

Abstract
The International Union of Pure and Applied Chemistry (IUPAC) code specified nearly 25 years ago provides a nomenclature for incompletely specified nucleic acids. However, no system currently exists that allows for the informatics representation of the relative abundance at polymorphic nucleic acids (e.g. single nucleotide polymorphisms) in a single specified character, or a string of characters. Here, I propose such an information code as a natural extension to the IUPAC nomenclature code, and present some potential uses and limitations to such a code. The primary anticipated use of this extended nomenclature code is to assist in the representation of the rapidly growing space of information in human genetic variation.
Contact: johnsonad2@nhlbi.nih.gov
Supplementary information: Supplementary data are available at Bioinformatics online.
1 INTRODUCTION
The International Union of Pure and Applied Chemistry (IUPAC) code specified nearly 25 years ago provides a nomenclature for incompletely specified nucleic acids (Cornish-Bowden, 1985). The correct origin of the code is with the International Union of Biochemistry and Molecular Biology (IUBMB) but it has become more widely known as the IUPAC code. The IUPAC code, presented in Table 1, has been applied in a wide-ranging manner, contributing to many biologically and chemically meaningful representations, including: (i) recognition sequences (e.g. restriction enzymes, protein and RNA binding sites, consensus signals); (ii) codon degeneracy; (iii) sequence base calling ambiguity; (iv) representation of ancestral states in phylogenetics; and (v) to a vast extent in the fields of genetics and genomics in representing polymorphic nucleic acids, e.g. single nucleotide polymorphisms (SNPs). However, no system currently exists that allows for the informatics representation of the relative abundance at polymorphic nucleic acids (e.g. SNPs) in a single specified character or a string of characters. Here I propose such an information code, displayed in Table 2, as a natural extension to the IUPAC nomenclature code, and present some potential uses and limitations to such a code. The original IUPAC code remains useful in all its previous applications and is also compatible as a subset of the extended code proposed here. The extended IUPAC code allows for new nucleic acid representations, in single characters or character strings, with potential applications in genetics, cross-species or cross-strain comparison, sequence alignment, bioinformatics, genome assembly, database design and querying and chemical sequencing and synthesis. The primary anticipated use of this extended nomenclature code is to assist in the representation of the rapidly growing space of information on human genetic variation.
Table 1.
IUPAC code for incomplete nucleic acid specification
| Symbol | Mnemonic | Translation |
|---|---|---|
| A | A (adenine) | |
| C | C (cytosine) | |
| G | G (guanine) | |
| T | T (thymine) | |
| U | U (uracil) | |
| R | puRine | A or G (purines) |
| Y | pYrimidine | C or T/U (pyrimidines) |
| M | aMino group | A or C |
| K | Keto group | G or T/U |
| S | Strong interaction | C or G |
| W | Weak interaction | A or T/U |
| H | not G | A, C or T/U |
| B | not A | C, G or T/U |
| V | not T/U | A, C or G |
| D | not C | A, G or T/U |
| N | aNy | A, C, G or T/U |
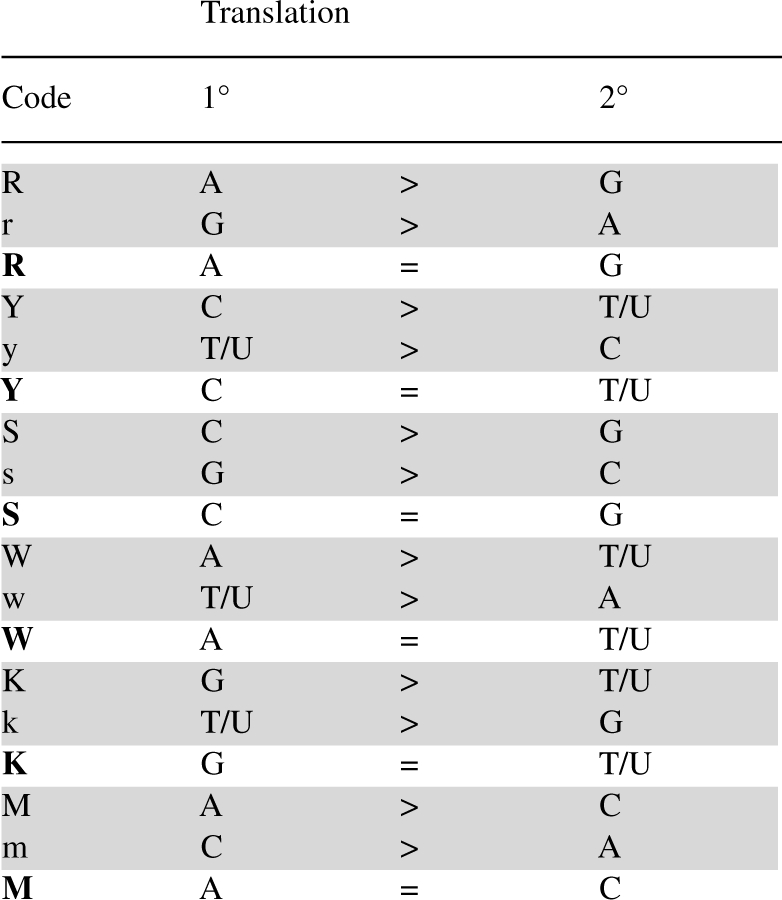
Table 2.
