

Abstract
Growing appreciation for astrocytes as active participants in nervous system development, neurovascular metabolic coupling, and neurological disease progression has stimulated recent investigation into specific astrocyte-secreted proteins that may mediate these functions. The current work utilized SILAC-generated isotope reference proteomes to quantify relative protein abundances between the astrocyte proteome and secretome. Multidimensional GeLC-MS/MS analysis of astrocyte conditioned media and cell lysates resulted in the relative quantification of 516 proteins, 92 of which were greater than 1.5-fold enriched in astrocyte-conditioned media (ACM). Eighty of the ACM-enriched proteins had N-terminal signal peptides, comprising well known classically secreted proteins, such as apolipoprotein E and SPARC, and several cathepsins that localize to endosomal/lysosomal compartments. The remaining twelve ACM-enriched proteins, such as vimentin, ferritins, and histones, lacked N-terminal signal peptides. Also, 47 proteins contained predicted N-terminal signal peptides but were not enriched in ACM (< 1.5-fold), 25 of which were localized to ER, Golgi, or mitochondria membrane-bound compartments. Overall, by combining quantitative proteomics with subcellular localization prediction, an informative description of protein distribution can be obtained, providing insights into protein secretion.
Keywords: astrocyte, protein secretion, nonconventional secretion, mass spectrometry, SILAC, secretory lysosomes
Introduction
For intact tissues and whole organisms, cells rarely operate autonomously, but rather in concert with and in response to the cellular physiology of their neighbors. While these responses can be generated by direct cell-cell coupling, such as the propagation of calcium waves through gap junctions in astrocytic cellular networks1, other responses are elicited as a result of secreted biomolecules. Stimulus-coupled neurotransmitter release is the prototypic intercellular signal in the brain, responsible for initiating activity-dependent synapse formation during development2 as well as synaptic remodeling after learning3. In addition, there is growing appreciation for the roles that secreted proteins play in nervous system function, particularly in distinct developmental stages or disease states4–10. However, understanding of the molecular pathways underlying protein secretion and subsequent initiation of intercellular signaling events is incomplete.
Recent studies suggest that astrocyte protein secretion may subserve a host of critical functions within the nervous system, including synapse formation and trophic support during development4, adult neurogenesis11, and immune response12, 13. In particular, in vitro and in vivo synapse formation was promoted by thrombospondins secreted by immature, but not mature astrocytes4. While astrocytes provide trophic, pro-survival support to neurons14, under certain cellular or physiological states such as those associated with disease, astrocytes can shift to a highly “reactive” phenotype15. Under these conditions, astrocytes secret pro-inflammatory mediators, such as cytokines and chemokines12, 13 resulting in increased levels of extracellular excitatory amino acids, such as glutamate, which may significantly impair neuronal survival16, 17. Despite the current knowledge of astrocyte-secreted proteins, prediction of classically secreted proteins has estimated that the mouse secretome may consistent of over one thousand proteins18, suggesting that most cellular secretomes are largely uncharacterized.
Towards a greater understanding of astrocyte protein secretion, several recent studies have investigated the astrocyte secretome using conditioned cell culture medium. These studies have identified several hundred proteins, including classically secreted and nonconventionally secreted proteins as well as cytosolic proteins19–21. Although the identification of classically secreted proteins can be supported by signal peptide prediction algorithms, the identification of novel, nonconventionally secreted proteins is more challenging, due in part to their poorly understood secretion mechanisms and lack of extensive training datasets for prediction algorithms22. A proteome-wide quantitative mass spectrometry-based approach to assess extracellular protein enrichment would not only facilitate the identification of proteins secreted by these alternative mechanisms, but also enable differentiation between secreted proteins and cytosolic contaminants.
This concept was recently demonstrated for the astrocyte secretome using proteomic approaches based on label-free, spectral counting analyses21, 23. In these studies, an enrichment index was defined for each protein, normalizing the extracellular abundance to intracellular steady-state expression level. Since label-free techniques based on protein spectral counts often have a limited dynamic range, metabolic/isotope labeling techniques can also be used as a viable alternative in some model systems. These strategies provide more accurate quantification at low signal-to-noise and reduce errors introduced during sample preparation prior to mass spectrometry analysis24, 25. For example, a quantitative proteomics approach was employed to study astrocyte protein secretion by differential amidination of lysine residues using isotope-coded S-methyl thioimidate reagents26. Yet, metabolic stable isotope labeling strategies such as SILAC have not been demonstrated in primary astrocytes. SILAC is now routinely used in transformed cell lines to assess relative changes in protein expression as a function of temporal and stimulus-dependent variables27. More recently, SILAC has been demonstrated in non-transformed cells, such as embryonic stem cells28 and primary neurons29. In the current work, we performed SILAC in primary astrocyte cultures, achieving at least 98 % incorporation of heavy isotope labeling in the astrocyte proteome and secretome. Generation of isotope-labeled reference proteomes permitted the evaluation of astrocyte protein secretory pathways by quantitative analysis of protein abundance within and between the astrocyte secretome and intracellular proteome (see Fig. 1).
Figure 1.
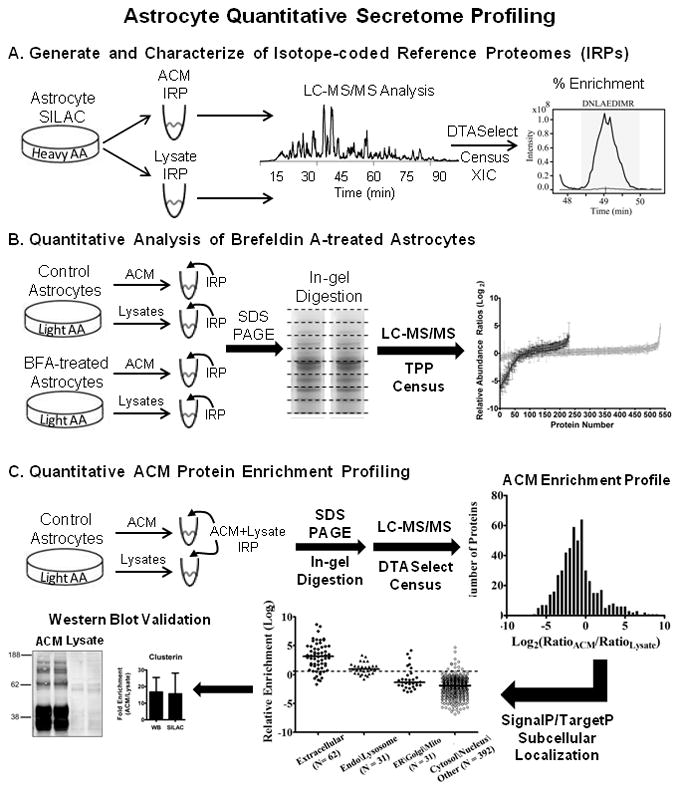
Experimental design and computational analysis of astrocyte protein secretion. (A) Metabolic labeling of proteins by isotope-enriched amino acids (heavy AA) was performed in primary mouse cortical astrocytes cultures by a SILAC-based strategy. After culturing for 20 days in vitro, astrocyte-conditioned media (ACM) and cellular lysates were collected. Protein aliquots were digested in-solution with trypsin and analyzed by LC-MS/MS. Peptide-spectrum matching was performed by SEQUEST/DTASelect, followed by automated reconstruction of extracted ion chromatograms (XIC) and filtering of SILAC peptide/protein ratios by Census quantitative analysis software. Isotope-coded reference proteomes (IRPs) had light-to-heavy (L/H) peptide ratios of ≤ 0.02, corresponding to an enrichment of at least 98 percent. (B) In separate experiments, control and brefeldin A-treated astrocytes were cultured with natural isotope abundance amino acids (light AA). After collection of ACM and cellular lysates, respective IRPs were mixed in known amounts with each sample and analyzed by multidimensional GeLC-MS/MS (SDS-PAGE/In-gel digestion/LC-MS-MS). MS and MS/MS data were analyzed by the TransProteomic Pipeline (TPP) and Census as above to calculate L/H protein ratios. Respective protein ratios calculated for BFA samples were normalized to control protein ratios. (C) A complementary approach to assess the extent of protein secretion was performed by quantifying the relative enrichment of protein in ACM versus cellular lysates. Control ACM and cellular lysates were collected and mixed with a “whole cell” ACM+Lysate IRP. Samples were analyzed by GeLC-MS/MS and the DTASelect/Census quantitative analysis workflow to assemble an ACM protein enrichment profile. Quantified proteins and their respective ratios were evaluated by SignalP/TargetP subcellular localization prediction algorithms. Western blot and LC-MRM-MS/MS analysis (not shown) were also used to independently validate SILAC L/H protein ratios.
