

Abstract
BACKGROUND
Traditionally, acromegaly evaded diagnosis until in its clinically obvious later stages when treatment is more difficult. Over the last 25 years diagnostic tests have improved, but whether clinical disease detection also improved was unknown so we tested if disease severity at diagnosis had changed from 1981 to 2006.
METHODS
Data on 324 consecutive acromegaly patients presenting from 1981–2006 at two New York City hospitals were collected by retrospective review (n=324) and by interview (n=200). The main complaint, acromegaly-associated co-morbidities, signs, symptoms, healthcare providers visited, pre-operative growth hormone (GH) and insulin-like growth factor I (IGF-I) levels and pituitary tumor size at diagnosis were compared in patients presenting in the earlier vs. later halves of the time period.
RESULTS
Times from symptom onset to diagnosis were 5.9 yr. (early) vs. 5.2 yr. (late)(p=ns). At diagnosis, 96% of early and late groups had facial feature changes and/or hand/foot enlargement. Co-morbidities included hypertension (HTN) 37 % (early) vs. 36% (late), carpal tunnel syndrome (24 vs. 24%), sleep apnea (13 vs. 29%)(p <0.01), osteoarthritis (25 vs. 23%), and diabetes mellitus (DM) (18 vs.15%); each patient had 1.2 (early) vs. 1.3 (late) (p=0.53) co-morbidities. Groups were similar in signs, symptoms, tumor size, GH and IGF-I.
CONCLUSIONS
Clinical, biochemical and tumor size characteristics at diagnosis of acromegaly patients were unchanged from 1981–2006. Most patients still have marked manifestations of acromegaly at diagnosis suggesting that acromegaly remains clinically under-recognized. Healthcare professionals should more commonly consider acromegaly, which can lead to earlier diagnosis and better treatment outcome.
Keywords: Acromegaly, pituitary tumor, growth hormone, insulin-like growth factor 1
INTRODUCTION
Acromegaly is a rare disease that is due in almost all cases to a GH secreting pituitary tumor 1. Excess GH and the resultant elevations of insulin-like growth factor I (IGF-I), the biochemical hallmarks of this disease 1, 2, produce its characteristic multi-system, often disfiguring, physical manifestations as well as its clinically significant co-morbidities including diabetes mellitus, hypertension, arthritis, sleep apnea and cardiovascular disease 3–8. When inadequately treated, acromegaly impairs patients' quality of life 9 and leads to a 2–5 fold increase in mortality rate 5, 9–12.
Acromegaly has long been known for its insidious nature and long delay from onset of symptoms to diagnosis 13–15. The reasons why acromegaly has traditionally been under-recognized are unclear, but could include its slowly progressive course allowing its changes to go un-noticed by the patient, family members or physicians, as well as the overlap of many of its co-morbidities with common disorders. Patients typically visit a number of healthcare providers prior to diagnosis.
As effective therapies can now prevent disease progression and return lifespan to normal early recognition of acromegaly is essential 5, 16, 17. Recent developments including highly sensitive biochemical markers, both GH and IGF-I 18, 19, and MRI scans to identify small tumors are available for diagnosing acromegaly. Biochemical methods have thus clearly improved and should allow for the diagnosis to be made earlier, but it is unknown if the factor critical to beginning the evaluation for acromegaly, an initial clinical suspicion by a healthcare professional, occurs at an earlier disease stage in more recent times. An analysis of changes over time in the clinical characteristics at diagnosis of acromegaly could be used to measured if the disease is being detected earlier. Therefore, in order to determine whether or not the severity of acromegaly at the time of its clinical recognition and thus its diagnosis had decreased in recent years, we evaluated 324 consecutive patients diagnosed with acromegaly from 1981–2006 in New York City, comparing the clinical and biochemical characteristics at diagnosis of those patients presenting in the earlier vs. later halves of this time period.
SUBJECTS AND METHODS
Study Subjects
The study cohort consisted of 324 consecutive patients who presented over a 26 year period from January 1981 to December 2006 for surgery for acromegaly, with two New York City based neurosurgeons, KDP (n=270, 1981–2006) and JNB (n=54, 1992–2006). All patients had pathologic confirmation of a GH-secreting pituitary tumor.