Extended IUPAC code for 2 nt combinations
 |
1.1 IUPAC code for incomplete nucleic acid specification
The IUPAC code is a 16-character code which allows the ambiguous specification of nucleic acids (Table 1). The code can represent states that include single specifications for nucleic acids (A, G, C, T/U) or allows for ambiguity among 2, 3 or 4 possible nucleic acid states. The IUPAC code is, in principle, case insensitive, but its established uses generally default to the capital case.
1.2 Extended IUPAC code for specification of relative nucleic acid prevalence
The extension of the standard IUPAC code is a character case-sensitive code that includes all 16 original IUPAC characters with similar meanings intact (Tables 2 and 3). This code relies on the use of character case information, additional available alphabetic characters, bolding and underlining to specify the ordering of all possible combinations and interrelationships containing 2 (Table 2) or 3 (Table 3) nucleic acid states. Specification of all possible orderings for four nucleic acids in a single character is not possible in the alphabetic system, since this requires 24 unique characters or more if interrelationships are considered, and only ‘P’/‘p’ and ‘Q’/‘q’ remain unspecified here. Instead, the extended code makes use of additional ASCII characters to represent states for four nucleic acids (Supplementary Table S1). The system for 2 nt combinations is relatively straightforward with bold case in instances where the two bases are represented in equal proportion (Table 2).
Table 3.
Extended IUPAC code for 3 nt combinations
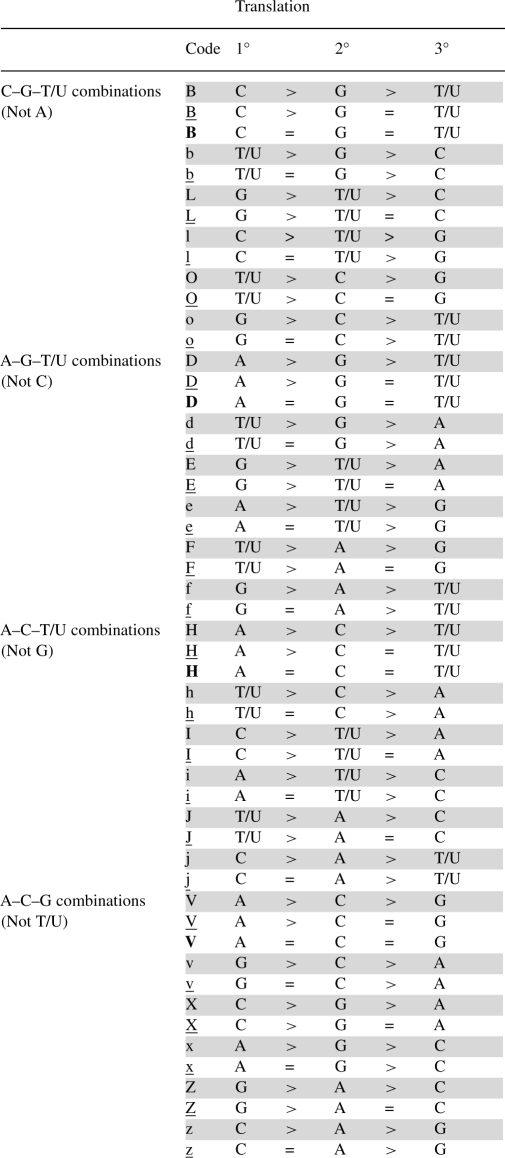 |
There are four classes of 3 nt groupings possible. Within each class, there are 13 possible relationships among the three nucleotides (represented in each of four subparts of Table 3). Bold case is again reserved for equality among all three states. The most common expected states where there will be differential states or counts for all three nucleotides are highlighted in gray. In uncommon cases where there is equality for two of the nucleotides, this is indicated using the corresponding underline character. While the code is more complex and less readable than previous human codes, the ability to specify all states means that it will be relatively straightforward to develop programmatic translation tables for process like strand flipping or rapid summarization of data across many sequences.
