

Abstract
Matrix metalloproteinases (MMPs) emerge as modulators of neuropathic pain. Because myelin protects Aβ afferents from ectopic hyperexcitability and nociception from innocuous mechanical stimuli (or mechanical allodynia), we analyzed the role of MMPs in the development of mechanical allodynia through myelin protein degradation after rat and MMP-9−/− mouse L5 spinal nerve crush (L5 SNC). MMPs were shown to promote selective degradation of myelin basic protein (MBP), with MMP-9 regulating initial Schwann cell-mediated MBP processing after L5 SNC. Acute and long-term therapy with GM6001 (broad-spectrum MMP inhibitor) protected from injury-induced MBP degradation, caspase-mediated apoptosis, macrophage infiltration in the spinal nerve and inhibited astrocyte activation in the spinal cord. The effect of GM6001 therapy on attenuation of mechanical allodynia was robust, immediate and sustained through the course of L5 SNC. In conclusion, MMPs mediate the initiation and maintenance of mechanical nociception through Schwann cell-mediated MBP processing and support of neuroinflammation.
Keywords: Neuropathic pain, MBP, Myelin, Allodynia, Schwann cell, Apoptosis
Introduction
After nerve damage, compromised myelin integrity of Aβ afferents promotes mechanical nociception through exposure of axonal plasma membrane to Schwann cell-derived nociceptive stimuli (Devor, 2006), such as the proinflammatory cytokines TNF-α and IL-1β (Stoll et al., 2002; Myers et al., 2006). TNF-α and IL-1β can directly stimulate neuronal hyperexcitability of A and C fibers (Sorkin et al., 1997; Schafers and Sorkin, 2008) and promote neuroinflammation and activation of neuropathic cascades, in part through the induction of matrix metalloproteinases (MMPs) in denervated Schwann cells (Shubayev et al., 2006; Chattopadhyay et al., 2007).
MMPs are a family of zinc and calcium-dependent extracellular proteases, comprising collagenases, gelatinases, stromelysins and MT-MMPs, that remodel a plethora of extracellular and cell surface receptors, ligands, adhesion and structural proteins (Page-McCaw et al., 2007). MMPs contribute to neurodegenerative disorders via degradation of neurovascular barriers, facilitation of immune cell migration and demyelination (Redford et al., 1997; Hughes et al., 1998; Rosenberg, 2002; Yong, 2005; Kieseier et al., 2006). MMPs have been implicated in regulating neurovascular permeability and demyelination in patients with symptomatic neuropathy (Leppert et al., 1999; Mawrin et al., 2003; Renaud et al., 2003; Teles et al., 2007).
Our focus has been on identifying the mechanisms that initiate neuropathic cascades at the site of nerve injury. Of specific interest are the early-gene MMP family members, such as MMP-9, induced by over 100-fold within 24 h of nerve injury in denervated myelinated Schwann cells, preceding neuropathological evidence of degeneration (La Fleur et al., 1996; Kherif et al., 1998; Ferguson and Muir, 2000; Shubayev and Myers, 2000, 2002; Platt et al., 2003; Shubayev et al., 2006). Earlier mechanistic studies of MMP action in neuropathic pain have identified their critical importance to regulation of local and central cytokine networks during macrophage recruitment (Shubayev et al., 2006) and glial activation (Shubayev and Myers, 2002; Shubayev et al., 2006; Chattopadhyay et al., 2007; Kawasaki et al., 2008).
Facilitation of macrophage infiltration by MMPs is believed to contribute to their role in demyelination (Rosenberg, 2002; Kieseier et al., 2006). Interestingly, MMP-9 promotes macrophage recruitment into the injured nerve (Shubayev et al., 2006) without altering their ability to phagocytose myelin (Siebert et al., 2001), suggesting that the role of MMP-9 in peripheral demyelination (Kieseier et al.,1999, Kiefer et al., 2001, Siebert et al., 2001) is independent of macrophage recruitment. Indeed, MMPs promote demyelination through degradation of myelin basic protein (MBP) in the CNS (Chandler et al., 1995), and sciatic nerves of MMP-9 knockout (MMP-9−/−) mice preserve MBP at 10 days after crush (Chattopadhyay et al., 2007). Peripheral demyelination is a multi-stage process orchestrated by Schwann cells, resident and hematogenous macrophages (Stoll et al., 1989; Bruck, 1997; Myers et al., 2006).
We here tested whether MMPs contribute to the initial Schwann cell-mediated MBP degeneration associated with myelinated (mechanosensory Aβ) fibers and the development of mechanical nociception using L5 spinal nerve crush (L5 SNC), a model of robust mechanical allodynia. The accumulation of undegraded MBP during Schwann cell-derived demyelination was observed in nerves of MMP-9−/− mice. During rat L5 SNC, we identified MMP-9 as the main MBP-degrading protease to be induced in myelinating Schwann cells, and determined that daily i.p. therapy with broad-spectrum GM6001 (Ilomastat) resulted in immediate and sustained attenuation of mechanical allodynia and MBP preservation, prevented macrophage influx, spinal glial activation and promoted cell survival.
Results
Early induction in MBP-degrading MMP-9 after L5 SNC
MMP-2, -7 and -9 degrade MBP in vitro (Chandler et al., 1995). We analyzed the changes in their expression after rat L5 SNC. The focus was made on the early time-points (10 min–24 h), corresponding to Schwann cell-mediated demyelination (Stoll et al., 1989). To our knowledge, there have been no reports on MMP mRNA expression within hours after any type of nerve injury.
MMP-9 mRNA expression was induced 1 and 24 h after L5 SNC, whereas MMP-2 and MMP-7 were not significantly changed at either time-point (Fig.1, A). Gelatinolytic activity for MMP-2 (or gelatinase A, 72 kDa) and MMP-9 (or gelatinase B, 92 kDa) demonstrated undetectable MMP-9 in uninjured nerve and its significant increase 1 d after L5 SNC in the spinal nerve and the corresponding L5 DRG, but not control L4 DRG. Mild MMP-2 activity was constituently expressed in uninjured nerve and was not significantly induced at this time-point. No MMP activity was detected in the segmental (L4/5) spinal cord 1 d after L5 SNC.
Fig. 1.
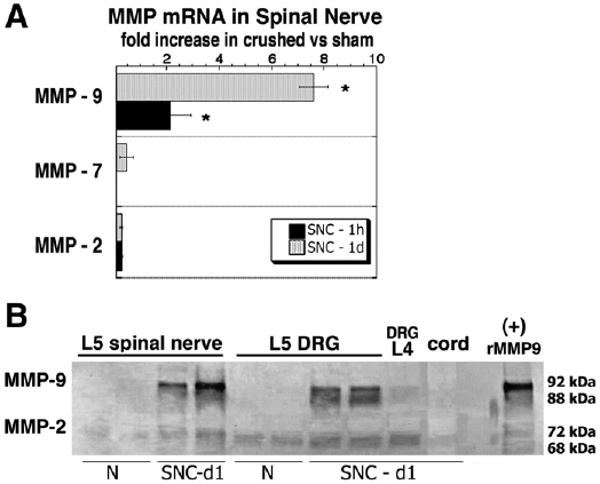
Immediate MMP induction after L5 spinal nerve crush. (A) Real-time Taqman qPCR for MBP-degrading MMPs after L5 spinal nerve crush. Data represents mean fold increase±SEM in crushed vs sham nerves of N=4/group; One-way ANOVA and Tukey–Kramer post-hoc test (*, p<0.05). (B) Gelatin zymography (inverted image) for MMP-9 (92 kDa pro-form, 88 kDa active) and MMP-2 (72 kDa pro-form, 68 kDa active) after L5 SNC. Increased MMP-9 activity in observed in spinal nerve and corresponding DRG at day 1 after crush (SNC-d1) in contrast to control normal (N) tissues. Constituent but low-level MMP-2 activity is detectable in all tissue. No activity is noted in the control L4 DRG or the segmental L5 spinal cord (representative duplicate samples of N=3/group). Recombinant human MMP-9 (10 ng) was used as positive control.
MMPi therapy and MMP-9−/− protect from MBP processing
MBP is a myelin protein expressed in peripheral nerve by myelinating Schwann cells (Scherer and Salzer, 2001). It is degraded by MMPs, producing products ranging from 25 to 6 kDa, with higher molecular weight MBP isoforms (25 and 21.5 kDa) corresponding to proteolytically unprocessed MBP (Chandler et al., 1995, 1996). The changes in the accumulation of unprocessed MBP were analyzed in spinal nerves of rats undergoing L5 SNC after systemic acute (10 μg/kg, i.p. for 1 day) and extended (10 μg/kg, i.p. daily for 12 days) GM6001 therapy, and in the spinal nerves of MMP-9−/− mice after 1 day of L5 SNC (Figs. 2, A–B). A statistically significant accumulation of unprocessed MBP was found one day after GM6001 therapy relative to vehicle and in MMP-9−/− compared to control wild-type nerves. Long-term GM6001 therapy produced even more protection from accumulation of MBP relative to vehicle at 12 days after L5 SNC. To control whether the finding was specific to MBP, we tested the levels of myelin protein zero (P0), the main peripheral myelin protein (Scherer and Salzer, 2001), demonstrating no significant change after MMPi therapy or MMP-9 gene deletion.