Study Design
A retrospective chart review of the neurosurgeons' charts of all 324 patients and additional in person interviews and examinations of 200 of them pre- and/or post-operatively (by PUF) were conducted. All available clinical, biochemical, radiographic and pathological data were collected including the time from onset of signs or symptoms to diagnosis, the main complaint bringing the patient to medical attention, symptoms and co-morbid diseases present at or prior to diagnosis, healthcare providers visited prior to diagnosis, GH and IGF-I levels at diagnosis and pituitary tumor size.
Patients were divided into two groups based on their date of surgery; an early group who had transsphenoidal pituitary surgery from 1/1981 – 12/1994 (n=108) and a later group from 1/1995 – 12/2006 (n=216). Interviews and examinations were conducted in 55% of the early vs. 56% of the later group (p=0.93). Prior non-curative pituitary surgery had been done elsewhere in 5 patients in the early and 3 in the late group. Prior ineffective medical therapy had been given to 11 patients; bromocriptine to 5 in the early and 1 in the late group and octreotide to 3 in the early and 2 in the late groups. All signs and symptoms and other data are those related to the initial presentation of acromegaly.
Preoperative GH levels measured with one polyclonal radioimmunoassay (RIA) assay were compared in subsets of the cohort, an earlier group, surgery from 1/1981 – 4/1991 (n=61) and a later group, surgery from 5/1992 – 11/2003 (n=32).
This study was approved by the Institutional Review Boards of Columbia University Medical Center and Mt. Sinai School of Medicine. Informed consent was obtained from those who participated in the in-person evaluation.
Biochemical Measurements
GH was measured by RIA using a polyclonal antiserum raised in rabbits immunized with purified human GH as previously described (18). The coefficients of variation are 6 % (intra-assay) and 8% (inter-assay). The assay has a sensitivity of 0.5 μg/L 20. Serum IGF-I levels were measured in clinical laboratories that employed different assays over the 26 years of this study so IGF-I data are expressed as a percentage of the upper limit of normal for the assay used.
Statistical Analysis
Symptom or co-morbidity prevalence and type of healthcare professional seen in early vs. late groups were compared by chi2 test. GH levels in earlier vs. later subsets were compared by ANOVA. Early vs. late groups differences in age, time to diagnosis and composite score for co-morbidities were tested by the Student t-test. Data are given as mean ± SE in early vs. late groups, respectively, unless otherwise specified. A p-value <0.05 was considered significant.
RESULTS
Patient Characteristics
The early group (n=108) had a mean age of 43 ± 1.3 yrs. (range 20 – 72 yrs.) and was 44% male and 56% female. The late group (n=216) had a mean age of 47 ± 0.9 yrs. (range 16 – 85 yrs.) and was 55% male and 45% female.
Main Complaint Leading to Diagnosis
The main presenting complaints that brought the patients to medical attention are shown in Table 1. Evaluations for acromegaly were initiated after facial features changes (15.2%), hand/foot enlargement (16.8%) or both (22.8%) were noted by the patient, their family, friends or a physician. These changes were more likely to have been noticed by others (58%) than the patient (42%)(p=0.0052). 6.7% of patients were diagnosed when medical professionals that they visited for an unrelated reason noted features of acromegaly. Facial feature changes included a spectrum of coarsening, deepening of the nasolabial folds, thickening of the soft tissues, enlargement of the lips or nose and in severe cases of the forehead bones (frontal bossing) or lower jaw (prognathism). Hand/foot enlargement was typically noted as a puffiness of the soft tissues often noticed by the need to enlarge rings, shoe size and width. The acromegaly work up was initiated in others because of neurological or visual complaints (18.4%), including headaches (8.2%), vision or visual field deficits (5.7%), vertigo and syncope or symptoms of gonadal dysfunction (8.6%)(30% males, 70% female) such as oligo/amenorrhea, decreased libido, galactorrhea, hirsuitism(females), gynecomastia, and infertility or arthritis (5.1%). Snoring, sinus problems, hyperhidrosis, weight gain, fatigue, abnormal thyroid function tests and enlargement of the salivary glands each brought < 2% of patients to medical attention. Acromegaly was diagnosed after the incidental discovery of an adenoma on brain imaging that was done for an unrelated complaint, such as after trauma, in 3.5% of patients.
Table 1.
