

Abstract
A mononuclear Ni(II) complex having an acireductone type ligand, and supported by the bnpapa (N,N-bis((6-neopentylamino-2-pyridyl)methyl-N-((2-pyridyl)methyl)amine ligand, [(bnpapa)Ni(PhC(O)C(OH)C(O)Ph)]ClO4 (14), has been prepared and characterized by elemental analysis, 1H NMR, FTIR, and UV-vis. To gain insight into the 1H NMR features of 14, the air stable analog complexes [(bnpapa)Ni(CH3C(O)CHC(O)CH3)]ClO4 (16) and [(bnpapa)Ni(ONHC(O)CH3)]ClO4 (17) were prepared and characterized by X-ray crystallography, 1H NMR, FTIR, UV-vis, mass spectrometry, and solution conductivity measurements. Compounds 16 and 17 are 1:1 electrolyte species in CH3CN. 1H and 2H NMR studies of 14, 16, and 17 and deuterated analogs revealed that the complexes having six-membered chelate rings for the exogenous ligand (14 and 16) do not have a plane of symmetry within the solvated cation and thus exhibit more complicated 1H NMR spectra. Compound 17, as well as other simple Ni(II) complexes of the bnpapa ligand (e.g. [(bnpapa)Ni(ClO4)(CH3CN)]ClO4 (18) and [(bnpapaNi)2(μ-Cl)2](ClO4)2 (19)), exhibit 1H NMR spectra consistent with the presence of a plane of symmetry within the cation. Treatment of [(bnpapa)Ni(PhC(O)C(OH)C(O)Ph)]ClO4 (14) with O2 results in aliphatic carbon-carbon bond cleavage within the acireductone-type ligand and the formation of [(bnpapa)Ni(O2CPh)]ClO4 (9), benzoic acid, benzil, and CO. Use of 18O2 in the reaction gives high levels of incorporation (>80%) of one labeled oxygen atom into 9 and benzoic acid. The product mixture and level of 18O incorporation in this reaction is different than that exhibited by the analog supported the hydrophobic 6-Ph2TPA ligand, [(6-Ph2TPA)Ni(PhC(O)C(OH)C(O)Ph)]ClO4 (2). We propose that this difference is due to variations in the reactivity of bnpapa- and 6-Ph2TPA-ligated Ni(II) complexes with triketone and/or peroxide species produced in the reaction pathway.
Introduction
Interest in how the secondary environment of a metal center influences the reactivity of metal-bound ligands stems from the possible roles of secondary interactions in metal-catalyzed reactions in biological systems.1 In the acireductone dioxygenases Ni(II)-ARD and Fe(II)-ARD, which contain the same protein component, it is proposed that differences in the secondary environment surrounding the divalent metal center influence the coordination mode of the acireductone substrate and the regioselectivity of the oxidative carbon-carbon bond cleavage reaction.1 Specifically, in Ni(II)-ARD the positioning of a tryptophan side chain (W162) is suggested to promote the formation of a six-membered chelate ring in the enzyme substrate adduct (Scheme 1(top)).2 Reaction of this adduct with O2 results in oxidative cleavage of the C(1)-C(2) and C(2)-C(3) bonds and the formation of CO and carboxylate products. For Fe(II)-ARD, a more open active site is suggested to enable the formation of a five-membered chelate enediolate structure, within which only C(1)-C(2) bond cleavage occurs upon reaction with O2 (Scheme 1(bottom)). For both Ni(II)-ARD and Fe(II)-ARD secondary hydrogen bonding interactions involving an arginine residue are suggested to stabilize enolate/enediolate coordination.
Scheme 1.

Interest in reactions of relevance to Ni(II)-ARD also stems from the fact that carbon monoxide is a molecule of current interest in biological systems. In humans, CO is primarily produced via the oxidative breakdown of heme catalyzed by heme oxygenases.3 While carbon monoxide is typically viewed as a toxic gas, recent studies have shown that this small molecule can have beneficial health effects. The signaling mechanism of CO is similar to that of NO in that it activates soluble guanylyl cyclase to produce cyclic guanylyl monophosphate (cGMP).3 This compound is involved in pathways pertinent to smooth muscle relaxation, platelet inhibition, and cell growth and differentiation. As these factors are related to several diseases, recent studies have focused on efforts toward the development of CO-releasing compounds for use as therapeutic agents.4 The majority of the chemical compounds tested so far have been transition metal carbonyl species5, although a main group CO-releasing compound (Na2[H3BCO2]) has been recently reported.4 We are interested in the CO release properties of acireductones and flavonolates, both of which possess three consecutive carbon centers having an oxygen substitutent. In fungal and bacterial systems, quercetin dioxygenases catalyze CO release from a metal-coordinated flavonolate in a 2,4-dioxygenolytic ring cleavage reaction.6–14 In a broad sense, we are interested in understanding in detail the chemical factors that modulate the CO-release reactivity of metal-coordinated acireductone and flavonolate species, with a long-term goal of producing new types of CO-releasing compounds containing these novel structural motifs.
Our laboratory has reported the only examples to date of synthetic complexes of relevance to substrate- and product carboxylate-bound forms of Ni(II)-ARD.15–18 We recently reported that use of a chelate ligand having a mixed hydrophobic/hydrogen bond donor secondary environment produced the first example of a Ni(II) enediolate complex of an acireductone-type ligand (1, Figure 1(top)).18 Somewhat surprisingly, this complex was originally generated under similar conditions to those used to produce the mononuclear Ni(II) enolate complex [(6-Ph2TPA)Ni(PhC(O)C(OH)C(O)Ph)]ClO4 (2, Figure 1(bottom)). These combined results indicate that the presence of a hydrogen bond donor in the secondary environment influences the coordination chemistry of an acireductone-type ligand.
Figure 1.

Supporting chelate ligands used to isolate Ni(II) enediolate (top) and enolate (bottom) complexes of relevance to Ni(II)-ARD.
In terms of carboxylate chemistry, we discovered that the secondary environment of the chelate ligand affects the coordination chemistry of Ni(II)-carboxylate species that are generated in O2-dependent ARD-type reactions. As shown in Scheme 2(top), a Ni(II)-carboxylate ligand in a hydrophobic microenvironment undergoes a shift from bidentate to monodentate in the presence of water (conversion of compounds 3–5 to 6–8).17 No evidence was found for this type of carboxylate shift chemistry in bnpapa-ligated Ni(II) carboxylate complexes (9–11, Scheme 2 (bottom)). In these analogs, the hydrogen bond donor appendages interact with one oxygen atom of the bidentate carboxylate. In a Ni(II) benzoate complex supported by a chelate ligand containing both hydrophobic phenyl appendages and a hydrogen bond donor, [(6-NA-6-Ph2TPA)Ni(O2CPh)(H2O)]ClO4 (12), a bidentate coordinated carboxylate ligand was also identified.18
Scheme 2.

From our initial studies, it is clear that the ligand environment of a Ni(II) center influences the chemistry of an acireductone-type ligand. In the studies outlined herein, we have further evaluated the influence of the chelate ligand environment by preparing and characterizing the enolate complex [(bnpapa)Ni(PhC(O)C(OH)C(O)Ph)]ClO4 (14) as an analog to the 6-Ph2TPA-supported 2 (Figure 1). Complex 14 has been characterized by elemental analysis, 1H NMR, FTIR, and UV-vis. Comparative O2 reactivity studies for 2 and 14 have been performed under conditions wherein the inorganic and organic byproducts were identified. Interestingly, these reactions differ in the specific mixture of products that is generated and in the level of 18O incorporation in the benzoate/benzoic acid products. We propose that these differences are due to variations in the reactivity of bnpapa- and 6-Ph2TPA ligated Ni(II) complexes with triketone and peroxide/hydroperoxide species produced in the reaction pathway.
