

Abstract
Recent work suggests that diabetes affects processing of peripheral, spinal and supraspinal signals in the spinal cord. However, there is little evidence for spinal cord lesions that would account for alterations in behavioral responses induced by experimental diabetes. Therefore, we assessed the expression of proteins that might affect neuronal cytoskeletal stability and thus promote dendritic and synaptic reorganization in diabetic rats. Expression of ILK, PINCH, PI3K, GSK-3β, tau, MAP2, synaptophysin and drebrin in the lumbar spinal cord of non-diabetic and streptozotocin-diabetic rats was assessed by western-blot analysis and immunocytochemistry after 8 and 20 weeks of diabetes. The impact of diabetes on the proteins studied was duration-dependent with changes observed after 20 but not 8 weeks of diabetes. ILK and PINCH proteins levels were significantly decreased and both colocalized to neurons and oligodendrocytes. PI3K protein levels were also significantly decreased, while GSK-3β activity tended to be increased. Phosphorylation of tau and MAP2A/B protein expression were significantly increased, and expression of synaptophysin and drebrin were reduced in diabetic rats. Decreased ILK and PINCH as well as alterations of components of related signaling pathways are associated with tau hyperphosphorylation, MAP2 overexpression and reduction of synaptic proteins in the spinal cord of diabetic rats, suggesting that ILK and PINCH contribute to stabilization of axonal and dendritic structures. However, these changes are not likely the cause of altered behavioral responses in diabetic rats that occur after short-term diabetes, but may contribute to structural changes occurring in long-term diabetes.
Keywords: Diabetes, ILK, PINCH, Streptozotocin, Spinal cord
1. Introduction
Neurological complications of both type 1 and type 2 diabetes mellitus affect up to 50% of those afflicted with diabetes (Thomas PK, 1993) and are associated with a variety of factors, the most important being poor glycemic control and duration of diabetes (Shaw et al., 2003). Of those patients with diabetic neuropathy, 10-20% experience abnormal sensations (Boulton et al., 1983; Partanen et al., 1995), including spontaneous pain, paresthesia, dysethesia, hyperalgesia and allodynia, which can have a severe impact on the quality of life (Ahroni et al., 1994). Clinical observations and studies have not yet established the pathogenesis of painful diabetic neuropathy, leading to the increased use of experimental animal models of diabetes to investigate potential mechanisms. While animals are not well suited to study spontaneous pain, they are appropriate models for investigating altered behavioral responses to thermal, tactile and chemical stimuli, such as heat-induced hyper or hypoalgesia, touch-evoked allodynia and formalin-induced hyperalgesia (Calcutt, 2002). These studies have established the importance of both primary afferent activity and spinal sensitization as components of pain states, and have generated interest in the spinal cord as a site processing pain-generated peripheral, spinal and supraspinal signals.
Histological evidence of neuropathy, radiculopathy and myelopathy in diabetic patients has been well documented (Mizisin AP, 2007). Moreover, MRI scans detect spinal cord atrophy not only in subjects with clinically detectable diabetic polyneuropathy, but also in those with sub-clinical diabetic polyneuropathy (Eaton et al., 2001; Selvarajah et al., 2006), consistent with subtle and early occurring, diabetes-induced changes in spinal cord. In contrast to diabetic patients, overt structural injury has, at best, been infrequently documented in the peripheral neuraxis and spinal cord of diabetic rodents (Mizisin AP, 2007). While reduced perikaryal volume of both sensory and motor neurons has been reported in streptozotocin-diabetic rats (Sidenius and Jakobsen, 1980), there is no evidence of degenerative changes culminating in neuronal loss in the dorsal root ganglia or spinal cord gray matter. Presently, there is little support in the literature for spinal lesions that would account for the changes in behavioral responses to thermal, tactile and chemical stimuli observed in experimental diabetes. However, imaging of the spinal cord of streptozotocin-diabetic rats during sensory stimulation shows decreased blood-oxygen-level-dependent fMRI activity compared to non-diabetic rats, which may be indicative of changes in synapses between primary afferents and second-order neurons (Malisza et al., 2009). Thus, in the spinal cord of diabetic rats, it is possible that there are ultrastructural correlates to altered sensation in the form of dendritic and synaptic reorganization or changes in the expression and distribution of synaptic proteins, as has been reported in the hippocampus of streptozotocin-diabetic rats (Grillo et al., 2005; Magarinos and McEwen, 2000). Changes in ultrastructure or protein expression may translate to alterations in receptors or channels responsible for spinal sensitization (Mizisin et al, 2008) or disinhibition (Jolivalt et al. 2008).
Dendritic formation, induced by growth factors and neuronal activity, requires signaling through phosphoinositide 3-kinase (PI3K) (Delcommenne et al., 1998), integrin-linked kinase (ILK) and glycogen-synthase kinase 3β (GSK-3β) (Naska et al., 2006). ILK interacts with transmembrane β1 and β3 integrin receptors through its COOH terminus, with the Particularly Interesting New Cystine-Histine rich protein (PINCH) (Rearden, 1994) via its ankyrin-repeat domain, and with phosphatidylinositol phosphates via the PH domain motif (Delcommenne et al., 1998; Hannigan et al., 1996; Hannigan and Dedhar, 1997). ILK-PINCH interactions link integrins and growth factors to intracellular signals that regulate actin assembly (Attwell et al., 2003; Filipenko et al., 2005; Qian et al., 2005; Wu and Dedhar, 2001; Yamaji et al., 2004) and ILK-GSK3 interactions promote inactivation of GSK3 and cytoskeletal stability (Naska et al., 2006). Activators of PI3K and thus modulators of ILK activity include growth factors, such as NGF and insulin, cytokines and extracellular matrix (Downward, 1998; King et al., 1997; Kok et al., 2009). Given the insulin deficiency in type 1 diabetes, impaired signaling through ILK and/or PINCH pathways might affect neuronal cytoskeletal stability and thus promote dendritic and synaptic reorganization, possible correlates to the altered behavioral responses to thermal, tactile and chemical stimuli observed in diabetic rodents. Therefore, we examined the expression of ILK and PINCH, and related upstream (PI-3K) and downstream (GSK-3β) signaling pathways, as well as markers of the neuronal cytoskeleton (tau and MAP2), and dendritic and synaptic organization (drebrin and synaptophysin) in the lumbar spinal cord of non-diabetic and streptozotocin-diabetic rats.
2. Results
2.1. Animals
Eight and 20-week diabetic rats exhibited hyperglycemia 4 days after streptozotocin injection and at sacrifice (8 weeks: non-diabetic: 152±6 mg/dl and diabetic: >600 mg/dl; 20 weeks: non-diabetic: 112±20 mg/dl and diabetic: 480±19 mg/dl) and had significantly (p<0.05) lower body weight than age-matched non-diabetic rats (8 weeks: non-diabetic: 261±3g and diabetic: 203±8g; 20 weeks: non-diabetic: 284±6 g and diabetic: 224±10 g).