Main presenting complaint that led to the work up and diagnosis of acromegaly in our cohort.
| (%)* | |
|---|---|
| Change in face noted by patient | 6.3 |
| Change in face noted by other person | 8.9 |
| Enlarging hands/feet noted by patient | 10.5 |
| Enlarging hands/feet noted by other person | 6.3 |
| Concurrent changes in face & hands/feet noted by patient | 6.3 |
| Concurrent changes in face & hands/feet noted by other person | 16.8 |
| Visual or neurological complaint | 18.1 |
| Symptoms of gonadal dysfunction | 8.6 |
| Arthritis | 5.1 |
| Others | 13 |
Percentages of the total cohort (n=324) with each presenting complaint. Percentages in the early vs. late groups did not differ.
There were no significant differences in the presenting complaints that brought patients to medical attention in the early vs. late groups of patients.
Earliest Sign or Symptom of acromegaly
The earliest sign or symptom that was recalled by the patient as being related to acromegaly are reported in Table 2. The most common were enlargement of the hands, feet and/or facial changes including changes in the facial features, jaw, bite or teeth changes. The percentages of each symptom did not differ in the early vs. late groups.
Table 2.
Earliest sign or symptom as recalled by the patient to be related to acromegaly.
| Sign or Symptom | %* |
|---|---|
| Hand/foot enlargement | 30 |
| Jaw enlargement/bite or teeth spacing changes | 9.8 |
| Facial feature changes** | 8.7 |
| Facial feature changes** & hand/foot enlargement | 8.7 |
| Joint pain | 6.5 |
| Menstrual irregularity/secondary amenorrhea | 6.1 |
| Carpal tunnel syndrome | 4.7 |
| Hyperhidrosis | 4 |
| Headache | 4 |
| Fatigue | 2.9 |
| Gonadal dysfunction/infertility - males | 2.5 |
| Snoring | 2.2 |
| Diabetes mellitus | 1.8 |
| Sleep apnea | 1.8 |
| Arthritis | 1.4 |
| Hirsutism/Acne | 1.1 |
| Hypertension | 1.1 |
| Hyperprolactinemia | <1.0 |
| Vision problems | <1.0 |
| Sinus problems | <1.0 |
| Kidney stones | <1.0 |
| Increased height | <1.0 |
| Skin tags | <1.0 |
Data on the earliest sign or symptom recalled by the patient were available in 85% of the cohort. Percentages did not differ significantly in the early vs. late groups.
Indicates the % of facial feature other than jaw, bite or teeth changes such as coarsening of the features.
Symptoms Present at Diagnosis
The most common signs and symptoms present at diagnosis are given in Table 3. These did not necessarily bring the patient to medical attention, but were elicited upon questioning of the patient at diagnosis. Nearly all patients, 96.3% (early) vs. 95.6% (late) (p=ns), had hand and/or foot enlargment and/or facial feature changes at presentation. The examining physician noted “obvious” physical signs of acromegaly in 59.2%(early) vs. 55.2% (late) (p=ns) of patients. Fatigue (26 vs. 44%; p=0.002) and snoring (25 vs. 60%; p=0.0001), were more prevalent in the later group.
Table 3.
Signs and symptoms present at diagnosis (as elicited from the patient) in earlier vs. later groups of the acromegaly cohort.
