 0.05 versus sham-operated diabetic rats).
Discussion
The JAK/STAT pathway regulates a wide range of genes involved in cell proliferation, inflammation, and fibrosis and is an important mechanism by which hyperglycemia contributes to nephropathy associated with diabetes.9,12,28 In this study, we show that SOCS proteins, negative regulators of the JAK/STAT pathway, are induced by hyperglycemia in renal cells. Furthermore, SOCS proteins reduce harmful JAK/STAT-mediated cell responses in the diabetic kidney, thus suggesting the potential benefit of SOCS to halt the progression of diabetic nephropathy.
In agreement with previous reports in renal diseases,13,20,25,26,28 we observed JAK2, STAT1 and STAT3 activation in diabetic rats. However, this is the first time that the expression of negative regulators SOCS has been demonstrated in an in vivo setting of diabetic nephropathy. Thus, SOCS1 and SOCS3 expression was found increased in renal samples from diabetic patients and STZ-induced diabetic rats when compared with control subjects. Histologic examination revealed that SOCS proteins are produced by glomerular and tubular cells, thus indicating that both renal compartments are involved in the JAK/STAT-mediated responses of diabetic kidney.
In this study, we also demonstrate activation of JAK/STAT/SOCS axis by hyperglycemia in vitro. Consistent with previous data,6,10,13,26,27 HG increased the tyrosine phosphorylation of JAK/STAT members in human MCs and HK2 cells. Furthermore, HG induced transcriptional activation of STAT1, STAT2, and STAT3. Along with STAT activation, HG transiently induced SOCS expression (gene and protein) in renal cells; SOCS1 expression was always preceded by SOCS3 induction. This suggests a dual negative regulatory system of SOCS in HG-stimulated renal cells, similar to the control of cytokine signaling17: SOCS3 first modulating the STAT-mediated response and SOCS1 as a regulator of activated pathways after longer exposure to hyperglycemia.
Previous findings suggest that the JAK/STAT pathway, especially the JAK2/STAT1/STAT3-dependent axis, contributes to HG-mediated renal cell responses, including enhanced expression of genes involved in leukocyte infiltration, cell growth, and fibrosis.6,9,11,22,28 Moreover, diverse approaches including JAK2 inhibition, STAT1 antisense oligonucleotides, STAT3 gene knockdown, and some drugs have been proven to counteract the harmful effects of JAK/STAT activation in cultured renal cells6,10,13,18,22,29 and in the development of diabetes in vivo.13,25,26,28 Recently, Shi et al.27 reported that SOCS1 inhibits JAK/STAT activation and gene expression by hyperglycemia in human MCs. In agreement with this, we demonstrate that SOCS1 and SOCS3 overexpression (plasmid and adenovirus vector systems) effectively prevented the JAK2/STAT1/STAT3 phosphorylation and the STAT transcriptional activity induced by HG in human MCs and tubular cells. However, we cannot discard that in addition to SOCS, other intracellular mechanisms (e.g., the constitutive inhibitors protein tyrosine phosphatases and protein inhibitors of activated STAT12,13,16) may act in conjunction or independently in controlling the JAK/STAT pathway in renal cells.