2.2. Actin levels in diabetic rat spinal cord
For western-blot analysis, the intensity of the bands corresponding to each protein of interest was normalized against the intensity of the band corresponding to actin. However, because actin is a structural protein that might be affected by diabetes, we confirmed that actin levels were unchanged in 20-week diabetic and age-matched non-diabetic rat spinal cord by normalizing the data to the intensity of the band corresponding to cyclophilin B, a protein whose DRG mRNA is unaffected by diabetes (Dr. D. Tomlinson personal communication). The ratio of actin/cyclophilin in spinal cord samples was 100±3.1% for non-diabetic and 99.9±2.5% for diabetic rats.
2.3. PINCH and ILK in diabetic rat spinal cord
PINCH 38kDa and PINCH 70kDa are both present in rat lumbar spinal cord and their protein levels were significantly reduced by 42% (p<0.001) and 45% (p<0.001), respectively, in spinal cord from 20-week diabetic rats, compared to that from non-diabetic rats (Fig. 1A). No significant changes of PINCH 38kDa and PINCH 70kDa protein levels were observed in lumbar spinal cord from 8-week diabetic rats (data not shown). Similarly, ILK protein levels were significantly decreased by 19% (p<0.01) in 20-week diabetic rat lumbar spinal cord compared to the control cord (Fig. 1B), whereas in 8-week diabetic rats, ILK protein levels were unchanged (data not shown).
Fig. 1.
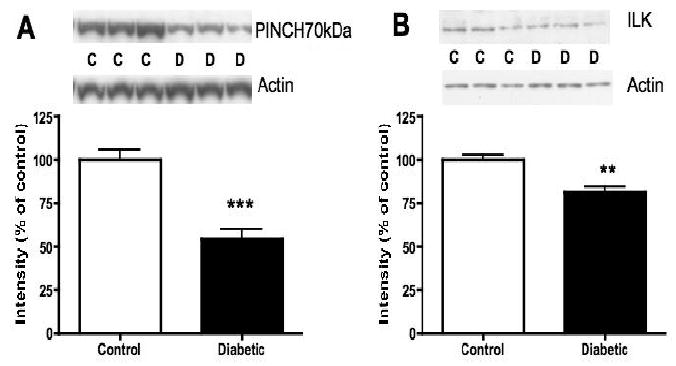
Protein levels of PINCH and ILK in lumbar spinal cord of non-diabetic control (C) and 20-week diabetic (D) rats. A: Intensity of bands corresponding to PINCH 70kDa normalized to the intensity of bands corresponding to actin. B: Intensity of bands corresponding to ILK normalized to the intensity of bands corresponding to actin. Data are represented as mean + SEM, n=6, **p<0.01, ***p<0.001 using unpaired, two-tailed t-test.
Immunocolocalization studies showed that PINCH immunoreactivity is present in neurons throughout gray matter (Fig. 2A-C) and in oligodendrocytes throughout white matter (Fig. 2D-F), but not in astrocytes and microglia (data not shown). Double immunostaining revealed co-localization of ILK and PINCH in neurons throughout gray matter (Fig. 3A-C) and oligodendrocytes throughout white matter (Fig. 3D-F).
Fig. 2.
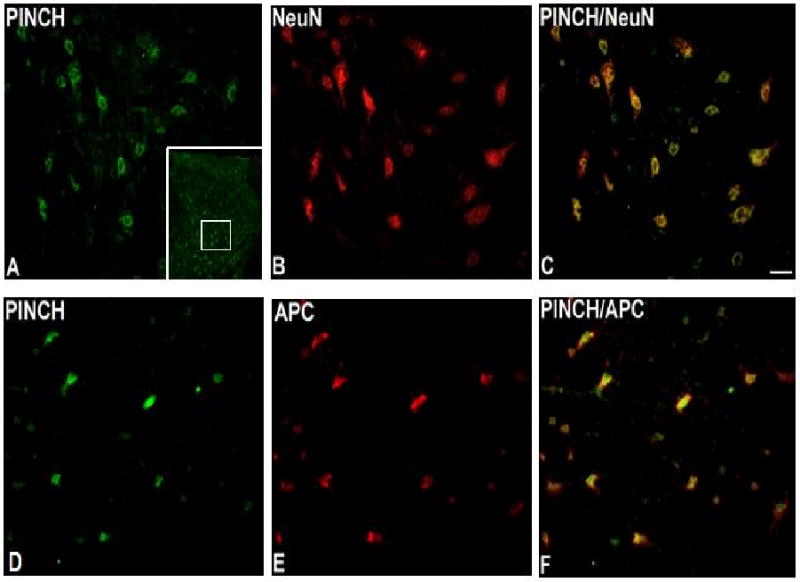
Immunoreactivity of PINCH in lumbar spinal cord. PINCH is coexpressed with NeuN in dorsal horn gray matter (A, B, C) and with APC in white matter (D, E, F), localizing PINCH in spinal cord to neurons and oligodendrocytes. Bar = 40μm. Inset in A: low-power view of the dorsal horn immunostained for PINCH, with the approximate location of higher-power images delimited by the white square.
Fig. 3.
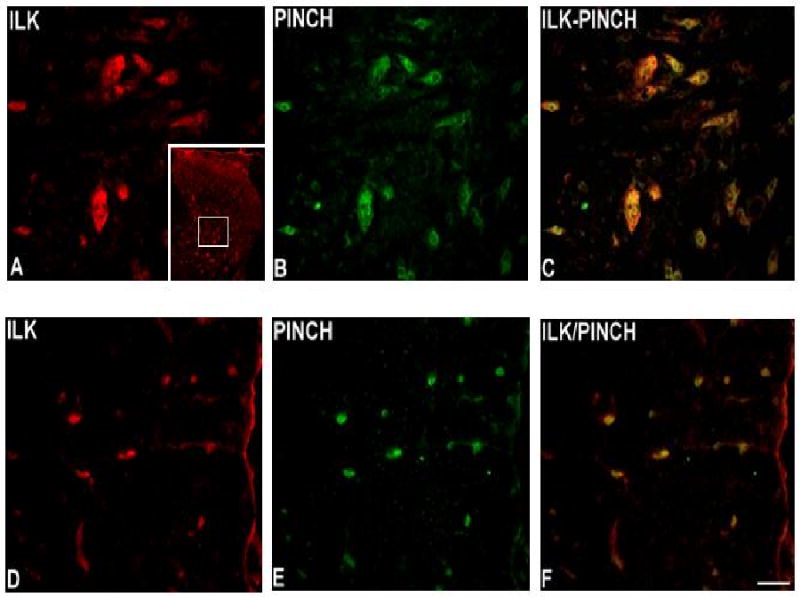
Immunoreactivity of ILK in lumbar spinal cord. ILK is present in dorsal horn gray matter (A) and in white matter (D). Similarly, PINCH immunoreactivity is present in dorsal horn gray matter (B) and in white matter (E). Note the coexpression of ILK and PINCH in both gray matter (C) and white matter (F). Bar = 40μm. Inset in A: low-power view of the dorsal horn immunostained for ILK, with the approximate location of higher-power images delimited by the white square.