| Sign or Symptom | Early (%) | Late (%) | P-value |
|---|---|---|---|
| Hand/foot enlargement | 84 | 87 | 0.61 |
| Facial feature changes | 83 | 85 | 0.79 |
| Hyperhidrosis | 52 | 56 | 0.32 |
| Headache | 46 | 47 | 0.94 |
| Joint pain | 39 | 42 | 0.66 |
| Gonadal dysfunction | 36 | 32 | 0.53 |
| Snoring | 25 | 60 | <0.01 |
| Fatigue | 26 | 44 | <0.01 |
| Macroglossia | 19 | 27 | 0.18 |
| Increased spacing between teeth | 19 | 26 | 0.21 |
| Vision problems | 18 | 21 | 0.52 |
| Weight gain | 16 | 16 | 0.96 |
| Deepening voice | 11 | 8 | 0.54 |
| Paresthesias | 9 | 19 | 0.03 |
| Acne | 8 | 9 | 0.95 |
| Hirsuitism | 9 | 8 | 0.94 |
| Sinus problems | 4 | 5 | 0.92 |
| Hair loss | 1 | 6 | 0.07 |
| Mood instability | 3 | 2 | 0.89 |
| Increased height | 3 | 1 | 0.43 |
| Increased muscle mass | 1 | 3 | 0.38 |
Co-morbidities Present at Diagnosis
The acromegaly-associated co-morbidities present at diagnosis are shown in Figure 1. The most common were: HTN (37 vs. 36%)(early vs. late)(p=ns), carpal tunnel syndrome (24 vs. 24%) (p=ns), osteoarthritis (25 vs. 23%)(p=ns), diabetes mellitus (18 vs. 15%)(p=ns), and sleep apnea (13 vs. 29%)(p=0.0026). On average each patient had 1.2 (early) vs. 1.3 (late) (p=0.53) of these major co-morbidities. Also present at or prior to diagnosis were goiter (15 vs. 12%)(p=0.6), malignancy (7.4 vs. 9.2%)(p=0.16), kidney stones (8 vs. 8%)(p=1.0), colon polyps (10 vs. 23%)(p=0.0188) and skin tags (19 vs. 46%)(p=0.0003). Thus, a history of sleep apnea (as diagnosed by a physician with or without a sleep study), skin tags or colon polyps was more common at diagnosis in the later group. The specific types of carcinomas that had been diagnosed in the early group were basal cell (n=2), papillary thyroid (n=1), colon (n=1), bladder (n=1), prostate (n=1) and melanoma (n=1). Specific carcinomas in the later group were breast (n=7), melanoma (n=2), non Hodgkins Lymphoma (n=1), papillary thyroid (n=1), hairy cell leukemia (n=1), prostate (n=2), osteochondroma (n=1), testicular seminoma (n=1) and one patient had papillary thyroid as well as breast (n=1) and another had breast, ovarian as well uterine cancer (n=1).
Figure 1.

Co-morbidities Present at Diagnosis in Early vs. Late Group of the Acromegaly Cohort.
Healthcare Providers Visited Prior to or Who Made the Diagnosis
Many patients visited a number of healthcare providers for various complaints related to acromegaly prior to their diagnosis. The specific types of physicians visited by the patients as well as the specific type of healthcare provider that made the diagnosis of acromegaly are shown in Table 4. About one half of the patients were diagnosed by a primary care provider. The types of physicians visited did not differ significantly in the early vs. late groups except that gastroenterologists were more commonly visited, in most cases for colonoscopies, in the later (23%) than earlier (10%) group (p=.02).
Table 4.
Types of healthcare professionals visited by the patient for complaints or illnesses related to acromegaly prior to their diagnosis. Types of healthcare professionals who diagnosed the patient with acromegaly.
| Visited by Patient Prior to Diagnosis | Diagnosed Patient* | |
|---|---|---|
| %** | %** | |
| Primary care/ Internist | 60 | 51 |
| Endocrinologist | 22 | 14.5 |
| Gastroenterologist | 15 | 1.0 |
| Gynecologist | 13 | 8 |
| Dentist | 9.9 | 7 |
| Orthopedist | 9.9 | <1.0 |
| Oncologist | 7.4 | <1.0 |
| Ophthalmologist | 5.5 | 5.4 |
| Otolaryngologist | 4.9 | 1.9 |
| Pulmonary - sleep specialist | 4.9 | 1.5 |
| Neurologist | 4.4 | 3.4 |
| Urologist | 3.7 | 1.5 |
| Rheumatologist | 3.3 | 2.3 |
| Cardiologist | 2.7 | <1.0 |
| Dermatologist | 2.4 | <1.0 |
| Endocrine surgeon | 2.4 | <1.0 |
| General surgeon | 1.5 | - |
| Pediatrician | 1.0 | 1.1 |
| Plastic surgeon | <1.0 | - |
| Psychiatrist | <1.0 | - |
Data on the types of physicians who made the diagnosis of acromegaly were available on 80 % of the cohort.
Percentages of patients who visited or were diagnosed by each type of healthcare professional. Percentages in the early vs. late groups did not differ.
Tumor Size
The mean pituitary tumor size was 15.6 ± 0.84 mm (early) vs. 15.4 ± 0.53 mm (late) groups (p=0.87). Tumor size distributions were the same in early vs. late groups (Figure 2). Macroadenomas (≥ 1 cm) were present in 79.6% (early) and 79.5% (late) (p=0.98).
Figure 2.