Excessive production of cytokines and growth factors is involved in the pathogenesis of diabetic nephropathy, with cell growth and fibrosis being the major contributors to the pathologic changes associated with this disease.2–4 JAK/STAT pathway activation mediates the mitogenic and fibrotic actions of cytokines, angiotensin II, and hyperglycemia in the kidney.12 Moreover, several groups, including ours, have previously demonstrated that the SOCS family regulates signaling by cytokines, angiotensin II, immune complexes, and HG in human MCs.14,18,20,27 In this work, we confirm that the SOCS endogenous pathway controls JAK/STAT-dependent events in HG-stimulated MCs; however, this is interestingly the first description of the involvement of SOCS1 and SOCS3 in the control of tubular cell responses to hyperglycemia, including JAK/STAT activation, cytokine and matrix protein expression, and cell growth. This reinforces the concept that, in addition to glomerular cells, tubular cells are a primary target of hyperglycemia and chronic exposure to elevated glucose levels contributes to the tubulointerstitial changes seen in overt diabetic nephropathy.2,6,8
Although diabetic nephropathy is traditionally considered a nonimmune disease, accumulating evidence indicates that immunoinflammatory mechanisms play a crucial role in its development and progression. In this sense, macrophages have been implicated in the pathogenesis of diabetic nephropathy by direct interaction with mesangial and tubular cells or by releasing factors involved in cell proliferation and matrix production.3 Moreover, expression of genes involved in the recruitment, infiltration, and activation of leukocytes has been observed in experimental diabetic nephropathy in parallel with disease progression and in cultured renal cells under hyperglycemic conditions4,30,31; their inhibition abolished diabetes-induced renal damage.32,33 Our study demonstrates that SOCS are able to inhibit the renal expression of STAT-dependent genes, including adhesion molecules, chemokines, and cytokines, thus unveiling the importance of the JAK/STAT/SOCS axis in inflammatory responses to hyperglycemia.
Evidence is emerging for the involvement of SOCS in inflammatory diseases (e.g., rheumatoid arthritis, cerebral ischemia, heart failure, hypertension, and vascular and renal injury).15,20,34–36 A recent report correlates the increased expression of SOCS1 and SOCS3 in mononuclear cells with decreased renal function in chronic kidney disease patients, thus proposing SOCS as a new cardiovascular risk marker in these patients.37 Moreover, SOCS3 expression increased in insulin-sensitive tissues from type 2 diabetic patients, although it remains to be established whether this is a cause or a consequence of insulin resistance.38 Different studies demonstrated that inhibition of SOCS expression leading to sustained STAT activation contributes to the progression of chronic inflammatory diseases.18,20,36,39–41 By contrast, transgenic expression of SOCS1 in β cells protects from T cell-mediated damage in diabetic mice,42 whereas adenovirus-mediated SOCS expression reduces arthritis15,34,43 and innate immune responses.44 Recently, a mimetic peptide containing the kinase inhibitory region of SOCS1 was proven to effectively protect against virus infection.45 On the basis of these observations and our in vitro data, we speculate that SOCS are induced in the diabetic kidney as a compensatory but not sufficient mechanism to suppress renal damage. Therefore, strategies to upregulate SOCS expression in the kidney may have therapeutic value for experimental diabetic nephropathy. Because transient expression and immune response are the major limitations of adenovirus-mediated gene therapy in chronic diseases,43,44,46 we studied the effects of a single-treatment strategy on early diabetic nephropathy. Transgene overexpression was detected up to 7 days after adenovirus injection, thereafter gradually diminishing within the next 4 weeks, a normal phenomenon related to the one-time gene transfection in vivo.46 Meanwhile, the transient expression of SOCS in diabetic rats exerts a beneficial effect on renal damage that transcends the duration of adenovirus-mediated SOCS expression. In fact, renal delivery of SOCS1 and SOCS3 attenuated STAT1 and STAT3 activity, ameliorated albuminuria and renal lesions, and reduced the expression of STAT-dependent genes involved in leukocyte infiltration, cell proliferation, and fibrosis during diabetic nephropathy. SOCS gene therapy also reduced the glomerular hyperfiltration associated with early diabetic nephropathy, probably by decreasing hyperplasia and hypertrophy, components of the increased glomerular volume that are altered by hyperglycemia via the JAK/STAT pathway.6,12,21,23
In summary, our data provide direct evidence for linkages between the JAK/STAT/SOCS axis and hyperglycemia-induced cell responses in the diabetic kidney. Moreover, the SOCS family of intracellular inducible proteins plays a critical role in the regulation of inflammation, abnormal cell proliferation, and matrix synthesis at early stages of diabetic nephropathy. Compensatory suppression of the JAK/STAT pathway by increasing SOCS may have a therapeutic potential in this disease.