Experimental Methods
Chemicals and Reagents
All reagents were purchased from Sigma-Aldrich (St. Louis, MO) unless otherwise stated. Custom Minimal Essential Media (MEM) lacking natural abundance L-leucine and L-lysine was purchased from AthenaES (Baltimore, MD). [13C6-15N2]-lysine and [13C6-15N1]-leucine were purchased from Cambridge Isotope Laboratories (Boston, MA). Primary antibodies used were: rabbit anti-apolipoprotein E (1:2000; Meridian Life Science, Inc, Saco, ME), goat anti-clusterin (1:2000; R&D Systems, Inc., Minneapolis, MN), rabbit anti-cyclophilin B (1:300; Abcam, Cambridge, MA), rabbit anti-GAPDH (1:10,000; Abcam, Cambridge, MA), goat anti-MIF (1:2000; Abcam, Cambridge, MA), SPARC (1:2000; Abcam, Cambridge, MA), and goat anti-alpha-tubulin (1:100; Santa Cruz Biotechnology, Inc., Santa Cruz, CA).
Astrocyte culture and media conditioning
Cortical astrocyte cultures were prepared from neonatal CD-1 mice (Charles River, Wilmington, MA) on postnatal day 1 as previously described19 but with minor modifications. Neonatal cortices were triturated in Minimal Essential Media (Invitrogen, Carlsbad, CA) supplemented with 10% fetal bovine serum (Hyclone), sodium pyruvate (1 mM), L-glutamine (2 mM), D-glucose (42 mM), sodium bicarbonate (14 mM), penicillin (100 U/ml), streptomycin (100μg/ml), fungizone (2.5 μg/ml) and plated at 3 cortices per T-75 vent-cap flask (Corning, Corning, NY). Mixed cortical cultures were raised for 10 days in 37°C and 5% CO2 with media change every 3–4 days. Cultures were then washed with cold EBSS and separated from neurons and microglia by shaking overnight at 37°C. Adherent cells were trypsinized (0.25%) and seeded in 100 mm Petri dishes (Corning) at 5 × 106 cells/plate (5 ml). Forty-two hours after plating, cells were washed three times with EBSS and then with serum-free media for 6 hours. Washing media was replaced with fresh serum-free media containing 1 μg/ml of brefeldin A or DMSO vehicle. Cell viability was quantified by trypan blue exclusion. For brefeldin A treatments, astrocyte-conditioned media (ACM) was collected after 24 hours, while all other experiments were conducted for 7 days. ACM was pooled between three culture dishes (15 mL) and centrifuged at 500 × g for 5 min to remove cell debris. The protein fraction (> 3 kDa) was obtained by 30-fold concentration by ultrafiltration of ACM at 4°C using CentriPrep Ultracel YM-3 filters (Millipore, Billerica, MA). Filtrates were then adjusted with protease inhibitors, 2μg/ml aprotinin, 3.3 μg/ml bestatin, 3.3 μg/ml E-64, and aliquoted and stored at −80 °C. Cell pellets were lysed by homogenization in 50 mM HEPES-NaOH, pH 7.2, containing 50 mM NaCl, 1 mM EDTA, 1% Triton-X100, and protease inhibitors (as above), incubated on ice for 20 min, and centrifuged at 20,000 × g for 20 min at 4 °C. Protein concentration of conditioned media and soluble lysates was determined by the Bradford method.
Astrocyte Stable Isotope Labeling by Amino Acids in Cell Culture (SILAC)
SILAC labeling of primary astrocytes was used to generate isotope reference proteomes for both intracellular and extracellular proteomes. Astrocytes were cultured by the protocol described above, except for the modification of culture medium and culture flasks. Minimal essential media devoid of natural abundance lysine and leucine was supplemented as above and also repleted with the respective isotope-labeled amino acid analogs. In preparation for media conditioning, enriched astrocytes were seeded in T-175 flasks at 12.7 × 106 cells/plate (20 mL) in serum-containing heavy isotope medium. Forty-eight hours later, serum was withdrawn as described above, and media conditioning was performed for 7 days. Collection and processing of media and cells were performed as described above, except ACM was concentrated about 300-fold. From this protocol, one can expect about 1.5 mg of heavy labeled ACM and 25 mg of heavy labeled cell lysates. IRPs were spiked into non-labeled (light) samples at a nominal protein ratio of 1:1 or 2:3 (light:heavy). Given a single GeLC-MS/MS analysis was performed with between 50 and 100 μg of total protein, 1.5 mg of heavy labeled ACM is sufficient for about 25 to 50 experiments.
GeLC-MS/MS analysis
The protein fraction obtained from ACM was analyzed by GeLC-MS/MS as described previously30 with modification. Concentrated ACM or cell lysates were spiked with appropriate isotope reference proteomes (total of 20 – 50 μg), mixed with 6X LDS sample buffer, and resolved on NuPAGE 10% Bis-Tris gels (Invitrogen, Carlsbad, CA) by electrophoresis in MOPS running buffer until the dye front reached either 1.6 or 3.2 cm. Proteins were visualized by Colloidal blue (Invitrogen, Carlsbad, CA) and each lane was cut into uniform (2 mm) slices using a MEF-1.5 Gel Cutter (The Gel Company, San Francisco, CA). Individual gel slices were cut into 1 × 1 mm cubes and digested in-gel with trypsin as previously described31. Tryptic digests were analyzed on either an LTQ or hybrid LTQ-Orbitrap mass spectrometer (Thermofisher Scientific, San Jose, CA) coupled with a NanoLC pump (Eksigent Technologies, Livermore, CA) and autosampler. Tryptic peptides were separated by reverse phase (RP)-HPLC on a nanocapillary column, 75 μm id × 20 cm ProteoPep (New Objective, Woburn, MA, USA). Mobile phase A consisted of 1% methanol/0.1% formic acid and mobile phase B of 1% methanol/0.1% formic acid/79% acetonitrile. Peptides were eluted into the mass spectrometer at 300 nL/min with each RP-LC run comprising a 15 min sample load at 3 % B and a 90 min linear gradient from 5 to 45 % B. The mass spectrometer was set to repetitively scan m/z from 300 to 1700 (R = 60,000 for LTQ-Orbitrap) followed by data-dependent MS/MS scans on the five most abundant ions, with a minimum signal of 1000, isolation width of 2.0, normalized collision energy of 28, and waveform injection and dynamic exclusion enabled. FTMS full scan AGC target value was 1e6, while MSn AGC was 5e3, respectively. FTMS full scan maximum fill time was 500 ms, while ion trap MSn fill time was 50 ms; microscans were set at one. A hybrid LTQ-Orbitrap was utilized for all SILAC experiments with additional instrument parameters as follows: a reject mass list containing 272 fully tryptic peptide m/z values from bovine serum albumin; a 5 ppm reject mass width; FT preview mode; charge state screening, and monoisotopic precursor selection were all enabled with rejection of unassigned and 1+ charge states.
Protein identification and validation
Srf files were generated from MS/MS spectra extracted from the RAW data file by Extract_msn (Bioworks 3.3.1; intensity threshold of 1000; minimum ion count 30). Srf files generated from LC-MS/MS runs belonging to the same biological samples were submitted to Sorcerer-SEQUEST (ver. 4.0.3, rev 11; SagenResearch, San Jose, CA). Database searching was performed against a UniProt database (Release 14.6) containing Mus musculus sequences from SwissProt and TrEMBL plus common contaminants, which were then reversed and appended to the forward sequences (121,248 total sequences). The database was indexed with the following parameters: mass range of 600 – 4200 Da, semi-tryptic cleavages with a maximum of 2 missed cleavage sites and static modification of cysteine by S-carbamidomethylation (+57 amu). Database searching was performed with the following parameters: precursor tolerance, 30 ppm; fragment tolerance, 1.0 amu; variable modification of methionine (+16 amu), and for SILAC experiments, variable modification of leucine (+7.017 amu) and lysine (+8.014 amu). The maximum number of variable modifications per instance and per peptide was 3 and 4, respectively. 13C isotope mass check was enabled. SEQUEST sequence-to-spectrum assignments were analyzed by Scaffold (Proteome Software, Portland, OR), the TransProteomic Pipeline (TPP ver. 4.0.2), or DTASelect (ver. 2.0). For all analyses, reverse database hits were used to control error rate at the peptide and protein level to less than 1 %. The following command-line parameters were used for modeling and filtering of sequence-to-spectrum assignments by DTASelect: -s 100 -d 0.1 -Smn 7 -e CON_ --iso --sp --mass --ionstat --trypstat. Peptides were assembled into protein groups to satisfy rules of parsimony requiring at least two unique peptides.
Census quantitative isotope labeling analysis
The isotope labeling feature of Census (ver. 1.44) was used for automated computation and filtering of extracted ion chromatograms and peptide ratios, respectively32. Individual isotope extraction of SILAC pairs was performed with a mass tolerance of 30 ppm and atom percent excess of 0.98. Default values for filtering of peptide ratios were used, except an outlier p-value of 0.2 was selected. Proteins with 2 or more unique, quantified spectra were retained. Protein ratio calculation was repeated for additional sample sets and normalized ratios were obtained by dividing the two experimental conditions to obtain relative fold differences in protein abundance. These normalized protein ratios were utilized for downstream functional and statistical analyses.
