

Abstract
Comparison between a series of pyrrolo[2,3-d]pyrimidines with and without the 2-amino group is presented in order to determine the validity of our hypothesis that inclusion of this group improves potency against receptor tyrosine kinases (RTK). The 2-amino analogs were better against epidermal growth factor receptor (EGFR) and platelet derived growth factor-β (PDGFR-β) in whole cell inhibition assays and in the A431 cytotoxicity assay compared to the 2-desamino analogs. However, the 2-desamino analogs were more potent inhibitors against vascular endothelial growth factor-2 (VEGFR-2) than the corresponding 2-amino compounds. In addition, none of the 2-desamino compounds exhibited better anti-angiogenic activity in the chorioallantoic membrane (CAM) assay as compared to the standard and were only micromolar inhibitors. This study validates our original hypothesis that the inclusion of a 2-amino group in pyrrolo[2,3-d]pyrimidines improves multiple RTK inhibition and antiangiogenic activity.
Receptor tyrosine kinases (RTKs) are a subfamily of protein tyrosine kinases, which play key roles in tumor growth, survival and dissemination.1 A variety of growth factors particularly vascular endothelial growth factor (VEGF), epidermal growth factor (EGF), platelet derived growth factor (PDGF) and their receptors are overexpressed in several tumors. These growth factors and their receptors are directly or indirectly involved in the growth and metastasis of tumors.2
Angiogenesis is the formation of new blood vessels from existing vasculature and is essential for both physiological and pathological processes. It is a complex cascade that is tightly regulated by proangiogenic and antiangiogenic factors.3 Members of the VEGF family are the predominant stimulators of angiogenesis and mediation of VEGF expression is one of the main mechanisms by which tissue vasculature is controlled under normal physiologic conditions.4–6 In majority of cancers, in addition to this PDGF subfamilies appear to play essential roles in all stages of tumor angiogenesis and are able to form autocrine loops, which mediate cancer cell growth and survival, and drive hematologic malignancies.7 Angiogenesis is a pivotal step in the transition of some solid tumors from a dormant state to a malignant state; it also provides a metastatic pathway for solid tumors.8 In addition, angiogenesis contributes to the development of hematologic malignancies, particularly multiple myeloma, leukemia, and lymphoma. Inhibition of tumor angiogenesis affords attractive targets for the development of antitumor agents.
A multifaceted approach that targets multiple signaling pathways has been shown to be more effective than the inhibition of a single target.9–11 The most important consequence of inhibiting multiple RTKs would be to retard tumor resistance by blocking potential “escape routes.”12
Several small molecule inhibitors of RTK targeting the ATP binding site of tyrosine kinases are currently used or are in clinical trials as antitumor agents (Figure 1).13, 14
Figure 1.

Structures of RTK inhibitors and standards used in the assays
Gangjee et al.15 designed a series of 2-amino-4-(m-bromoanilino)-6-substituted pyrrolo[2,3-d]pyrimidines as multiple RTK inhibitors and antiangiogenic agents. A key aspect of this study was the inclusion of a 2-amino group on the pyrrolo[2,3-d]pyrimidine scaffold which was anticipated to utilize an additional hydrogen-bond binding site in the Hinge region (ATP binding site) compared to 2-desamino analogs. ATP, which lacks this 2-NH2 moiety, does not use this site. The RTK inhibitors in the literature, which contain 6-6 and 6-5 bicyclic ring scaffolds usually do not have a 2-amino group to exploit this H-bonding site.
Recently Gangjee et al.16 reported a series of N4-(substitutedphenyl)-6-(2-phenylethyl)-7H-pyrrolo[2,3-d]pyrimidine-2,4-diamines (compounds 12–18, Figure 2) as RTK inhibitors. In this series the nature and substitutions on the anilino moiety determines both the selectivity and potency against a variety of RTKs in whole cell assays.
Figure 2.