Fig. 2.
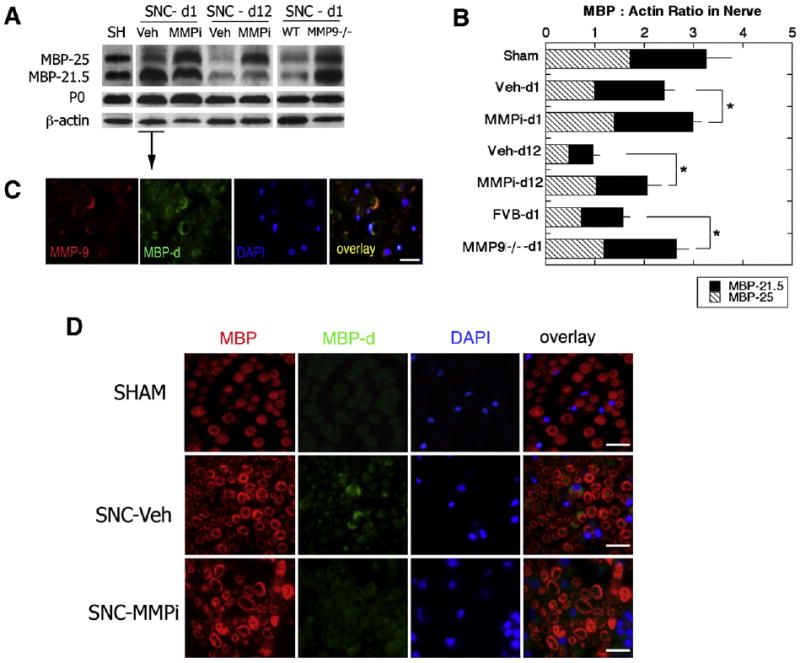
MBP protection in spinal nerve after GM6001 therapy (MMPi) and MMP-9 gene deletion (MMP-9−/−). (A) Undegraded MBP (25 and 21.5 kDa) and P0 (30 kDa) in spinal nerves after 1 day (SNC-d1) and 12 days (SNC-d12) of rat L5 SNC after daily GM6001 (MMPi) or vehicle (Veh) therapy, or MMP-9−/− or wild-type (WT) mice at 1 day after L5 SNC. β-actin was used to control protein loading. (B) Densitometry of A. representing mean MBP to β-actin ratios±SEM in MMPi vs vehicle or MMP-9−/− vs WT mice of N=4/group; Student's t-test (*, p<0.05). (C) Immunofluorescence for the degraded MBP (MBP-d, green), MMP-9 (red) and DAPI nuclear stain (blue) in SNC-d1 (vehicle-treated) spinal nerves demonstrate co-localization of MMP-9 with the degraded MBP in myelinating Schwann cells (crescent structures). (D) Immunofluorescence for total MBP (MBP, red), MBP-d (green) and DAPI (blue) demonstrate SNC-induced increase in MBP degradation in vehicle-treated nerves that was inhibited after MMPi therapy. C–D: scale bar= 20 μm.
Dual-immunofluorescence using an antibody against MMP-9, and specifically, degraded MBP, showed their co-localization in myelinated Schwann cells and myelin sheaths 1 day after rat L5 SNC (C). Myelinated Schwann cells were identified morphologically by the characteristic crescent structure and 1:1 axon to Schwann cell ratio. The levels of degraded MBP were increased after L5 SNC of vehicle-treated rats relative to sham but were reduced after GM6001 therapy (D). Immunoreactivity to total MBP was used for control (D).
MMP inhibition alleviates neuropathic behavior
Myelin integrity protects Aβ fibers from mechanical allodynia (Devor, 2006), particularly after spinal nerve injury (Liu et al., 2000). After L5 SNC, mechanical allodynia develops rapidly and lasts for weeks (Campana and Myers, 2003; Kobayashi et al., 2007). We here tested the effect of daily i.p. GM6001 therapy on mechanical withdrawal thresholds using the von Frey test after L5 SNC (Fig. 3). A robust increase in injury-induced mechanical allodynia was evident after vehicle treatment in the L5 SNC group (L5 SNC–Vehicle) relative to sham (SHAM–Vehicle). GM6001 therapy (L5 SNC–MMPi) elevated the withdrawal thresholds after its initiation relative to vehicle (L5 SNC–Vehicle) immediately and its effect maintained throughout the therapy. At 10–12 days, the thresholds of GM6001 therapy and sham became not statistically significant, indicating MMPi-induced recovery from mechanical allodynia.
Fig. 3.
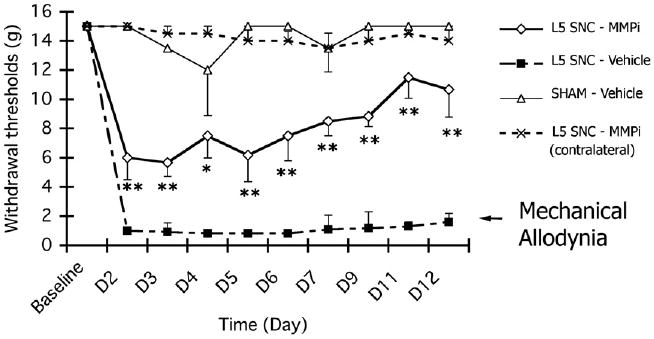
GM6001 therapy attenuates mechanical allodynia. von Frey mechanical withdrawal thresholds after L5 SNC or sham operation and daily, i.p. GM6001 (MMPi) or vehicle therapy. All animals received 3 consecutive days of baseline testing before operation and therapy. Thereafter, L5 SNC–Vehicle group displayed an immediate and robust mechanical allodynia, as indicated by withdrawal threshold of 0.4–2 g for the course of 12 days. In contrast, sham-operated, vehicle-treated group (SHAM–Vehicle) displayed no mechanical sensitivity to von Frey stimuli of below 8-15 g. GM6001 attenuated allodynia immediately after its administration (L5 SNC–MMPi) and at 10–12 days its values became statistically not significant from SHAM-vehicle group, pointing at complete attenuation of mechanical allodynia. Control withdrawal thresholds of L5 SNC–MMPi group in a contralateral, uninjured hindpaw displayed no mechanical allodynia. Data represents the mean withdrawal thresholds (gram force)±SEM of N=8/group. One-way ANOVA and Dunnett's post-hoc of MMPi vs. vehicle ipsilateral to L5 SNC (**, p<0.01 and *, p<0.05). Legends: -△-SHAM–Vehicle group, ipsilateral; -■-L5 SNC–Vehicle group, ipsilateral; -◇-L5 SNC–MMPi group, ipsilateral; -X-L5 SNC–MMPi group, contralateral.
MMP inhibition reduces spinal glia activation
Recent data implicated MMPs in the maintenance of neuropathic pain through activation of spinal cord astrocytes after spinal nerve ligation (Kawasaki et al., 2008). Astrocyte activation associated with neuropathic pain is reliably measured by increased GFAP levels in the ipsilateral dorsal horn of the spinal cord 3 days after L5 spinal nerve damage (Colburn et al., 1999). Thus, following daily i.p. GM6001 therapy for 3 days after L5 SNC, GFAP levels were measured in the ipsilateral dorsal horn by western blotting (Fig. 4, A) and immunofluorescence (Fig. 4, B). An L5 SNC-induced increase in GFAP levels was observed relative to sham, and its levels were reduced by 40% in GM6001– relative to vehicle-treated groups (A). Morphologically, astrocytes showed somatic hypertrophy characteristic of an activated phenotype in the SNC–Vehicle group compared to the dormant phenotype of sham (B). GM6001 therapy notably reversed somatic hypertrophy of astrocytes and their overall morphology resembled that of sham (SNC– MMPi, B). These data suggest that MMP action in neuropathic pain relates to induction of spinal glial activation associated with L5 SNC, which is consistent with the earlier report of spinal nerve ligation (Kawasaki et al., 2008).
Fig. 4.