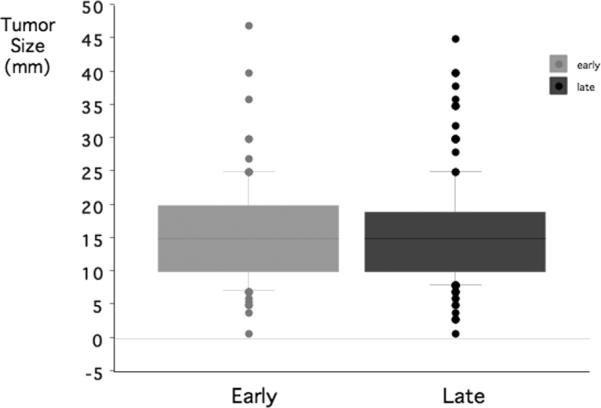
The distribution of pituitary tumor sizes in the early vs. later group of the cohort Horizontal lines of the box plots indicate the 10th, 25th, 50th, 75th and 90th percentiles of the distributions and tumers < 10th or > 90th percentiles are individual data points.
Preoperative GH and IGF-I Levels
The distribution of preoperative random GH levels was similar in the earlier vs. later subsets of the cohort (Figure 3). Serum IGF-I levels were performed in 57% of the earlier group (because the test was new and of limited availability) vs. 90% of the later group. IGF-I levels at diagnosis were 3.14 vs. 3.42 (p=ns) times the upper limit of normal in the early vs. late groups.
Figure 3.

Preoperative Growth Hormone Leavels in Early vs. Late Groups of the Acromegaly Cohort.
Time to Diagnosis from Sign or Symptom Onset
The time from the onset of signs or symptoms reported by the patient to diagnosis did not differ in the early 5.9 ± 0.58 yrs. (range 0–25 yrs.) vs. the late 5.2 ± 0.37 yrs. (range 0–36 yrs.)(p=0.316) groups.
DISCUSSION
Our analysis of the presentations of 324 acromegaly patients, the largest single-city consecutive cohort reported, demonstrates that the prevalence of signs, symptoms and associated co-morbidities, tumor size and preoperative GH levels did not change over the 26 years that these patients were diagnosed. Most patients still present with many manifestations and co-morbidities consistent with the disease being advanced at diagnosis and suggesting that acromegaly continues to be under-recognized and thus under-diagnosed.
The route to ultimate diagnosis of acromegaly varies. In our cohort, 56% of patients came to medical attention after the characteristic changes in facial features or extremities were noted and most had a complaint referable to acromegaly that led to their diagnosis. Some, however, were diagnosed “incidentally” when a physician visited for an unrelated reason noted features consistent with acromegaly (6.7%) or when the pituitary tumor was found during head imaging done for another reason (3.5%). Other studies that used a broader definition for “incidental” diagnosis reported a higher percentage of such patients 14 15, 21. The earliest manifestations of acromegaly recalled by our patients were most commonly hand, feet and/ or facial changes including those of the features, jaw, bite or teeth. These data should be interpreted cautiously, however, as they could have been biased by patient recall.
Regardless of their path to diagnosis, nearly all our patients had many characteristic signs and symptoms and comorbidities of acromegaly at diagnosis and these were equally prevalent over the 26 years studied. In recent years a history of fatigue and snoring were more often elicited, potentially reflecting a greater general awareness of their association with acromegaly. In recent years sleep apnea and skin tags were more common. The higher prevalence of sleep apnea may be due to increased reporting, as its diagnosis and treatment increased in general during the study period and only recently became clearly recognized as a complication of acromegaly 22, 23. About one-third had a history of gonadal dysfunction and this did not differ over time. Other studies have reported menstrual dysfunction in 56–72% of females 15, 22.
We also compared GH levels and tumor size at diagnosis in early vs. later groups of our cohort. Our study provided a unique opportunity to examine pre-operative GH levels in one cohort over many years because we used the same GH assay over this time, which was not the case in other studies 17. Preoperative GH levels, in general, correlate with tumor size and inversely with likelihood of surgical cure 24, 25 as well as with success of medical therapies 26. These levels were similar in earlier vs. later groups and about 25% in both groups had levels in the two highest quintiles, > 40 μg/L, suggestive of a long delay from disease onset to diagnosis and conferring a worse outcome of therapy. IGF-I levels in this series should be interpreted cautiously because these were measured in a much smaller percentage of the earlier group and could only be presented as a percentage of the upper normal limit. Macroadenomas were present in similarly large percentages of the early and late groups as in other reports 3, 27, 28. Tumor size also correlates negatively with likelihood of surgical cure 24. These objective data also suggest that disease severity at diagnosis has not changed over time.