2 DISCUSSION
The development of an extended IUPAC code was primarily motivated by informatics issues within the realm of databases and research relating to genetic variability. It is predicted that in the near future full genome sequencing of many humans may be feasible and cost-effective, and that patients may approach clinicians, counselors and researchers for interpretations of their primary sequence data. While much attention has been paid to developing technologies to make such a situation feasible, little attention has been paid to the informatics challenges that would accompany it. Assembling and storing this volume of data is feasible with current computing standards, although perhaps not trivial when many genomes are considered (e.g. the 1000 Genomes Project, www.1000genomes.org). However, we currently have an incomplete and evolving picture of human genetic variation. A recent survey of CLIA-tested variants indicates that the majority of these are not precisely mapped or represented against the human genome or within the major databases of genetic variation (Johnson et al., 2010). At the same time, the reference human genome sequence which is used in a wide range of informatics tools (e.g. BLAST) contains nucleic acids at some positions that are found at the lowest abundance in the general population, with potential implications for processes that rely on the reference sequence. Furthermore, as sequencing efforts and deposition in databases like GenBank continue we will continue to discover and align more variation, particularly variants that are rare or private to particular subsets of individuals. Thus, we would be hard pressed given current standards and available databases to rapidly provide an individual with a nearly complete picture of where their personal sequence deviated from some reference group or even to provide a concise informational representation of a ‘reference group’ sequence itself. The realities of the coming informatics challenges of individual human genomes are highlighted in the recent releases of four full human genome sequences (Bentley et al., 2008; Levy et al., 2007; Wang et al., 2008; Wheeler et al., 2008).
While the current IUPAC code allows incomplete nucleic acid specification, it requires additional information such as allele strand and relative allele frequencies for many analyses. This information is by necessity disconnected from the primary sequence information, or requires the specification of multiple, and sometimes many sequences. This often complicates algorithmic processes involving sequence data or genetic analysis, for instance by necessitating the integration of information on the flanking sequence, strand orientation and relative allele abundances. This has important implications for instance in imputation methods which estimate unknown genotypes given prior known patterns, which can be confounded to some degree by problems with strand orientation and incompletely specified nucleic acids (de Bakker et al., 2008; Franke et al., 2008). The alternative system presented here could assist by providing an unambiguous code within primary polymorphic sequences, and may also facilitate a wide range of potential applications.
2.1 Advantages and applications of an extended IUPAC code
Allowing for an extended IUPAC code for the representation of sequences has a number of potential advantages both in the representation and presentation of polymorphic sequences, and in potential future bioinformatics and analytic applications, with a number of simulated examples presented in Figure 1. Using the extended IUPAC code it would be possible to create a single reference sequence that summarizes the dominant and recessive allele codings across a group of sequences with storage savings proportional to N, where N = the number of sequences summarized. This would make it feasible to summarize genetic variation and SNP annotations for instance across 1000 individual genomes. Combined with other strategies for compression and representation of genome sequences, individual personal genome sequences and summaries for meaningful reference sequence groups (Fig. 1E) could realistically be e-mailed or transferred on portable media (Brandon et al., 2009; Christley et al., 2009). Lean storage formats should also facilitate the ability to rapidly determine where a de novo sequence differs from a known reference summary of genetic variants for a group of sequences of interest (Fig. 1A and B).
Fig. 1.

Simulated example uses for an extended IUPAC code relating to modified reference genome sequences (A), biological classification (B), preferred sequence representation (C), sequence assembly (D) and population-level comparisons (E).