In the course of this research, as a means of gaining additional insight in the spectroscopic and solution properties of [(bnpapa)Ni(PhC(O)C(OH)C(O)Ph)]ClO4(14), we have also prepared and characterized mononuclear bnpapa-supported Ni(II) acetoacetonato and hydroxamato complexes, as well as perchlorate and chloro derivatives. Preparation of this family of complexes has enabled a structural comparison with a similar family of 6-Ph2TPA-supported mononuclear Ni(II) complexes. This comparison has provided insight into the structural factors that may be responsible for the differences in O2 reactivity between [(bnpapa)Ni(PhC(O)C(OH)C(O)Ph)]ClO4(14) and [(6-Ph2TPA)Ni(PhC(O)C(OH)C(O)Ph)]ClO4 (2) noted above.
Experimental
General Methods
All reagents and solvents were obtained from commercial sources and were used as received unless otherwise noted. Solvents were dried according to published procedures and were distilled under N2 prior to use.19 Air sensitive reactions were performed in the MBraun Unilab glovebox or a Vacuum Atmospheres MO-20 glovebox under a N2 atmosphere. The ligand bnpapa and the Ni(II) complex [(6-Ph2TPA)Ni(PhC(O)C(OH)C(O)Ph)]ClO4 (2) were prepared according to literature procedures.15,17
Physical Methods
1H NMR spectra of Ni(II) complexes were obtained using a Bruker ARX-400 spectrometer as previously described.20 Chemical shifts (in ppm) are referenced to the residual solvent peak(s) in CHD2CN (1H, 1.94 (quintet) ppm). UV-vis spectra were collected on a HP8453A spectrometer at ambient temperature. FTIR spectra were recorded on a Shimadzu FTIR-8400 spectrometer as KBr pellets. Conductance measurements were made at 22(1) °C using an YSI model 31A conductivity bridge with a cell having a cell constant of 1.0 cm−1 and using Me4NClO4 as a 1:1 electrolyte standard. Preparation of the acetonitrile and standard solutions for conductance measurements and subsequent data analysis were performed as previously described.21 Mass spectrometry data was obtained at the Mass Spectrometry Facility, Department of Chemistry, University of California, Riverside. Elemental analyses were performed by Atlantic Microlabs, Inc., Norcross, Georgia, or Canadian Microanalytical Service, Inc., British Columbia.
Caution! Perchlorate salts of metal complexes with organic ligands are potentially explosive. Only small amounts of material should be prepared, and these should be handled with great care.22
[(bnpapa)Ni(PhC(O)C(OH)C(O)Ph)]ClO4 (14)
Under a N2 atmosphere, equimolar amounts of bnpapa (0.273 mmol) and Ni(ClO4)2·6H2O (0.273 mmol) were mixed in acetonitrile and stirred until the ligand had fully dissolved. Me4NOH·5H2O (0.301 mmol) was then added to the mixture, which was stirred for additional 15 minutes, resulting in a dark green solution. The solution was added to solid 2-hydroxy-1,3-diphenylpropan-1,3-dione23 (0.301 mmol) and the mixture was stirred for 4 h, yielding a very dark orange/red solution. The solvent was then removed under reduced pressure. The solid was dissolved in CH2Cl2 and the solution was filtered through a celite plug. The filtrate was concentrated and brought to dryness under vacuum, leaving a dark orange/red solid, which was washed with Et2O and dried under vacuum. Yield: 83%. Anal. Calcd for C43H51N6O7ClNi: C, 60.19; H, 5.99; N, 9.79. Found: C, 60.52; H, 5.88; N, 9.57. UV-vis [CH3CN, nm (ε, M−1cm−1)] 393(10,000); FTIR: (KBr, cm−1) 3420 (br s, νNH), 1094 (νClO4), 621 (νClO4).
[(bnpapa)Ni(CH3C(O)CHC(O)CH3)]ClO4 (16)
A solution of Ni(ClO4)2·6H2O (0.15 mmol) in CH3CN (~1 mL) was added to solid bnpapa (0.15 mmol). The resulting mixture was stirred for 20 min at room temperature. The solution was then transferred to a vial containing Me4NOH·5H2O (0.15 mmol). To this mixture 2,4-pentanedione (0.15 mmol) was added and the resulting solution was stirred overnight. The solvent was then removed under reduced pressure. The remaining solid was dissolved in CH2Cl2 (~5 mL) and the solution was filtered through a celite/glass wool plug. Pentane diffusion into CH2Cl2 yielded purple crystals suitable for X-ray crystallography. The crystals were crushed and dried under vacuum prior to elemental analysis. Yield: 46%. Anal. Calcd for C33H47ClN6NiO6: C, 55.21; H, 6.60; N, 11.71. Found: C, 55.28; H, 6.55; N, 11.70. FTIR (KBr, cm−1) ~3300 (s, νNH), 1089 (νClO4), 622 (νClO4). UV-vis [CH3CN, nm (ε, M−1cm−1)] 244(31600), 305(17800), 543(sh), 966(19); FAB-MS [m/z (relative intensity)]: 617 ([M – ClO4]+, 100%).
[(bnpapa)Ni(ONHC(O)CH3)]ClO4 (17)
A solution of Ni(ClO4)2·6H2O (0.11 mmol) in CH3CN (~1 mL) was added to solid bnpapa (0.11 mmol). The resulting mixture was stirred for 5 min at the room temperature. The solution was then transferred to a vial containing Me4NOH·5H2O (0.12 mmol) and acetohydroxamic acid (0.12 mmol), and the resulting solution was stirred for 1 h. The solvent was then removed under reduced pressure. The remaining solid was dissolved in CH2Cl2 (~5 mL) and the solution was filtered through a celite/glass wool plug. Diffusion of diethyl ether into a CH2Cl2/MeOH/iPrOH (1:0.5:1) solution of the compound yielded purple crystals suitable for single crystal X-ray crystallography. These crystals were crushed and dried under vacuum prior to elemental analysis. Yield: 60%. Anal. Calcd for C30H44N7NiO6Cl·0.25AHA·0.35CH2Cl2: C, 51.93; H, 6.34; N, 13.12. Found: C, 51.55; H, 6.41; N, 13.63. The presence of acetohydroxamic acid (AHA) in the crystalline sample was confirmed by X-ray crystallography. The presence of CH2Cl2 in the elemental analysis sample was confirmed by 1H NMR. FTIR (KBr, cm−1) ~3288 (s, νNH), 1103 (νClO4), 621 (νClO4); UV-vis [CH3CN, nm (ε, M−1cm−1)] 247(25800), 325(10400), 548(sh), 977(32); ESI/APCI-MS [m/z (relative intensity)]: 592.2914 ([M – ClO4]+, 100%).
[(bnpapa)Ni(ClO4)(CH3CN)]ClO4 (18)
A solution of Ni(ClO4)2·6H2O (0.07 mmol) in CH3CN (~1 mL) was added to solid bnpapa (0.07 mmol). The resulting mixture was stirred for 20 min at room temperature. The solvent was then removed under reduced pressure and the residue was dissolved in CH2Cl2. Pentane diffusion yielded purple crystals suitable for X-ray crystallography. The crystals were crushed and dried under vacuum prior to elemental analysis. Yield: 54%. Anal. Calcd for C30H43Cl2N7NiO8·1/2CH2Cl2: C, 45.69; H, 5.53; N, 12.23. Found: C, 45.32; H, 5.60; N, 12.09. UV-vis [CH3CN, nm (ε, M−1cm−1)] 318(10300), 553(15), 931(17); FTIR (KBr, cm−1) ~3400 (s, νNH), 1103 (νClO4), 624 (νClO4); FAB-MS [m/z (relative intensity)]: 617 ([M – ClO4 – CH3CN]+, 100%).