The delay in diagnosis was long, an average of 5.3 ± 4 yrs from symptom onset, with no change over time. In most prior studies mean times from symptoms to diagnosis are 4 to 8 years 15, 22, 29, but in a recent series this was 2.5 years, potentially reflecting a difference in that study's cohort 21. The time of true disease onset is unknown, but likely precedes symptom onset by years. The reasons for the delay in diagnosis are not clear. Although acromegaly cannot be detected from routine laboratory testing and all patients must have some sign or symptom prompting the appropriate biochemical work up, the high prevalence of obvious physical manifestations and years of symptoms prior to diagnosis could reflect the failure of health care professionals to recognize this disease early. Our patients typically visited a number of physicians prior to diagnosis, and were diagnosed, as in another series, most commonly by primary care physicians 21. Although nearly ubiquitous are changes in facial and acral features 15, 21, 22, 29, 30 these may be attributed to “aging” 15 and inconsistently lead to a work up. The breadth and heterogeneity of the signs and symptoms with which patients present could contribute to the delay. Many co-morbidities of acromegaly are common in the general population and thus alone would not trigger an evaluation for acromegaly.
Our study was in part a retrospective review that relied on patient recall. However, the subjective data are supported by the objective tumor size, GH and physical finding data. We also cannot exclude that patient referral patterns to our surgeons have not changed over time. However, our cohort represents the complete experiences of two hospitals that were positioned as both local and tertiary referral centers for pituitary tumors similarly over the study period.
Our study is novel in that we examined one very large consecutive, single city series and because we were able to compare large groups of patients diagnosed up through recent years. Although some of our data are similar to prior reports, the latter's limitations need to be considered. One series included only patients enrolled in multi-center octreotide trials 29, which does not allow for determination of whether the high prevalence of signs represented a bias due to selection for patients with more difficult to control disease. Another recent series reported data from only half the center's cohort 21. Also, most previously reported patients were diagnosed over 30 years ago15. Given these limitations a reappraisal of the currently presentations of acromegaly was needed.
In conclusion, the clinical characteristics at diagnosis of patients with acromegaly did not change from 1981–2006 suggesting that clinical recognition of acromegaly has not significantly improved over the last 25 years. As the likelihood of successful therapy relates to tumor size and disease severity, earlier recognition and initiation of treatment is needed. Increased awareness by healthcare professionals, especially primary care physicians, of acromegaly presentations is needed. Educational programs should be targeted at early recognition of acromegaly with the hope that these will lead to earlier diagnosis.
Acknowledgment
Supported by NIH grants DK064720 and DK073040 to PUF. TJR was supported by a grant from Dr. and Mrs. Leo Guthart.
Footnotes
Conflicting interests: nothing to declare.