A number of additional practical applications of the extended IUPAC code are possible. The extended code could also be used to specify the relative allele frequencies for triallelic SNPs in databases (e.g. dbSNP), where the third allele is often but not always relatively uncommon. This has possible important implications for sequence-based analyses and genotyping assay design (e.g. Huebner et al., 2007). There are also potential clinical implications when diagnostic laboratories rely on reference sequences to design custom genotyping assays or sequencing primers. For example, the cystic fibrosis gene CFTR and breast cancer genes BRCA1 and BRCA2 contain hundreds of known disease-causing mutations and hundreds more common variants, including triallelic SNPs. These gene regions are routinely sequenced in diagnostics and research, but the many variants in these gene regions can make interpretations challenging and have the potential to confound sequence assay probes. The construction of important gene reference sequences (e.g. CFTR) using the extended IUPAC code could facilitate better assay design by enabling design algorithms to avoid polymorphic regions or preferentially select a probe that will contain the most common allele in a given population. The construction of disease- or condition-specific IUPAC masks for groups of important variants (Fig. 1B) could also facilitate initial categorization and visualization of de novo sequences relative to summaries of what is known.
With the ability to represent the relative abundance of polymorphic genetic sequences in a single character or string, a number of additional potential applications include: (i) the less ambiguous specification of alleles relative to their genomic strand without additional information (Fig. 1A); (ii) the facile and unambiguous summarization of relative abundances of nucleic acids among meaningful groups (e.g. patients versus controls, tumors of specific types, tumor cell subtypes within single tumors, biological strains with different virulence characteristics, population groupings) (Fig. 1B and E); (iii) simplified sequence specification of polymorphisms within genomes or sequence databases with ramifications for sequence alignment (e.g. BLAST) and the assembly and analysis of de novo sequence data (Fig. 1D); (iv) in phylogenetics applications, particularly in the representation of proportions associated with ancestral states (Lewis, 2001), and the resolution of non-binary nodes; (v) representation and analysis of differing sequences in triploid genomes; and (vi) the ability to specify preferred differential nucleic acid states in a high-throughput environment [e.g. probe design where SNPs may cause problems (Koboldt et al., 2006), preferred recognition sequences, sequence tag or SELEX analysis] (Fig. 1C). The code has the additional advantage of being fairly straightforward to operationalize and is an inclusive extension of the IUPAC code that is already used.
2.2 Potential limitations of an extended IUPAC code
While the extended IUPAC code provides a less ambiguous code than the standard IUPAC code, it remains a relatively ambiguous code with a number of implications. First, the application of such a code certainly does not replace the need for more detailed specification of relative nucleic acid abundances in many cases. For instance, two different polymorphisms encoding G > A transitions would similarly be represented as ‘r’ even if the observed population allele frequencies are G (50.1%) > A (49.9%) and G (99.9%) > A (0.1%), respectively. Further, in the case of specifying preferred sequence recognition sites for transcription factor binding, a position weight matrix (PWM) would provide more detailed and quantitative information than an extended IUPAC coding. Such PWMs are useful in deriving graphical representations of relative preferences. These are widely applied extensions of an original sequence logos proposal for consensus sequence representation (Scheinder and Stephens, 1990). Thus, most systems or databases that might employ the extended IUPAC code would likely do so while still retaining more detailed information (e.g. information about flanking sequences and population allele frequencies). A further limitation is that this code by itself provides no assistance in addressing more complex variants such as insertions, deletions, repeats and copy number variants.
A potential pitfall to the use of an extended IUPAC code is that software may not be equipped to handle, may ignore or may otherwise employ case-specific character information. In most cases, it is expected that software will simply ignore case sensitivity resulting in little difference. However, where useful, software could be reconfigured to recognize whether unambiguous sequence, standard IUPAC coding or extended IUPAC coding is employed if the user specifies it. The difference between standard and extended IUPAC coding may not be computer auto-detectable, particularly for short sequences, and thus software should default to the standard IUPAC coding unless otherwise specified. Interpretations that default to the standard IUPAC code will likely perform as always since the two codes are essentially compatible. The format of nucleic acid files or database entries could be specified in any representation. For example, in the instance of FASTA type nucleic acid sequences, format could be specified in the header line, as ‘FORMAT=SEQ’, ‘FORMAT=IUPAC’ or ‘FORMAT=IUPACX’ or file naming conventions could be represented as ‘.FASTA’, ‘.FASTI’ and ‘.FASTX’, respectively.