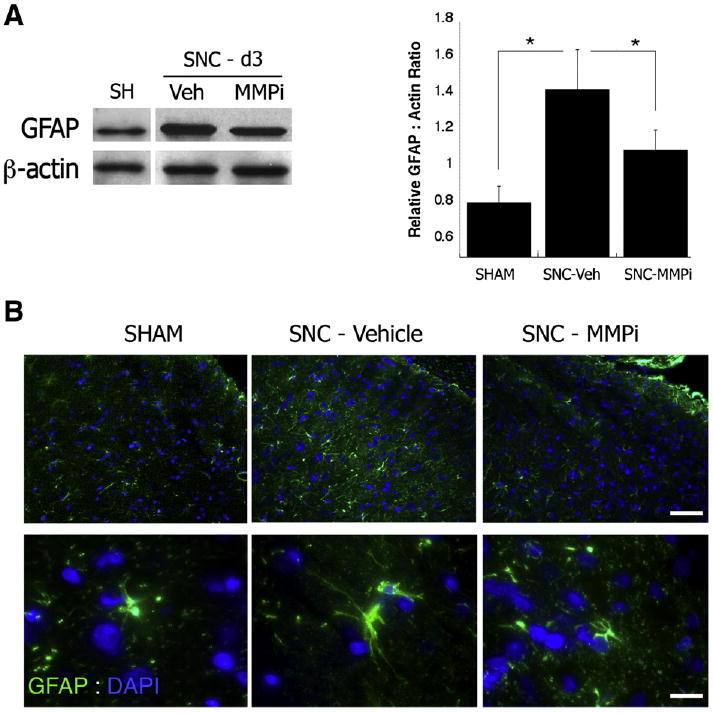
MMPi therapy suppresses spinal glial activation. (A) Western blot for GFAP (40 kDa) in the ipsilateral dorsal horn 3 days after L5 SNC. β-actin was used as loading control. The graph represents densitometry of the mean GFAP to β-actin ratios±SEM of N=3/group. Note a 40% decline in GFAP after daily i.p GM6001 therapy for 3 days (MMPi) relative to vehicle; Student's t-test (*, p<0.05). (B) Immunofluorescence for GFAP (green) and DAPI (blue) in ipsilateral dorsal horn spinal cord 3 days after L5 SNC, demonstrating somal hypertrophy of activated astrocytes caused by nerve injury in vehicle-treated cord, that was normalized after MMPi therapy. Scale bar: Top panel=100 μm, lower panel=20 μm.
MMP inhibition promotes cell survival
Nuclear DAPI stain that accompanied the MBP and GFAP immunofluorescence above showed a visible increase in cell content of the analyzed spinal nerve and ipsilateral dorsal horn. Because MMPi therapy inhibits immune cell infiltration associated with nerve injury (Kieseier et al., 2006; Shubayev et al., 2006), the data obtained in the nerve itself was initially surprising. Therefore, we conducted additional experiments, such as morphometric quantification of the DAPI-positive nuclear profiles in the spinal nerve, L5 DRG and ipsilateral dorsal horn (Fig. 5), and of the macrophage (CD68-immunoreactive) profiles in the spinal nerve (Fig. 6). Both analyses were done at 3 d after L5 SNC, the time of active macrophage infiltration into the nerve (Bruck, 1997), before and after daily i.p. GM6001 therapy.
Fig. 5.
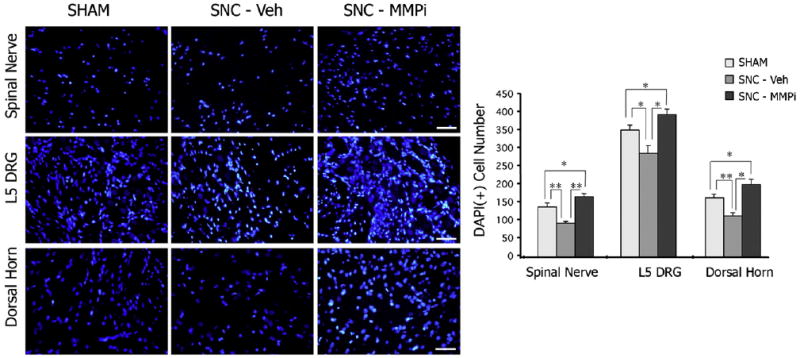
MMPi therapy increases DAPI counts. Nuclear DAPI stain (blue, A) in spinal nerve, DRG and ipsilateral dorsal horn spinal cord at 3 days after L5 SNC, and morphometry of DAPI-positive profiles (graph) after daily i.p. GM6001 (MMPi) or vehicle therapy for 3 days performed at objective magnification ×40. The mean±SEM of total of 16 fields in N=3/group; One-way ANOVA and Dunnett's post-hoc (*, p<0.05). Note that injury reduced the overall nuclear count and that MMPi therapy produced increase in DAPI-positive profiles in the nerve, DRG and spinal cord. Scale bar=80 μm.
Fig. 6.
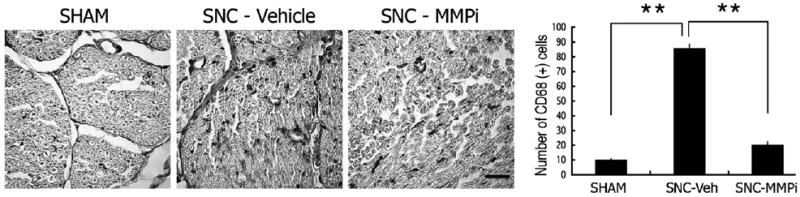
MMPi therapy reduced macrophage content. Immunohistochemistry for CD68 (macrophages, black) in spinal nerves 3 days after L5 SNC. Injury-induced increase in macrophage infiltration is seen in SNC-vehicle tissue compared to sham-operated nerves, which was inhibited after daily i.p. GM6001 therapy for 3 days (SNC-MMPi). Morphometry of CD68-positive immunohistochemical profiles (graph) represents the mean of N=3/group, 4 fields per N (graph)±SEM, demonstrating an 8-fold increase in macrophage numbers 3 days after L5 SNC in vehicle-treated tissue and only a 2-fold increase MMPi-treated tissue, corresponding to a 4-fold decline in macrophage number after MMPi therapy; Student's t-test (*, p<0.05). Scale bar=40 μm.
DAPI-positive nuclear profiles were significantly reduced in the injured spinal nerve, L5 DRG and ipsilateral dorsal horn after L5 SNC (SNC–Vehicle group) relative to sham (Fig. 5). Interestingly, GM6001 therapy (SNC–MMPi) universally increased cell content in each of the analyzed compartments relative to vehicle (SNC–Vehicle). About an 8-fold increase in infiltration of CD68-positive macrophages was observed in the L5 SNC–Vehicle group relative to sham (Fig. 6). GM6001 therapy (SNC–MMPi) demonstrated only a 2-fold increase relative to sham, corresponding to a statistically significant 4-fold decline in MMPi vs. vehicle treatment.
Because MMP inhibition protects neuronal cells from apoptosis (Gu et al., 2002, 2005; Manabe et al., 2005), we assessed the changes in the L5 SNC-induced apoptosis by caspase 3 immunoblotting after acute GM6001 therapy for one day (SNC-d1) or long-term daily i.p. therapy for 12 days (SNC-d12) after L5 SNC (Fig. 7). An injury-induced increase in caspase 3 was observed in nerve and DRG, and MMPi therapy significantly reduced the levels of caspase 3 relative to vehicle at both 1 and 12 days after L5 SNC. The changes in caspase 3 levels of the ipsilateral dorsal horn were not statistically significant after L5 SNC and GM6001 therapy at the analyzed time-points. Tissues from sham animals from 12 d after operation are used for densitometry measurement and are presented in the western blots.
Fig. 7.
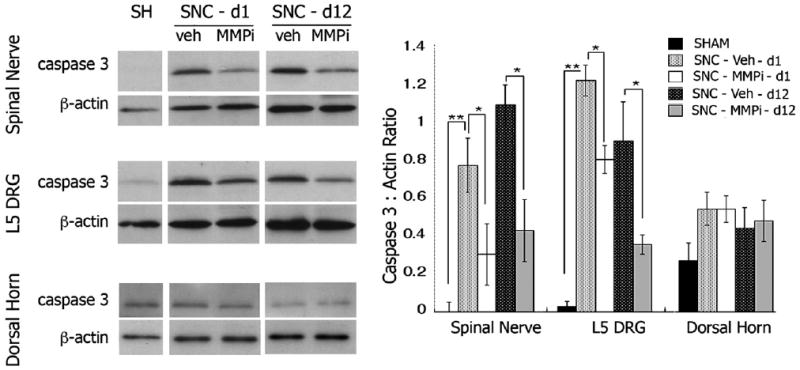
MMPi therapy protects from injury-induced apoptosis. Western blot for caspase 3 after L5 SNC and densitometry of mean caspase 3 to β-actin ratios±SEM in N=3/group (graph). MMPi therapy reduced caspase 3 levels in nerve and DRG at 1 and 12 days of daily administration; One-way ANOVA and Tukey–Kramer post-hoc (*, p<0.05).
Discussion
Myelin protects mechanosensory Aβ fibers, and myelin degradation promotes susceptibility of their axonal plasma membrane to nociceptive stimuli, leading to ectopic depolarization and mechanical allodynia (Devor, 2006). The involvement of Aβ fibers in the development of mechanical allodynia is particularly prominent after spinal nerve injury originating close to the neuronal soma of DRG (Liu et al., 2000), such as L5 SNC used here. Earlier studies on the mechanisms of MMP action in neuropathic pain have focused on their relationship with cytokines in promoting blood–nerve permeability (Myers et al., 2006) and central glial activation (Kawasaki et al., 2008).