2-amino16 and 2-desaminopyrrolo[2,3-d]pyrimidine RTK inhibitors
It was important to validate our hypothesis regarding the inclusion of a 2-amino group to increase binding and potency of RTK inhibitors.17 This was especially crucial because most of the marketed tyrosine kinase inhibitors such as 1 (gefitinib), 2 (erlotinib) and 6 (lapatinib) including the substrate ATP lack any substitution at their corresponding 2-position. Hence compounds 19–25 (Figure 2) were designed as 2-desamino analogs of 12–18, in order to compare their activities as RTK inhibitors. Substitutions on the 4-anilino ring were kept same as 12–18 from our previous report16 to allow us for one-to-one comparison between 2-amino analogs (12–18) and 2-desamino analogs (19–25).
Superimposition of 12 (Figure 3) and 19 (Figure 4) on to ATP from its crystal structure with EGFR (PDB id: 2gs619) using MOE2007.0918 showed that the pyrrolo[2,3-d]pyrimidine ring fits in the same region as the adenine ring of ATP. The N-3 and 4-NH moieties of both 12 and 19 are within H-bond distances (≈ 3Å) from the backbone NH of Met769 and backbone carbonyl of Gln767 respectively in the Hinge region of the binding pocket. These are important binding sites for ATP and ATP-competitive inhibitors and serve to anchor the heterocyclic portion of the molecule and appropriately orient the other parts of the molecule in the ATP binding site. In addition to this, the 2-NH2 group of 12 (Figure 3) is only 2.5 Å away from the backbone carbonyl of Met769 and hence can form an additional H-bond. For the 2-desamino analog 19 (Figure 4), however, there is no group at the 2-position to provide an H-bond with EGFR. The molecular modeling studies provided further credence to our hypothesis. The distance of the C-2 of 19 and backbone carbonyl of Met769 is ≈ 3.5 Å (Figure 4). Similarly ATP does not use this H-bonding site. Hence the inclusion of a 2-amino group in our pyrrolo[2,3-d]pyrimidine analogs was also expected to perhaps improve selectivity. Thus 19–25 were designed so as to compare their biological activities with 12–18.
Figure 3.
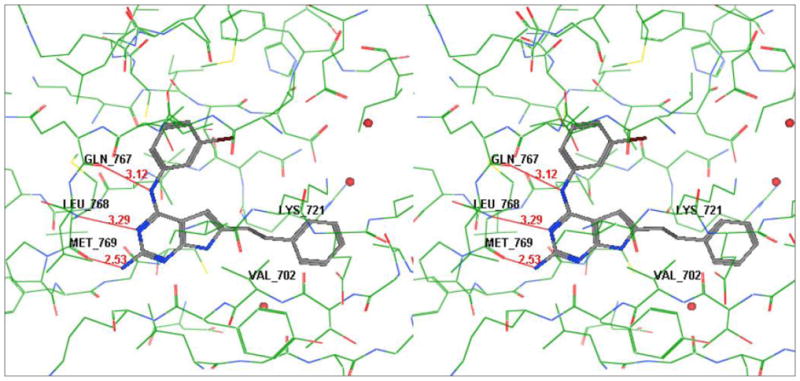
Stereoview of compound 12 in the active site of EGFR [Superimposed on ATP from its crystal structure (pdb id: 2gs6)]; generated using MOE2007.0818 (Hydrogens removed for clarity)
Figure 4.