LC-MRM-MS/MS analysis of SILAC pairs
This approach was used to determine the degree of isotope incorporation into the astrocyte proteome and to validate SILAC ratios obtained by proteome-wide SILAC quantification. Data-independent analysis of light and heavy isotope-labeled peptides was conducted using a “pseudo-MRM” approach on an LTQ XL mass spectrometer (Thermofisher Scientific, San Jose, CA). The RP-LC gradient was identical to the method described above. The mass spectrometer was set to repetitively perform data-independent MS2 acquisition on specific light and heavy-labeled SILAC precursor masses selected empirically based on MS/MS spectra acquired in data-dependent experiments, which allowed the selection of optimal precursor-product ion transitions (e.g. prominent y-ions at an m/z greater than the precursor, which often represented N-terminal to Pro or C-terminal to Asp/Glu fragment). Between 4 and 8 precursor masses (2 – 4 SILAC pairs) were monitored in a single segment, with an isolation width of 3.0, normalized collision energy of 28, 2 microscans, and accumulation time and target value of 100 ms and 5e3, respectively. Xcalibur was used to reconstruct precursor-product ion chromatograms. SILAC peptide pair ratios were computed by area under the curve (AUC) measurements. MRM protein ratios were calculated as the average of four SILAC peptide ratios (two precursors per protein × two product ions per precursor).
Western blot analysis
Protein concentration of ACM and cellular lysates were measured using the Bradford reagent (Bio-rad, Hercules, CA). Thirty micrograms of ACM or cellular lysates from two independent biological replicates were denatured in LDS (6X) sample buffer by heating at 70 °C for 10 min, then separated by 1D SDS-PAGE. Proteins were transferred to PVDF membranes (Millipore, Billerica, MA) at 20V overnight at 25 °C and then visualized with Ponceau S to verify equal protein loading. Ponceau S was destained briefly with 0.01 N NaOH, rinsed with water, and then incubated in blocking buffer (BBT; Odyssey blocking buffer mixed 1:1 with TBS) for 1 hour at 25 °C. Membranes were then incubated with primary antibody (see Chemicals and Reagents) in blocking buffer adjusted to contain 0.1% Tween-20 (BBT-t) for 1 hour at 25 °C. Membranes were then washed in TBS containing 0.1% Tween-20 (TBS-t) and incubated with appropriate secondary antibodies conjugated to Alexa Fluor 680 or Alexa Fluor 800 (1:15000, Invitrogen, Carlsbad, CA) for 1 hour in BBT-t. Membranes were washed in TBS-t and visualized using the Odyssey Infrared Imaging system (Licor Biosciences, Lincoln, NE). Quantification was performed by densitometry using the Odyssey system software (ver. 2.1).
Computational and functional gene ontology analysis
For computational analysis of classically secreted proteins, FASTA sequences were submitted to SignalP33 to predict proteins containing signal peptides, followed by TargetP34, 35 to predict the localization (Extracellular, Mitochondria, Other). For prediction of nonconventionally secreted proteins, SecretomeP 2.022 was used. FatiGO36 provide gene ontology annotation.
Statistical analysis of normalized protein ratios
Histograms were constructed and statistical analyses were performed using GraphPad Prism 5 (GraphPad Software, Inc., San Diego, CA). For brefeldin A experiments, significantly altered protein ratios were determined from large-scale protein profiling experiments using the complementary error function (erfc) within Microsoft Excel (see Eq. 1), which determines the probability that each individual protein ratio is not significantly different from the average protein ratio37. The average ratio and other parameters (shown below) were calculated from a Gaussian least-squares nonlinear regression of the distribution of protein ratios. Gaussian fits were constructed using the normal distribution equation provided in GraphPad Prism with automatic outlier detection enabled.
| (Eq. 1) |
where Δlog rp = 0.4343 (Δrp/rp), Δlog(r0) = 0.4343 (Δr0/r0), rp and Δrp = protein abundance ratio and its associated error, r0 and Δr0 = average curve fit ratio and its associated error, and σ = mean error of the background distribution.
Results and Discussion
Generation and characterization of isotope-coded reference proteomes (IRPs)
Previous work established astrocytes as a viable model for the study of protein secretion under various culture conditions19–21, 38. To further understand and define the pathways of astrocyte protein secretion, we employed a modified SILAC approach in primary mouse astrocyte cultures. In this approach, isotope reference proteomes (IRPs) were generated in independent experiments and then added to samples collected from natural abundance culture conditions. This strategy has been previously used to uncover novel biomarkers and pharmacological targets for leukemias with MLL translocations39. Importantly, no significant changes in preparation of enriched astrocyte cultures were performed as compared to our previous work19, except for the addition of heavy-labeled [13C6-15N2]-lysine and [13C6-15N1]-leucine to medium that was devoid of their light isotope analogs. Mixed glial/neuronal cortical cultures were established from cortices dissected from a total of 20 P1 neonatal CD-1 mice and maintained in heavy labeled medium from in vitro day 0 until the end of the experiment at day 20. At 11 days in vitro, cells were washed, trypsinized and combined into a single cell stock to establish enriched astrocyte cultures (> 95%). After 48 hours, astrocytes were changed to serum-free heavy isotope-containing media and remained without media change for an additional 7 days. No differences in rate of proliferation, gross morphology (by light microscopy), or cell viability were observed (data not shown). Astrocyte-conditioned media (ACM) was collected and concentrated, while adherent cells were collected and lysed. The heavy labeled ACM IRP yielded about 1.5 mg of protein, while the astrocyte lysate IRP yielded approximately 25 mg of protein.
Data-dependent and -independent mass spectrometric analyses were performed on each IRP to determine the atom percent excess of stable isotope incorporation. Protein aliquots (25 μg) from each IRP were in-solution trypsin digested. Peptides (5 μg) were separated by reverse-phase LC and analyzed by either data-dependent MS/MS on an LTQ-Orbitrap or by a data-independent “pseudo-MRM” approach on an LTQ mass spectrometer. For the former approach, 1085 and 2067 unique peptides were identified from ACM and lysate IRPs, respectively, with less than 1 % of unique peptides containing a light leucine or lysine. Atom percent excess was quantified by Census, a software tool that provides automated reconstruction of light/heavy (SILAC) extracted ion chromatograms and computation of SILAC peptide ratios (see Experimental Procedures). For greater than 98% of the leucine and/or lysine-containing peptides, the light component was below the limit of quantification. In contrast, vimentin was identified in ACM by 30 unique peptides and had three SILAC peptide pairs that could be quantified. As shown in figure 2, extracted ion chromatograms from two of the SILAC peptide pairs, EEAESTLQSFR and DNLAEDIMR, resulted in SILAC ratios of 0.02 (1:50), corresponding to an estimated atom percent excess of about 98%. The level of isotope enrichment was also supported by data-independent mass spectrometric analysis. In these experiments, 16 SILAC peptide pairs were selected for MS/MS analysis (2 unique peptides from 4 ACM proteins and 4 cell lysate proteins). Precursor-product ion chromatograms for the 2 peptide pairs from GAPDH showed greater than 98 % incorporation (data not shown), while for the remaining 14 peptides, the light component was not detected. Collectively, these data demonstrated that culturing primary mouse astrocytes in isotope-containing medium for 20 days in vitro achieved at least 98 % incorporation for the majority of the intracellular and extracellular proteome.
Figure 2.

Extracted ion chromatograms of vimentin peptides from ACM isotope reference proteome. Extracted ion chromatograms (XICs) were constructed by the Census quantitation tool with automatic thresholds for acceptance of light/heavy extracted ion chromatograms set at a correlation coefficient of 0.7 and outlier detection value of 0.2. XICs correspond to tryptic peptides assigned to vimentin, (A) EEAESTLQSFR and (B) DNLAEDIMR, which were identified in the ACM isotope reference proteome. Comparing linear regression analysis of XIC pairs show a light (gray trace) to heavy (black trace) ratio of 0.02, which approximates 98 % isotopic enrichment. The time window over which the regression analysis was performed is indicated by the gray shaded rectangle. A small vertical line denotes the MS/MS event from which the peptide was identified and the XIC reconstructed.
Previous SILAC studies conducted in primary neurons demonstrated a maximum of 80 % incorporation29. The difference in isotope incorporation between neurons and astrocytes likely resulted from the ability of P1 astrocytes to maintain their proliferative state when cultured under serum conditions. Primary astrocytes cultures established from 20 neonatal mouse brains, maintained in heavy isotope-containing media and conditioned for 7 days, generated isotope reference proteomes (IRPs) that yielded sufficient protein to conduct about 75 large-scale relative quantitative experiments. Since the same IRP was used as an internal standard for all samples, variability introduced due to culturing under heavy-labeled conditions is largely eliminated in the normalized protein abundance ratio.