[(bnpapaNi)2(μ-Cl)2](ClO4)2 (19)
A solution of Ni(ClO4)2·6H2O (0.14 mmol) in MeOH (~1 mL) was added to a solution of bnpapa (0.14 mmol) in MeOH (~1 mL). The resulting mixture was stirred for 20 min at room temperature. The solution was then transferred to a vial containing Me4NCl·5H2O (0.14 mmol) and the resulting mixture was stirred for 1 h. The solvent was then removed under reduced pressure. The remaining solid was dissolved in CH2Cl2 (~5 mL), and the solution was filtered through a celite/glass wool plug. Diffusion of diethyl ether into a CH2Cl2 solution of the compound yielded green crystals suitable for X-ray crystallography. The crystals were crushed and dried under vacuum prior to elemental analysis. Yield: 33%. Anal. Calcd for C28H40Cl2N6NiO4: C, 51.40; H, 6.16; N, 12.84. Found: C, 51.15; H, 6.29; N, 12.88. FTIR (KBr, cm−1) ~3340 (s, νNH), 1095 (νClO4), 621 (νClO4); UV-vis [CH3CN, nm (ε, M−1cm−1)] 325(15900), 600(31), 1032(33); ESI/APCI-MS [m/z (relative intensity)]: 553.2348 ([M – 2ClO4]2+, 100%).
O2 Reactivity of 14: Product Isolation and 18O Labeling Studies
Complex 14 (0.101 mmol) was dissolved in 10 mL of acetonitrile and O2 was bubbled through the solution for ~1 min. The reaction was then left stirring overnight at ambient temperature. Over the course of this time, the color changed from deep orange/red to pale yellow. CO production was verified by the PdCl2 method.24 The solvent was removed under reduced pressure and the resulting solid was dissolved in hexanes:ethyl acetate (4:1) and the solution was passed through a silica plug. This produced a yellow filtrate containing the organic products from the reaction. Ni(II) complexes that adhered to the silica gel were then eluted using acetonitrile, followed by methanol. As determined by TLC, 1H NMR, and GC-MS, the organic fraction contained primarily benzoic acid (11 mg overall yield – 91% based on production of one equivalent of free benzoic acid per enolate ligand; sample is 88–90% benzoic acid based on 1H NMR), with a small amount of benzil (PhC(O)C(O)Ph; 10–12%) and a trace amount of the ester PhC(O)OCH2C(O)Ph present. The ester was identified via independent synthesis.25 This compound is an isomer of 2-hydroxy-1,3-diphenylpropan-1,3-dione23, and the reaction leading to ester formation will be discussed in detail elsewhere. In each reaction, two Ni(II) complexes were eluted from the column, [(bnpapa)Ni(O2CPh)]ClO4 (9) and [(bnpapa)Ni(PhC(O)C(O)CHC(O)Ph)]ClO4 (15), the latter of which was present in a very low amount (<5%) and was identified via mass spectrometry (ESI-MS [m/z (relative intensity)]: 769.3376 (M – ClO4)+, 100%)). The formation of 15 is related to the trace ester formation in the reaction and will be discussed in detail elsewhere. The yield of the metal complexes was ~88% by mass following the column (based on stoichiometric formation of [(bnpapa)Ni(O2CPh)]ClO4 (9)). Thus, from this data, the formation of [(bnpapa)Ni(O2CPh)]ClO4 (9) was judged as quantitative. Use of 18O2 in the reaction of 14 with O2 produces benzoic acid with 81% incorporation of one labeled oxygen atom. No 18O incorporation was found in benzil or the ester PhC(O)OCH2C(O)Ph, as determined by GC-MS. Mass spectral analysis of the metal complexes indicated 87% 18O incorporation into one oxygen atom of the benzoate ligand of 9. No 18O incorporation was found in 15.
Reanalysis of O2 reactivity of [(6-Ph2TPA)Ni(PhC(O)C(OH)C(O)Ph)]ClO4 (2)
Compound 2 was treated with an excess of O2 in a reaction very similar to that outlined above using 14. An identical work-up procedure was also performed. The organic products generated were benzoic acid and benzil in a reproducible ~75:25 ratio. A trace amount of the ester PhC(O)OCH2C(O)Ph is also generated, as determined by GC-MS and 1H NMR. The total yield of organic products was ~96% based on the proposed formation of one equivalent of free benzoic acid per enolate ligand of 2. As previously reported, the Ni(II) complex, [(6-Ph2TPA)Ni(O2Ph)]ClO4 (3) is produced in this reaction.16 A trace amount of the enolate complex [(6-Ph2TPA)Ni(PhC(O)C(O)CHC(O)Ph)]ClO416 is also generated. The reaction leading to the formation this complex will be discussed in detail elsewhere. The total mass of complexes isolated suggests a ~70% yield based on the stoichiometric formation of 3. However, a control experiment using [(6-Ph2TPA)Ni(O2Ph)]ClO4 (3) indicated that ~30% of the material is lost in the column purification process used to isolate the metal complexes from the reaction mixture. Thus, the formation of 3 is nearly quantitative in the O2 reaction of 2. Use of 18O2 in the reaction produced free benzoic acid with 59% 18O incorporation in one oxygen atom position, and the benzoate complex 3 having 64% incorporation in one oxygen atom. The level of isotope incorporation into 3 is slightly higher than that previously reported (~50%) based on multiple 18O reactions.16
18O2 reactivity of [Me4N][PhC(O)C(OH)C(O)Ph]
We have previously reported that treatment of the salt [Me4N][PhC(O)C(OH)C(O)Ph] with O2 results in the formation of tetramethylammonium benzoate, benzoic acid and CO.16 We have repeated this reaction using 18O2 and have found by mass spectrometry that the benzoate/benzoic acid contains ~65% 18O incorporation when generated from Me4NOH·5H2O and PhC(O)C(OH)C(O)Ph in dry CH3CN. Pre-drying of the Me4NOH·5H2O under vacuum for 24 h prior to use in the reaction produced a slightly higher level of 18O incorporation (72%) in the benzoate/benzoic acid product.
X-ray Crystallography
A single crystal of each compound 16, 17·1/4AHA (AHA = acetohydroxamic acid), 18·CH2Cl2, and 19·2CH2Cl2 was mounted on a glass fiber with traces of viscous oil and then transferred to a Nonius KappaCCD diffractometer equipped with Mo Kα radiation (λ = 0.71073 Å) for data collection. For unit cell determination, ten frames of data were collected at 103(1) K or 150(1) K with an oscillation range of 1 deg/frame and an exposure time of 20 sec/frame. Final cell constants were determined from a set of strong reflections from the actual data collection. All reflections were indexed, integrated, and corrected for Lorentz polarization and absorption effects using DENZO-SMN and SCALEPAC.26 The structures were solved by a combination of direct and heavy-atom methods using SIR 97. All of the non-hydrogen atoms were refined with anisotropic displacement coefficients. Unless otherwise stated, all hydrogen atoms were assigned isotropic displacement coefficients U(H) = 1.2U(C) or 1.5U(Cmethyl), and their coordinates were allowed to ride on their respective carbons using SHELXL97.27 Complex 16 crystallizes in the space group P-1, with two carbon atoms of one neopentyl group exhibiting disorder over two positions. Complex 17·1/4AHA crystallizes in the space group C2/c with three oxygen atoms of the perchlorate anion and the carbon atoms of one nepentyl group exhibiting disorder. Complex 18·CH2Cl2 crystallizes in the space group P21/c. The CH2Cl2 solvate molecule exhibits disorder. Complex 19·2CH2Cl2 crystallizes in the space group P21/n, with one CH2Cl2 exhibiting disorder in the position of one chlorine atom.