Concise Methods
Reagents
D-glucose, d-mannitol, and STZ were obtained from Sigma Chemical (St. Louis, MO). Primary antibodies for immunohistochemistry included SOCS1, SOCS3, and TGFβ1 (Santa Cruz Biotechnology, Santa Cruz, CA); phosphotyrosine (P)-STAT1 (Biosource International, Camarillo, CA); and P-STAT3 (Cell Signaling, Beverly, MA). Primary antibodies for Western blot included SOCS1 and P-STAT1 (Zymed Laboratories, South San Francisco, CA); SOCS3, P-JAK2, and P-STAT3 (Santa Cruz); fibronectin (Chemicon, Temecula, CA); MCP-1 (Peprotech, Rocky Hill, NJ); and α-tubulin (Sigma).
Construction of Adenoviral Vectors
The first generation of adenoviruses regulated by the cytomegalovirus promoter containing mouse SOCS1 and SOCS3 genes (Ad-S1 and Ad-S3) were prepared using the AdEasyTM vector system (Qbiogene, Inc., Carlsbad, CA). In brief, mouse SOCS1 and SOCS3 cDNAs were excised from the p513HA vector20 and inserted into pShuttle-cytomegalovirus transfer vector. The resulting plasmids were cotransformed into bacteria together with pAdEasy-1 (E1 and E3 deleted). The recombinant adenoviral constructs were cleaved with PacI and transfected into HEK293 cells to produce viral particles. Purification and titration of each virus were made using Adeno-X Virus Kits (BD Biosciences Clontech, Palo Alto, CA). Ad-GFP and Ad-null were used as control vectors.
Cell Cultures
Human MCs were cultured in RPMI 1640 with 25 mM HEPES (pH 7.4) supplemented with 10% FCS, 100 U/ml penicillin, 100 mg/ml streptomycin, and 2 mM glutamine (Life Technologies, Paisley, Scotland, United Kingdom) and 5 μg/ml transferrin, 5 μg/ml bovine insulin, and 5 ng/ml sodium selenite (Sigma), as described.18,20 The HK2 cell line (American Type Culture Collection, Rockville, MD) was cultured in RPMI 1640 with 10% FCS. Quiescent, subconfluent cells were stimulated in culture medium supplemented with d-glucose (final concentration 30 mM) using d-mannitol as an osmolarity control. Transient expression of SOCS1 and SOCS3 proteins in cells was performed by plasmid transfection (S1wt, S3wt, and control vector p513)18,21 and adenovirus infection (Ad-S1, Ad-S3, and Ad-null; multiplicity of infection = 40) for 24 hours before stimulation.
Luciferase Assay
Cells were cotransfected with luciferase plasmids containing the STAT1, STAT1/2, and STAT3 binding sites and the pRenilla-TK vector and then stimulated for an additional 24 hours. Dual luciferase activity was assayed using a luminometer, and data were normalized for protein content.18,20
Cell Proliferation Assay
Quiescent cells on 96-well plates were incubated under hyperglycemic conditions for 48 hours. Cell proliferation was measured by colorimetric assay.21
Human Renal Biopsies
Kidney samples were obtained from patients with type 2 diabetes mellitus (n = 5) that were submitted to renal biopsy because of nephrotic proteinuria.30 Clinical data at the biopsy are as follows: mean age = 59 years (range 50 to 65 years); mean serum creatinine = 2.31 mg/dl (range 1.2 to 3.4 mg/dl); mean proteinuria = 7.6 g/d (range 2.3 to 10.5 g/d). Biopsies showed a clear picture of diffuse or nodular glomerulosclerosis with a marked tubulointerstitial involvement. Samples from minimal change disease patients were used as a control group (n = 5).