2.4. PI3K/GSK3 signaling pathway in diabetic rat spinal cord
Although there were no changes after 8 weeks of diabetes (data not shown), PI3K protein levels were significantly reduced by 21% (p<0.005) in 20-week diabetic rat lumbar spinal cord, compared to non-diabetic spinal cord (Fig. 4A). Phosphorylation of GSK3β, a downstream element of the PI3K pathway, was reduced by 10% (p=0.054) at the serine 9 inactivating site. Reduced phosphorylation of GSK3β represents increased activity of this kinase, as confirmed by the 16% (p=0.053) increase of GSK3β phosphorylation at the tyrosine 216 activating site (Fig. 4B), while total GSK3 protein levels were unchanged.
Fig. 4.
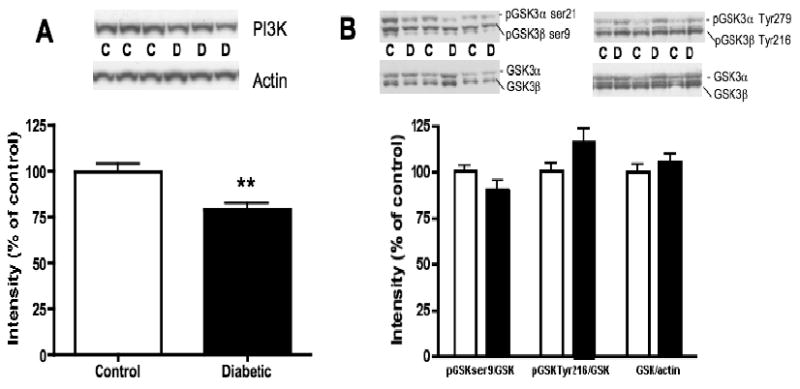
Protein levels of PI3K and phosphorylation levels of GSK3 in lumbar spinal cord of non-diabetic control (C) and 20-week diabetic (D) rats. A: Intensity of bands corresponding to PI3K normalized to the intensity of bands corresponding to actin. B: Intensity of bands corresponding to phosphorylated GSK3 at ser9 (inactivating site) and at Tyr 216 (activating site) normalized to the intensity of bands corresponding to total GSK3. Bands corresponding to total GSK3 were normalized to bands corresponding to actin. Data are represented as mean + SEM, n=6, **p<0.01 using unpaired, two-tailed t-test.
2.5. Microtubule-associated proteins in diabetic rat spinal cord
In lumbar spinal cord from 20-week diabetic rats, western-blot analysis of tau showed a 55% increase (p<0.05) of its phosphorylation at threonine 231, a site phosphorylated by GSK3, compared to cord from non-diabetic rats (Fig. 5A). A 3-fold increase (p<0.05) in Microtubule-Associated Protein 2A/B (MAP2A/B) expression was also observed in lumbar spinal cord from 20-week diabetic rats, compared to that from non-diabetic rats (Fig. 5B), whereas in 8-week diabetic rats, MAP 2A/B protein levels were undetectable in both non-diabetic and diabetic rat spinal cord homogenates (data not shown). MAP 2C/D proteins were detectable after 8 and 20 weeks of diabetes but expression levels were not different between non-diabetic and diabetic rats at both time points (data not shown).
Fig. 5.
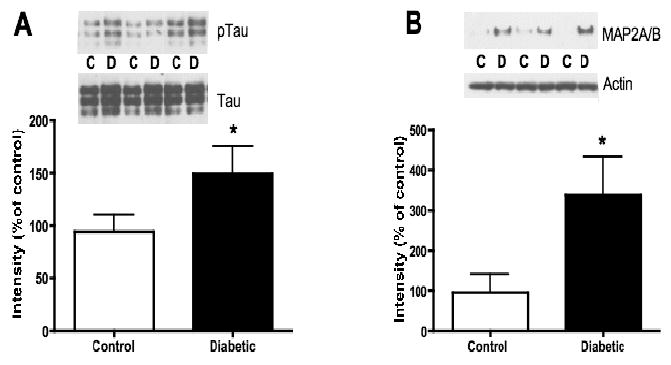
Phosphorylation levels of tau and protein level of MAP2A/B in lumbar spinal cord of non-diabetic control (C) and 20-week diabetic (D) groups. A: Intensity of bands corresponding to phosphorylated tau (threonine 231) normalized to the intensity of bands corresponding to total tau. B: Intensity of bands corresponding to MAP2A/B normalized to the intensity of bands corresponding to actin. Data are represented as mean + SEM, n=6, *p<0.05 using unpaired, two-tailed t-test.
2.6. Synaptic proteins in diabetic rat spinal cord
Compared to non-diabetic rats, the presynaptic protein, synaptophysin, was significantly (p<0.05) reduced by 31% in 20-week diabetic rat lumbar spinal cord (Fig. 6A). A 17% reduction (p=0.060) in drebrin, a postsynaptic dendritic spine protein, was also observed in lumbar spinal cord from 20-week diabetic rats, compared to non-diabetic rats (Fig. 6B). Synaptophysin and drebrin protein levels were similar in non-diabetic and 8-week diabetic rat spinal cord (data not shown).
Fig. 6.
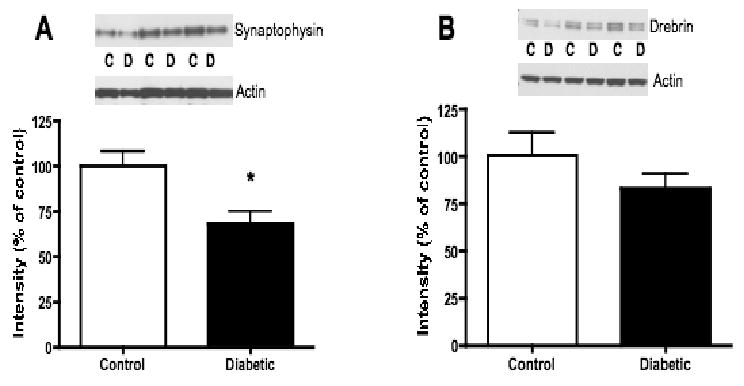
Protein levels of synaptophysin and drebrin in lumbar spinal cord of non-diabetic control (C) and 20-week diabetic (D) groups. A: Intensity of bands corresponding to synaptophysin normalized to the intensity of bands corresponding to actin. B: Intensity of bands corresponding to drebrin normalized to the intensity of bands corresponding to actin. Data are represented as mean + SEM, n=6, *p<0.05 using unpaired, two-tailed t-test.