Results
Preparation and Characterization of [(bnpapa)Ni(PhC(O)C(OH)C(O)Ph)]ClO4 (14)
Treatment of the bnpapa chelate ligand with equimolar amounts of Ni(ClO4)2·6H2O, Me4NOH·5H2O, and 2-hydroxy-1,3-diphenylpropan-1,3-dione in CH3CN, followed by work up using CH2Cl2, yielded a deep orange/red solid. Unfortunately, repeated attempts to produce crystals of this complex suitable for single crystal X-ray crystallography were unsuccessful. Instead, the orange/red solid was characterized by elemental analysis, UV-vis, FTIR, and 1H NMR. The elemental analysis data is consistent with the proposed formulation. The π→π* transition of the coordinated enolate ligand of 14 is found at 393 nm (ε ~ 10,000 M−1cm−1). The wavelength and intensity of this absorption feature is generally similar to that reported for [(6-Ph2TPA)Ni((PhC(O)C(OH)C(O)Ph)]ClO4 (2; λmax = 399 nm (ε ~ 6,800 M−1cm−1)).16 A broad absorption feature at ~3420 cm−1 in the infrared spectrum of 14 is consistent with hydrogen bonding interactions involving the neopentylamine groups of the bnpapa chelate ligand. Based on structural studies of air stable analogs (vida infra), we propose that this hydrogen bonding involves an oxygen atom of the chelating enolate ligand that is positioned trans to the tertiary amine nitrogen donor of the chelate ligand.
Air Stable Analogs
To aid in the interpretation of the 1H NMR spectroscopic features of 14, air stable analog complexes containing a bidentate acetoacetonato ([(bnpapa)Ni(CH3C(O)CHC(O)CH3)]ClO4 (16)) or hydroxamato ([(bnpapa)Ni(CH3C(O)NHO)]ClO4 (17)) ligand were prepared and comprehensively characterized. For solution conductivity and spectroscopic studies, the perchlorate complex [(bnpapa)Ni(ClO4)(CH3CN)]ClO4 (18) and chloride complex [(bnpapaNi)2(μ-Cl)2](ClO4)2 (19) were also prepared and characterized. Drawings of the cationic portions of these complexes are given in Scheme 3.
Scheme 3.

X-ray Crystallography
The cationic portions of 16 and 17 are shown in Figure 2, and those of 18 and 19 are shown in Figure 3. Details of the data collection and refinement are given in Table 1. Selected bond distances and angles are given in Table 2. The mononuclear 16 and 17 each contain a pseudo-octahedral Ni(II) center with the oxygen donor atoms of the chelate ligand positioned trans to N(3) and N(6). The respective Ni-O/N bonds of the primary coordination sphere are similar in these complexes, with the most noticeable difference in bond lengths being that the Ni-N interactions involving the neopentylamine-appended pyridyl donors are ~0.02 Å longer in 16 than those found in the hydroxamato complex (17). In terms of bond angles, a more acute O(1)-Ni(1)-O(2) angle in 17 (81.86(10)°) than that found in 16 (94.98(3)°) is due to the difference in chelate ring size of the bidentate O,O-donor ligands. In both complexes, an oxygen atom of the bidentate chelate ligand accepts two hydrogen bonds from the neopentyl amine groups of the supporting chelate ligand. These interactions are characterized by N(H)....O distances of 2.8–2.9 Å.
Figure 2.

Thermal ellipsoid drawings of the cationic portions of 16 and 17. All ellipsoids are drawn at the 50% probability level. Hydrogen atoms, other than the neopentyl amine N-H hydrogen atoms, and the hydroxyamato N-H hydrogen in 17, have been omitted for clarity.
Figure 3.

Thermal ellipsoid drawings of the cationic portions of 18 and 19. All ellipsoids are drawn at the 50% probability level. Hydrogen atoms other than the neopentyl amine N-H hydrogen atoms have been omitted for clarity.
Table 1.
Summary of X-ray data collection and refinementa
| 16 | 17·1/4AHA | 18·CH2Cl2 | 19·2CH2Cl2 | |
|---|---|---|---|---|
| Empirical formula | C33H47ClN6NiO6 | C30.50H45.25ClN7.25NiO6.5 | C31H45Cl4N7NiO8 | C58H84Cl8N12Ni2O8 |
| Mr | 717.93 | 711.25 | 844.25 | 1478.39 |
| Crystal system | triclinic | monoclinic | monoclinic | monoclinic |
| Space group | P-1 | C2/c | P21/c | P21/n |
| a/Å | 8.9810(2) | 32.3396(8) | 8.8221(2) | 10.9123(2) |
| b/Å | 10.9647(2) | 11.4649(3) | 20.0037(4) | 18.9857(3) |
| c/Å | 18.2657(4) | 19.4692(3) | 21.7170(4) | 16.9677(3) |
| α/° | 102.6548(12) | 90 | 90 | 90 |
| β/° | 96.3498(11) | 93.1236(14) | 91.7754(11) | 96.6495(10) |
| γ/° | 93.1764(13) | 90 | 90 | 90 |
| V/Å3 | 1738.34(6) | 7207.9(3) | 3830.66(14) | 3491.68(10) |
| Z | 2 | 8 | 4 | 2 |
| Dc/Mg m−3 | 1.372 | 1.312 | 1.464 | 1.406 |
| T/K | 150(1) | 103(1) | 150(1) | 150(1) |
| Color | purple | purple | purple | green |
| Crystal size/mm | 0.38 × 0.30 × 0.28 | 0.20 × 0.20 × 0.15 | 0.38 × 0.38 × 0.33 | 0.38 × 0.30 × 0.25 |
| μ/(mm−1) | 0.687 | 0.644 | 0.842 | 0.904 |
| 2θ max(°) | 55.00 | 55.04 | 54.96 | 54.96 |
| Completeness to θ(%) | 97.9 | 99.5 | 99.2 | 99.8 |
| Reflections collected | 12170 | 15128 | 14723 | 15105 |
| Independent reflections | 7825 | 8259 | 8717 | 7984 |
| Rint | 0.0219 | 0.0401 | 0.0255 | 0.0200 |
| Variable parameters | 577 | 486 | 469 | 422 |
| R1/wR2b | 0.0491/0.1289 | 0.0620/0.1452 | 0.0636/0.1571 | 0.0432/0.1038 |
| Goodness-of-fit (F2) | 1.043 | 1.040 | 1.042 | 1.081 |
| Δρmax/min/e Å−3 | 0.758/−0.778 | 0.790/−0.626 | 0.783/−1.250 | 0.499/−0.530 |
Diffractometer: Nonius KappaCCD; Radiation used: Mo Kα (λ = 0.71073 Å).
R1 = Σ||Fo| − |Fc||/Σ ||Fo|; wR2 = [Σ[w(Fo2−Fc2)2]/[Σ(Fo2)2]]1/2, where w = 1/[σ2(Fo2) + (aP)2 + bP].
Table 2.