Experimental Diabetic Nephropathy
Diabetes was induced in male Wistar rats (220 to 250 g) by intraperitoneal injection of STZ (50 mg/kg in 10 mM citrate buffer, pH 4.5) as described.31 Blood glucose levels were measured 48 hours after STZ injection. Only animals with glucose levels >350 mg/dl were included in the studies. To prevent animal death, 2 U of insulin were administered weekly 7 days after STZ injection (blood glucose levels > 400 mg/dl). After 6 months, diabetic rats (n = 5) were killed and renal samples were processed. Age-matched nondiabetic rats were used as a control group (n = 5).
Renal Delivery of SOCS-Expressing Adenovirus
The adenovirus-mediated transduction in vivo was performed by injection into the left renal vein. In brief, rats were anesthetized and the left kidney was exposed by midline incision. Adenoviruses (1 × 109 viral particles per rat) were infused into the renal vein over 7 minutes and then blood flow to the kidney was restored. For assessing the efficiency and localization of adenovirus, initial experiments were performed in control rats infused with Ad-GFP, Ad-null, Ad-S1, and Ad-S3 (n = 6 per group) and kidneys were removed after 3, 5, and 7 days of delivery. For studying the effects of SOCS on diabetic nephropathy, 3 weeks after the STZ injection diabetic rats were randomly distributed into four groups (n = 12 per group): sham operation, control adenovirus (Ad-null), SOCS1 adenovirus (Ad-S1), and SOCS3 adenovirus (Ad-S3). Nondiabetic rats were used as a control group (n = 12). Four weeks after injection, blood and 24-hour urine samples were collected and the animals were killed. After saline perfusion, kidneys were dissected, weighed, and processed for histology and RNA expression. Creatinine clearance was calculated from the formula of UV/P × 1/1440 minutes, where U represents the urine concentration, V is the urine volume, and P is the plasma concentration. The results were then corrected for body weight. The concentration of urine albumin was measured by ELISA (Cell Trend, Luckenwalde, Germany) and values were normalized by creatinine concentration. All studies were performed in accordance to the European Union normative.
Histology and Immunohistochemistry
Cryostat sections of unfixed, snap-frozen specimens from Ad-GFP-injected rats were analyzed under the fluorescence microscope. Paraffin-embedded kidney sections (5-μm thick) were used for histologic analysis. Renal lesions in periodic acid–Schiff and Masson's trichrome stained sections were scored on a scale of 0 to 3 according to the extent of glomerular changes (hypertrophy, hypercellularity, and mesangial expansion) and tubular dilation as follows: 0 = absent, 1 = mild, 2 = moderate, and 3 = severe. Renal fibrosis was examined by polarized light microscopy after picrosirius red staining. Immunodetection of proteins (SOCS1, SOCS3, P-STAT1, P-STAT3, and TGFβ1) and macrophages (CD68) in renal samples was performed by an indirect immunoperoxidase technique. Positive staining (15 fields per kidney section, ×200) was quantified using Image Pro-Plus analysis software, and positive area was expressed as a percentage of the total area. CD68+ cells were expressed as a percentage of the total glomerular cells and number of interstitial cells per square millimeter. Glomerular area measurement (n = 50 per rat) was used to calculate glomerular volume by the formula VolG = mean area1.5 × 1.38/1.1.
mRNA Expression
Total RNA from cells and tissues was extracted and gene expression was analyzed by real-time PCR on a TaqMan ABI 7500 sequence detection system (Applied Biosystem, Foster City, CA). The expression of target genes was analyzed in duplicate and normalized to housekeeping 18S transcripts.
Western Blot
Cytosolic proteins from cells and tissues were resolved on SDS-PAGE gels, transferred onto polyvinylidene fluoride membranes, and immunoblotted with antibodies against SOCS1, SOCS3, P-JAK2, P-STAT1, P-STAT3, and fibronectin. After visualizing with an enhanced chemiluminescence system, membranes were reblotted for α-tubulin (loading control).
Statistics
Data are presented as mean ± SEM or representative experiment, when indicated. Statistical analysis was performed by one-way ANOVA. P < 0.05 was considered statistically significant.
Disclosures
None.