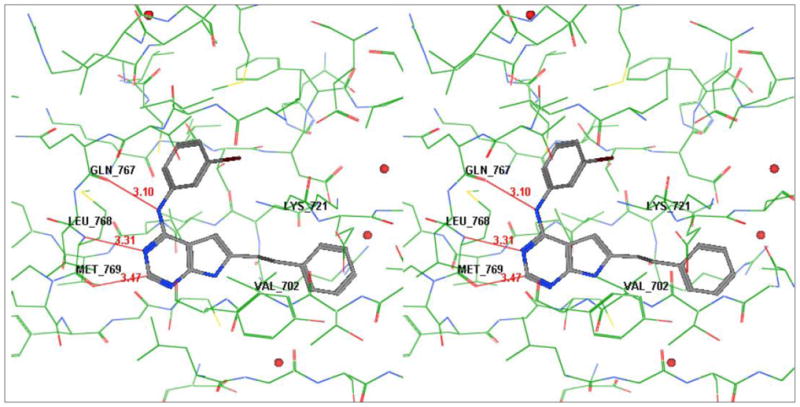
Stereoview of compound 19 in active site of EGFR [Superimposed on ATP from its crystal structure (pdb id: 2gs6)]; generated using MOE2007.08.118 (Hydrogens removed for clarity)
The synthesis of 19–25 is shown in Scheme 1. Intermediate 2716 (obtained from phenyl propionic acid in two steps) was treated with the free base of ethoxycarbonylacetamidine hydrochloride 2820, 21 (obtained from ethyl cyanoacetate) at reflux to afford the pyrrole 29.22 Pyrrolo[2,3-d]pyrimidine 30 was synthesized from 29 by cyclization with formamide.23 Chlorination of 30 with phosphorous oxychloride afforded 31. Nucleophilic displacement of the 4-chloro moiety of 31 with appropriate anilines 32a–g in isopropyl alcohol at reflux for 4 h afforded 19–24 (catalytic amounts of conc HCl were added at the start of the reaction) and 25 (no conc HCl needed).
Scheme 1. Synthesis of 19–25.

Reagents and conditions: (a) Triethylamine, Ethyl acetate, reflux, 25min; (b) Na, MeOH, Formamide, 150°C, 12 h.; (c) Phosphorous oxychloride, reflux, 3h; (d) Isopropanol, 2–3 drops concd HCl (except for 25), reflux
RTK inhibitory activity of the compounds 19–25 were evaluated using human tumor cells known to express high levels of EGFR, VEGFR-1, VEGFR-2 and PDFGR-β using a phosphotyrosine ELISA cytoblot.24, 25 Compounds known to inhibit a particular RTK were used as positive controls for these assays. Whole cell assays were used for RTK inhibitory activity since these assays afford more meaningful results for translation to in vivo studies. The effect of compounds on cell proliferation was measured using inhibition of the growth of A431 cancer cells known to overexpress EGFR. EGFR has been shown to play a role in the overall survival of the cells.26 Cell proliferation was assessed using CYQUANT®, a DNA intercalating dye that has been shown to give a linear approximation of cell number.27 Finally, the effect of selected compounds on blood vessel formation was assessed using the chicken embryo chorioallantoic membrane (CAM) assay, a standard test for angiogenesis.28 In this assay, purified angiogenic growth factors are placed locally on a vascularized membrane of a developing chicken embryo together with possible inhibitors. Digitized images of the vasculature are taken at 48 h after growth factor administration and the number of vessels per unit area evaluated as a measure of vascular density.
Since the IC50 values of RTK inhibitors vary under different assay conditions, we used a standard (control) compound (Figure 1) in each of the evaluations. For EGFR the standard was 7 (PD153035); for VEGFR-1 the standard was 8 (CB676475); for VEGFR-2 and the CAM angiogenesis assay the standard was 9 (semaxanib); for PDGFR-β the standard was 10 (AG1295); for the cytotoxicity study against the growth of A431 cells in culture the standard was 11 (cisplatin).
It is important to note that this study involves the use of cellular assays since we believe that the data from such assays can be extrapolated more accurately for the selection of candidates for in vivo studies than isolated kinase inhibitory studies. However, direct comparison of activities of different compounds becomes difficult, since these may involve other factors such as cellular transport and metabolism.
Removal of the 2-amino group was detrimental for the inhibitory activity against EGFR (with the exception of 25). Compounds 19–24 were inactive in the EGFR inhibition assay (IC50 >200μM); while 25 was about 280-fold less active as compared to the standard 7. In comparison the corresponding 2-amino compounds 12 and 15–17 showed significant potency against EGFR. Compounds 19 and 24 showed significant loss of activities as compared to the corresponding 2-amino analogs (12 and 17).
Against VEGFR-1, 20 and 23 showed slightly better potencies than the corresponding 2-amino compounds 13, 16 and 18; while 19 showed lower VEGFR-1 inhibition than the corresponding 2-amino analog. Compounds 21, 22 and 24 were inactive against VEGFR-1.