3. Discussion
Here we have examined the impact of diabetes on the expression of ILK, PINCH, components of signaling pathways involved in regulating neuronal cytoskeletal stability (PI-3K and GSK-3β), and markers of the neuronal cytoskeleton (tau and MAP2) as well as dendritic and synaptic organization (synaptophysin and drebrin) in the spinal cord. PINCH and ILK were colocalized in neurons and oligodendrocytes of lumbar spinal cord, consistent with previous studies where ILK immunoreactivity was demonstrated in neuronal cell bodies and dendrites (Mills et al., 2003), and in oligodendrocytes in white matter tracts (Chun et al., 2003). While diabetes did not alter the cellular distribution of PINCH and ILK in the lumbar spinal cord, expression of these proteins was significantly reduced in insulin-deficient diabetic rats, as we have shown previously for other proteins, such as the potassium-chloride transporter KCC2 (Jolivalt et al., 2008b). The impact of diabetes on the proteins studied was duration dependent with changes in protein expression observed only after 20 weeks of hyperglycemia, suggesting these changes are not likely to account for altered behavioral responses to thermal, tactile and chemical stimuli that appear after 4-8 weeks of diabetes. Several studies have suggested that streptozotocin diabetes induces presynaptic and postsynaptic neuronal modifications in rat hippocampus (Biessels et al., 1998; Grillo et al., 2005; Magarinos et al., 1997; Magarinos and McEwen, 2000; Magarinos et al., 2001), which may be indicative of ongoing changes in synaptic plasticity. Our current study documents changes in dendritic (MAP2, drebrin) and synaptic (synaptophysin) proteins in the spinal cord of streptozotocin-diabetic rats, indicating possible diabetes-induced alterations in spinal cord plasticity.
The basis for the diabetes-induced reduction in ILK and PINCH observed here in the lumbar spinal cord is unknown, although altered neurotrophic support and/or oxidative stress may be involved. Growth factor support, in particular insulin, is deficient in type 1 diabetes (Sima, 2003) and might lead, via a reduced stimulation of the PI3K pathway, to reduced ILK activity (Attwell et al., 2003; Delcommenne et al., 1998; Mills et al., 2003). Attenuated PI-3K pathway activity is not only reduced in diabetic spinal cord, but also in the vagus nerve and vagus afferent neurons in 16-week diabetic rats (Cai et al., 2003) as well as in the brain of diabetic mice, where reductions result from impaired insulin signaling via the insulin receptor pathway (Jolivalt et al., 2008a). Increased oxidative stress plays also an important role in diabetic neuropathy (Pop-Busui et al., 2006). As oxidative stress impacts ILK expression in neuronal cells after ischemia (Saito et al., 2006), the oxidative stress associated with diabetes may also contribute to a reduction of ILK in the spinal cord of diabetic rats observed in this study.
Although ILK (Chun et al., 2003; Pereira et al., 2009) and integrins (Baron et al., 2005; Relvas et al., 2001) are essential for myelin formation during development, a function for ILK and integrin related to myelination in adult nerve has not been evaluated. While autopsy studies of diabetic patients reveal demyelination in white matter of the spinal cord (Deng et al., 2002; Reske-Nielsen and Lundbaek, 1968; Slager and Webb, 1973; Slager, 1978), demyelination in the cord is not a feature of experimental diabetes (Mizisin AP, 2007), prompting us to focus on other downstream pathways as well as markers of the neuronal cytoskeleton, and dendritic and synaptic organization. Among several other targets, GSK3 is a downstream effector of ILK. A reduced level of ILK in diabetic rat spinal cord may lead to a reduced inactivating phosphorylation of GSK3, resulting in its activation. Active GSK3 phosphorylates, among other proteins, the microtubule-associated protein, tau, resulting in its impaired capacity to bind to axonal microtubules ((Hong et al., 1997; Hong and Lee, 1997). Consistent with a reduced ILK expression and with our findings in the brain of STZ-diabetic mice (Jolivalt et al., 2008a), GSK3 activity tends to be increased and tau phosphorylation is increased in 20-week diabetic rat lumbar spinal cord. Our data are supported by in vitro studies showing increased GSK3 activity leading to tau hyperphosphorylation after inhibition of ILK in neuronal cells (Mills et al., 2003) or in a dominant-negative ILK neuroblastoma cell line (Ishii et al., 2003).
The demonstration that ILK-GSK3 pathway is central to the regulation of dendrite formation (Naska et al., 2006) prompted us to study dendritic and synaptic proteins in the spinal cord of diabetic rats. Complementary to tau's role in neuronal plasticity, high molecular weight MAP2 (MAP2A/B) promotes assembly and stability of microtubules in dendrites (Caceres et al., 1983; Matus, 1988; Matus, 1994). As in rat hippocampus (Grillo et al., 2005), MAP2 protein expression is significantly increased in lumbar spinal cord of diabetic rats, consistent with an increased expression of genes related to cytoskeletal organization and biogenesis, and microtubule-based processes in diabetic rat DRG (Price et al., 2006). Similar to the plastic reorganization that takes place in the hippocampus of diabetic rats (Grillo et al., 2005), the increased expression of MAP2 may be indicative of an early phase of dendritic retraction, or defective organization of the cytoskeleton in the spinal cord of diabetic rats. In addition to their related sequences, MAP2 can be sequestered by hyperphosphorylated tau, as shown in Alzheimer's disease (Alonso et al., 1997), and thus detached from microtubules. Sequestration of MAP2 may contribute to increased microtubule dynamics and thereby induce aberrant remodeling of dendrites, as suggested by the reduced complexity of the apical dendritic arborization in the hippocampus of streptozotocin-diabetic rats (Magarinos et al., 2001) and as described in the brain in early Alzheimer's disease (Arendt, 2001).
Changes in neuroplasticity in diabetic rat lumbar spinal cord are also supported by a reduction of drebrin protein levels, which may contribute to destabilization of the cytoskeleton, as this protein is involved in the control of actin dynamics (Hayashi et al., 1996; Shirao and Obata, 1985). Synaptophysin protein levels, a marker of synaptic integrity, are decreased in lumbar spinal cord from 20-week diabetic rats, as in the brain of genetically aged diabetic rats (Li et al., 2007) and diabetic mice displaying cognitive deficits (Francis et al., 2008). In contrast, synaptophysin mRNA levels are significantly increased in early diabetes in the brain and DRG of rats (Grillo et al., 2005; Price et al., 2006). This difference disappeared after 8 weeks of diabetes (Price et al., 2006), suggesting an early compensatory increase in synaptophysin protein levels followed by a failure to compensate that, with the changes in other protein levels demonstrated in this study, may lead to a loss of synapse integrity in the diabetic rat spinal cord.