A potential confounding case could arise when polymorphic nucleic acids are specified within repeat masked sequences which may subsequently be translated to lower case. In such cases, it is suggested that the repeat masking program should flip the IUPAC case as it does with the other characters except for N/n (whether standard or extended).
3 CONCLUSION
While the use of an extended IUPAC code is not appropriate in many cases and is not intended to replace the need for detailed primary sequence databases, it may provide more efficient representations of consensus sequences for a variety of applications. In particular, the application of such a code may allow simplified representations of polymorphic nucleic acids within, and among, species, and other meaningful groupings. It may not be appropriate to have one reference omnibus representation of the human genome as we gain understanding of more individual DNA sequences worldwide; however, the availability of accurate summarizations of meaningful but differing groups of sequences will likely remain useful to researchers well into the future for both descriptive and applied purposes. Given the coming explosive growth in DNA sequencing, the extended IUPAC code may have its greatest use in summarizing and providing rapid representations of and identification of polymorphic nucleic acids among large groups of sequences.
Supplementary Material
ACKNOWLEDGEMENTS
I gratefully acknowledge Richard Lathe, Athel Cornish-Bowden, Robert Handsaker, Ilene Mizrachi, Farhat Habib, Dan Janies, Soumya Raychaudhuri and Wolfgang Sadee for their discussions and comments on the manuscript.
Funding: American Heart Association Pre-doctoral Fellowship (AHA0515157B to A.D.J.) and an NIH/NHLBI Post-doctoral IRTA Fellowship (to A.D.J.).
Conflict of Interest: none declared.
REFERENCES
- Cornish-Bowden A. Nomenclature for incompletely specified bases in nucleic acid sequences: recommendations 1984. Nucleic Acids Res. 1985;13:3021–3030. doi: 10.1093/nar/13.9.3021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bentley DR, et al. Accurate whole human genome sequencing using reversible terminator chemistry. Nature. 2008;456:53–59. doi: 10.1038/nature07517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandon MC, et al. Data structures and compression algorithms for genomic sequence data. Bioinformatics. 2009;25:1731–1738. doi: 10.1093/bioinformatics/btp319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christley S, et al. Human genomes as email attachments. Bioinformatics. 2009;25:274–275. doi: 10.1093/bioinformatics/btn582. [DOI] [PubMed] [Google Scholar]
- de Bakker PI, et al. Practical aspects of imputation-driven meta-analysis of genome-wide association studies. Hum. Mol. Gen. 2008;17:R122–R128. doi: 10.1093/hmg/ddn288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franke L, et al. Detection, imputation, and association analysis of small deletions and null alleles on oligonucleotide arrays. Am. J. Hum. Genet. 2008;82:1316–1333. doi: 10.1016/j.ajhg.2008.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huebner C, et al. Triallelic single nucleotide polymorphisms and genotyping errors in genetic epidemiology studies: MDR1 (ABCB1) G2677/T/A as an example. Canc. Epi. Bio. Prev. 2007;16:1185–1192. doi: 10.1158/1055-9965.EPI-06-0759. [DOI] [PubMed] [Google Scholar]
- Johnson AD, et al. CLIA-tested genetic variants on commercial SNP arrays: potential for incidental findings in genome-wide association studies. Genet. Med. 2010 doi: 10.1097/GIM.0b013e3181e1e2a9. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koboldt DC, et al. Distribution of human SNPs and its effect on high-throughput genotyping. Hum. Mut. 2006;27:249–254. doi: 10.1002/humu.20286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy S, et al. The diploid genome sequence of an individual human. PLoS Biol. 2007;5:e524. doi: 10.1371/journal.pbio.0050254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis PO. A likelihood approach to estimating phylogeny from discrete morphological character data. Systematic Biol. 2001;50:913–925. doi: 10.1080/106351501753462876. [DOI] [PubMed] [Google Scholar]
- Schneider TD, Stephens RM. Sequence logos: a new way to display consensus sequences. Nucleic Acids Res. 1990;18:6097–6100. doi: 10.1093/nar/18.20.6097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, et al. The diploid genome sequence of an Asian individual. Nature. 2008;456:60–65. doi: 10.1038/nature07484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheeler DA, et al. The complete genome of an individual by massively parallel DNA sequencing. Nature. 2008;452:872–876. doi: 10.1038/nature06884. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.