SILAC quantitative analysis of brefeldin A-induced changes in protein abundance
As a proof-of-concept, IRPs were used to quantify alterations in the astrocyte proteome and secretome following treatment with brefeldin A (BFA), which blocks anterograde ER-to-Golgi vesicular transport at least in part via inhibition of the small GTPase, Arf1p, preventing COPI coat protein assembly on transport vesicles40, 41. BFA treatment was selected as a proof-of-concept for several reasons: (1) the inhibitory effects of BFA on classical, ER-to-Golgi protein secretion have been well-documented40, 41, (2) predictive capabilities of N-terminal signal peptides for classical protein secretion are specific and sensitive33, 35, and (3) the treatment conditions and effects of BFA on primary astrocyte cultures have been previously documented, which showed effective inhibition of classical protein secretion in astrocytes38. Consistent with work by Lafon-Cazal et. al., BFA exposure at 1 μg/mL for 24 hours did not significantly impair cell viability (data not shown). ACM and soluble intracellular protein lysates isolated from control and BFA-treated astrocytes (10 μg) were mixed with respective isotope reference proteomes at a 1:1 ratio, separated by SDS-PAGE, and digested in-gel with trypsin. Peptides (~2.5 μg) from each gel slice were analyzed by LC-MS/MS using an LTQ-Orbitrap XL mass spectrometer.
SEQUEST sequence-to-spectrum assignments were generated and probabilistic validation of peptides was performed by PeptideProphet42. Peptide assignments were filtered by probability to achieve an error rate of less than 1 % and then analyzed by Census. In total, 231 and 535 normalized protein ratios (RatioBFA/RatioCtl) from ACM and cellular lysates were calculated, respectively (Fig. 3A). Previous large-scale protein expression profiling studies have used different statistical approaches to identify proteins that exhibit differential expression or abundance24, 37. One approach, implemented by Li and colleagues in the ASAPRatio algorithm37, modeled a best-fit Gaussian curve to the distribution of protein ratios and then computed the complementary error function to test the null hypothesis that each protein ratio was not significantly different from the mean curve fit ratio (see Experimental Procedures).
Figure 3.

Brefeldin A-induced changes in relative protein abundance. Relative protein abundance ratios were calculated by normalizing protein ratios calculated in the BFA-treated astrocyte sample to the corresponding protein ratio from the control sample. Gaussian curve fit parameters; mean ratio (r0), standard deviation of the mean ratio (SD), and the background distribution (σ) were calculated for protein ratio distributions. (A) Individual log2 protein ratios from ACM (black; N = 231) and cell lysates (gray; N=535) plotted in order of increasing relative abundance. (B) Histogram of BFA-induced changes in cell lysates as a function of log2 normalized protein ratio. Protein ratio distribution demonstrated good fit to the Gaussian curve (r0 ± SD = 1.28 ± 0.01, σ = 1.28). (C) Significantly altered cell lysate protein ratios (black) versus non-significant protein ratios (white) calculated using the complementary error function (p < 0.05) and Gaussian curve fit values. Scaling of the y-axis has been modified to emphasis the significantly altered (black) data points. (D) Histogram of BFA-induced changes in relative protein abundance of ACM proteins. Curve fit values for ACM protein ratios were r0 ± SD = 1.97 ± 0.10, σ = 1.75. (E) Significantly altered cell lysate protein ratios (black) versus non-significant ratios (white) calculated using the complementary error function (p < 0.05) and Gaussian curve fit values.
Using this approach, the distribution of cellular lysate protein ratios showed a good fit to the Gaussian curve and resulted in a mean curve fit ratio close to the expected 1:1 (1.28 ± 0.01) (Fig. 3B). Statistical analysis identified 21 out of 535 proteins that were significantly altered due to BFA treatment (p < 0.05) (Fig. 3C and Supplemental Table 1). Proteins such as coatomer subunit beta and gamma as well as RAP guanine nucleotide exchange factor 2 were down-regulated due to BFA treatment (see Supplemental Table 1). In contrast, the relative intracellular abundance of several classically secreted proteins was found significantly increased, including cathepsin L1 (9.4-fold), apolipoprotein E (21-fold), and thrombospondin-1 (28-fold). Also, the ATP-binding cassette sub-family A member 1 (ABCA1) was increased five-fold, consistent with its functional relationship between apolipoprotein E lipidation and promotion of mature lipoparticle secretion from astrocytes43 as well as macrophages44. Moreover, the glucose-regulated proteins, grp78 and grp94, were 3.4±1.5-fold (p = 0.06) and 2.32±1.05-fold (p = 0.25) increased after BFA treatment, respectively. Though these changes did not reach statistical significance (p > 0.05), upregulation of glucose-regulated proteins is a known consequence of ER stress and the unfolded protein response45. The observation that BFA exposure induced only a modest upregulation of these glucose-regulated proteins, while robustly inhibiting classical protein secretion, provided evidence that the BFA treatment conditions were appropriate. Overall, the proteins identified as differentially expressed in the intracellular proteome due to BFA treatment were consistent with disruption of the classical ER-Golgi trafficking system, including downregulation of several vesicular transport proteins as well as increased abundance of classically secreted proteins, which likely resulted from their secretion being inhibited.
Next, the concomitant BFA-induced protein abundance changes were assessed in ACM. In contrast to cellular lysate protein ratios, the distribution of ACM protein ratios deviated from normality (Fig. 3D). This result was not unexpected given that a significant proportion of proteins identified in ACM contain an N-terminal signal peptide and would be potential targets of BFA-mediated inhibition. Although the curve-fit values derived from the ACM distribution exhibited a wider distribution about the mean (σ) than the cellular lysate distribution (compare Fig. 2B vs 3D), the strong inhibitory effect of BFA on classical secretion permitted the identification of 55 proteins that were significantly decreased in ACM after BFA treatment (p < 0.05) (Fig. 3E and Supplemental Table 2). Significantly, 51 of these proteins possessed an N-terminal signal peptide. On the other hand, 49 signal peptide-containing proteins were not significantly inhibited after BFA treatment. Several possible reasons could account for this observation. Certain signal peptide-containing proteins, such as CD45 and cystic fibrosis transmembrane conductance regulator (CFTR)46–48, are secreted by Golgi- or COPII-independent mechanisms and would be unaffected by BFA treatment. Alternatively, not all luminal or integral membrane proteins (which possess signal peptides) may actively accumulate in conditioned media under these experimental conditions. Also, deviation of the protein ratio distribution from normality and larger distribution about the mean (σ) may have contributed to reduced sensitivity. This highlights a challenge for differential quantitative proteomic studies where a large proportion of the proteome is significantly altered. For these cases, development and validation of more sophisticated statistical analyses will be useful for improving sensitivity and accuracy while maintaining low false discovery rates. Despite these drawbacks, the data support the use of SILAC-generated isotope reference proteomes as a tool for quantification of relative protein abundance within the astrocyte secretome and intracellular proteome.
Protein enrichment profile of ACM from naïve primary astrocyte cultures
Using this SILAC-based analysis, a quantitative protein enrichment profile of astrocyte conditioned media under basal culture conditions was constructed. The extent of protein enrichment in ACM was calculated by comparing the relative abundance ratios between the extracellular and intracellular compartments. For this purpose, a “whole cell” IRP was generated by mixing isotope-coded reference proteins from both the intracellular and extracellular proteomes. This “whole cell” IRP was then mixed with ACM and cell lysates samples obtained from the same astrocyte culture after 7 days of conditioning in light isotope media. Equal aliquots (100 μg) of ACM and cell lysate samples were split between two lanes, resolved by SDS-PAGE, and digested in-gel with trypsin. Extracted peptides corresponding to gel slices from replicate lanes were pooled and analyzed by LC-MS/MS. SEQUEST sequence-to-spectrum assignments were processed by DTASelect49 for protein identification (see Supplemental Data) and then by Census32 for quantitative analysis, resulting in the relative quantification of 516 proteins. Normalized protein ratios (RatioACM/RatioLysate) represent the fold difference in abundance between the extracellular and intracellular compartment. The distribution of protein enrichment ratios spanned at least 4 orders of magnitude (Fig. 4A) and included cytosolic proteins such as filamins and ribosomal subunits that were 20-fold to 50-fold more abundant in cell lysates, as well as secreted proteins such as SPARC and alpha-2-macroglobulin, which were 20- to 50-fold more abundant in ACM (see Supplemental Table 3). An additional 109 protein were excluded from the ACM enrichment profile as their ratios were calculated to infinity (i.e. the calculated cellular lysate protein ratio was 0). At least a subset of these excluded proteins could be secreted since 75% possessed a signal peptide and all were confidently identified by isotope reference peptides in both samples. This suggests that their low intracellular abundance may have hindered quantification or that increased sample complexity in cellular lysates could have contributed to poor quantification, especially at low signal-to-noise.
Figure 4.