References
- 1.Melmed S. Medical progress: Acromegaly. N Engl J Med. 2006;355:2558–2573. doi: 10.1056/NEJMra062453. [DOI] [PubMed] [Google Scholar]
- 2.Freda PU. Current concepts in the biochemical assessment of the patient with acromegaly. Growth Horm IGF Res. 2003;13:171–184. doi: 10.1016/s1096-6374(03)00029-7. [DOI] [PubMed] [Google Scholar]
- 3.Beauregard C, Truong U, Hardy J, Serri O. Long-term outcome and mortality after transsphenoidal adenomectomy for acromegaly. Clin Endocrinol (Oxf) 2003;58:86–91. doi: 10.1046/j.1365-2265.2003.01679.x. [DOI] [PubMed] [Google Scholar]
- 4.Mestron A, Webb SM, Astorga R, Benito P, Catala M, Gaztambide S, Gomez JM, Halperin I, Lucas-Morante T, Moreno B, Obiols G, de Pablos P, Paramo C, Pico A, Torres E, Varela C, Vazquez JA, Zamora J, Albareda M, Gilabert M. Epidemiology, clinical characteristics, outcome, morbidity and mortality in acromegaly based on the Spanish Acromegaly Registry (Registro Espanol de Acromegalia, REA) Eur J Endocrinol. 2004;151:439–446. doi: 10.1530/eje.0.1510439. [DOI] [PubMed] [Google Scholar]
- 5.Swearingen B, Barker FG, 2nd, Katznelson L, Biller BM, Grinspoon S, Klibanski A, Moayeri N, Black PM, Zervas NT. Long-term mortality after transsphenoidal surgery and adjunctive therapy for acromegaly. J Clin Endocrinol Metab. 1998;83:3419–3426. doi: 10.1210/jcem.83.10.5222. [DOI] [PubMed] [Google Scholar]
- 6.Ezzat S, Strom C, Melmed S. Colon polyps in acromegaly. Ann Intern Med. 1991;114:754–755. doi: 10.7326/0003-4819-114-9-754. [DOI] [PubMed] [Google Scholar]
- 7.Ron E, Gridley G, Hrubec Z, Page W, Arora S, Fraumeni JF., Jr. Acromegaly and gastrointestinal cancer. Cancer. 1991;68:1673–1677. doi: 10.1002/1097-0142(19911015)68:8<1673::aid-cncr2820680802>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- 8.Colao A, Vitale G, Pivonello R, Ciccarelli A, Di Somma C, Lombardi G. The heart: an end-organ of GH action. Eur J Endocrinol. 2004;151(Suppl 1):S93–101. doi: 10.1530/eje.0.151s093. [DOI] [PubMed] [Google Scholar]
- 9.Webb SM, Badia X, Surinach NL. Validity and clinical applicability of the acromegaly quality of life questionnaire, AcroQoL: a 6-month prospective study. Eur J Endocrinol. 2006;155:269–277. doi: 10.1530/eje.1.02214. [DOI] [PubMed] [Google Scholar]
- 10.Alexander L, Appleton D, Hall R, Ross WM, Wilkinson R. Epidemiology of acromegaly in the Newcastle region. Clin Endocrinol (Oxf) 1980;12:71–79. doi: 10.1111/j.1365-2265.1980.tb03135.x. [DOI] [PubMed] [Google Scholar]
- 11.Bengtsson BA, Eden S, Ernest I, Oden A, Sjogren B. Epidemiology and long-term survival in acromegaly. A study of 166 cases diagnosed between 1955 and 1984. Acta Med Scand. 1988;223:327–335. doi: 10.1111/j.0954-6820.1988.tb15881.x. [DOI] [PubMed] [Google Scholar]
- 12.Dekkers OM, Biermasz NR, Pereira AM, Romijn JA, Vandenbroucke JP. Mortality in acromegaly: a metaanalysis. J Clin Endocrinol Metab. 2008;93:61–67. doi: 10.1210/jc.2007-1191. [DOI] [PubMed] [Google Scholar]
- 13.Pearce JM. Pituitary tumours and acromegaly (Pierre Marie's disease) J Neurol Neurosurg Psychiatry. 2002;73:394. doi: 10.1136/jnnp.73.4.394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nabarro JD. Acromegaly. Clin Endocrinol (Oxf) 1987;26:481–512. doi: 10.1111/j.1365-2265.1987.tb00805.x. [DOI] [PubMed] [Google Scholar]
- 15.Molitch ME. Clinical manifestations of acromegaly. Endocrinol Metab Clin North Am. 1992;21:597–614. [PubMed] [Google Scholar]
- 16.Bates AS, Van't Hoff W, Jones JM, Clayton RN. An audit of outcome of treatment in acromegaly. Q J Med. 1993;86:293–299. [PubMed] [Google Scholar]