In the VEGFR-2 inhibition assay, compounds 19–24 showed improved activity compared to the corresponding 2-amino derivatives (12–17). Compound 25 was equipotent with the corresponding 18. Compounds 21, 22 and 25 were only 2–3-fold less potent than the standard 9. All the compounds showed 2-digit micromolar inhibition against VEGFR-2 except 19; which was only 10-fold less potent than the standard 9.
In the PDGFR-β inhibition assay, 20 and 23 were inactive; while corresponding 2-amino compounds 13 and 16 exhibited 2- to 3-digit micromolar inhibition against PDGFR-β. Additionally compounds 21 and 24 were inactive. Compound 19 was equipotent to the corresponding 2-amino analog 12; while 22 was more potent than its corresponding 2-amino analog 15.
In the A431 cytotoxicity assay all of the 2-desamino compounds 19–25 showed diminished potency compared to the corresponding 2-amino analogs 12–18. Compounds 19–23 were inactive in this assay. Compound 24 was the best compound in the 2-desamino series; however it was about 300-fold less active than the corresponding 2-amino compound 17 and 6-fold less active than the standard 11.
In the CAM angiogenesis inhibitory assay all the 2-desamino compounds showed one to two-digit micromolar inhibition of angiogenesis and were significantly less active than the standard 9. Compounds 20, 21, 23 and 24 were significantly less potent than corresponding 2-amino analogs (13, 14, 16 and 17). The biggest difference was seen for 21 (186-fold less potent than 14) and for 23 (163-fold less potent than 17). Compounds 19, 22 and 25 were better in potency than corresponding 2-amino analogs (12, 15 and 18).
In summary, removal of the 2-amino group in N4-(substitutedphenyl)-6-(2-phenylethyl)-7H-pyrrolo[2,3-d]pyrimidine-2,4-diamines led to a significant loss of activity against EGFR (except 25), PDGFR-β (except 19 and 22) and in the A431 cytotoxicity assay (in all seven cases). Interestingly, removal of the 2-amino group was conducive for the inhibition of VEGFR-2, (in five of seven cases). In the CAM angiogenesis assay, however, the 2-desamino compounds (19–25) showed only one to two-digit digit micromolar inhibition whereas the 2-amino analogs (14 and 16) showed nanomolar inhibition16 lending credence to our hypothesis15 of the importance of the 2-amino group for antiangiogenic activity. The results of this study also indicate that the inclusion a 2-amino group in the marketed RTK inhibitors such as 1, 2 and 6 may improve their potency and perhaps their activity against multiple RTKs.
Supplementary Material
Table 1.
IC50 values (μM) of kinase inhibition, A431 cytotoxicity and inhibition of the CAM assay.
| Compd # | R | EGFR Inhibitiona | VEGFR-1 Inhibitiona | VEGFR-2 Inhibitiona | PDGFR-β Inhibitiona | A431 Cytotoxicitya | CAM Angiogenesis Inhibitiona |
|---|---|---|---|---|---|---|---|