We demonstrate that MMPs promote peripheral myelin damage through MBP degradation in a neuropathic model of L5 SNC, and that broad-spectrum MMP inhibition attenuates the initiation and the maintenance of mechanical allodynia. Our findings are summarized in Fig. 8. Although not in a model of neuropathic pain, MMP's role in MBP degradation of the CNS has been established (Chandler et al., 1995; Yong, 2005). Differential targeting of MBP and P0 by MMPs is a novel and intriguing finding, as the two proteins coordinate during myelin formation to regulate myelin thickness (Martini and Schachner, 1997), which is enhanced in crushed sciatic nerves after MMP-9 inhibition (Siebert et al., 2001). Although this study did not directly determine that retained myelin integrity reduced ectopic discharge, myelin protects from nociceptive hyperexcitibility, in part, by inhibition of sodium channel insertion into the axonal plasma membrane (Devor, 2006).
Fig. 8.
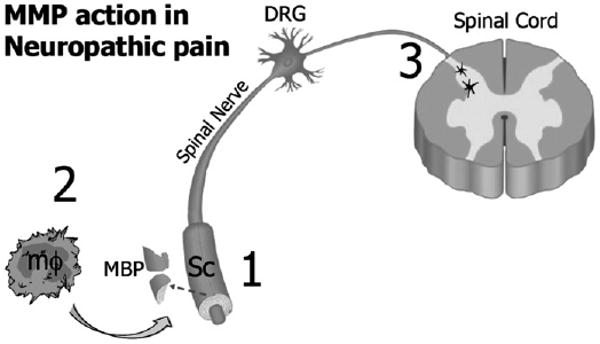
Model diagram of MMP action in peripheral demyelination and mechanical nociception after nerve injury: (1) Early-gene MMPs, such as MMP-9, are induced in myelinated Schwann cells (Sc) within 1 day after injury by the action of proinflammatory cytokines (Shubayev et al., 2006; Chattopadhyay et al., 2007). They selectively degrade myelin basic protein (MBP) in myelinated Sc at 1 day of injury, contributing to Sc-mediated demyelination (Fig. 2). Myelin degradation exposes axonal plasma membrane of mechanosensory myelinated Aβ fibers for ectopic hyperexcitability to Sc-derived nociceptive stimuli (Liu et al., 2000; Devor, 2006). (2) MMPs promote macrophage infiltration (Fig. 6) and (Shubayev et al., 2006), known to promote axonal demyelination, degeneration and neuropathic pain (Myers et al., 2006). In addition, MMPs directly contribute to the late-phase demyelination by processing MBP (Fig. 2). (3) MMPs promote activation of central glia, such as astrocytes, (Fig. 4) and (Kawasaki et al., 2008), however the exact mechanisms of this action is not yet understood. Astrocytic activation is required for the maintenance of the peripheral neuropathic pain (Wieseler-Frank et al., 2004). Thus, based on 1–3, broad-spectrum MMP inhibition is effective in attenuation of acute and persistent mechanical nociception (Fig. 3). Abbreviations: MMP – matrix metalloproteinase, Sc – Schwann cell, MBP – myelin basic protein, mf – macrophage, DRG – dorsal root ganglia.
In the course of peripheral nerve damage, myelin degradation is controlled by Schwann cells, resident and hematogenous macrophages (Myers et al., 2006). At 24 h after injury, when resident macrophages show no signs of activation and hematogenous macrophages have not yet infiltrated (Stoll et al., 1989; Bruck, 1997), Schwann cells are in charge of myelin degradation (Fernandez-Valle et al., 1995). We find this initial Schwann cell-mediated demyelination to be regulated by MMP-9 after L5 SNC. Specifically, induced before 24 h, MMP-9 co-localized with the degraded MBP in myelinating Schwann cells, and MMP-9 gene deletion preserved MBP from degradation 24 h after SNC. Ischemic brains of MMP-9−/− mice also show preserved MBP after 24 h of cerebral artery occlusion (Asahi et al., 2001). Augmented MBP levels in MMP-9−/− mice compared to GM6001 therapy may relate to the developmental role of MMP-9 in myelination (Larsen et al., 2006), or to species-related post-translational modification of MBP, such as degree of phosphorylation and myelin lipid binding, that determine MBP susceptibility to MMP digestion (D'Souza and Moscarello, 2006). Since sciatic nerves of MMP-9−/− mice accumulate MBP at later times (10 d) after crush (Chattopadhyay et al., 2007), it remains likely that MMP-9 contributes to the later stages of peripheral demyelination as well.
Lack of MMP-2 and MMP-7 expression at one day of L5 SNC does not discount the possibility that these and other MBP-degrading MMPs contribute to the sustained attenuation of mechanical allodynia and lasting MBP protection observed after long-term GM6001 therapy. In fact, the concept of distinct but complementary roles of the early- and late-gene MMPs in the development of neuropathic pain (Shubayev and Myers, 2002; Shubayev et al., 2006), as well as MMP activity pattern after other types of nerve injury (Shubayev and Myers, 2000; Hughes et al., 2002; Platt et al., 2003), are consistent with those reported here. Particularly in strong support for our data, MMP-9 has been correlated to the initiation of neuropathic pain, and MMP-2 to its maintenance, while intrathecal administration of either recombinant protein was sufficient to induce mechanical allodynia (Kawasaki et al., 2008).
Other studies have established the importance of MMPs in pain-related behavior. MT5-MMP−/− mice failed to develop mechanical allodynia after partial sciatic nerve ligation (Komori et al., 2004) and MMP-9−/− mice showed reduced spontaneous pain scores after sciatic nerve crush (Chattopadhyay et al., 2007). TAPI (an inhibitor to TACE, a member of metalloprotease family of ADAMs related to MMPs), attenuated mechanical allodynia and thermal hyperalgesia after chronic sciatic nerve constriction (Sommer et al., 1997). Hyperalgesia caused by intraplantar endotoxin declined with MMPi pre-treatment (Talhouk et al., 2000), implicating MMPs in the development of inflammatory pain, such as seen during arthritis. The anti-nociceptive action of minocycline has been linked to downregulation of cytokine and MMP expression (Ledeboer et al., 2005).
Importantly, GM6001 is specific to MMPs with broad-spectrum action to a range of family members at varying Ki (Fingleton, 2007), specified in the Experimental methods. The regimen of its administration used here has shown to be effective in reversing experimental autoimmune encephalomyelitis (Hartung and Kieseier, 2000), improving neurological function after spinal cord injury (Noble et al., 2002), reducing oxidative stress and brain edema during stroke (Wang and Tsirka, 2005), attenuating traumatic brain damage (Sifringer et al., 2007) and promoting survival from NMDA-induced death of retinal ganglion cells in vivo (Manabe et al., 2005).
An incidental finding was that GM6001 therapy increased cell content in the injured nerve and the associated DRG and spinal cord. Because MMPs regulate cell migration, death and proliferation (Page-McCaw et al., 2007), and each of these processes follow a distinct spatial and temporal pattern after L5 SNC, several potential mechanisms deserve consideration. In nerve, MMP-9 promotes macrophage recruitment and its inhibition results in reduced macrophage content (Shubayev et al., 2006). Thus, we here demonstrated the relevance of this mechanism in the L5 SNC after broad-spectrum MMPi therapy. Although the exact mechanisms of MMP-mediated macrophage recruitment remain to be identified, MMPs promote blood–nerve barrier permeability (Kieseier et al., 2006), focal adhesion remodeling of migrating immune cells, and their chemotaxis via chemokine release (Murphy and Gavrilovic, 1999; VanSaun and Matrisian, 2006). It is noteworthy that MMP-9 inhibition after sciatic nerve crush did not impact the ability of macrophages to phagocytize myelin (Siebert et al., 2001), consistent with our current finding of direct MMP-9 effect on myelin degradation rather than that consequent to macrophage infiltration phagocytotic activity.
In spite of the increased macrophage infiltration, the overall cell content after L5 SNC was reduced. Consistently, the augmented neuronal and glial loss after L5 SNC has been reported (Degn et al., 1999; Campana and Myers, 2003; Hammond et al., 2004) and is supported here by the finding of increased caspase 3 levels. GM6001-mediated reduction of caspase 3 and increase in cell content implicates it in protecting from injury-induced apoptosis. Indeed, MMPi therapy promotes survival of many neuronal phenotypes (Lee et al., 2004; Kauppinen and Swanson, 2005; Kim et al., 2005; Manabe et al., 2005), including primary DRG neurons (Nguyen et al., 2007) relevant to this study. Among the mechanisms of GM6001 and SB-3CT (specific MMP-9 and MMP-2 inhibitor) protection from neuronal apoptosis is inhibition of iNOS-mediated S-nitrosylation of pro-MMP-9 (Gu et al., 2002), preventing MMP-9-mediated laminin cleavage (Gu et al., 2005). While apoptosis is believed to contribute to the development of neuropathic pain after spinal nerve injury via replenishment of the proinflammatory cytokine milieu (Campana and Myers, 2003) and direct targeting of inhibitory spinal interneurons (Scholz et al., 2005), it is conceivable that MMPi effects on cell survival and nociception are regulated independently.