Quantification and validation of relative protein enrichment in ACM. (A) Histogram of 516 relative enrichment ratios, expressed as normalized log2 ratios of ACM relative to cell lysates. Positive values represent proteins that were enriched in ACM, while negative values represent proteins that were enriched in cell lysates. Protein ratios greater than 1.5-fold were considered significantly enriched in ACM. (B) Comparison of enrichment ratios from global SILAC MS analysis to abundance ratios for the same proteins calculated by targeted MRM-MS/MS analysis. Six proteins that ranged from −6 to +6 log2-fold enriched in ACM were selected for MRM-MS/MS analyses. A dashed linear line represents the ideal 1:1 correlation. (C) Protein ratios from (A) were grouped by subcellular localization. A protein ratio of 1.5-fold is indicated by the dashed horizontal line, while solid horizontal lines represent median relative enrichment ratios. (D) Protein aliquots (30 μg) from ACM and cellular lysates (two independent biological replicates) were analyzed by Western blot (WB) for selected proteins that spanned the ACM enrichment profile distribution in (A). For proteins with more than one significant band (apolipoprotein E and clusterin), individual bands within each lane were quantified and summed before enrichment ratios were calculated. Bar graphs compare the average enrichment ratio calculated from WB densitometry analysis (N = 2) to the enrichment ratio calculated by SILAC. Error bars reflect ±S.D. SILAC standard deviation based on individual SILAC peptide ratios (see values in Table 1 and Supplemental Table 3). (D) Western blot analysis for apolipoprotein E, SPARC, and clusterin, which showed significant enrichment in ACM. (E) Western analysis for macrophage inhibitory factor (MIF) and cyclophilin B (peptidylprolyl isomerase B). Both have N-terminal signal peptides, but were not enriched in ACM. Enrichment ratios determined by WB for abundant cytosolic proteins, alpha-tubulin and GAPDH, were in agreement with SILAC analysis.
Next, MRM quantitative MS analysis was used to estimate the linear range of ACM enrichment ratios calculated by SILAC analysis, as it provides improved selectivity and sensitivity compared to non-targeted, global approaches based on precursor (MS1) ion abundance. Six proteins were selected, GAPDH, triosphosphate isomerase, glyoxylase domain-containing protein 4, alpha-N-acetylglucosaminidase, histone H4, and SPARC, which ranged in relative enrichment from −6 to +6 (log2 units). Light and heavy SILAC peptide pairs were selected empirically based on MS and MS/MS spectra from previous data-dependent experiments, facilitating the selection of optimal precursor-product ion transitions. Light-to-heavy peptide ratios were calculated from data-independent acquisition of MS/MS spectra and reconstruction of precursor-product ion chromatograms by Xcalibur. MRM protein enrichment ratios were then calculated as the average ratio from two unique peptides and two distinct precursor-product ion transitions per peptide. As shown in figure 4B, SILAC enrichment ratios were in good agreement with targeted MRM ratios between −4 and +4 log2 units, while for ratios outside this range, enrichment ratios measured by SILAC were systematically greater than those measured by MRM (Fig. 4B). Furthermore, both global and targeted mass spectrometry-based determination of extracellular enrichment ratios were largely consistent with Western blot analyses of two independent biological replicates for classically secreted proteins, clusterin and cyclophilin B, as well as MIF, a nonconventional secreted protein, and two cytosolic proteins (GAPDH and alpha-tubulin) (Fig. 4D and E). Relative enrichment measured by Western for the classically secreted proteins, apolipoprotein E and SPARC, were less than those calculated by SILAC (Fig. 4C, top), which was not unexpected given that these ratios were likely outside the linear range of quantification (see Fig. 4B, log2 ratio > 4).
Examination of subcellular localization as a function of ACM enrichment revealed a significant clustering of enrichment ratios according to predicted localization, facilitating the identification of protein enrichment ratios that differed from the mean ratio within each subcellular compartment (Fig. 4C). Proteins with a 1.5-fold or greater enrichment ratio were selected as significantly enriched in ACM (Table 1) as this represented two times the median of the relative standard deviation of enrichment ratios (RSD = 25 %; N = 516). As expected, the extracellular/membrane protein group had the largest proportion of significantly enriched ratios (N = 52/62; median = 8.92-fold) (Fig. 4C). These proteins also showed the widest range of enrichment ratios, consisting of constitutively secreted proteins with large (> 50-fold) enrichment ratios such as extracellular superoxide dismutase and osteopontin, as well as secreted proteins with enrichment ratios less than one, such as mesencephalic astrocyte-derived neurotrophic factor/protein ARMET (0.32-fold) and gelsolin (0.48-fold) (see Supplemental Table 3). Interestingly, gelsolin can exist in three different isoforms derived from alternative mRNA splicing50, 51. One isoform lacks a signal peptide and functions predominantly in the cytosol as an actin-binding protein. The other two isoforms are identical in sequence except for the addition of an N-terminal signal peptide. Upon secretion, these proteins retain their actin-binding ability and may function in actin filament clearance after tissue injury50. This suggests that secreted proteins with enrichment ratios significantly less than one may have dual extra/intracellular function or could function in stimulus-dependent, regulated secretory pathways.
Table 1.
Proteins with significant enrichment in ACM.
| Accession | Protein name | Ratio1 | SD2 | Pep Count3 | SL4 |
|---|---|---|---|---|---|
| P08226 | Apolipoprotein E | 414.00 | 244.00 | 38/6 | EM |
| O09164 | Extracellular superoxide dismutase [Cu-Zn] | 327.00 | 50.00 | 15/1 | EM |
| P10923 | Osteopontin | 187.25 | 93.50 | 4/1 | EM |
| Q8R422 | CD109 antigen | 76.00 | 22.50 | 50/1 | EM |
| Q00493 | Carboxypeptidase E | 74.33 | 26.33 | 46/1 | EM |
| P07214 | SPARC | 67.00 | 35.93 | 36/5 | EM |
| A2APM1 | CD44 antigen | 65.33 | 4.33 | 3/1 | EM |
| P47867 | Secretogranin-3 | 64.00 | 16.00 | 29/1 | EM |
| Q3UGY5 | Fibronectin 1 | 57.67 | 87.06 | 119/6 | EM |
| Q6GQT1 | Alpha-2-macroglobulin-P | 45.60 | 28.62 | 140/9 | EM |
| P02463 | Collagen alpha-1(IV) chain | 41.89 | 12.13 | 9/2 | EM |
| O88307 | Sortilin-related receptor | 31.45 | 5.76 | 42/6 | EM |
| P43025 | Tetranectin | 30.62 | 5.62 | 8/1 | EM |
| P29533 | Vascular cell adhesion protein 1 | 30.15 | 4.23 | 21/1 | EM |
| Q921T4 | Phospholipase A2, group VII (Platelet- activating factor acetylhydrolase, plasma) | 29.70 | 4.80 | 43/1 | EM |
| Q80YX1 | Tenascin | 27.30 | 10.10 | 10/1 | EM |
| Q9Z0J0 | Epididymal secretory protein E1 | 22.00 | 12.65 | 19/7 | EM |
| Q80W15 | Insulin-like growth factor-binding protein- like 1 | 21.33 | 2.83 | 14/1 | EM |