In summary, reduction of ILK and PINCH protein levels in diabetic spinal cord may lead to tau hyperphosphorylation, MAP2 overexpression, and reduction of synaptic proteins, suggesting a role for ILK-PINCH in the stabilization of axonal and dendritic structures. Presently it is not clear if these changes in protein expression precede, are co-incident with or are a consequence of alterations in synaptic ultrastrastructure. Further studies using insulin treatment and inhibitors of the signaling pathways explored here are clearly warranted. Nevertheless, altered expression of the proteins studied after 20 weeks of diabetes is not likely to account for the abnormal behavioral responses to thermal, tactile and chemical stimuli that appear after 4 to 8 weeks of diabetes. Finally, insulin-deficient diabetes may impair neuronal plasticity at the spinal cord level by precipitating long-term structural alterations in patients with well-established diabetes.
4. Experimental procedures
4.1. Animals
The study was performed using adult female Sprague-Dawley rats (Harlan Industries, San Diego, CA). Animals were housed 2-3 per cage with free access to food and water, and maintained in a vivarium approved by the American Association for the Accreditation of Laboratory Animal Care. The study was carried out according to protocol approved by the Institutional Animal Care and Use Committee of the University of California, San Diego in accordance with the National Institutes for Health guide, ‘Principles of Laboratory Animal Care, 1985 revised version’.
4.2. Induction of diabetes
All procedures have been described in detail elsewhere (Calcutt, 2004). Briefly, insulin-deficient diabetes was induced following an overnight fast by a single intra-peritoneal injection (50 mg/kg dissolved in 0.9% sterile saline) of streptozotocin (Sigma, St. Louis. MO, USA). Hyperglycemia (>15 mmol/l or >270 mg/dl) was confirmed 4 days after streptozotocin injection and at the end of the study using a strip-operated reflectance meter in a blood sample obtained by tail prick. All streptozotocin-treated rats exhibited blood glucose levels higher than 270 mg/dl on these two occasions. Animals were observed daily and weighed regularly during the 8- or 20-week study period. The 8-week time point was chosen to represent an early stage of diabetes where tactile allodynia and formalin hyperalgesia are present, while the 20-week time-point was chosen to represent a more chronic diabetes where all these behaviors, including thermal hypoalgesia, are well-established.
4.3. Western blotting
After 8 or 20 weeks of diabetes, rats were sacrificed by decapitation under isoflurane anesthesia. Spinal cords were obtained by hydraulic extrusion with saline and the lumbar enlargements were homogenized in buffer (50 mM Tris-HCl pH7.4, 150 mM NaCl, 0.5% Triton X, 1 mM EDTA, protease inhibitor cocktail), centrifuged (13 000 ×g, 4°C) and stored in aliquots at -80°C. Lumbar spinal cord was chosen because sensory afferents innervating the hindlimbs and feet synapse in the lumbar dorsal horn. Protein concentration was assessed using the bicinchoninic acid method (BCA protein assay kit, Pierce, Rockford, IL, USA). Five to 20 μg of the clear extract was prepared in Laemmli LDS sample buffer (Invitrogen, Carlsbad, CA, USA). A fraction of the crude homogenate was boiled for 5 minutes under detergent-free conditions and insoluble material removed from the heat-stable supernatant by centrifugation for 30 minutes. Heat-stable supernatants (5μg protein) were prepared with an equal volume of Laemmli SDS sample buffer for western-blotting of tau. Proteins were then separated on 4-12% Bis-Tris gels (Novex, Invitrogen, Carlsbad, CA, USA) and immunoblotted on nitrocellulose. Membranes were incubated with antibodies to ILK, PINCH, PI-3K, phosphorylated GSK3 α/β, phosphorylated GSK3 α/β activating site, GSK3 α/β, phosphorylated tau and Tau-5, MAP2, synaptophysin, drebrin, cyclophilin B, or actin (Table 1), followed by incubation with horseradish peroxidase-linked anti-rabbit or anti-mouse secondary antibody (1:10 000, Santa Cruz Biotechnology, Santa Cruz, CA, USA).
Table 1.
Primary antibodies used for western blotting and immunocytochemistry.
| Antibody | Dilution | Protein/Marker | Source |
|---|---|---|---|
| Western blotting | |||
| Anti-ILK (rabbit polyclonal) | 1:1000 | ILK | Upstate, Temecula, CA |
| Anti-PI-3K (rabbit polyclonal) | 1:5000 | PI-3K p85 subunit | Upstate, Temecula, CA |
| Anti-phosphorylated GSK3 α/β | 1:1000 | GSK3 α/β phospho Ser21/Ser9 | Cell Signaling Technology, Beverly, MA |
| Anti-phosphorylated GSK3 α/β activating site | 1:1000 | GSK3 α/β activating site phospho Tyr279/Tyr216 | Abcam Inc., Cambridge, MA |
| Anti-phosphorylated tau/Tau-5 | 1:3000 | Tau/tau-5 phosph Thr231 | Biosource, Camarillo, CA |
| Anti-GSK3 α/β | 1:10000 | GSK3 α/β | Upstate, Temecula, CA |
| Anti-MAP2 | 1:500 | MAP2 | Cell Signaling Technology, Beverly, MA |
| Anti-Synaptophysin | 1:10000 | Synaptic organization | Chemicon International, Temecula, CA |
| Anti-Drebrin | 1:1000 | Dendrite organization | Abcam Inc., Cambridge, MA |
| Anti-Actin | 1:2000 | Actin | Sigma, St. Louis, MO |
| Anti-Cyclophilin | 1:5000 | Cyclophilin | Abcam Inc., Cambridge, MA |
| Immunocytochemistry | |||
| Anti-ILK (mouse monoclonal) | 1:10 | ILK | Upstate, Temecula, CA |
| Anti-PINCH (rabbit polyclonal) | 1:500 | PINCH | Ann Rearden |
| Anti-NeuN (rat monoclonal MAB377) | 1:2000 | Neurons | Chemicon International, Temecula, CA |
| Anti-GFAP (mouse monoclonal MAB360) | 1:2000 | Astrocytes | Chemicon International, Temecula, CA |
| Anti-APC-Ab7 (OP80) | 1:500 | Oligodendrocytes | Calbiochem, San Diego, CA |
| Anti-CD11b (ARS1122) | 1:50 | Microglia | Biosource, Camarillo, CA |
Blots were developed using an enhanced-chemiluminescence western-blot protocol (Amersham Pharmacia Biotech, Little Chalfont, UK). For sequential analysis of western-blot membranes, previously bound antibodies were removed with stripping buffer (0.2-mol/l glycine, pH 2.5, 0.05% Tween 20). Quantification of immunoreactivity was performed by densitometric scanning using Quantity One software (BioRad, San Diego, CA, USA). For each animal, band intensities were normalized by calculating the ratio of the intensity of the band corresponding to the protein of interest to the density of the band corresponding to actin. To allow grouping of samples run on different gels, actin-normalized densitometric measures of band density for each animal were expressed as a percentage of the group mean of all samples from control rats present on the same gel.