The central (spinal cord) effects of GM6001 therapy may relate to its systemic delivery or neuroplasticity of spinal nerve injury, such as caspase-mediated apoptosis (Scholz et al., 2005) and glial proliferation in the ipsilateral dorsal horn (Echeverry et al., 2008). GM6001 did not affect caspase 3 levels of dorsal horn, suggesting that the cell increase there is not related to suppression of apoptosis. MMPs inhibit glial proliferation by producing anti-mitotic fragments of fibronectin (Muir and Manthorpe, 1992), suggesting that MMP inhibition may promote glial survival.
In conclusion, MMPs emerge as modulators of neuropathic pain, and MMP-9 acts as the initiator of the neuropathic cascade (Shubayev and Myers, 2002; Kawasaki et al., 2008). In addition to the earlier identified mechanisms of MMP action in neuropathic pain through activation of the neuroimmune cytokine network at the injury site and in the central glia, we here found that MMPs degrade peripheral myelin MBP and established the value of broad-spectrum MMPi therapy in immediate and sustained mechanical nociception. Because the modalities of MMPs in damaged nerve are profound in that MMP gene deletion can virtually abolish axonal degeneration (Shubayev et al., 2006) and neuropathic pain (Komori et al., 2004; Kawasaki et al., 2008), we find particular importance in identifying both, specific and overlapping roles of individual MMPs.
Experimental methods
Reagents and antibodies
Antibodies for immunodetection: mouse anti-human MMP-9 (Calbiochem, 1:1000), mouse anti-human myelin basic protein (MBP, clone 22, AbD Serotec, 1:500), rabbit anti-degraded MBP (MBP-d, Chemicon, 1:1000), mouse anti-rat CD68 (clone ED1, AbD Serotec, 1:500), rabbit anti-myelin protein zero (P0, Protein Tech Group, IL, USA, 1:1000), mouse anti-glial fibrillary acidic protein (GFAP, Chemicon, 1:1000 for western blot), rabbit anti-GFAP (DAKO, Carpinteria, CA, 1:1000, for immunofluorescence), rabbit anti-caspase 3 antibody (Cell Signaling, 1: 1000), mouse anti-β-actin (Sigma, 1:10,000); 4′-6-Diamidino-2-phenylindole (DAPI, Molecular Probes, 1:20,000). Primers and Taqman probes were obtained for rat MMP-9 (Biosearch Technologies, Novato, CA), MMP-2 (Roche, Palo Alto, CA) and MMP-7 (ABI, New York, NY).
MMP inhibitor therapy
The broad-spectrum MMP inhibitor, GM6001 (Ilomastat, or N-[(2R)-2-(hydrox-amidocarbonylmethyl)-4-methylpentanoyl]-l-tryptophan methylamide, Chemicon, Temecula, CA) has reported Ki values of 0.4 nM for MMP-1, 27 nM for MMP-3, 0.5 nM for MMP-2, 0.1 nM for MMP-8, 0.2 nM for MMP-9. GM6001 or vehicle (dimethyl sulfoxide in ethanol) were prepared according to the manufacturer's instructions and administered at 10 mg/kg/day by intraperitoneal (i.p.) injection immediately after producing L5 SNC and then once daily thereafter, as identified in Table 1. GM6001 i.p. therapy at 10–100 mg/kg/day has been shown efficacious in numerous rodent models of neuronal degeneration (Gijbels et al., 1994; Hartung and Kieseier, 2000; Noble et al., 2002; Manabe et al., 2005; Wang and Tsirka, 2005; Sifringer et al., 2007).
Table 1.
Experimental schedule for MMPi therapy studies after rat L5 SNC
| 24h | |||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| −3d | −2d | −1d | 0 | 1h | 1d | 2d | 3d | 4d | 5d | 6d | 7d | 8d | 9d | 10d | 11d | 12d | |
| Surgery | * | ||||||||||||||||
| Therapy | * | * | * | * | * | * | * | * | * | * | * | * | * | * | |||
| von Frey test | * | * | * | * | * | * | * | * | * | * | |||||||
| Tissue harvest | * | * | * | * | * | ||||||||||||
Surgery (L5 SNC or sham) performed in rats is identified as point 0. Therapy (GM6001 or vehicle) was administered daily i.p. for the course of the study. von Frey test was performed for 3 days before surgery to record baseline mechanical withdrawal thresholds, and then every other day thereafter. The L5 spinal nerve, DRG and ipsilateral spinal cord dorsal horn tissues were harvested and analyzed at 0–24 h for the early MMP mRNA expression. The changes in cellular mechanisms (macrophage and DAPI nuclear cell counts and astrocytic activation) were evaluated at 3 d after surgery and/or therapy. Changes in target molecular mechanisms of MMP action (MBP, caspase 3) were evaluated after acute (1 day) and long-term (12 days) MMPi therapy.
Animals and surgeries
Adult female Sprague–Dawley rats (200–250 g, Harlan Labs), female MMP-9 knockout mice (MMP-9 −/−, FVB.Cg-Mmp9tm1Tvu/, Jackson Labs, Bar Harbor, ME) and control wild-type FVB/NJ mice (Jackson Labs) were housed in plastic cages in a temperature-controlled environment with 12:12 light:dark cycle and free access to food and water. Animals were anesthetized with 4% isofluorane (IsoSol; Vedco, St. Joseph, MO). The L5 spinal nerve crush (L5 SNC) was performed as we previously described (Campana and Myers, 2003; Kobayashi et al., 2007). Briefly, through a midline incision, the thoracolumbar fascia was incised just left of the spinous processes of the 5th and 6th lumbar (L5/6) vertebrae. The sacrospinalis muscle was gently moved laterally to expose the facet joint between L5 and L6 vertebrae. The L5 nerve root, DRG and spinal nerve were exposed by L5/6 facetectomy using great care to avoid trauma to the neurological tissue. The spinal L5 nerve 2 mm distal to the DRG was crushed once for 3 s using fine, smooth-surface forceps. Control animals underwent sham operation in which the spinal nerve was exposed but not crushed; naïve animals received no surgery. Animals were sacrificed at 1 h, 1 d, 3 d or 12 d after L5 SNC by an overdose of intraperitoneal rodent anesthesia cocktail containing sodium pentobarbital (Nembutal, 50 mg/ml; Abbott Labs, North Chicago, IL), diazepam (5 mg/ml, Steris Labs, Phoenix, AZ) and saline (0.9%, Steris Labs) in a volume proportion of 1:1:2, respectively, followed by intracardiac Euthasol (Virbac, Fort Worth, TX). All procedures were performed according to protocols approved by the VA Healthcare System Committee on Animal Research and the NIH Guidelines for Animal Use.
von Frey test for mechanical allodynia
Sensitivity to non-noxious mechanical stimuli was measured by the von Frey test (Chaplan et al., 1994), as we described previously (Igarashi et al., 2000; Schafers et al., 2003; Kobayashi et al., 2007). Briefly, rats were acclimated to being on a suspended 6-mm wire grid and the plantar surface of the hindpaw was stimulated within the spinal nerve innervation area using calibrated von Frey filaments (Stoelting, Wood Dale, IL). Stimuli were applied through the mesh, perpendicular to the mid-paw plantar surface for 4–6 s with buckling forces between 0.41–15.2 g force sequentially with ascending filament stiffness until paw withdrawal response, determined as the lowest gram force that provoked a paw withdrawal at least twice in 10 applications. Stimuli were separated by several seconds or until the animal was calm with both hindpaws placed on the grid. Testing was performed in both hindpaws for three days before L5 SNC or sham operation (baseline) and every other day thereafter, as specified in Table 1. Statistical analyses were done by one-way analyses of variance (ANOVA) and Dunnett's post-hoc test using SPSS 16.0 software (SPSS Inc, Chicago, IL).