Selected Bond Distances (Å) and Angles (°)
| 16 | 17·1/4AHA | 18·CH2Cl2 | 19·2CH2Cl2 | |
|---|---|---|---|---|
| Ni(1)-O(1) | 2.0162(18) | 2.018(2) | 2.177(2) | |
| Ni(1)-O(2) | 2.0183(17) | 2.058(2) | ||
| Ni(1)-N(2) | 2.1509(19) | 2.137(3) | 2.151(3) | 2.1998(19) |
| Ni(1)-N(3) | 2.0762(19) | 2.076(3) | 2.055(3) | 2.0648(19) |
| Ni(1)-N(4) | 2.154(2) | 2.134(3) | 2.164(3) | 2.2267(19) |
| Ni(1)-N(6) | 2.061(2) | 2.048(3) | 2.042(3) | 2.038(2) |
| Ni(1)-N(7) | 2.049(3) | |||
| Ni(1)-Cl(1) | 2.4284(6) | |||
| Ni(1)-Cl(1)#1 | 2.4485(6) | |||
| O(1)-Ni(1)-O(2) | 90.94(8) | 81.86(10) | ||
| O(1)-Ni(1)-N(6) | 172.86(8) | 94.53(10) | 90.23(12) | |
| O(2)-Ni(1)-N(6) | 95.76(8) | 174.68(10) | ||
| O(1)-Ni(1)-N(3) | 89.89(8) | 178.78(10) | 176.14(10) | |
| O(2)-Ni(1)-N(3) | 178.63(7) | 99.35(11) | ||
| N(6)-Ni(1)-N(3) | 83.46(8) | 84.26(11) | 85.97(13) | 85.13(8) |
| O(1)-Ni(1)-N(2) | 91.27(7) | 99.44(11) | 99.80(10) | |
| O(2)-Ni(1)-N(2) | 99.02(7) | 89.69(10) | ||
| N(6)-Ni(1)-N(2) | 90.13(7) | 87.04(11) | 88.72(11) | 88.00(7) |
| N(3)-Ni(1)-N(2) | 79.86(7) | 80.67(11) | 80.84(11) | 79.27(7) |
| O(1)-Ni(1)-N(4) | 88.86(8) | 100.25(12) | 98.42(10) | |
| O(2)-Ni(1)-N(4) | 99.99(8) | 91.27(10) | ||
| N(6)-Ni(1)-N(4) | 87.55(8) | 93.24(11) | 89.12(11) | 90.16(7) |
| N(3)-Ni(1)-N(4) | 81.12(8) | 79.68(11) | 80.84(11) | 79.58(7) |
| N(2)-Ni(1)-N(4) | 160.98(8) | 160.22(11) | 161.66(11) | |
| N(6)-Ni(1)-N(7) | 179.59(13) | |||
| N(7)-Ni(1)-N(3) | 94.09(12) | |||
| N(7)-Ni(1)-N(2) | 90.89(11) | |||
| N(7)-Ni(1)-N(4) | 91.29(11) | |||
| N(7)-Ni(1)-O(1) | 89.71(11) | |||
| N(6)-Ni(1)-Cl(1) | 97.34(6) | |||
| N(3)-Ni(1)-Cl(1) | 177.53(6) | |||
| N(2)-Ni(1)-Cl(1) | 100.65(5) | |||
| N(4)-Ni(1)-Cl(1) | 100.48(5) | |||
| N(6)-Ni(1)-Cl(1)#1 | 177.63(6) | |||
| N(3)-Ni(1)-Cl(1)#1 | 93.76(6) | |||
| N(2)-Ni(1)-Cl(1)#1 | 89.74(5) | |||
| N(4)-Ni(1)-Cl(1)#1 | 91.70(5) | |||
| Cl(1)-Ni(1)-Cl(1)#1 | 83.77(2) | |||
| Ni(1)-Cl(1)-Ni(1)#1 | 96.23(2) |
Solution Conductivity Properties
The acetoacetonato complex 16 and the hydroxamato complex 17 both are 1:1 electrolytes in CH3CN, as determined by variable concentration conductance measurements.21 Onsager plots for these complexes (Figure 4) exhibit slopes similar to the 1:1 standard Me4NClO4, and a notably different slope than that exhibited by [(bnpapa)Ni(ClO4)(CH3CN)](ClO4)2 (18 (Figure 3(top)) or [(bnpapaNi)2(μ-Cl)2](ClO4)2 (19, Figure 3(bottom)), both of which are 1:2 electrolyte species in CH3CN.
Figure 4.
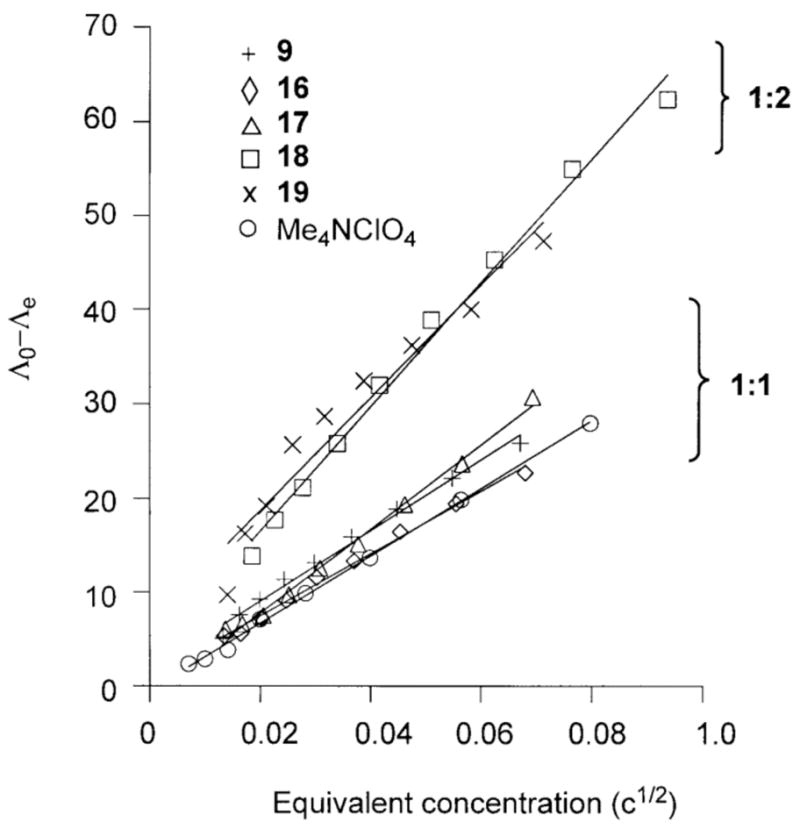
Onsager plots of the solution conductivity properties of 9, 16–19, and the 1:1 standard Me4NClO4 in CH3CN at 22(1) °C.
1H NMR Spectra
The 1H NMR features of analytically pure [(bnpapa)Ni(CH3C(O)CHC(O)CH3)]ClO4 (16) and [(bnpapa)Ni(ONHC(O)CH3)]ClO4 (17) in CD3CN at 22(1) °C in the region of 20–80 ppm are shown in Figure 5(b) and (c). These are compared to the 1H NMR spectral features of the O2-sensitive enolate complex [(bnpapa)Ni(PhC(O)C(OH)C(O)Ph)]ClO4 (14, Figure 5(a)). The resonances in the 20–80 ppm region include those of pyridyl β-H ring protons and a portion of the benzylic proton resonances.20
Figure 5.

1H NMR spectral features of (a) 14, (b) 16, and (c) 17 in the region of 20–80 ppm. Spectra obtained in CD3CN at 22(1) °C.
The 1H NMR spectra of 14 and 16 (Figure 5(a) and (b)) are similar, both having a more complicated appearance than that of the hydroxamato compound 17. Based on the conductivity data presented above for the air stable 16 and 17, which are both 1:1 electrolyte species in acetonitrile, the greater complexity of the 1H NMR spectrum of 16, relative to the spectral features of 17, is not due to the formation of multinuclear species in solution. 2H NMR studies of analogs of 14, 16, and 17 supported by a version of the bnpapa chelate ligand (Figure S1) having deuterium atoms at the benzylic positions, as well as partial deuteration of the β-H positions of the neopentylamine-appended pyridyl rings, indicated the presence of three unique benzylic proton resonances for 14 and 16 in the region of 165–175 ppm. This is consistent with no plane of symmetry within the cation.20 However, 17 exhibits only one benzylic proton resonance in the same region, which is consistent with the presence of a plane of symmetry containing the unsubstituted pyridyl donor and the hydroxamato ligand. The factors that govern the subtle differences in solution structure of the cationic portions of 14, 16, and 17 are not entirely clear, but appear to relate to the chelate ring size of the O,O-donor ligand coordinated to the Ni(II) center. Specifically, the compounds having a six-membered chelate O,O donor lack a plane of symmetry in the solution form of the cation. We note that the 1:2 electrolyte compounds [(bnpapa)Ni(ClO4)(CH3CN)](ClO4)2 (18) or [(bnpapaNi)2(μ-Cl)2](ClO4)2 (19) both exhibit 1H NMR spectra similar to 17 wherein an effective plane of symmetry renders the two neopentylamine-appended pyridyl donors equivalent in solution (Figure S2). Ni(II) carboxylate complexes of the general formula [(bnpapa)Ni(O2CR)]ClO4 (R = Ph, (CH2)2SCH3, and H) also exhibit 1H NMR spectra that are generally similar to the spectrum found for the hydroxamato complex 17.17 In terms of comparing chelate ligand environments, the mononuclear Ni(II) complexes [(6-Ph2TPA)Ni(PhC(O)C(OH)C(O)Ph)]ClO4 (2) and [(6-Ph2TPA)Ni(RC(O)CHC(O)R)]ClO4 (R = Ph, CH3) exhibit spectra which indicate the presence of a plane of symmetry within the cation.15,16 Thus, the bnpapa-ligated Ni(II) six-membered ring enolate complexes 14 and 16 exhibit unique solution 1H NMR properties relative to their hydrophobic analogs.