4.4. Immunocytochemistry
Non-diabetic and diabetic rats were anesthetised by an intraperitoneal injection (2 ml/kg) of a cocktail containing pentobarbital (12.5mg/ml) and diazepam (1.25mg/ml) in sterile saline. While deeply anesthetized, cardiac perfusion was performed using 50 ml of 0.9% saline, followed by 500 ml of 4% paraformaldehyde in 0.1 M phosphate buffered saline (PBS) (pH 7.4). Immediately after the perfusion, lumbar spinal cord was dissected and post-fixed in 4% paraformaldehyde at 4°C for 3 hours, cryoprotected by immersion in 0.1 M PBS (pH 7.4) containing 30% sucrose for 48 hours at 4°C, embedded in OCT medium, and stored at -20°C.
To minimize effects resulting from variability in stain intensity, non-diabetic and diabetic tissues were stained side-by-side at the same time. Floating 14-μm-thick sections of spinal cords were immunostained prior to mounting on superfrost slides. The presence and cellular distribution of PINCH and ILK in non-diabetic and diabetic animals was compared using immunofluorescence. After washing with 0.05 M Tris buffer solution (TBS) (pH 7.6), sections were blocked with 10% normal goat serum (NGS) containing 0.3% Triton-X. Thereafter, sections were incubated with primary antibodies to ILK and PINCH (Table 1) diluted in a medium containing 5% NGS and 0.3% Triton-X overnight at 4°C. Sections were then incubated with goat anti-mouse Alexa-594 (1:500; Molecular Probes, Eugene, OR, USA) or goat anti-rabbit Alexa-488 (1:500; Molecular Probes, Eugene, OR, USA) for 2 hours. All sections were mounted in anti-fade mounting medium (Molecular Probes), cover-slipped and examined using fluorescence microscopy (Olympus BX41).
To identify the ILK- and PINCH-immunoreactive cell types in spinal cord, double immunofluorescent labeling was performed with antibodies against NeuN, GFAP, APC-Ab7 and CD11b, markers of neurons, astrocytes, oligodendrocytes, and microglia, respectively (Table 1). Double immunofluorescent labeling was also performed to identify the co-expression of ILK and PINCH (Table 1). After overnight incubation with ILK antibodies at 4°C, sections were next incubated with goat anti-mouse Alexa-594 (1:500; Molecular Probes, Eugene, OR) for 2 hours. Sections were then incubated with the PINCH antibody overnight at 4°C, followed by incubation with goat anti-rabbit Alexa-488 (1:500; Molecular Probes).
4.5. Statistical analysis
Data are expressed as group mean ± SEM and differences between non-diabetic and diabetic groups were analyzed by two-tailed, unpaired t-tests.
Acknowledgments
This work was supported by the JDRF (CGJ and APM) and a NIH grant DK078374 (APM). The helpful assistance of Mario Brock with immunocytochemistry is gratefully acknowledged.
Abbreviations
- DRG
dorsal root ganglia
- fMRI
functional magnetic resonance imaging
- GSK-3β
glycogen-synthase kinase 3β
- ILK
integrin-linked kinase
- MAP2
microtubule-associated protein 2
- PINCH
Particularly Interesting New Cystine-Histine rich protein
- PI3K
phosphoinositide 3-kinase
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ahroni JH, Boyko EJ, Davignon DR, Pecoraro RE. The health and functional status of veterans with diabetes. Diabetes Care. 1994;17:318–21. doi: 10.2337/diacare.17.4.318. [DOI] [PubMed] [Google Scholar]
- Alonso AD, Grundke-Iqbal I, Barra HS, Iqbal K. Abnormal phosphorylation of tau and the mechanism of Alzheimer neurofibrillary degeneration: sequestration of microtubule-associated proteins 1 and 2 and the disassembly of microtubules by the abnormal tau. Proc Natl Acad Sci U S A. 1997;94:298–303. doi: 10.1073/pnas.94.1.298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arendt T. Disturbance of neuronal plasticity is a critical pathogenetic event in Alzheimer's disease. Int J Dev Neurosci. 2001;19:231–45. doi: 10.1016/s0736-5748(01)00007-7. [DOI] [PubMed] [Google Scholar]
- Attwell S, Mills J, Troussard A, Wu C, Dedhar S. Integration of cell attachment, cytoskeletal localization, and signaling by integrin-linked kinase (ILK), CH-ILKBP, and the tumor suppressor PTEN. Mol Biol Cell. 2003;14:4813–25. doi: 10.1091/mbc.E03-05-0308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baron W, Colognato H, ffrench-Constant C. Integrin-growth factor interactions as regulators of oligodendroglial development and function. Glia. 2005;49:467–79. doi: 10.1002/glia.20132. [DOI] [PubMed] [Google Scholar]
- Biessels GJ, Kamal A, Urban IJ, Spruijt BM, Erkelens DW, Gispen WH. Water maze learning and hippocampal synaptic plasticity in streptozotocin-diabetic rats: effects of insulin treatment. Brain Res. 1998;800:125–35. doi: 10.1016/s0006-8993(98)00510-1. [DOI] [PubMed] [Google Scholar]
- Boulton AJ, Armstrong WD, Scarpello JH, Ward JD. The natural history of painful diabetic neuropathy--a 4-year study. Postgrad Med J. 1983;59:556–9. doi: 10.1136/pgmj.59.695.556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caceres A, Payne MR, Binder LI, Steward O. Immunocytochemical localization of actin and microtubule-associated protein MAP2 in dendritic spines. Proc Natl Acad Sci U S A. 1983;80:1738–42. doi: 10.1073/pnas.80.6.1738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai J, Ahmad S, Jiang WG, Huang J, Kontos CD, Boulton M, Ahmed A. Activation of vascular endothelial growth factor receptor-1 sustains angiogenesis and Bcl-2 expression via the phosphatidylinositol 3-kinase pathway in endothelial cells. Diabetes. 2003;52:2959–68. doi: 10.2337/diabetes.52.12.2959. [DOI] [PubMed] [Google Scholar]
- Calcutt NA. Potential mechanisms of neuropathic pain in diabetes. Int Rev Neurobiol. 2002;50:205–28. doi: 10.1016/s0074-7742(02)50078-7. [DOI] [PubMed] [Google Scholar]