- 17.Kauppinen-Makelin R, Sane T, Reunanen A, Valimaki MJ, Niskanen L, Markkanen H, Loyttyniemi E, Ebeling T, Jaatinen P, Laine H, Nuutila P, Salmela P, Salmi J, Stenman UH, Viikari J, Voutilainen E. A nationwide survey of mortality in acromegaly. J Clin Endocrinol Metab. 2005;90:4081–4086. doi: 10.1210/jc.2004-1381. [DOI] [PubMed] [Google Scholar]
- 18.Freda PU, Post KD, Powell JS, Wardlaw SL. Evaluation of disease status with sensitive measures of growth hormone secretion in 60 postoperative patients with acromegaly. J Clin Endocrinol Metab. 1998;83:3808–3816. doi: 10.1210/jcem.83.11.5266. [DOI] [PubMed] [Google Scholar]
- 19.Freda PU, Reyes CM, Nuruzzaman AT, Sundeen RE, Bruce JN. Basal and glucose-suppressed GH levels less than 1 microg/L in newly diagnosed acromegaly. Pituitary. 2003;6:175–180. doi: 10.1023/b:pitu.0000023424.72021.e2. [DOI] [PubMed] [Google Scholar]
- 20.Frantz AG, Rabkin MT. Human Growth Hormone. Clinical Measurement, Response to Hypoglycemia and Suppression by Corticosteroids. N Engl J Med. 1964;271:1375–1381. doi: 10.1056/NEJM196412312712701. [DOI] [PubMed] [Google Scholar]
- 21.Nachtigall L, Delgado A, Swearingen B, Lee H, Zerikly R, Klibanski A. Changing patterns in diagnosis and therapy of acromegaly over two decades. J Clin Endocrinol Metab. 2008;93:2035–2041. doi: 10.1210/jc.2007-2149. [DOI] [PubMed] [Google Scholar]
- 22.Holdaway IM, Rajasoorya C. Epidemiology of acromegaly. Pituitary. 1999;2:29–41. doi: 10.1023/a:1009965803750. [DOI] [PubMed] [Google Scholar]
- 23.Pekkarinen T, Partinen M, Pelkonen R, Iivanainen M. Sleep apnoea and daytime sleepiness in acromegaly: relationship to endocrinological factors. Clin Endocrinol (Oxf) 1987;27:649–654. doi: 10.1111/j.1365-2265.1987.tb02947.x. [DOI] [PubMed] [Google Scholar]
- 24.Freda PU, Wardlaw SL, Post KD. Long-term endocrinological follow-up evaluation in 115 patients who underwent transsphenoidal surgery for acromegaly. J Neurosurg. 1998;89:353–358. doi: 10.3171/jns.1998.89.3.0353. [DOI] [PubMed] [Google Scholar]
- 25.Kreutzer J, Vance ML, Lopes MB, Laws ER., Jr. Surgical management of GH-secreting pituitary adenomas: an outcome study using modern remission criteria. J Clin Endocrinol Metab. 2001;86:4072–4077. doi: 10.1210/jcem.86.9.7819. [DOI] [PubMed] [Google Scholar]
- 26.Bevan JS, Atkin SL, Atkinson AB, Bouloux PM, Hanna F, Harris PE, James RA, McConnell M, Roberts GA, Scanlon MF, Stewart PM, Teasdale E, Turner HE, Wass JA, Wardlaw JM. Primary medical therapy for acromegaly: an open, prospective, multicenter study of the effects of subcutaneous and intramuscular slow-release octreotide on growth hormone, insulin-like growth factor-I, and tumor size. J Clin Endocrinol Metab. 2002;87:4554–4563. doi: 10.1210/jc.2001-012012. [DOI] [PubMed] [Google Scholar]
- 27.Arita K, Kurisu K, Tominaga A, Eguchi K, Iida K, Uozumi T, Kasagi F. Mortality in 154 surgically treated patients with acromegaly--a 10-year follow-up survey. Endocr J. 2003;50:163–172. doi: 10.1507/endocrj.50.163. [DOI] [PubMed] [Google Scholar]
- 28.Fukuda I, Hizuka N, Murakami Y, Itoh E, Yasumoto K, Sata A, Takano K. Clinical features and therapeutic outcomes of 65 patients with acromegaly at Tokyo Women's Medical University. Intern Med. 2001;40:987–992. doi: 10.2169/internalmedicine.40.987. [DOI] [PubMed] [Google Scholar]
- 29.Ezzat S, Forster MJ, Berchtold P, Redelmeier DA, Boerlin V, Harris AG. Acromegaly. Clinical and biochemical features in 500 patients. Medicine (Baltimore) 1994;73:233–240. [PubMed] [Google Scholar]
- 30.Klijn JG, Lamberts SW, de Jong FH, van Dongen KJ, Birkenhager JC. Interrelationships between tumour size, age, plasma growth hormone and incidence of extrasellar extension in acromegalic patients. Acta Endocrinol (Copenh) 1980;95:289–297. doi: 10.1530/acta.0.0950289. [DOI] [PubMed] [Google Scholar]