Supplementary Material
Acknowledgments
This study was supported by grants from Ministry of Science (SAF2005/05857, SAF2007/63648, SAF2009/11794), Ministry of Health (Instituto de Salud Carlos III, Red RECAVA RD06/0014/0035), Fundacion Ramon Areces, and Comunidad de Madrid (S2006/GEN-0247) in Spain and Fondecyt (1080083) in Chile. We thank Drs. H.M. Wilson and A.J. Rees (Aberdeen University, United Kingdom), Dr. A.H. Baker (Glasgow University, United Kingdom), and Dr. N. Vilaboa (Hospital La Paz, Madrid) for their help with adenovirus preparation and S. Carrasco and T. Carrizosa for skilled technical assistance.
Footnotes
Published online ahead of print. Publication date available at www.jasn.org.
See related editorial, “A New Pair of SOCS for Diabetic Nephropathy,” on pages 723–724.
Supplemental information for this article is available online at http://www.jasn.org/.
REFERENCES
- 1. Forbes JM, Fukami K, Cooper ME: Diabetic nephropathy: Where hemodynamics meets metabolism. Exp Clin Endocrinol Diabetes 115: 69–84, 2007. [DOI] [PubMed] [Google Scholar]
- 2. Schrijvers BF, De Vriese AS, Flyvbjerg A: From hyperglycemia to diabetic kidney disease: The role of metabolic, hemodynamic, intracellular factors and growth factors/cytokines. Endocr Rev 25: 971–1010, 2004. [DOI] [PubMed] [Google Scholar]
- 3. Williams MD, Nadler JL: Inflammatory mechanisms of diabetic complications. Curr Diab Rep 7: 242–248, 2007. [DOI] [PubMed] [Google Scholar]
- 4. Navarro-Gonzalez JF, Mora-Fernandez C: The role of inflammatory cytokines in diabetic nephropathy. J Am Soc Nephrol 19: 433–442, 2008. [DOI] [PubMed] [Google Scholar]
- 5. Ha H, Yu MR, Choi YJ, Kitamura M, Lee HB: Role of high glucose-induced nuclear factor-kappaB activation in monocyte chemoattractant protein-1 expression by mesangial cells. J Am Soc Nephrol 13: 894–902, 2002. [DOI] [PubMed] [Google Scholar]
- 6. Huang JS, Chuang LY, Guh JY, Huang YJ, Hsu MS: Antioxidants attenuate high glucose-induced hypertrophic growth in renal tubular epithelial cells. Am J Physiol Renal Physiol 293: F1072–F1082, 2007. [DOI] [PubMed] [Google Scholar]
- 7. Weigert C, Sauer U, Brodbeck K, Pfeiffer A, Haring HU, Schleicher ED: AP-1 proteins mediate hyperglycemia-induced activation of the human TGF-beta1 promoter in mesangial cells. J Am Soc Nephrol 11: 2007–2016, 2000. [DOI] [PubMed] [Google Scholar]
- 8. Nangaku M: Mechanisms of tubulointerstitial injury in the kidney: Final common pathways to end-stage renal failure. Intern Med 43: 9–17, 2004. [DOI] [PubMed] [Google Scholar]
- 9. Brosius FC, III: New insights into the mechanisms of fibrosis and sclerosis in diabetic nephropathy. Rev Endocr Metab Disord 9: 245–254, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wang X, Shaw S, Amiri F, Eaton DC, Marrero MB: Inhibition of the JAK/STAT signaling pathway prevents the high glucose-induced increase in tgf-beta and fibronectin synthesis in mesangial cells. Diabetes 51: 3505–3509, 2002. [DOI] [PubMed] [Google Scholar]
- 11. Amiri F, Shaw S, Wang X, Tang J, Waller JL, Eaton DC, Marrero MB: Angiotensin II activation of the JAK/STAT pathway in mesangial cells is altered by high glucose. Kidney Int 61: 1605–1616, 2002. [DOI] [PubMed] [Google Scholar]