- Calcutt NA. Modeling diabetic sensory neuropathy in rats. Methods Mol Med. 2004;99:55–65. doi: 10.1385/1-59259-770-x:225. [DOI] [PubMed] [Google Scholar]
- Chun SJ, Rasband MN, Sidman RL, Habib AA, Vartanian T. Integrin-linked kinase is required for laminin-2-induced oligodendrocyte cell spreading and CNS myelination. J Cell Biol. 2003;163:397–408. doi: 10.1083/jcb.200304154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delcommenne M, Tan C, Gray V, Rue L, Woodgett J, Dedhar S. Phosphoinositide-3-OH kinase-dependent regulation of glycogen synthase kinase 3 and protein kinase B/AKT by the integrin-linked kinase. Proc Natl Acad Sci U S A. 1998;95:11211–6. doi: 10.1073/pnas.95.19.11211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng JT, Sutherland C, Brautigan DL, Eto M, Walsh MP. Phosphorylation of the myosin phosphatase inhibitors, CPI-17 and PHI-1, by integrin-linked kinase. Biochem J. 2002;367:517–24. doi: 10.1042/BJ20020522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Downward J. Signal transduction. New exchange, new target. Nature. 1998;396:416–7. doi: 10.1038/24743. [DOI] [PubMed] [Google Scholar]
- Eaton SE, Harris ND, Rajbhandari SM, Greenwood P, Wilkinson ID, Ward JD, Griffiths PD, Tesfaye S. Spinal-cord involvement in diabetic peripheral neuropathy. Lancet. 2001;358:35–6. doi: 10.1016/S0140-6736(00)05268-5. [DOI] [PubMed] [Google Scholar]
- Filipenko NR, Attwell S, Roskelley C, Dedhar S. Integrin-linked kinase activity regulates Rac- and Cdc42-mediated actin cytoskeleton reorganization via alpha-PIX. Oncogene. 2005;24:5837–49. doi: 10.1038/sj.onc.1208737. [DOI] [PubMed] [Google Scholar]
- Francis GJ, Martinez JA, Liu WQ, Xu K, Ayer A, Fine J, Tuor UI, Glazner G, Hanson LR, Frey WH, 2nd, Toth C. Intranasal insulin prevents cognitive decline, cerebral atrophy and white matter changes in murine type I diabetic encephalopathy. Brain. 2008;131:3311–34. doi: 10.1093/brain/awn288. [DOI] [PubMed] [Google Scholar]
- Grillo CA, Piroli GG, Wood GE, Reznikov LR, McEwen BS, Reagan LP. Immunocytochemical analysis of synaptic proteins provides new insights into diabetes-mediated plasticity in the rat hippocampus. Neuroscience. 2005;136:477–86. doi: 10.1016/j.neuroscience.2005.08.019. [DOI] [PubMed] [Google Scholar]
- Hannigan GE, Leung-Hagesteijn C, Fitz-Gibbon L, Coppolino MG, Radeva G, Filmus J, Bell JC, Dedhar S. Regulation of cell adhesion and anchorage-dependent growth by a new beta 1-integrin-linked protein kinase. Nature. 1996;379:91–6. doi: 10.1038/379091a0. [DOI] [PubMed] [Google Scholar]
- Hannigan GE, Dedhar S. Protein kinase mediators of integrin signal transduction. J Mol Med. 1997;75:35–44. doi: 10.1007/s001090050084. [DOI] [PubMed] [Google Scholar]
- Hayashi K, Ishikawa R, Ye LH, He XL, Takata K, Kohama K, Shirao T. Modulatory role of drebrin on the cytoskeleton within dendritic spines in the rat cerebral cortex. J Neurosci. 1996;16:7161–70. doi: 10.1523/JNEUROSCI.16-22-07161.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong M, Chen DC, Klein PS, Lee VM. Lithium reduces tau phosphorylation by inhibition of glycogen synthase kinase-3. J Biol Chem. 1997;272:25326–32. doi: 10.1074/jbc.272.40.25326. [DOI] [PubMed] [Google Scholar]
- Hong M, Lee VM. Insulin and insulin-like growth factor-1 regulate tau phosphorylation in cultured human neurons. J Biol Chem. 1997;272:19547–53. doi: 10.1074/jbc.272.31.19547. [DOI] [PubMed] [Google Scholar]
- Ishii T, Furuoka H, Muroi Y, Nishimura M. Inactivation of integrin-linked kinase induces aberrant tau phosphorylation via sustained activation of glycogen synthase kinase 3beta in N1E-115 neuroblastoma cells. J Biol Chem. 2003;278:26970–5. doi: 10.1074/jbc.M304113200. [DOI] [PubMed] [Google Scholar]
- Jolivalt CG, Lee CA, Beiswenger KK, Smith JL, Orlov M, Torrance MA, Masliah E. Defective insulin signaling pathway and increased glycogen synthase kinase-3 activity in the brain of diabetic mice: parallels with Alzheimer's disease and correction by insulin. J Neurosci Res. 2008a;86:3265–74. doi: 10.1002/jnr.21787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jolivalt CG, Lee CA, Ramos KM, Calcutt NA. Allodynia and hyperalgesia in diabetic rats are mediated by GABA and depletion of spinal potassium-chloride co-transporters. Pain. 2008b;140:48–57. doi: 10.1016/j.pain.2008.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King WG, Mattaliano MD, Chan TO, Tsichlis PN, Brugge JS. Phosphatidylinositol 3-kinase is required for integrin-stimulated AKT and Raf-1/mitogen-activated protein kinase pathway activation. Mol Cell Biol. 1997;17:4406–18. doi: 10.1128/mcb.17.8.4406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kok K, Geering B, Vanhaesebroeck B. Regulation of phosphoinositide 3-kinase expression in health and disease. Trends Biochem Sci. 2009;34:115–27. doi: 10.1016/j.tibs.2009.01.003. [DOI] [PubMed] [Google Scholar]
- Li ZG, Zhang W, Sima AA. Alzheimer-like changes in rat models of spontaneous diabetes. Diabetes. 2007;56:1817–24. doi: 10.2337/db07-0171. [DOI] [PubMed] [Google Scholar]
- Magarinos AM, Verdugo JM, McEwen BS. Chronic stress alters synaptic terminal structure in hippocampus. Proc Natl Acad Sci U S A. 1997;94:14002–8. doi: 10.1073/pnas.94.25.14002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magarinos AM, McEwen BS. Experimental diabetes in rats causes hippocampal dendritic and synaptic reorganization and increased glucocorticoid reactivity to stress. Proc Natl Acad Sci U S A. 2000;97:11056–61. doi: 10.1073/pnas.97.20.11056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magarinos AM, Jain K, Blount ED, Reagan L, Smith BH, McEwen BS. Peritoneal implantation of macroencapsulated porcine pancreatic islets in diabetic rats ameliorates severe hyperglycemia and prevents retraction and simplification of hippocampal dendrites. Brain Res. 2001;902:282–7. doi: 10.1016/s0006-8993(01)02400-3. [DOI] [PubMed] [Google Scholar]