Based on the structural and solution features of the air stable acetoacetonateo complex 16, and the similarity of the 1H NMR spectrum of this complex to the spectrum of [(bnpapa)Ni(PhC(O)C(OH)C(O)Ph)]ClO4 (14), we propose that the O2-reactive enolate complex 14 has a structure that is similar to the analog supported by 6-Ph2TPA (Figure 6), but is distorted such that no plane of symmetry relates the neopentylamine pyridyl donors in the solution form of the cation.
Figure 6.

(a) Proposed structure of [(bnpapa)Ni(PhC(O)C(OH)C(O)Ph)]ClO4 (14). (b) Structure of [(6-Ph2TPA)Ni(PhC(O)C(OH)C(O)Ph)]ClO4(2) as determined by X-ray crystallography.15
Reaction of 14 with O2
Exposure of a CH3CN solution of 14 to O2 results in an immediate color change from deep orange/red to yellow. CO production in this reaction was identified qualitatively using the PdCl2 method.24 The inorganic and organic products of the reaction were separated by column chromatography and identified (Scheme 4). One equivalent of benzoic acid is generated along with a small amount of benzil (PhC(O)C(O)Ph; ratio of benzoic acid to benzil is ~90:10) and a trace amount of the ester PhC(O)OCH2C(O)Ph, the latter of which was identified via independent synthesis.25 The reaction leading to ester formation will be described elsewhere. The monobenzoate complex [(bnpapa)Ni(O2CPh)]ClO4 (9) is produced in nearly quantitative yield in the reaction, with a trace amount of a second Ni(II) complex, [(bnpapa)Ni(PhC(O)C(O)CHC(O)Ph]ClO4 (15), being identified by mass spectrometry. The production of 15 is related to the presence of the ester PhC(O)OCH2C(O)Ph in the reaction and will be discussed elsewhere. Use of 18O2 in the reaction with 14 produces benzoic acid with 81% 18O incorporation in one oxygen atom position. No 18O incorporation was found in benzil or the ester PhC(O)OCHsC(O)Ph, as determined by GC-MS. Mass spectral analysis of the Ni(II) products indicated that the benzoate complex 9 had 87% 18O incorporation in one oxygen atom of the benzoate ligand. No 18O incorporation was found in 15. We note that the level of 18O incorporation for the reaction involving 14 is slightly higher than that reported for the Ni(II)-ARD enzymatic reaction (77.5%).28
Scheme 4.

Major products identified in reaction of 14 and 2 with O2; results of reactions performed using 18O2.
The work-up procedure used to isolate the organic and inorganic products of the reaction of 14 with O2 is slightly different in terms of solvents used from the procedure that we previously reported for identification of the products of the reaction of [(6-Ph2TPA)Ni(PhC(O)C(OH)C(O)Ph)]ClO4 (2) with O2.16 The new conditions are more amenable to the isolation of free benzoic acid in the reaction mixture. Therefore, the reaction involving 2 was repeated under conditions identical with those employed for [(bnpapa)Ni(PhC(O)C(OH)C(O)Ph)]ClO4 (14), with the results shown in Scheme 4. CO production was again identified qualitatively using the PdCl2 method.24 In this reaction a ~75:25 mixture of benzoic acid and benzil is produced, along with a trace amount of the ester PhC(O)OCH2C(O)Ph. The Ni(II)-containing products are [(6-Ph2TPA)Ni(O2CPh)]ClO4 (3) and a trace amount of [(6-Ph2TPA)Ni(PhC(O)C(O)CHC(O)Ph)]ClO4 (20).16,17 Use of 18O2 in the reaction with 2 yielded benzoic acid with 59% 18O incorporation in one oxygen atom position, and the benzoate complex 3 having 64% incorporation in one oxygen atom. We note that the level of isotope incorporation in 3 is slightly higher that than previously reported (~50%), but is similar to that found for the O2 reaction of the salt [Me4N][PhC(O)C(OH)C(O)Ph], which decomposes upon reaction with 18O2 to give [Me4N][O2CPh] and benzoic acid (~65% incorporation of one 18O into carboxylate group) and CO. Thus, the reaction involving 2 gives a mixture of organic products (benzoic acid and benzil), but with a similar level of 18O incorporation to that found for the O2 reaction of [Me4N][PhC(O)C(OH)C(O)Ph].
Overall, the studies outlined above revealed two interesting differences in the O2 reactivity of [(bnpapa)Ni(PhC(O)C(OH)C(O)Ph)]ClO4 (14) and [(6-Ph2TPA)Ni(PhC(O)C(OH)C(O)Ph)]ClO4 (2). First, the O2 reaction involving the bnpapa-ligated complex 14 produces less of the side product benzil, Second, the reaction involving 14 has a higher level of 18O incorporation in the benzoic acid/benzoate products. These combined results provide evidence that the chelate ligand environment is influencing the aliphatic carbon-carbon bond cleavage reaction pathway.
Comparison of the Secondary Environments of bnpapa- and 6-Ph2TPA-ligated Ni(II) Complexes
To date, we have structurally characterized four bnpapa- and thirteen 6-Ph2TPA-ligated mononuclear Ni(II) complexes. Shown in Tables 3 and 4 are bond distances and angles involving the neopentyl-appended pyridyl donors in the bnpapa complexes and the phenyl-appended pyridyl donors in the 6-Ph2TPA derivatives. Several conclusions can be drawn from comparison of the microenvironments in these two families of complexes. First, the neopentylamine amine-appended pyridyl donors in the bnpapa ligand, which have a substituent that is more electron donating and less sterically demanding relative to the phenyl substitutents of the 6-Ph2TPA ligand, have slightly shorter Ni-N distances (overall average of bnpapa complexes 2.14 Å; 6-Ph2TPA complexes 2.22 Å). There is also a more linear N-Ni-N bond angle (overall average of bnpapa complexes 161°; 6-Ph2TPA complexes 154°) between the substituted pyridyl groups. Second, the phenyl-appended pyridyl donors of 6-Ph2TPA exhibit more variation in terms of Ni-N distances (~0.12 Å) in response to the presence of different exogenous ligands. This is not surprising in that aryl-appended pyridyl donors of chelate ligands have been shown to fully dissociate from a metal center in response to exogenous ligand coordination.29 Third, the microenvironment of bnpapa-ligated complexes is more compact, as determined by comparison of the distance between the two neopentylamine nitrogen atoms (~5.6 Å) versus that of the ipso carbons of the phenyl appendages of 6-Ph2TPA (~6.4 Å).
Table 3.