- 12. Marrero MB, Banes-Berceli AK, Stern DM, Eaton DC: Role of the JAK/STAT signaling pathway in diabetic nephropathy. Am J Physiol Renal Physiol 290: F762–F768, 2006. [DOI] [PubMed] [Google Scholar]
- 13. Shi YH, Zhao S, Wang C, Li Y, Duan HJ: Fluvastatin inhibits activation of JAK and STAT proteins in diabetic rat glomeruli and mesangial cells under high glucose conditions. Acta Pharmacol Sin 28: 1938–1946, 2007. [DOI] [PubMed] [Google Scholar]
- 14. O'Sullivan LA, Liongue C, Lewis RS, Stephenson SE, Ward AC: Cytokine receptor signaling through the JAK-STAT-SOCS pathway in disease. Mol Immunol 44: 2497–2506, 2007. [DOI] [PubMed] [Google Scholar]
- 15. Yoshimura A, Naka T, Kubo M: SOCS proteins, cytokine signaling and immune regulation. Nat Rev Immunol 7: 454–465, 2007. [DOI] [PubMed] [Google Scholar]
- 16. Rakesh K, Agrawal DK: Controlling cytokine signaling by constitutive inhibitors. Biochem Pharmacol 70: 649–657, 2005. [DOI] [PubMed] [Google Scholar]
- 17. Wormald S, Zhang JG, Krebs DL, Mielke LA, Silver J, Alexander WS, Speed TP, Nicola NA, Hilton DJ: The comparative roles of suppressor of cytokine signaling-1 and -3 in the inhibition and desensitization of cytokine signaling. J Biol Chem 281: 11135–11143, 2006. [DOI] [PubMed] [Google Scholar]
- 18. Hernandez-Vargas P, Lopez-Franco O, Sanjuan G, Ruperez M, Ortiz-Munoz G, Suzuki Y, Guado-Roncero P, Perez-Tejerizo G, Blanco J, Egido J, Ruiz-Ortega M, Gomez-Guerrero C: Suppressors of cytokine signaling regulate angiotensin II-activated Janus kinase-signal transducers and activators of transcription pathway in renal cells. J Am Soc Nephrol 16: 1673–1683, 2005. [DOI] [PubMed] [Google Scholar]
- 19. Soriano SF, Hernanz-Falcon P, Rodriguez-Frade JM, De Ana AM, Garzon R, Carvalho-Pinto C, Vila-Coro AJ, Zaballos A, Balomenos D, Martinez A, Mellado M: Functional inactivation of CXC chemokine receptor 4-mediated responses through SOCS3 up-regulation. J Exp Med 196: 311–321, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Gomez-Guerrero C, Lopez-Franco O, Sanjuan G, Hernandez-Vargas P, Suzuki Y, Ortiz-Munoz G, Blanco J, Egido J: Suppressors of cytokine signaling regulate Fc receptor signaling and cell activation during immune renal injury. J Immunol 172: 6969–6977, 2004. [DOI] [PubMed] [Google Scholar]
- 21. Ortiz-Munoz G, Martin-Ventura JL, Hernandez-Vargas P, Mallavia B, Lopez-Parra V, Lopez-Franco O, Munoz-Garcia B, Fernandez-Vizarra P, Ortega L, Egido J, Gomez-Guerrero C: Suppressors of cytokine signaling modulate JAK/STAT-mediated cell responses during atherosclerosis. Arterioscler Thromb Vasc Biol 29: 525–531, 2009. [DOI] [PubMed] [Google Scholar]
- 22. de Prati AC, Ciampa AR, Cavalieri E, Zaffini R, Darra E, Menegazzi M, Suzuki H, Mariotto S: STAT1 as a new molecular target of anti-inflammatory treatment. Curr Med Chem 12: 1819–1828, 2005. [DOI] [PubMed] [Google Scholar]
- 23. Grote K, Luchtefeld M, Schieffer B: JANUS under stress—Role of JAK/STAT signaling pathway in vascular diseases. Vascul Pharmacol 43: 357–363, 2005. [DOI] [PubMed] [Google Scholar]