- Malisza KL, Jones C, Gruwel ML, Foreman D, Fernyhough P, Calcutt NA. Functional magnetic resonance imaging of the spinal cord during sensory stimulation in diabetic rats. J Magn Reson Imaging. 2009;30:271–6. doi: 10.1002/jmri.21856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matus A. Microtubule-associated proteins: their potential role in determining neuronal morphology. Annu Rev Neurosci. 1988;11:29–44. doi: 10.1146/annurev.ne.11.030188.000333. [DOI] [PubMed] [Google Scholar]
- Matus A. Stiff microtubules and neuronal morphology. Trends Neurosci. 1994;17:19–22. doi: 10.1016/0166-2236(94)90030-2. [DOI] [PubMed] [Google Scholar]
- Mills J, Digicaylioglu M, Legg AT, Young CE, Young SS, Barr AM, Fletcher L, O'Connor TP, Dedhar S. Role of integrin-linked kinase in nerve growth factor-stimulated neurite outgrowth. J Neurosci. 2003;23:1638–48. doi: 10.1523/JNEUROSCI.23-05-01638.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizisin AP, Jolivalt C, Calcutt NA. Spinal cord, structure and function in diabetes. In: Veves A MR, editor. Diabetic neuropathy. Clinical management. 2nd. Humana Press; Totowa: 2007. pp. 165–185. [Google Scholar]
- Naska S, Park KJ, Hannigan GE, Dedhar S, Miller FD, Kaplan DR. An essential role for the integrin-linked kinase-glycogen synthase kinase-3 beta pathway during dendrite initiation and growth. J Neurosci. 2006;26:13344–56. doi: 10.1523/JNEUROSCI.4462-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Partanen J, Niskanen L, Lehtinen J, Mervaala E, Siitonen O, Uusitupa M. Natural history of peripheral neuropathy in patients with non-insulin-dependent diabetes mellitus. N Engl J Med. 1995;333:89–94. doi: 10.1056/NEJM199507133330203. [DOI] [PubMed] [Google Scholar]
- Pereira JA, Benninger Y, Baumann R, Goncalves AF, Ozcelik M, Thurnherr T, Tricaud N, Meijer D, Fassler R, Suter U, Relvas JB. Integrin-linked kinase is required for radial sorting of axons and Schwann cell remyelination in the peripheral nervous system. J Cell Biol. 2009;185:147–61. doi: 10.1083/jcb.200809008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pop-Busui R, Sima A, Stevens M. Diabetic neuropathy and oxidative stress. Diabetes Metab Res Rev. 2006;22:257–73. doi: 10.1002/dmrr.625. [DOI] [PubMed] [Google Scholar]
- Price SA, Zeef LA, Wardleworth L, Hayes A, Tomlinson DR. Identification of changes in gene expression in dorsal root ganglia in diabetic neuropathy: correlation with functional deficits. J Neuropathol Exp Neurol. 2006;65:722–32. doi: 10.1097/01.jnen.0000228199.89420.90. [DOI] [PubMed] [Google Scholar]
- Qian Y, Zhong X, Flynn DC, Zheng JZ, Qiao M, Wu C, Dedhar S, Shi X, Jiang BH. ILK mediates actin filament rearrangements and cell migration and invasion through PI3K/Akt/Rac1 signaling. Oncogene. 2005;24:3154–65. doi: 10.1038/sj.onc.1208525. [DOI] [PubMed] [Google Scholar]
- Rearden A. A new LIM protein containing an autoepitope homologous to “senescent cell antigen”. Biochem Biophys Res Commun. 1994;201:1124–31. doi: 10.1006/bbrc.1994.1822. [DOI] [PubMed] [Google Scholar]
- Relvas JB, Setzu A, Baron W, Buttery PC, LaFlamme SE, Franklin RJ, ffrench-Constant C. Expression of dominant-negative and chimeric subunits reveals an essential role for beta1 integrin during myelination. Curr Biol. 2001;11:1039–43. doi: 10.1016/s0960-9822(01)00292-5. [DOI] [PubMed] [Google Scholar]
- Reske-Nielsen E, Lundbaek K. Pathological changes in the central and peripheral nervous system of young long-term diabetics. II. The spinal cord and peripheral nerves. Diabetologia. 1968;4:34–43. doi: 10.1007/BF01241031. [DOI] [PubMed] [Google Scholar]
- Selvarajah D, Wilkinson ID, Emery CJ, Harris ND, Shaw PJ, Witte DR, Griffiths PD, Tesfaye S. Early involvement of the spinal cord in diabetic peripheral neuropathy. Diabetes Care. 2006;29:2664–9. doi: 10.2337/dc06-0650. [DOI] [PubMed] [Google Scholar]
- Shaw PJ, Zimmet PZ, Gries FA, Ziegler D. Epidemiology of diabetic neuropathy. In: Gries FA, Cameron NE, Low PA, Ziegler D, editors. Textbook of diabetic neuropathy. Thieme; Stuttgart: 2003. pp. 64–82. [Google Scholar]
- Shirao T, Obata K. Two acidic proteins associated with brain development in chick embryo. J Neurochem. 1985;44:1210–6. doi: 10.1111/j.1471-4159.1985.tb08745.x. [DOI] [PubMed] [Google Scholar]
- Sidenius P, Jakobsen J. Reduced perikaryal volume of lower motor and primary sensory neurons in early experimental diabetes. Diabetes. 1980;29:182–6. doi: 10.2337/diab.29.3.182. [DOI] [PubMed] [Google Scholar]
- Slager UT, Webb AT. Pathologic findings in the spinal cord. Arch Pathol. 1973;96:388–94. [PubMed] [Google Scholar]
- Slager UT. Diabetic myelopathy. Arch Pathol Lab Med. 1978;102:467–9. [PubMed] [Google Scholar]
- Thomas PK, Tomlinson D. Diabetic and hypoglycemic neuropaty. In: Dyck TP, J P, Griffin JN, Low PA, Podusto JF, editors. Peripheral neuropathy. 3rd. WB Saunders; Philadelphia: 1993. pp. 1219–1250. [Google Scholar]
- Wu C, Dedhar S. Integrin-linked kinase (ILK) and its interactors: a new paradigm for the coupling of extracellular matrix to actin cytoskeleton and signaling complexes. J Cell Biol. 2001;155:505–10. doi: 10.1083/jcb.200108077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaji S, Suzuki A, Kanamori H, Mishima W, Yoshimi R, Takasaki H, Takabayashi M, Fujimaki K, Fujisawa S, Ohno S, Ishigatsubo Y. Affixin interacts with alpha-actinin and mediates integrin signaling for reorganization of F-actin induced by initial cell-substrate interaction. J Cell Biol. 2004;165:539–51. doi: 10.1083/jcb.200308141. [DOI] [PMC free article] [PubMed] [Google Scholar]