Structural properties of bnpapa-ligated mononuclear Ni(II) complexes
| Complex | N(2)-Ni-N(4) (°)a | Ni-N(2), Ni-N(4)a (Å) | N(H)…N(H) (Å) | Reference |
|---|---|---|---|---|
| [(bnpapa)Ni(ClO4)(CH3CN)]ClO4 | 161.66(11) | 2.151(3), 2.164(3) | 5.7 | this work |
| [(bnpapa)Ni(ONHC(O)CH3)]ClO4 | 160.22(11) | 2.137(3), 2.134(3) | 5.5 | this work |
| [(bnpapa)Ni(CH3C(O)CHC(O)CH3)]ClO4 | 160.98(8) | 2.1509(19), 2.154(2) | 5.6 | this work |
| [(bnpapa)Ni(O2Ph)]ClO4a | 161.78(12) | 2.125(3), 2.136(3) | 5.6 | 17 |
N(2) and N(4) are the neopentylamine-appended pyridyl donors of the bnpapa ligand.
Data reported for one of two crystalline forms of the complex that were previously reported.
Table 4.
Structural properties of 6-Ph2TPA-ligated mononuclear Ni(II) complexes
| Complex | N(3)-Ni-N(4) (°)a | Ni-N(3), Ni-N(4) (Å)a | C(Ph)..C(Ph)b (Å) | Reference |
|---|---|---|---|---|
| [(6-Ph2TPA)Ni(CH3CN)(CH3OH)](ClO4)2 | 158.59(16) | 2.218(5), 2.200(5) | 6.3 | 20 |
| [(6-Ph2TPA)Ni(ONHC(O)CH3)]ClO4 | 147.19(7) | 2.229(2), 2.263(2) | 6.4 | 20 |
| [(6-Ph2TPA)Ni(CH3C(O)CHC(O)CH3)]ClO4 | 150.41(8) | 2.217(2), 2.221(2) | 6.4 | 16 |
| [(6-Ph2TPA)Ni(PhC(O)C(O)CHC(O)Ph)]ClO4 | 158.40(11) | 2.209(3), 2.333(3) | 6.5 | 16 |
| [(6-Ph2TPA)Ni(PhC(O)C(OH)C(O)Ph)]ClO4 | 157.04(8) | 2.271(2), 2.323(2) | 6.6 | 15 |
| [(6-Ph2TPA)Ni-Cl(CH3CN)]ClO4 | 157.67(10) | 2.081(3), 2.213(2) | 6.4 | 20 |
| [(6-Ph2TPA)Ni(O2CPh)]ClO4 | 149.83(6) | 2.1458(16), 2.234(16) | 6.2 | 16 |
| [(6-Ph2TPA)Ni(O2CPh)(H2O)]ClO4 | 158.28(10) | 2.216(2), 2.241(2) | 6.4 | 17 |
| [(6-Ph2TPA)Ni(O2C(CH2)2SCH3)]ClO4 | 141.57(10) | 2.233(3), 2.193(3) | 6.3 | 17 |
| [6-Ph2TPA)Ni(O2(CH2)2SCH3)(H2O)]ClO4 | 159.33(12) | 2.240(3), 2.186(3) | 6.3 | 17 |
| [(6-Ph2TPA)Ni(O2CCH2SCH3)]ClO4 | 148.73(8) | 2.198(2), 2.165(2) | 6.3 | 17 |
| [(6-Ph2TPA)Ni(O2CCH2SCH3)(H2O)]ClO4 | 150.97(7) | 2.1884(17), 2.2050(17) | 6.4 | 17 |
| [(6-Ph2TPA)Ni(O2CH)(H2O)]ClO4 | 158.42(11) | 2.194(3), 2.218(3) | 6.3 | 17 |
N(3) and N(4) are the phenyl-appended pyridyl donors of the 6-Ph2TPA ligand.
Ipso carbon atoms of phenyl appendages.
Discussion
As shown in Scheme 1, a key issue in the chemistry of acireductone dioxygenases is how the coordination mode of the acireductone substrate may influence the reaction with O2. Pochapsky and coworkers have suggested, on the basis of NMR experiments, that differences in the secondary environment in Ni(II)- versus Fe(II)-containing ARD enzyme may be responsible for inducing different coordination modes of the acireductone substrate.1 Specifically, in Ni(II)-ARD the side chain of the tryptophan residue is positioned such that the binding pocket for the acireductone is smaller than that found in Fe(II)-ARD. This is proposed to induce the formation of a six-membered chelate ring structure, which leads to 1,3-dioxygenolytic bond cleavage and CO production. We note that in both Ni(II)- and Fe(II)-containing acireductone dioxygenases an arginine residue is positioned within the active site, likely within hydrogen-bonding distance od the coordinated acireductone.1
As an approach toward evaluating the influence of the secondary environment on the chemistry of Ni(II) complexes having an acireductone-type ligand, we have prepared and characterized [(6-Ph2TPA)Ni(PhC(O)C(OH)C(O)Ph)]ClO4 (2), which has a hydrophobic microenvironment, and [(bnpapa)Ni(PhC(O)C(OH)C(O)Ph)]ClO4 (14), which contains a hydrogen-bond donor secondary environment. Based on the results of analytical and spectroscopic studies, we propose that the coordination mode of the acireductone-type ligand in these complexes is similar, with each having a six-membered chelate ring. However, the solutions structures of 2 and 14 differ in terms of the presence of a plane of symmetry that relates the modified pyridyl donors, with the hydrogen bond donor pocket lacking such symmetry. Treatment of each complex with O2 results in CO production and aliphatic carbon-carbon bond cleavage, with the major products in each reaction being a mononuclear Ni(II) benzoate complex, benzoic acid, and CO. In both reactions, the formation of the side product benzil was also identified. The yield of this byproduct is higher in the reaction involving [(6-Ph2TPA)Ni(PhC(O)C(OH)C(O)Ph)]ClO4 (2). Notably, this reaction exhibits a level of 18O incorporation in the benzoate/benzoic acid products that is ~20% lower than the reaction involving 14. A possible rationale for the differences in product distribution and 18O incorporation between the reactions involving 2 and 14 could be that differing pathways for carbon-carbon bond cleavage may be operative as outlined in Scheme 5. Pathway A involves a cyclic peroxide resulting from two-electron oxidation of the coordinated acireductone analog. Carbon-carbon bond cleavage in this type of structure would produce two equivalents of benzoate/benzoic acid, with each product having one labeled oxygen atom if the reaction were performed using 18O2. The production of benzoic acid in high yield in the reaction involving [(bnpapa)Ni(PhC(O)C(OH)C(O)Ph)]ClO4 (14), and the observed high level of 18O incorporation, suggests that this pathway could possibly be operative for this reaction. However, the production of a small amount of benzil in the reaction involving 14 suggests that another type of reactivity is also occurring. For both complexes, we propose that benzil is generated via a pathway B type reaction sequence (Scheme 5). Specifically, two electron oxidation of the coordinated acireductone type ligand could produce 1,3-diphenyltriketone and a solvated Ni(II) complex of the chelate ligand. It has been previously shown that 1,3-diphenyltriketone will undergo phenyl or benzoyl migration in the presence of a Lewis acids such as AlCl3 to give benzil and carbon monoxide.30 If this type of reaction pathway is occurring with the complexes described herein, the secondary environment of the [(bnpapa)Ni(sol)2]2+ and [(6-Ph2TPA)Ni(sol)2]2+ cations (sol = solvent; e.g. CH3CN) could influence interactions between the Ni(II) center and the 1,3-diphenyltriketone. For example, the larger pocket for the 6-Ph2TPA-ligated Ni(II) center could be a key factor in promoting Lewis acid activation of a carbonyl moiety of the triketone, which is necessary for the migration reaction leading to benzil production.30 This is a possible rationale for the higher amount of benzil produced in the reaction involving 2. In terms of the lower level of 18O incorporation found this reaction, triketones are known to hydrate easily at the central carbonyl, and this may be a pathway for the introduction of unlabeled oxygen atoms into the benzoate/benzoic acid products.31 The observation that the salt [Me4N][PhC(O)CH(OH)C(O)Ph] undergoes reaction with 18O2 to produce a similar level of 18O incorporation (~65%) in the benzoic acid product as is found for the reaction involving 2 suggests that both may proceed via a similar reaction pathway. 18O incorporation into the carbon-carbon bond cleavage products could occur via reaction of the free triketone with hydroperoxide to form a five-membered cyclic peroxide species from which carbon-carbon bond cleavage could occur. Pochapsky has previously shown that treatment of 2,3,4-pentanetrione with H2O2 results in the formation of CO and two equivalents acetic acid.32 No isotope labeling studies were reported for this reaction.