- 24. Berthier CC, Zhang H, Schin M, Henger A, Nelson RG, Yee B, Boucherot A, Neusser MA, Cohen CD, Carter-Su C, Argetsinger LS, Rastaldi MP, Brosius FC, Kretzler M: Enhanced expression of Janus kinase-signal transducer and activator of transcription pathway members in human diabetic nephropathy. Diabetes 58: 469–477, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Banes AK, Shaw S, Jenkins J, Redd H, Amiri F, Pollock DM, Marrero MB: Angiotensin II blockade prevents hyperglycemia-induced activation of JAK and STAT proteins in diabetic rat kidney glomeruli. Am J Physiol Renal Physiol 286: F653–F659, 2004. [DOI] [PubMed] [Google Scholar]
- 26. Banes-Berceli AK, Shaw S, Ma G, Brands M, Eaton DC, Stern DM, Fulton D, Caldwell RW, Marrero MB: Effect of simvastatin on high glucose- and angiotensin II-induced activation of the JAK/STAT pathway in mesangial cells. Am J Physiol Renal Physiol 291: F116–F121, 2006. [DOI] [PubMed] [Google Scholar]
- 27. Shi Y, Zhang Y, Wang C, Du C, Zhao S, Qi Z, Zhang Q, Duan H: Suppressor of cytokine signaling-1 reduces high glucose-induced TGF-beta1 and fibronectin synthesis in human mesangial cells. FEBS Lett 582: 3484–3488, 2008. [DOI] [PubMed] [Google Scholar]
- 28. Lu TC, Wang ZH, Feng X, Chuang PY, Fang W, Shen Y, Levy DE, Xiong H, Chen N, He JC: Knockdown of Stat3 activity in vivo prevents diabetic glomerulopathy. Kidney Int 76: 63–71, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Neria F, Castilla MA, Sanchez RF, Gonzalez Pacheco FR, Deudero JJ, Calabia O, Tejedor A, Manzarbeitia F, Ortiz A, Caramelo C: Inhibition of JAK2 protects renal endothelial and epithelial cells from oxidative stress and cyclosporin A toxicity. Kidney Int 75: 227–234, 2009. [DOI] [PubMed] [Google Scholar]
- 30. Mezzano S, Aros C, Droguett A, Burgos ME, Ardiles L, Flores C, Schneider H, Ruiz-Ortega M, Egido J: NF-kappaB activation and overexpression of regulated genes in human diabetic nephropathy. Nephrol Dial Transplant 19: 2505–2512, 2004. [DOI] [PubMed] [Google Scholar]
- 31. Sanchez-Nino MD, Sanz AB, Ihalmo P, Lassila M, Holthofer H, Mezzano S, Aros C, Groop PH, Saleem MA, Mathieson PW, Langham R, Kretzler M, Nair V, Lemley KV, Nelson RG, Mervaala E, Mattinzoli D, Rastaldi MP, Ruiz-Ortega M, Martin-Ventura JL, Egido J, Ortiz A: The MIF receptor CD74 in diabetic podocyte injury. J Am Soc Nephrol 20: 353–362, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Okada S, Shikata K, Matsuda M, Ogawa D, Usui H, Kido Y, Nagase R, Wada J, Shikata Y, Makino H: Intercellular adhesion molecule-1-deficient mice are resistant against renal injury after induction of diabetes. Diabetes 52: 2586–2593, 2003. [DOI] [PubMed] [Google Scholar]
- 33. Chow FY, Nikolic-Paterson DJ, Ozols E, Atkins RC, Rollin BJ, Tesch GH: Monocyte chemoattractant protein-1 promotes the development of diabetic renal injury in streptozotocin-treated mice. Kidney Int 69: 73–80, 2006. [DOI] [PubMed] [Google Scholar]
- 34. Shouda T, Yoshida T, Hanada T, Wakioka T, Oishi M, Miyoshi K, Komiya S, Kosai K, Hanakawa Y, Hashimoto K, Nagata K, Yoshimura A: Induction of the cytokine signal regulator SOCS3/CIS3 as a therapeutic strategy for treating inflammatory arthritis. J Clin Invest 108: 1781–1788, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Adhikari N, Charles N, Lehmann U, Hall JL: Transcription factor and kinase-mediated signaling in atherosclerosis and vascular injury. Curr Atheroscler Rep 8: 252–260, 2006. [DOI] [PubMed] [Google Scholar]