| 19 (12) | 3′-Br | >200 (0.3 ±0.042) | 129.4 ± 30.1 (45.1 ±8.9) | 112.3 ± 18.1 (>50) | 8.5 ± 2.4 (11.8 ±3.7) | >500 (28.6 ±8.2) | 1.93 ± 0.09 (19.5 ±5.1) |
| 20 (13) | 3′-ethynyl | >200 (>200) | 142.3 ± 23.5 (>200) | 49.4 ± 8.0 (>200) | >500 (100.4 ±22.1) | >500 (8.5 ±1.1) | 5.12 ± 0.46 (1.1 ±0.07) |
| 21 (14) | 3′-CF3 | >200 (>200) | >200 (>200) | 22.1 ± 4.1 (>200) | >500 (>500) | >500 (4.6 ±0.67) | 5.63 ± 0.67 (0.03 ±0.004) |
| 22 (15) | 3′-Br, 4′-F | >200 (22.8 ±4.7) | >200 (>200) | 25.2 ± 5.2 (132.1 ±16.7) | 192.4 ± 20.8 (>500) | >500 (6.8 ±0.71) | 2.14 ± 0.39 (5.9 ±0.7) |
| 23 (16) | 3′-CF3, 4′-F | >200 (122 ±30.1) | 164.3 ± 18.1 (>200) | 57.5 ± 7.1 (>200) | >500 (90.0 ±17.8) | >500 (9.8 ±1.0) | 19.6 ± 5.9 (0.12 ±0.12) |
| 24 | 2′-F, 4′-Cl | >200 | >200 | 40.5 ± 9.3 | >500 | 43.0 | 36.9 ± 3.2 |
| (17) | (1.32 ±0.09) | (>200) | (>200) | (>500) | (1.4 ±0.12) | (8.6 ±7.6) | |
| 25 (18) | 3′,4′-(C2H3N) | 56.1 ± 8.7 (>200) | 223.8 ± 41.3 (>200) | 31.6 ± 8.8 (24.9 ±4.1) | 104.2 ± 16.7 (77.4 ± 9.1) | 81.9 ± 10.1 (5.5 ± 1.4) | 20.1 ± 4.8 (50.6 ±18.8) |
| 7 | 0.2 ±0.04 | 12.6 ±2.9 | |||||
| 8 | 17.7 ±5.5 | ||||||
| 9 | 10.6 ±2.7 | 19.2 ±1.1 | 0.085 ±0.0031 | ||||
| 10 | 6.5 ±1.3 | ||||||
| 11 | 7.65 ±1.4 |
Values in parentheses are for the corresponding 2-amino compounds.16 All the values are means of three experiments; standard deviation follows the mean value.
Acknowledgments
This work was supported, in part, by the National Institutes of Health, National Cancer Institute Grant CA98850 (A.G.).
The authors thank Dr. Hiteshkumar D. Jain for providing intermediate 28.
Abbreviations
- RTK
receptor tyrosine kinase
- EGF
epidermal growth factor receptor
- VEGF
vascular endothelial growth factor
- PDGF
platelet derived growth factor
- EGFR
epidermal growth factor receptor
- VEGFR
vascular endothelial growth factor receptor
- PDGFR
platelet derived growth factor receptor
- CAM
chorioallantoic membrane
- ATP
adenine triphosphate
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.Bridges AJ. Chem Rev. 2001;101:2541. doi: 10.1021/cr000250y. [DOI] [PubMed] [Google Scholar]
- 2.Herbst RS, Fukuoka M, Baselga J. Nat Rev Cancer. 2004;4:956. doi: 10.1038/nrc1506. [DOI] [PubMed] [Google Scholar]
- 3.Ellis LM. Hematol Oncol Clin North Am. 2004;18:1007. doi: 10.1016/j.hoc.2004.06.002. [DOI] [PubMed] [Google Scholar]
- 4.Jain RK. Semin Oncol. 2002;29:3. doi: 10.1053/sonc.2002.37265. [DOI] [PubMed] [Google Scholar]
- 5.Ferrara N. Semin Oncol. 2002;29:10. doi: 10.1053/sonc.2002.37264. [DOI] [PubMed] [Google Scholar]
- 6.Ferrara N, Gerber HP, LeCouter J. Nat Med. 2003;9:669. doi: 10.1038/nm0603-669. [DOI] [PubMed] [Google Scholar]
- 7.Potapova O, Laird AD, Nannini MA, Barone A, Li G, Moss KG, Cherrington JM, Mendel DB. Mol Cancer Ther. 2006;5:1280. doi: 10.1158/1535-7163.MCT-03-0156. [DOI] [PubMed] [Google Scholar]
- 8.Folkman J. Annu Rev Med. 2006;57:1. doi: 10.1146/annurev.med.57.121304.131306. [DOI] [PubMed] [Google Scholar]