| Q9R0E1 | Procollagen-lysine,2-oxoglutarate 5- dioxygenase 3 | 18.40 | 3.02 | 20/2 | ER |
| P51655 | Glypican-4 | 16.91 | 17.02 | 34/2 | EM |
| Q06890 | Clusterin | 15.71 | 12.44 | 62/6 | EM |
| Q61592 | Growth arrest-specific protein 6 | 14.67 | 2.33 | 26/1 | EM |
| P21460 | Cystatin-C | 14.00 | 2.45 | 37/1 | EM |
| Q9WTR5 | Cadherin-13 | 13.27 | 2.27 | 5/1 | EM |
| Q02819 | Nucleobindin-1 | 12.29 | 2.22 | 22/6 | EM |
| Q3UHN9 | Bifunctional heparan sulfate N- deacetylase/N-sulfotransferase 1 | 12.13 | 2.33 | 6/2 | G |
| Q61147 | Ceruloplasmin | 12.00 | 14.40 | 37/2 | EM |
| Q61361 | Brevican core protein | 11.23 | 2.39 | 14/2 | EM |
| Q9WVJ3 | Plasma glutamate carboxypeptidase | 10.37 | 1.74 | 10/2 | EM |
| P30412 | Peptidyl-prolyl cis-trans isomerase C | 10.36 | 0.57 | 4/2 | EM |
| O70370 | Cathepsin S | 10.09 | 2.09 | 18/1 | EL |
| Q80XP1 | Complement component 3 | 9.75 | 11.25 | 130/5 | EM |
| Q91ZX7 | Prolow-density lipoprotein receptor-related protein 1 | 9.48 | 4.78 | 99/22 | EM |
| Q9QXA3 | Fat 1 cadherin (Fragment) | 9.40 | 1.52 | 64/2 | EM |
| Q99LJ1 | Tissue alpha-L-fucosidase | 8.56 | 1.73 | 8/2 | EL |
| P55066 | Neurocan core protein | 8.47 | 10.22 | 47/2 | EM |
| P97298 | Pigment epithelium-derived factor | 7.65 | 9.26 | 34/3 | EM |
| Q9ES89 | Exostosin-like 2 | 7.50 | 0.38 | 3/1 | ER |
| Q60847 | Collagen alpha-1(XII) chain | 7.25 | 16.12 | 109/4 | EM |
| P13595 | Neural cell adhesion molecule 1 | 7.00 | 1.71 | 10/1 | EM |
| Q99M71 | Mammalian ependymin-related protein 1 | 7.00 | 1.76 | 13/10 | EM |
| Q91VU0 | Protein FAM3C | 6.81 | 3.28 | 5/2 | EM |
| Q8BND5 | Sulfhydryl oxidase 1 | 5.88 | 1.08 | 27/1 | EM |
| P18242 | Cathepsin D | 5.25 | 8.52 | 42/8 | EL |
| Q8K2I4 | Beta-mannosidase | 5.03 | 1.31 | 5/1 | EL |
| Q91WP6 | Serine protease inhibitor A3N | 5.02 | 2.23 | 20/2 | EM |
| Q8BFR4 | N-acetylglucosamine-6-sulfatase | 4.92 | 1.18 | 18/4 | EL |
| Q8R464 | Cell adhesion molecule 4 | 3.83 | 0.50 | 9/1 | EM |
| B2RXS4 | Plexin B2 (MCG140951) (Plxnb2 protein) | 3.79 | 7.13 | 19/13 | EM |
| Q04857 | Collagen alpha-1(VI) chain | 3.76 | 0.68 | 12/1 | EM |
| Q8K479 | Complement C1q tumor necrosis factor- related protein 5 | 3.55 | 0.35 | 11/2 | EM |
| O88325 | Alpha-N-acetylglucosaminidase (Sanfilippo disease IIIB) | 3.50 | 0.47 | 16/7 | EL |
| Q9R0B9 | Procollagen-lysine,2-oxoglutarate 5- dioxygenase 2 | 3.44 | 0.64 | 14/2 | ER |
| Q9WUT8 | DSD-1-proteoglycan | 3.22 | 4.07 | 25/3 | EM |
| Q61207 | Sulfated glycoprotein 1 | 3.04 | 0.72 | 29/13 | EM |
| O88668 | Protein CREG1 | 2.95 | 0.71 | 5/2 | EM |
| Q197W7 | N-glycan processing alpha-mannosidase IIx | 2.94 | 0.60 | 3/2 | G |
| Q64191 | N(4)-(beta-N-acetylglucosaminyl)-L- asparaginase | 2.88 | 2.13 | 15/6 | EL |
| O88531 | Palmitoyl-protein thioesterase 1 | 2.79 | 0.41 | 17/12 | EL |
| P20060 | Beta-hexosaminidase subunit beta | 2.78 | 1.77 | 32/4 | EL |
| P70158 | Acid sphingomyelinase-like phosphodiesterase 3a | 2.77 | 0.36 | 8/3 | EM |
| Q9ET22 | Dipeptidyl-peptidase 2 | 2.70 | 0.66 | 9/4 | EL |
| O89017 | Legumain | 2.60 | 0.57 | 16/1 | EL |
| P17047 | Lysosome-associated membrane glycoprotein 2 | 2.55 | 1.53 | 5/4 | EL |
| Q3TCN2 | Putative phospholipase B-like 2 | 2.43 | 0.58 | 14/10 | EL |
| A2ARV4 | Low-density lipoprotein receptor-related protein 2 | 2.24 | 0.62 | 59/9 | EM |
| Q9EQH2 | Endoplasmic reticulum aminopeptidase 1 | 2.20 | 0.57 | 6/3 | ER |
| P10605 | Cathepsin B | 2.16 | 1.34 | 43/13 | EL |
| P50429 | Arylsulfatase B | 2.12 | 0.72 | 9/3 | EL |
| P01029 | Complement C4-B | 2.03 | 0.77 | 109/1 | EM |
| Q9WV54 | Acid ceramidase | 1.96 | 0.36 | 16/7 | EL |
| Q91XG3 | Hexosaminidase A | 1.93 | 1.36 | 13/3 | EL |
| Q60648 | Ganglioside GM2 activator | 1.87 | 0.40 | 20/6 | EL |
| P16675 | Lysosomal protective protein | 1.87 | 0.73 | 25/12 | EL |
| Q07797 | Galectin-3-binding protein | 1.78 | 0.22 | 21/1 | EM |
| O09159 | Lysosomal alpha-mannosidase | 1.69 | 0.21 | 23/1 | EL |
| O54782 | Epididymis-specific alpha-mannosidase | 1.64 | 0.89 | 21/4 | EM |
| P23780 | Beta-galactosidase | 1.55 | 0.38 | 10/1 | EL |
| P97290 | Plasma protease C1 inhibitor | 1.54 | 1.53 | 34/2 | EM |
| Q9WUU7 | Cathepsin Z | 1.53 | 0.48 | 15/3 | EL |
| P20152 | Vimentin | 27.43 | 19.36 | 61/8 | O |
| P62806 | Histone H4 | 16.17 | 4.64 | 9/7 | O |
| P27661 | Histone H2A.x | 6.82 | 1.93 | 4/6 | O |
| Q61599 | Rho GDP-dissociation inhibitor 2 | 4.80 | 2.56 | 2/2 | O |
| Q3UCL5 | Ferritin | 3.75 | 0.79 | 9/1 | O |
| Q61233 | Plastin-2 | 3.75 | 1.04 | 12/2 | O |
| P09528 | Ferritin heavy chain | 3.59 | 1.25 | 7/2 | O |
| P00493 | Hypoxanthine-guanine phosphoribosyltransferase | 2.63 | 0.60 | 9/6 | O |
| P80314 | T-complex protein 1 subunit beta | 2.40 | 0.39 | 5/2 | O |
| O88569 | Heterogeneous nuclear ribonucleoproteins A2/B1 | 2.06 | 0.65 | 3/2 | O |
| Q8CHQ7 | UPF0727 protein C6orf115 homolog | 2.01 | 0.01 | 2/2 | O |
| P10852 | 4F2 cell-surface antigen heavy chain | 1.48 | 0.34 | 9/2 | O |
Proteins are sorted in order of decreasing protein ratio, as well as by predicted classical and putative non-conventional section (designated by O in last column).
Protein ratio calculated from the intensity weighted average of individual peptide ratios. Ratio reflects the fold difference in protein abundance of ACM relative to cell lysates.
Standard deviation of the protein ratio, calculated by error propagation derived from individual peptide ratios for ACM and cell lysates. Standard deviations of zero reflected instances where unique peptides had the same L/H ratio (two significant figures).
Number of unique, quantified peptides used to calculate the protein ratio from ACM (left) and cell lysates (right) samples. A minimum of 2 unique, quantified peptides were required in at least ACM or cell lysates samples.
Predicted protein subcellular localization. Primary sequence analysis was performed by Signal P 3.0 to distinguish N-terminal signal peptide-containing proteins from non-signal peptide-containing proteins (classified as cytosolic), followed by TargetP to provide predicted subcellular localization. EM = Extracellular/Plasma membrane; EL=Endo/Lysosome; G = Golgi; M = Mitochondria; ER = Endoplasmic reticulum; O = Cytoplasm/Nucleus/Other
In the endosomal/lysosomal group (median enrichment = 1.96-fold), 22 out of 31 proteins were significantly enriched (Table 1), including cathepsin B (2.2-fold), D (5.25-fold), S (10.1-fold), and Z (1.5-fold), and beta-mannosidase (5.0-fold). Traditionally, possession of secretory lysosomes, which function primarily in immune defense, have been restricted to select cell types from the haematopoietic lineage (neutrophils, basophils, and cytotoxic T lymphocytes)52, 53. Although astrocytes are not derived from this lineage, the participation of astrocytes in CNS immune responses has been well studied13. Also, recent work has shown that cultured astrocytes spontaneously internalize styryl dyes into a lysosomal/endosomal vesicular pool, a subset of which undergo calcium-dependent exocytosis54. In addition, astrocytic ATP release, which subserves calcium wave propagation1 and modulation of synaptic transmission55 was found to occur from a lysosomal vesicular pool56.