Immunohistochemistry
Animals were perfused with fresh 4% paraformaldehyde in 0.2 M phosphate buffer and the tissues were removed, post-fixed, rinsed with phosphate buffer, followed by cryoprotection in 15–30% sucrose and frozen OCT embedding and cut into 10-μm-thick sections. For bright-light microscopy, endogenous peroxidase was blocked with 3% H2O2, followed by antigen retrieval (Dako, Carpinteria, CA) (5 min at 95 °C and 20 min at room temperature). Non-specific binding block with 5% normal serum from the animal that the secondary was made in, followed by primary antibody (above) incubation overnight at 4 °C, biotinylated secondary antibody against the primary antibody IgG (Vector) and avidin–biotin complex (ABC Elite, Vector) application. Sections were developed with 3,3′-diaminobenzidine (DAB, brown, Vector) and counterstained with methyl green (Fisher). For immunofluorescence, sections were immersed in 0.5% sodium borohydride followed by antigen retrieval and nonspecific binding block as described above, then primary antibody incubation overnight at 4 °C, goat anti-mouse 594 Alexa (Molecular Probes, 1:500, red) or goat anti-rabbit 488 Alexa (Molecular Probes, 1:500, green) for 1 h, and nuclear DAPI stain (Molecular Probes, 1:20,000, blue) for 5 min. Sections were mounted using Slowfade gold antifade reagent (Molecular Probes).
Morphometry
Immunoreactive and DAPI-positive profiles were quantified in the L5 spinal nerve at the crush site, ipsilateral L5 DRG and L4–L6 dorsal horn (lamina I and II) spinal cord. Four to eight areas were selected randomly per each animal in 3 animals per group by a blinded experimenter using Openlab 4.0 software (Improvision) followed by statistical analyses using SPSS 16.0 software.
Quantitative real-time PCR
Primers and Taqman probes were optimized as described (Shubayev et al., 2006). Nerve samples were stored in RNA-later (Ambion) at −20 °C, RNA was extracted with Trizol (Invitrogen), and treated with RNAse-free DNAse (Qiagen). The RNA purity was verified by OD260/280 absorption ratio of about 2.0. cDNA was synthesized using SuperScript first-strand RT-PCR kit (Invitrogen). Gene expression was measured by real-time qPCR (MX4000, Stratagene, La Jolla, CA) using 50 ng of cDNA and 2× Taqman Universal PCR Master Mix (Applied Biosystems) with a one-step program: 95 °C for 10 min, 95 °C for 30 s and 60 °C for 1 min for 50 cycles. Duplicate samples without cDNA (no-template control) for each gene showed no contaminating DNA. Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was used as a normalizer. Relative mRNA levels were quantified in four samples per group using the comparative Ct method (Livak and Schmittgen, 2001), and a fold change was determined by the MX4000 (Pfaffl, 2001).
Gelatin zymography
Nerve tissues were lysed in non-reducing, protease inhibitor-free Laemmli sample buffer (Biorad) and analyzed for gelatinolytic (MMP-2 and MMP-9) activity, as described (Shubayev and Myers, 2000; Shubayev et al., 2006). Samples were heated at 55 °C for 5 min and run on 10% SDS polyacrylamide gel containing 1 mg/ml of gelatin at 160 V for 90 min. The gels were washed in 2.5% Triton X-100, developed at 37 °C overnight in 50 mM Tris–HCl, 150 mM NaCl, 5 mM CaCl2, 1 μM ZnCl2, 0.2 mM sodium azide (pH 7.6) and stained with colloidal blue (Invitrogen), indicating gelatinolytic activity as a clear band on a dark background of undegraded gelatin. Inverted images are presented. Recombinant human MMP-9 (rMMP-9, Oncogene, 100 ng) was used as a positive control.
Western blotting
Neuronal tissues were snap frozen in liquid nitrogen and stored at −80 °C. Proteins were extracted using lysis buffer (50 mM Tris–HCl, pH 7.4, 1% NP 40, 150 mM NaCl, 1 mM EDTA, 1 mM PMSF, 1 μg/mL aprotinin and leupeptin, 1 mM sodium orthovanadate). Equal amounts of protein (30 to 50 μg, by Pierce BCA Protein Assay) corresponding to 10–30 μl of 1:50 tissue lysates were separated by SDS-PAGE using Laemmli system. Transfer to nitrocellulose membranes was done in Tris–Glycine transfer buffer (Invitrogen) at 200 mA for 1 h. The membranes were blocked with 5% non-fat milk (Biorad), incubated with primary antibodies in 5% BSA (see above) overnight at 4 °C, washed in TBS containing 0.05% Tween and incubated for 1 h at room temperature with HRP-conjugated anti-rabbit or anti-mouse secondary antibody (Cell Signaling; 1: 5000). The blots were developed using an enhanced chemiluminescence (ECL, Amersham).
Densitometry (optical density, OD) of the bands of interest was performed in 3–5 samples per group using NIH Image J 1.38 software, followed by statistical analyses using SPSS 16.0 software.
Acknowledgments
This work was supported by NIH/NINDS grant R21 NS060307-01 and the Department of Veterans Affairs Merit Review and Rehabilitation Research and Development Awards to Shubayev V.I.
Footnotes
Publisher's Disclaimer: This article appeared in a journal published by Elsevier. The attached copy is furnished to the author for internal non-commercial research and education use, including for instruction at the authors institution and sharing with colleagues.
Other uses, including reproduction and distribution, or selling or licensing copies, or posting to personal, institutional or third party websites are prohibited.
In most cases authors are permitted to post their version of the article (e.g. in Word or Tex form) to their personal website or institutional repository. Authors requiring further information regarding Elsevier's archiving and manuscript policies are encouraged to visit: http://www.elsevier.com/copyright
References
- Asahi M, Wang X, Mori T, Sumii T, Jung JC, Moskowitz MA, Fini ME, Lo EH. Effects of matrix metalloproteinase-9 gene knock-out on the proteolysis of blood–brain barrier and white matter components after cerebral ischemia. J Neurosci. 2001;21:7724–7732. doi: 10.1523/JNEUROSCI.21-19-07724.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruck W. The role of macrophages in Wallerian degeneration. Brain Pathol. 1997;7:741–752. doi: 10.1111/j.1750-3639.1997.tb01060.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campana WM, Myers RR. Exogenous erythropoietin protects against dorsal root ganglion apoptosis and pain following peripheral nerve injury. Eur J Neurosci. 2003;18:1497–1506. doi: 10.1046/j.1460-9568.2003.02875.x. [DOI] [PubMed] [Google Scholar]
- Chandler S, Coates R, Gearing A, Lury J, Wells G, Bone E. Matrix metalloproteinases degrade myelin basic protein. Neurosci Lett. 1995;201:223–226. doi: 10.1016/0304-3940(95)12173-0. [DOI] [PubMed] [Google Scholar]
- Chandler S, Cossins J, Lury J, Wells G. Macrophage metalloelastase degrades matrix and myelin proteins and processes a tumour necrosis factor-alpha fusion protein. Biochem Biophys Res Commun. 1996;228:421–429. doi: 10.1006/bbrc.1996.1677. [DOI] [PubMed] [Google Scholar]
- Chaplan SR, Bach FW, Pogrel JW, Chung JM, Yaksh TL. Quantitative assessment of tactile allodynia in the rat paw. J Neurosci Methods. 1994;53:55–63. doi: 10.1016/0165-0270(94)90144-9. [DOI] [PubMed] [Google Scholar]
- Chattopadhyay S, Myers RR, Janes J, Shubayev V. Cytokine regulation of MMP-9 in peripheral glia: implications for pathological processes and pain in injured nerve. Brain Behav Immun. 2007;21:561–568. doi: 10.1016/j.bbi.2006.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colburn RW, Rickman AJ, DeLeo JA. The effect of site and type of nerve injury on spinal glial activation and neuropathic pain behavior. Exp Neurol. 1999;157:289–304. doi: 10.1006/exnr.1999.7065. [DOI] [PubMed] [Google Scholar]
- D'Souza CA, Moscarello MA. Differences in susceptibility of MBP charge isomers to digestion by stromelysin-1 (MMP-3) and release of an immunodominant epitope. Neurochem Res. 2006;31:1045–1054. doi: 10.1007/s11064-006-9116-9. [DOI] [PubMed] [Google Scholar]