- 9.Faivre S, Djelloul S, Raymond E. Semin Oncol. 2006;33:407. doi: 10.1053/j.seminoncol.2006.04.005. [DOI] [PubMed] [Google Scholar]
- 10.Laird AD, Cherrington JM. Expert Opin Investig Drugs. 2003;12:51. doi: 10.1517/13543784.12.1.51. [DOI] [PubMed] [Google Scholar]
- 11.Tabernero J. Mol Cancer Res. 2007;5:203. doi: 10.1158/1541-7786.MCR-06-0404. [DOI] [PubMed] [Google Scholar]
- 12.de Jonge MJ, Verweij J. Eur J Cancer. 2006;42:1351. doi: 10.1016/j.ejca.2006.02.013. [DOI] [PubMed] [Google Scholar]
- 13.Ochs JS. Int J Radiat Oncol Biol Phys. 2004;58:941. [Google Scholar]
- 14.Wedge SR, Ogilvie DJ, Dukes M, Kendrew J, Curwen JO, Hennequin LF, Thomas AP, Stokes ES, Curry B, Richmond GH, Wadsworth PF. Cancer Res. 2000;60:970. [PubMed] [Google Scholar]
- 15.Gangjee A, Yang J, Ihnat MA, Kamat S. Bioorg Med Chem. 2003;11:5155. doi: 10.1016/j.bmc.2003.08.034. [DOI] [PubMed] [Google Scholar]
- 16.Gangjee A, Namjoshi OA, Yu J, Ihnat MA, Thorpe JE, Warnke LA. Bioorg Med Chem. 2008;16:5514. doi: 10.1016/j.bmc.2008.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Namjoshi OA, Gangjee A, Ihnat MA, Miller WT. Abstract of Papers, 97th Annual Meeting of the American Association of Cancer Research; Washington D.C. Philadelphia, PA: American Association of Cancer Research; 2006. Abstract 243. [Google Scholar]
- 18.Molecular Operating Environment (MOE), 2007.09. Chemical Computing Group
- 19.The Protein Data Bank. www.pdb.org.
- 20.Kobayashi T, Inoue T, Kita Z, Yoshiya H, Nishino S, Oizumi K, Kimura T. Chem Pharm Bull (Tokyo) 1995;43:788. doi: 10.1248/cpb.43.788. [DOI] [PubMed] [Google Scholar]
- 21.Kobayashi T, Inoue T, Nishino S, Fujihara Y, Oizumi K, Kimura T. Chem Pharm Bull (Tokyo) 1995;43:797. doi: 10.1248/cpb.43.797. [DOI] [PubMed] [Google Scholar]
- 22.Toja E, DePaoli A, Tuan G, Kettenring J. Synthesis. 1987:272. [Google Scholar]
- 23.Showalter HD, Bridges AJ, Zhou H, Sercel AD, McMichael A, Fry DW. J Med Chem. 1999;42:5464. doi: 10.1021/jm9903949. [DOI] [PubMed] [Google Scholar]
- 24.Fong TA, Shawver LK, Sun L, Tang C, App H, Powell TJ, Kim YH, Schreck R, Wang X, Risau W, Ullrich A, Hirth KP, McMahon G. Cancer Res. 1999;59:99. [PubMed] [Google Scholar]
- 25.Stockwell BR, Haggarty SJ, Schreiber SL. Chem Biol. 1999;6:71. doi: 10.1016/S1074-5521(99)80004-0. [DOI] [PubMed] [Google Scholar]
- 26.Schilder RJ, Hall L, Monks A, Handel LM, Fornace AJ, Jr, Ozols RF, Fojo AT, Hamilton TC. Int J Cancer. 1990;45:416. doi: 10.1002/ijc.2910450306. [DOI] [PubMed] [Google Scholar]
- 27.Wilson SM, Barsoum MJ, Wilson BW, Pappone PA. Cell Prolif. 1999;32:131. doi: 10.1046/j.1365-2184.1999.32230131.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Marks MG, Shi J, Fry MO, Xiao Z, Trzyna M, Pokala V, Ihnat MA, Li PK. Biol Pharm Bull. 2002;25:597. doi: 10.1248/bpb.25.597. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.