The median enrichment ratio for ER/Golgi/mitochondria-localized proteins was 0.39-fold (N = 31), while the remaining 392 proteins quantified did not contain N-terminal signal peptides and had a median enrichment ratio of 0.27-fold (Fig. 4C). When analyzed by SecretomeP, a computational prediction algorithm for nonconventional protein secretion22, 160 proteins were predicted to be nonconventionally secreted including known nonconventionally secreted proteins such as galectins, macrophage migration inhibitory factor, and acyl-CoA-binding protein57–60. A majority of these proteins (153) were less than 1.5-fold enriched in ACM, suggesting that under the current experimental conditions, nonconventional protein secretion did not significantly contribute to the overall abundance of astrocyte-secreted proteins quantified in ACM. Interestingly, 7 proteins, including vimentin (26-fold), and ferritin light (3.75-fold) and heavy (3.60-fold) chains were enriched in ACM (Table 1). MS/MS spectra and extracted ion chromatograms from selected proteins supported their identification and enrichment in ACM (see Supplemental Fig. 1 and 2). Vimentin has previously been characterized as a nonconventionally secreted protein from activated macrophages61 and also identified in astrocyte conditioned media by mass spectrometry-based proteomics19. The secretion of ferritin chains has recently been documented in macrophages, which was shown to provide a functional ferritin-iron source for cultured erthyroid precursor62. Although glial cells possess abundant ferritin and are able to accumulate iron, they lack transferrin receptors, which are thought to mediate much of brain-derived cellular import of iron63. Therefore, additional investigation into the role of astrocytes in brain iron homeostasis may be warranted.
Conclusions
Overall, this work demonstrated a SILAC-based quantitative mass spectrometry approach using isotope reference proteomes to quantify relative protein abundance changes within intracellular and extracellular astrocyte proteomes. The feasibility of SILAC quantitative MS for primary astrocytes was demonstrated by achieving stable isotope incorporation of 98 % or greater at 20 days in vitro and by quantification of protein abundance changes within intracellular and extracellular proteomes after brefeldin A treatment. These IRPs were used to assemble a quantitative protein enrichment profile of astrocyte-conditioned media collected from naïve astrocyte cultures. In combination with computational prediction algorithms and subcellular localization, the enrichment profile of proteins in astrocyte conditioned media revealed a large dynamic range, comprising highly abundant classically secreted proteins, proteins that may proceed by nonconventional secretion, proteins that function in regulated secretion, as well as less abundant, cytosolic contaminants. Interestingly, the identification and quantification of potential lysosomal-secreted proteins provides new molecular targets to investigate the potential role of secretory lysosomes in astrocyte biology. While the function of lysosomal-secreted proteins from astrocytes is unclear, one could speculate that lysosomal enzymes may function in the extracellular space for tissue remodeling following cellular injury. This approach can be applied to comprehensively define, at the molecular level, basal and stimulated extracellular enrichment profiles under various experimental conditions, which will further advance our understanding of secretory pathways encompassing both classical as well as nonconventional secretion.
Supplementary Material
Acknowledgments
We thank Jessica Lee in the CHOP Protein Core for her assistance in operating and maintaining the mass spectrometers. We also thank Sung Kyu (Robin) Park for his technical assistance and continued development with the Census quantitative analysis software. The work was supported by grants from the National Institutes of Health NIA AG13966 and ES013508 NIEHS Center of Excellence in Environmental Toxicology. HI is the Gisela and Dennis Alter Research Professor of Pediatric Neonatology at the Children’s Hospital of Philadelphia Research Institute.
Abbreviations
- ACM
astrocyte-conditioned media
- ALS
amyotrophic lateral sclerosis
- CNS
central nervous system
- GeLC-MS/MS
gel-liquid chromatography-tandem mass spectrometry
- LC-MRM-MS/MS
liquid chromatography-multiple reaction monitoring-tandem mass spectrometry
- NGF
nerve growth factor
Footnotes
Supporting Information Available: Table 1, a list of differentially expressed proteins in the astrocyte proteome after brefeldin A treatment; Table 2, a list of proteins with differential abundance in astrocyte-conditioned media after brefeldinA treatment; Table 3, a complete list of proteins identified and quantified in conditioned media of naïve astrocytes; Figures 1 and 2, Representative MS/MS spectra and extracted ion chromatograms from selected nonconventionally secreted proteins; Excel file with total number of identified proteins, including unique and redundant peptides, from astrocyte-conditioned media and cellular lysates. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Nedergaard M. Science. 1994;5154:1768–1771. doi: 10.1126/science.8134839. [DOI] [PubMed] [Google Scholar]
- 2.Zito K, Svoboda K. Neuron. 2002;6:1015–1017. doi: 10.1016/s0896-6273(02)00903-0. [DOI] [PubMed] [Google Scholar]
- 3.De Roo M, Klauser P, Garcia PM, Poglia L, Muller D. Prog Brain Res. 2008:199–207. doi: 10.1016/S0079-6123(07)00011-8. [DOI] [PubMed] [Google Scholar]
- 4.Christopherson KS, Ullian EM, Stokes CC, Mullowney CE, Hell JW, Agah A, Lawler J, Mosher DF, Bornstein P, Barres BA. Cell. 2005;3:421–433. doi: 10.1016/j.cell.2004.12.020. [DOI] [PubMed] [Google Scholar]
- 5.Glabinski AR, Tani M, Strieter RM, Tuohy VK, Ransohoff RM. Am J Pathol. 1997;2:617–630. [PMC free article] [PubMed] [Google Scholar]
- 6.Koistinaho M, Lin S, Wu X, Esterman M, Koger D, Hanson J, Higgs R, Liu F, Malkani S, Bales KR, Paul SM. Nat Med. 2004;7:719–726. doi: 10.1038/nm1058. [DOI] [PubMed] [Google Scholar]
- 7.Krumbholz M, Theil D, Cepok S, Hemmer B, Kivisakk P, Ransohoff RM, Hofbauer M, Farina C, Derfuss T, Hartle C, Newcombe J, Hohlfeld R, Meinl E. Brain. 2006;(Pt 1):200–211. doi: 10.1093/brain/awh680. [DOI] [PubMed] [Google Scholar]
- 8.Liauw J, Hoang S, Choi M, Eroglu C, Choi M, Sun GH, Percy M, Wildman-Tobriner B, Bliss T, Guzman RG, Barres BA, Steinberg GK. J Cereb Blood Flow Metab. 2008;10:1722–1732. doi: 10.1038/jcbfm.2008.65. [DOI] [PubMed] [Google Scholar]
- 9.Nagai M, Re DB, Nagata T, Chalazonitis A, Jessell TM, Wichterle H, Przedborski S. Nat Neurosci. 2007;5:615–622. doi: 10.1038/nn1876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Park JA, Lee HS, Ko KJ, Park SY, Kim JH, Choe G, Kweon HS, Song HS, Ahn JC, Yu YS, Kim KW. Glia. 2008;3:247–258. doi: 10.1002/glia.20600. [DOI] [PubMed] [Google Scholar]
- 11.Song H, Stevens CF, Gage FH. Nature. 2002;6884:39–44. doi: 10.1038/417039a. [DOI] [PubMed] [Google Scholar]
- 12.Babcock AA, Kuziel WA, Rivest S, Owens T. J Neurosci. 2003;21:7922–7930. doi: 10.1523/JNEUROSCI.23-21-07922.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dong Y, Benveniste EN. Glia. 2001;2:180–190. doi: 10.1002/glia.1107. [DOI] [PubMed] [Google Scholar]
- 14.Banker GA. Science. 1980;4458:809–810. doi: 10.1126/science.7403847. [DOI] [PubMed] [Google Scholar]
- 15.Pekny M, Nilsson M. Glia. 2005;4:427–434. doi: 10.1002/glia.20207. [DOI] [PubMed] [Google Scholar]