- Degn J, Tandrup T, Jakobsen J. Effect of nerve crush on perikaryal number and volume of neurons in adult rat dorsal root ganglion. J Comp Neurol. 1999;412:186–192. doi: 10.1002/(sici)1096-9861(19990913)412:1<186::aid-cne14>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- Devor M. Sodium channels and mechanisms of neuropathic pain. J Pain. 2006;7:S3–S12. doi: 10.1016/j.jpain.2005.09.006. [DOI] [PubMed] [Google Scholar]
- Echeverry S, Shi XQ, Zhang J. Characterization of cell proliferation in rat spinal cord following peripheral nerve injury and the relationship with neuropathic pain. Pain. 2008;135:37–47. doi: 10.1016/j.pain.2007.05.002. [DOI] [PubMed] [Google Scholar]
- Ferguson TA, Muir D. MMP-2 and MMP-9 increase the neurite-promoting potential of schwann cell basal laminae and are upregulated in degenerated nerve. Mol Cell Neurosci. 2000;16:157–167. doi: 10.1006/mcne.2000.0859. [DOI] [PubMed] [Google Scholar]
- Fernandez-Valle C, Bunge RP, Bunge MB. Schwann cells degrade myelin and proliferate in the absence of macrophages: evidence from in vitro studies of Wallerian degeneration. J Neurocytol. 1995;24:667–679. doi: 10.1007/BF01179817. [DOI] [PubMed] [Google Scholar]
- Fingleton B. Matrix metalloproteinases as valid clinical targets. Curr Pharm Des. 2007;13:333–346. doi: 10.2174/138161207779313551. [DOI] [PubMed] [Google Scholar]
- Gijbels K, Galardy RE, Steinman L. Reversal of experimental autoimmune encephalomyelitis with a hydroxamate inhibitor of matrix metalloproteases. J Clin Invest. 1994;94:2177–2182. doi: 10.1172/JCI117578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu Z, Kaul M, Yan B, Kridel SJ, Cui J, Strongin A, Smith JW, Liddington RC, Lipton SA. S-nitrosylation of matrix metalloproteinases: signaling pathway to neuronal cell death. Science. 2002;297:1186–1190. doi: 10.1126/science.1073634. [DOI] [PubMed] [Google Scholar]
- Gu Z, Cui J, Brown S, Fridman R, Mobashery S, Strongin AY, Lipton SA. A highly specific inhibitor of matrix metalloproteinase-9 rescues laminin from proteolysis and neurons from apoptosis in transient focal cerebral ischemia. J Neurosci. 2005;25:6401–6408. doi: 10.1523/JNEUROSCI.1563-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammond DL, Ackerman L, Holdsworth R, Elzey B. Effects of spinal nerve ligation on immunohistochemically identified neurons in the L4 and L5 dorsal root ganglia of the rat. J Comp Neurol. 2004;475:575–589. doi: 10.1002/cne.20209. [DOI] [PubMed] [Google Scholar]
- Hartung HP, Kieseier BC. The role of matrix metalloproteinases in autoimmune damage to the central and peripheral nervous system. J Neuroimmunol. 2000;107:140–147. doi: 10.1016/s0165-5728(00)00225-3. [DOI] [PubMed] [Google Scholar]
- Hughes PM, Wells GM, Clements JM, Gearing AJ, Redford EJ, Davies M, Smith KJ, Hughes RA, Brown MC, Miller KM. Matrix metalloproteinase expression during experimental autoimmune neuritis. Brain. 1998;121(Pt 3):481–494. doi: 10.1093/brain/121.3.481. [DOI] [PubMed] [Google Scholar]
- Hughes PM, Wells GM, Perry VH, Brown MC, Miller KM. Comparison of matrix metalloproteinase expression during Wallerian degeneration in the central and peripheral nervous systems. Neuroscience. 2002;113:273–287. doi: 10.1016/s0306-4522(02)00183-5. [DOI] [PubMed] [Google Scholar]
- Igarashi T, Kikuchi S, Shubayev V, Myers RR. 2000 Volvo Award winner in basic science studies: exogenous tumor necrosis factor-alpha mimics nucleus pulposus-induced neuropathology. Molecular, histologic, and behavioral comparisons in rats. Spine. 2000;25:2975–2980. doi: 10.1097/00007632-200012010-00003. [DOI] [PubMed] [Google Scholar]
- Kauppinen TM, Swanson RA. Poly(ADP-ribose) polymerase-1 promotes microglial activation, proliferation, and matrix metalloproteinase-9-mediated neuron death. J Immunol. 2005;174:2288–2296. doi: 10.4049/jimmunol.174.4.2288. [DOI] [PubMed] [Google Scholar]
- Kawasaki Y, Xu ZZ, Wang X, Park JY, Zhuang ZY, Tan PH, Gao YJ, Roy K, Corfas G, Lo EH, Ji RR. Distinct roles of matrix metalloproteases in the early- and late-phase development of neuropathic pain. Nat Med. 2008;14:331–336. doi: 10.1038/nm1723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kherif S, Dehaupas M, Lafuma C, Fardeau M, Alameddine HS. Matrix metalloproteinases MMP-2 and MMP-9 in denervated muscle and injured nerve. Neuropathol Appl Neurobiol. 1998;24:309–319. doi: 10.1046/j.1365-2990.1998.00118.x. [DOI] [PubMed] [Google Scholar]
- Kiefer R, Kieseier BC, Stoll G, Hartung HP. The role of macrophages in immune-mediated damage to the peripheral nervous system. Prog Neurobiol. 2001;64:109–127. doi: 10.1016/s0301-0082(00)00060-5. [DOI] [PubMed] [Google Scholar]
- Kieseier BC, Seifert T, Giovannoni G, Hartung HP. Matrix metalloproteinases in inflammatory demyelination: targets for treatment. Neurology. 1999;53:20–25. doi: 10.1212/wnl.53.1.20. [DOI] [PubMed] [Google Scholar]
- Kieseier BC, Hartung HP, Wiendl H. Immune circuitry in the peripheral nervous system. Curr Opin Neurol. 2006;19:437–445. doi: 10.1097/01.wco.0000245365.51823.72. [DOI] [PubMed] [Google Scholar]
- Kim YS, Kim SS, Cho JJ, Choi DH, Hwang O, Shin DH, Chun HS, Beal MF, Joh TH. Matrix metalloproteinase-3: a novel signaling proteinase from apoptotic neuronal cells that activates microglia. J Neurosci. 2005;25:3701–3711. doi: 10.1523/JNEUROSCI.4346-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi H, Kitamura T, Sekiguchi M, Homma MK, Kabuyama Y, Konno S, Kikuchi S, Homma Y. Involvement of EphB1 receptor/EphrinB2 ligand in neuropathic pain. Spine. 2007;32:1592–1598. doi: 10.1097/BRS.0b013e318074d46a. [DOI] [PubMed] [Google Scholar]
- Komori K, Nonaka T, Okada A, Kinoh H, Hayashita-Kinoh H, Yoshida N, Yana I, Seiki M. Absence of mechanical allodynia and Abeta-fiber sprouting after sciatic nerve injury in mice lacking membrane-type 5 matrix metalloproteinase. FEBS Lett. 2004;557:125–128. doi: 10.1016/s0014-5793(03)01458-3. [DOI] [PubMed] [Google Scholar]
- La Fleur M, Underwood JL, Rappolee DA, Werb Z. Basement membrane and repair of injury to peripheral nerve: defining a potential role for macrophages, matrix metalloproteinases, and tissue inhibitor of metalloproteinases-1. J Exp Med. 1996;184:2311–2326. doi: 10.1084/jem.184.6.2311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsen PH, DaSilva AG, Conant K, Yong VW. Myelin formation during development of the CNS is delayed in matrix metalloproteinase-9 and -12 null mice. J Neurosci. 2006;26:2207–2214. doi: 10.1523/JNEUROSCI.1880-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ledeboer A, Sloane EM, Milligan ED, Frank MG, Mahony JH, Maier SF, Watkins LR. Minocycline attenuates mechanical allodynia and proinflammatory cytokine expression in rat models of pain facilitation. Pain. 2005;115:71–83. doi: 10.1016/j.pain.2005.02.009. [DOI] [PubMed] [Google Scholar]
- Lee SR, Tsuji K, Lee SR, Lo EH. Role of matrix metalloproteinases in delayed neuronal damage after transient global cerebral ischemia. J Neurosci. 2004;24:671–678. doi: 10.1523/JNEUROSCI.4243-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leppert D, Hughes P, Huber S, Erne B, Grygar C, Said G, Miller KM, Steck AJ, Probst A, Fuhr P. Matrix metalloproteinase upregulation in chronic inflammatory demyelinating polyneuropathy and nonsystemic vasculitic neuropathy. Neurology. 1999;53:62–70. doi: 10.1212/wnl.53.1.62. [DOI] [PubMed] [Google Scholar]
- Liu CN, Wall PD, Ben-Dor E, Michaelis M, Amir R, Devor M. Tactile allodynia in the absence of C-fiber activation: altered firing properties of DRG neurons following spinal nerve injury. Pain. 2000;85:503–521. doi: 10.1016/S0304-3959(00)00251-7. [DOI] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- Manabe S, Gu Z, Lipton SA. Activation of matrix metalloproteinase-9 via neuronal nitric oxide synthase contributes to NMDA-induced retinal ganglion cell death. Invest Ophthalmol Vis Sci. 2005;46:4747–4753. doi: 10.1167/iovs.05-0128. [DOI] [PubMed] [Google Scholar]