- 36. Raghavendra R, V, Bowen KK, Dhodda VK, Song G, Franklin JL, Gavva NR, Dempsey RJ: Gene expression analysis of spontaneously hypertensive rat cerebral cortex following transient focal cerebral ischemia. J Neurochem 83: 1072–1086, 2002. [DOI] [PubMed] [Google Scholar]
- 37. Rastmanesh MM, Bluyssen HA, Joles JA, Boer P, Willekes N, Braam B: Increased expression of SOCS3 in monocytes and SOCS1 in lymphocytes correlates with progressive loss of renal function and cardiovascular risk factors in chronic kidney disease. Eur J Pharmacol 593: 99–104, 2008. [DOI] [PubMed] [Google Scholar]
- 38. Lebrun P, Van OE: SOCS proteins causing trouble in insulin action. Acta Physiol (Oxf) 192: 29–36, 2008. [DOI] [PubMed] [Google Scholar]
- 39. Suzuki A, Hanada T, Mitsuyama K, Yoshida T, Kamizono S, Hoshino T, Kubo M, Yamashita A, Okabe M, Takeda K, Akira S, Matsumoto S, Toyonaga A, Sata M, Yoshimura A: CIS3/SOCS3/SSI3 plays a negative regulatory role in STAT3 activation and intestinal inflammation. J Exp Med 193: 471–481, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Wong PK, Egan PJ, Croker BA, O'Donnell K, Sims NA, Drake S, Kiu H, McManus EJ, Alexander WS, Roberts AW, Wicks IP: SOCS-3 negatively regulates innate and adaptive immune mechanisms in acute IL-1-dependent inflammatory arthritis. J Clin Invest 116: 1571–1581, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Egan PJ, Lawlor KE, Alexander WS, Wicks IP: Suppressor of cytokine signaling-1 regulates acute inflammatory arthritis and T cell activation. J Clin Invest 111: 915–924, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Barral AM, Thomas HE, Ling EM, Darwiche R, Rodrigo E, Christen U, Ejrnaes M, Wolfe T, Kay TW, von Herrath MG: SOCS-1 protects from virally-induced CD8 T cell mediated type 1 diabetes. J Autoimmun 27: 166–173, 2006. [DOI] [PubMed] [Google Scholar]
- 43. Veenbergen S, Bennink MB, de Hooge AS, Arntz OJ, Smeets RL, van den Berg WB, van de Loo FA: Splenic suppressor of cytokine signaling 3 transgene expression affects T cell responses and prevents development of collagen-induced arthritis. Arthritis Rheum 58: 3742–3752, 2008. [DOI] [PubMed] [Google Scholar]
- 44. Sakurai H, Tashiro K, Kawabata K, Yamaguchi T, Sakurai F, Nakagawa S, Mizuguchi H: Adenoviral expression of suppressor of cytokine signaling-1 reduces adenovirus vector-induced innate immune responses. J Immunol 180: 4931–4938, 2008. [DOI] [PubMed] [Google Scholar]
- 45. Ahmed CM, Dabelic R, Waiboci LW, Jager LD, Heron LL, Johnson HM: SOCS-1 mimetics protect mice against lethal poxvirus infection: identification of a novel endogenous antiviral system. J Virol 83: 1402–1415, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Xiong J, Wang SM, Chen LH, Lin Y, Zhu YF, Ye CS: Elastic fibers reconstructed using adenovirus-mediated expression of tropoelastin and tested in the elastase model of abdominal aortic aneurysm in rats. J Vasc Surg 48: 965–973, 2008. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.