- 16.Abele AE, Scholz KP, Scholz WK, Miller RJ. Neuron. 1990;3:413–419. doi: 10.1016/0896-6273(90)90053-i. [DOI] [PubMed] [Google Scholar]
- 17.Ding S, Fellin T, Zhu Y, Lee SY, Auberson YP, Meaney DF, Coulter DA, Carmignoto G, Haydon PG. J Neurosci. 2007;40:10674–10684. doi: 10.1523/JNEUROSCI.2001-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Grimmond SM, Miranda KC, Yuan Z, Davis MJ, Hume DA, Yagi K, Tominaga N, Bono H, Hayashizaki Y, Okazaki Y, Teasdale RD RIKEN GER Group; GSL Members. Genome Res. 2003;6B:1350–1359. doi: 10.1101/gr.983703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Keene SD, Greco TM, Parastatidis I, Lee SH, Hughes EG, Balice-Gordon RJ, Speicher DW, Ischiropoulos H. Proteomics. 2009;3:768–782. doi: 10.1002/pmic.200800385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dowell JA, Johnson JA, Li L. J Proteome Res. 2009;8:4135–4143. doi: 10.1021/pr900248y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Moore NH, Costa LG, Shaffer SA, Goodlett DR, Guizzetti M. J Neurochem. 2009;4:891–908. doi: 10.1111/j.1471-4159.2008.05836.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bendtsen JD, Jensen LJ, Blom N, Von Heijne G, Brunak S. Protein Eng Des Sel. 2004;4:349–356. doi: 10.1093/protein/gzh037. [DOI] [PubMed] [Google Scholar]
- 23.Chen Y, Gu B, Wu S, Sun W, Ma S, Liu Y, Gao Y. J Mass Spectrom. 2009;3:397–403. doi: 10.1002/jms.1517. [DOI] [PubMed] [Google Scholar]
- 24.Oda Y, Huang K, Cross FR, Cowburn D, Chait BT. Proc Natl Acad Sci U S A. 1999;12:6591–6596. doi: 10.1073/pnas.96.12.6591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ong SE, Blagoev B, Kratchmarova I, Kristensen DB, Steen H, Pandey A, Mann M. Mol Cell Proteomics. 2002;5:376–386. doi: 10.1074/mcp.m200025-mcp200. [DOI] [PubMed] [Google Scholar]
- 26.Delcourt N, Jouin P, Poncet J, Demey E, Mauger E, Bockaert J, Marin P, Galeotti N. Mol Cell Proteomics. 2005;8:1085–1094. doi: 10.1074/mcp.M500040-MCP200. [DOI] [PubMed] [Google Scholar]
- 27.Ong SE, Mann M. Nat Chem Biol. 2005;5:252–262. doi: 10.1038/nchembio736. [DOI] [PubMed] [Google Scholar]
- 28.Graumann J, Hubner NC, Kim JB, Ko K, Moser M, Kumar C, Cox J, Scholer H, Mann M. Mol Cell Proteomics. 2008;4:672–683. [Google Scholar]
- 29.Spellman DS, Deinhardt K, Darie CC, Chao MV, Neubert TA. Mol Cell Proteomics. 2008;6:1067–1076. doi: 10.1074/mcp.M700387-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tang HY, Ali-Khan N, Echan LA, Levenkova N, Rux JJ, Speicher DW. Proteomics. 2005;13:3329–3342. doi: 10.1002/pmic.200401275. [DOI] [PubMed] [Google Scholar]
- 31.Speicher K, Kolbas O, Harper S, Speicher D. J Biomol Tech. 2000;2:74–86. [PMC free article] [PubMed] [Google Scholar]
- 32.Park SK, Venable JD, Xu T, Yates JR., 3rd Nat Methods. 2008;4:319–322. doi: 10.1038/nmeth.1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bendtsen JD, Nielsen H, von Heijne G, Brunak S. J Mol Biol. 2004;4:783–795. doi: 10.1016/j.jmb.2004.05.028. [DOI] [PubMed] [Google Scholar]
- 34.Emanuelsson O, Brunak S, von Heijne G, Nielsen H. Nat Protoc. 2007;4:953–971. doi: 10.1038/nprot.2007.131. [DOI] [PubMed] [Google Scholar]
- 35.Emanuelsson O, Nielsen H, Brunak S, von Heijne G. J Mol Biol. 2000;4:1005–1016. doi: 10.1006/jmbi.2000.3903. [DOI] [PubMed] [Google Scholar]
- 36.Al-Shahrour F, Diaz-Uriarte R, Dopazo J. Bioinformatics. 2004;4:578–580. doi: 10.1093/bioinformatics/btg455. [DOI] [PubMed] [Google Scholar]
- 37.Li XJ, Zhang H, Ranish JA, Aebersold R. Anal Chem. 2003;23:6648–6657. doi: 10.1021/ac034633i. [DOI] [PubMed] [Google Scholar]
- 38.Lafon-Cazal M, Adjali O, Galeotti N, Poncet J, Jouin P, Homburger V, Bockaert J, Marin P. J Biol Chem. 2003;27:24438–24448. doi: 10.1074/jbc.M211980200. [DOI] [PubMed] [Google Scholar]
- 39.Yocum AK, Busch CM, Felix CA, Blair IA. J Proteome Res. 2006;10:2743–2753. doi: 10.1021/pr060235v. [DOI] [PubMed] [Google Scholar]
- 40.Donaldson JG, Finazzi D, Klausner RD. Nature. 1992;6402:350–352. doi: 10.1038/360350a0. [DOI] [PubMed] [Google Scholar]
- 41.Helms JB, Rothman JE. Nature. 1992;6402:352–354. doi: 10.1038/360352a0. [DOI] [PubMed] [Google Scholar]
- 42.Keller A, Nesvizhskii AI, Kolker E, Aebersold R. Anal Chem. 2002;20:5383–5392. doi: 10.1021/ac025747h. [DOI] [PubMed] [Google Scholar]
- 43.Hirsch-Reinshagen V, Zhou S, Burgess BL, Bernier L, McIsaac SA, Chan JY, Tansley GH, Cohn JS, Hayden MR, Wellington CL. J Biol Chem. 2004;39:41197–41207. doi: 10.1074/jbc.M407962200. [DOI] [PubMed] [Google Scholar]
- 44.Von Eckardstein A, Langer C, Engel T, Schaukal I, Cignarella A, Reinhardt J, Lorkowski S, Li Z, Zhou X, Cullen P, Assmann G. FASEB J. 2001;9:1555–1561. doi: 10.1096/fj.00-0798com. [DOI] [PubMed] [Google Scholar]
- 45.Price BD, Mannheim-Rodman LA, Calderwood SK. J Cell Physiol. 1992;3:545–552. doi: 10.1002/jcp.1041520314. [DOI] [PubMed] [Google Scholar]
- 46.Fatal N, Karhinen L, Jokitalo E, Makarow M. J Cell Sci. 2004;(Pt 9):1665–1673. doi: 10.1242/jcs.01019. [DOI] [PubMed] [Google Scholar]
- 47.Baldwin TA, Ostergaard HL. J Biol Chem. 2002;52:50333–50340. doi: 10.1074/jbc.M209075200. [DOI] [PubMed] [Google Scholar]
- 48.Wang X, Matteson J, An Y, Moyer B, Yoo JS, Bannykh S, Wilson IA, Riordan JR, Balch WE. J Cell Biol. 2004;1:65–74. doi: 10.1083/jcb.200401035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cociorva DL, Tabb D, Yates JR. Curr Protoc Bioinformatics. 2007;(Unit 13.4) doi: 10.1002/0471250953.bi1304s16. [DOI] [PubMed] [Google Scholar]
- 50.Kwiatkowski DJ, Mehl R, Yin HL. J Cell Biol. 1988;2:375–384. doi: 10.1083/jcb.106.2.375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Vouyiouklis DA, Brophy PJ. J Neurochem. 1997;3:995–1005. doi: 10.1046/j.1471-4159.1997.69030995.x. [DOI] [PubMed] [Google Scholar]
- 52.Stinchcombe J, Bossi G, Griffiths GM. Science. 2004;5680:55–59. doi: 10.1126/science.1095291. [DOI] [PubMed] [Google Scholar]
- 53.Holt OJ, Gallo F, Griffiths GM. J Biochem. 2006;1:7–12. doi: 10.1093/jb/mvj126. [DOI] [PubMed] [Google Scholar]
- 54.Li D, Ropert N, Koulakoff A, Giaume C, Oheim M. J Neurosci. 2008;30:7648–7658. doi: 10.1523/JNEUROSCI.0744-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Haydon PG, Carmignoto G. Physiol Rev. 2006;3:1009–1031. doi: 10.1152/physrev.00049.2005. [DOI] [PubMed] [Google Scholar]
- 56.Zhang Z, Chen G, Zhou W, Song A, Xu T, Luo Q, Wang W, Gu XS, Duan S. Nat Cell Biol. 2007;8:945–953. doi: 10.1038/ncb1620. [DOI] [PubMed] [Google Scholar]
- 57.Seelenmeyer C, Stegmayer C, Nickel W. FEBS Lett. 2008;9:1362–1368. doi: 10.1016/j.febslet.2008.03.024. [DOI] [PubMed] [Google Scholar]
- 58.Merk M, Baugh J, Zierow S, Leng L, Pal U, Lee SJ, Ebert AD, Mizue Y, Trent JO, Mitchell R, Nickel W, Kavathas PB, Bernhagen J, Bucala R. J Immunol. 2009;11:6896–6906. doi: 10.4049/jimmunol.0803710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kinseth MA, Anjard C, Fuller D, Guizzunti G, Loomis WF, Malhotra V. Cell. 2007;3:524–534. doi: 10.1016/j.cell.2007.06.029. [DOI] [PubMed] [Google Scholar]
- 60.Nickel W, Rabouille C. Nat Rev Mol Cell Biol. 2009;2:148–155. doi: 10.1038/nrm2617. [DOI] [PubMed] [Google Scholar]
- 61.Mor-Vaknin N, Punturieri A, Sitwala K, Markovitz DM. Nat Cell Biol. 2003;1:59–63. doi: 10.1038/ncb898. [DOI] [PubMed] [Google Scholar]
- 62.Leimberg MJ, Prus E, Konijn AM, Fibach E. J Cell Biochem. 2008;4:1211–1218. doi: 10.1002/jcb.21499. [DOI] [PubMed] [Google Scholar]
- 63.Dringen R, Bishop GM, Koeppe M, Dang TN, Robinson SR. Neurochem Res. 2007;11:1884–1890. doi: 10.1007/s11064-007-9375-0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.