- Martini R, Schachner M. Molecular bases of myelin formation as revealed by investigations on mice deficient in glial cell surface molecules. Glia. 1997;19:298–310. [PubMed] [Google Scholar]
- Mawrin C, Brunn A, Rocken C, Schroder JM. Peripheral neuropathy in systemic lupus erythematosus: pathomorphological features and distribution pattern of matrix metalloproteinases. Acta Neuropathol. 2003;105:365–372. doi: 10.1007/s00401-002-0653-2. Berl. [DOI] [PubMed] [Google Scholar]
- Muir D, Manthorpe M. Stromelysin generates a fibronectin fragment that inhibits Schwann cell proliferation. J Cell Biol. 1992;116:177–185. doi: 10.1083/jcb.116.1.177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy G, Gavrilovic J. Proteolysis and cell migration: creating a path? Curr Opin Cell Biol. 1999;11:614–621. doi: 10.1016/s0955-0674(99)00022-8. [DOI] [PubMed] [Google Scholar]
- Myers RR, Campana WM, Shubayev VI. The role of neuroinflammation in neuropathic pain: mechanisms and therapeutic targets. Drug Discov Today. 2006;11:8–20. doi: 10.1016/S1359-6446(05)03637-8. [DOI] [PubMed] [Google Scholar]
- Nguyen HX, O'Barr TJ, Anderson AJ. Polymorphonuclear leukocytes promote neurotoxicity through release of matrix metalloproteinases, reactive oxygen species, and TNF-alpha. J Neurochem. 2007;102:900–912. doi: 10.1111/j.1471-4159.2007.04643.x. [DOI] [PubMed] [Google Scholar]
- Noble LJ, Donovan F, Igarashi T, Goussev S, Werb Z. Matrix metalloproteinases limit functional recovery after spinal cord injury by modulation of early vascular events. J Neurosci. 2002;22:7526–7535. doi: 10.1523/JNEUROSCI.22-17-07526.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Page-McCaw A, Ewald AJ, Werb Z. Matrix metalloproteinases and the regulation of tissue remodelling. Nat Rev Mol Cell Biol. 2007;8:221–233. doi: 10.1038/nrm2125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfaffl MW. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001;29:e45. doi: 10.1093/nar/29.9.e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Platt CI, Krekoski CA, Ward RV, Edwards DR, Gavrilovic J. Extracellular matrix and matrix metalloproteinases in sciatic nerve. J Neurosci Res. 2003;74:417–429. doi: 10.1002/jnr.10783. [DOI] [PubMed] [Google Scholar]
- Redford EJ, Smith KJ, Gregson NA, Davies M, Hughes P, Gearing AJ, Miller K, Hughes RA. A combined inhibitor of matrix metalloproteinase activity and tumour necrosis factor-alpha processing attenuates experimental autoimmune neuritis. Brain. 1997;120(Pt 10):1895–1905. doi: 10.1093/brain/120.10.1895. [DOI] [PubMed] [Google Scholar]
- Renaud S, Erne B, Fuhr P, Said G, Lacroix C, Steck AJ, Leppert D. Matrix metalloproteinases-9 and -2 in secondary vasculitic neuropathies. Acta Neuropathol. 2003;105:37–42. doi: 10.1007/s00401-002-0607-8. Berl. [DOI] [PubMed] [Google Scholar]
- Rosenberg GA. Matrix metalloproteinases in neuroinflammation. Glia. 2002;39:279–291. doi: 10.1002/glia.10108. [DOI] [PubMed] [Google Scholar]
- Schafers M, Sorkin L. Effect of cytokines on neuronal excitability. Neurosci Lett. 2008;437:188–193. doi: 10.1016/j.neulet.2008.03.052. [DOI] [PubMed] [Google Scholar]
- Schafers M, Sorkin LS, Geis C, Shubayev VI. Spinal nerve ligation induces transient upregulation of tumor necrosis factor receptors 1 and 2 in injured and adjacent uninjured dorsal root ganglia in the rat. Neurosci Lett. 2003;347:179–182. doi: 10.1016/s0304-3940(03)00695-5. [DOI] [PubMed] [Google Scholar]
- Scherer SS, Salzer JL. Axon-Schwann cell interactions during peripheral nerve degeneration and regeneration. In: Jessen KR, Richardson WD, editors. Glial Cell Development. Oxford Univ. Press; Oxford: 2001. [Google Scholar]
- Scholz J, Broom DC, Youn DH, Mills CD, Kohno T, Suter MR, Moore KA, Decosterd I, Coggeshall RE, Woolf CJ. Blocking caspase activity prevents transsynaptic neuronal apoptosis and the loss of inhibition in lamina II of the dorsal horn after peripheral nerve injury. J Neurosci. 2005;25:7317–7323. doi: 10.1523/JNEUROSCI.1526-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shubayev VI, Myers RR. Upregulation and interaction of TNFalpha and gelatinases A and B in painful peripheral nerve injury. Brain Res. 2000;855:83–89. doi: 10.1016/s0006-8993(99)02321-5. [DOI] [PubMed] [Google Scholar]
- Shubayev VI, Myers RR. Endoneurial remodeling by TNFalpha- and TNFalpha-releasing proteases. A spatial and temporal co-localization study in painful neuropathy. J Peripher Nerv Syst. 2002;7:28–36. doi: 10.1046/j.1529-8027.2002.02003.x. [DOI] [PubMed] [Google Scholar]
- Shubayev VI, Angert M, Dolkas J, Campana WM, Palenscar K, Myers RR. TNFalpha-induced MMP-9 promotes macrophage recruitment into injured peripheral nerve. Mol Cell Neurosci. 2006;31:407–415. doi: 10.1016/j.mcn.2005.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siebert H, Dippel N, Mader M, Weber F, Bruck W. Matrix metalloproteinase expression and inhibition after sciatic nerve axotomy. J Neuropathol Exp Neurol. 2001;60:85–93. doi: 10.1093/jnen/60.1.85. [DOI] [PubMed] [Google Scholar]
- Sifringer M, Stefovska V, Zentner I, Hansen B, Stepulak A, Knaute C, Marzahn J, Ikonomidou C. The role of matrix metalloproteinases in infant traumatic brain injury. Neurobiol Dis. 2007;25:526–535. doi: 10.1016/j.nbd.2006.10.019. [DOI] [PubMed] [Google Scholar]
- Sommer C, Schmidt C, George A, Toyka KV. A metalloprotease-inhibitor reduces pain associated behavior in mice with experimental neuropathy. Neurosci Lett. 1997;237:45–48. doi: 10.1016/s0304-3940(97)00813-6. [DOI] [PubMed] [Google Scholar]
- Sorkin LS, Xiao WH, Wagner R, Myers RR. Tumour necrosis factor-alpha induces ectopic activity in nociceptive primary afferent fibres. Neuroscience. 1997;81:255–262. doi: 10.1016/s0306-4522(97)00147-4. [DOI] [PubMed] [Google Scholar]
- Stoll G, Griffin JW, Li CY, Trapp BD. Wallerian degeneration in the peripheral nervous system: participation of both Schwann cells and macrophages in myelin degradation. J Neurocytol. 1989;18:671–683. doi: 10.1007/BF01187086. [DOI] [PubMed] [Google Scholar]
- Stoll G, Jander S, Myers RR. Degeneration and regeneration of the peripheral nervous system: from Augustus Waller's observations to neuroinflammation. J Peripher Nerv Syst. 2002;7:13–27. doi: 10.1046/j.1529-8027.2002.02002.x. [DOI] [PubMed] [Google Scholar]
- Talhouk RS, Hajjar L, Abou-Gergi R, Simaa'n CJ, Mouneimne G, Saade NE, Safieh-Garabedian B. Functional interplay between gelatinases and hyperalgesia in endotoxin-induced localized inflammatory pain. Pain. 2000;84:397–405. doi: 10.1016/s0304-3959(99)00238-9. [DOI] [PubMed] [Google Scholar]
- Teles RM, Antunes SL, Jardim MR, Oliveira AL, Nery JA, Sales AM, Sampaio EP, Shubayev V, Sarno EN. Expression of metalloproteinases (MMP-2, MMP-9, and TACE) and TNF-alpha in the nerves of leprosy patients. J Peripher Nerv Syst. 2007;12:195–204. doi: 10.1111/j.1529-8027.2007.00139.x. [DOI] [PubMed] [Google Scholar]
- VanSaun MN, Matrisian LM. Matrix metalloproteinases and cellular motility in development and disease. Birth Defects Res C Embryo Today. 2006;78:69–79. doi: 10.1002/bdrc.20061. [DOI] [PubMed] [Google Scholar]
- Wang J, Tsirka SE. Neuroprotection by inhibition of matrix metalloproteinases in a mouse model of intracerebral haemorrhage. Brain. 2005;128:1622–1633. doi: 10.1093/brain/awh489. [DOI] [PubMed] [Google Scholar]
- Wieseler-Frank J, Maier SF, Watkins LR. Glial activation and pathological pain. Neurochem Int. 2004;45:389–395. doi: 10.1016/j.neuint.2003.09.009. [DOI] [PubMed] [Google Scholar]
- Yong VW. Metalloproteinases: mediators of pathology and regeneration in the CNS. Nat Rev Neurosci. 2005;6:931–944. doi: 10.1038/nrn1807. [DOI] [PubMed] [Google Scholar]