Scheme 5.

Possible reaction pathways for oxidative carbon-carbon bond cleavage in the Ni(II) enolate complexes 2 and 14. The supporting chelate ligands have been omitted for clarity.
The higher level of 18O incorporation in the benzoate/benzoic acid products of the reaction involving 14 is interesting. Based on the results of our current experiments, it is unclear whether exclusively pathway B chemistry is occurring in the O2 reactions of 14 and 2, or whether a portion of the labeled product is generated via pathway A type reactivity. If exclusively pathway B chemistry is operative, an important distinction between the two supporting chelate ligand systems is that the bnpapa-ligated Ni(II) center may coordinate the hydroperoxide anion within the hydrogen bond donor pocket. The bnpapa chelate ligand has been previously used to stabilize mononuclear Zn(II) and Cu(II) hydroperoxide complexes.33,34 To date, similar complexes have not been reported using the 6-Ph2TPA ligand. Thus, differences in the anion coordination properties between the bnpapa and 6-Ph2TPA-supported complexes could contribute to the observed differences in reactivity between 2 and 14.
The results of this investigation have enabled the formulation of hypotheses regarding the reaction pathways of 2 and 14 with O2 (Scheme 5) that can now be tested in individual control reactions. For example, we are now performing experiments to examine the reactivity of [(6-Ph2TPA)Ni(CH3CN)(CH3OH)](ClO4)235 and [(bnpapa)Ni(ClO4)(CH3CN)]ClO4 (18) with 1,3-diphenyltriketone and H2O2 to evaluate the efficacy of pathway B chemistry leading to the observed products. Additionally, we are pursuing kinetic studies of the reactions of complexes 2 and 14 with O2, as well as examining the reactivity of [Me4N][PhC(O)C(OH)C(O)Ph] with O2 via computational methods to assess its reactivity relative to that found in the native C(1)-H acireductone substrate processed by the acireductone dioxygenases.36
Conclusions
Using the hydrogen-bond donor ligand bnpapa, a mononuclear Ni(II) complex of the formula [(bnpapa)Ni(PhC(O)C(OH)C(O)Ph)]ClO4 (14) has been isolated and characterized. Based on comparison of the spectroscopic features of this complex to those of air stable analogs, the structure of 14 is proposed to be generally similar to that previously reported for [(6-Ph2TPA)Ni(PhC(O)C(OH)C(O)Ph)]ClO4 (2). Notably, complexes 2 and 14 exhibit differences in their O2-dependent aliphatic carbon-carbon bond cleavage reactivity in terms of the product mixture generated and the level of 18O incorporation in the benzoate/benzoic acid products. Two possible reaction pathways leading to 18O incorporation into the benzoate/benzoic acid products are proposed, with one pathway involving the formation of a triketone and hydroperoxide anion. Comparison of the chemical features of several bnpapa- and 6-Ph2TPA-ligated complexes suggests that differences in the pocket size and anion binding properties for these two ligand types may be key factors in producing the observed differences in reaction products.
Supplementary Material
Acknowledgments
The authors thank the National Science Foundation (CHE-0848858) and the National Institutes of Health (1R15GM072509) for financial support of this research.
Footnotes
Supporting Information Available. X-ray crystallographic (CIF) files for 16, 17·1/4AHA, 18·CH2Cl2, and 19·2CH2Cl2; a drawing showing the level of 2H incorporation in the bnpapa ligand used for 2H NMR studies; drawings of the ~20–80 ppm region of the 1H NMR spectra of 18 and 19;. This material is available free of charge via the internet at http://pubs.acs.org.
References
- 1.Pochapsky TC, Ju T, Dang R, Beaulieu R, Pagani GM, OuYang B. In: Metal Ions in Life Sciences. Sigel A, Sigel H, Sigel RKO, editors. Wiley-VCH; Weinheim, Germany: 2007. pp. 473–498. [Google Scholar]
- 2.Ju T, Goldsmith RB, Chai SC, Maroney MJ, Pochapsky SS, Pochapsky TC. J Mol Biol. 2006;393:823–834. doi: 10.1016/j.jmb.2006.08.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mann BE, Motterlini R. Chem Commun. 2007:4197–4208. doi: 10.1039/b704873d. [DOI] [PubMed] [Google Scholar]
- 4.Motterlini R, Sawle P, Hammad J, Bains S, Alberto R, Foresti R, Green CJ. FASEB J. 2005;19:284–286. doi: 10.1096/fj.04-2169fje. [DOI] [PubMed] [Google Scholar]
- 5.Zhang WQ, Atkin AJ, Thatcher RJ, Whitwood AC, Fairlamb IJS, Lynam JM. Dalton Trans. 2009:4351–4358. doi: 10.1039/b822157j. and references cited therein. [DOI] [PubMed] [Google Scholar]
- 6.Oka T, Simpson FJ. Biochem Biophys Res Commun. 1971;43:1–5. doi: 10.1016/s0006-291x(71)80076-1. [DOI] [PubMed] [Google Scholar]
- 7.Oka T, Simpson FJ, Krishnamurty HG. Can J Microbiol. 1972;18:493–508. doi: 10.1139/m72-076. [DOI] [PubMed] [Google Scholar]
- 8.Hund HK, Breuer J, Lingens F, Huttermann J, Kappl R, Fetzner S. Eur J Biochem. 1999;263:871–878. doi: 10.1046/j.1432-1327.1999.00574.x. [DOI] [PubMed] [Google Scholar]
- 9.Tranchimand S, Ertel G, Gaydou V, Gaudin C, Tron T, Iacazio G. Biochemie. 2008;90:781–789. doi: 10.1016/j.biochi.2007.12.004. [DOI] [PubMed] [Google Scholar]
- 10.Kooter IM, Steiner RA, Dijkstra BW, van Noort PI, Egmond MR, Huber M. Eur J Biochem. 2002;269:2971–2979. doi: 10.1046/j.1432-1033.2002.02973.x. [DOI] [PubMed] [Google Scholar]
- 11.Fusetti F, Schroter KH, Steiner RA, van Noort PI. Structure. 2002;10:259–268. doi: 10.1016/s0969-2126(02)00704-9. [DOI] [PubMed] [Google Scholar]
- 12.Gopal B, Madan LL, Betz SF, Kossiakoff AA. Biochemistry. 2005;44:193–201. doi: 10.1021/bi0484421. [DOI] [PubMed] [Google Scholar]
- 13.Schaab MR, Barney BM, Francisco WA. Biochemistry. 2006;45:1035–1042. doi: 10.1021/bi051571c. [DOI] [PubMed] [Google Scholar]
- 14.Merkens H, Kappl R, Jakob RP, Schmid FX, Fetzner S. Biochemistry. 2008;47:12185–12196. doi: 10.1021/bi801398x. [DOI] [PubMed] [Google Scholar]
- 15.Szajna E, Arif AM, Berreau LM. J Am Chem Soc. 2005;127:17186–17187. doi: 10.1021/ja056346x. [DOI] [PubMed] [Google Scholar]
- 16.Szajna-Fuller E, Rudzka K, Arif AM, Berreau LM. Inorg Chem. 2007;46:5499–5507. doi: 10.1021/ic061177i. [DOI] [PubMed] [Google Scholar]
- 17.Szajna-Fuller E, Chambers BM, Arif AM, Berreau LM. Inorg Chem. 2007;46:5486–5498. doi: 10.1021/ic061316w. [DOI] [PubMed] [Google Scholar]
- 18.Rudzka K, Arif AM, Berreau LM. Inorg Chem. 2008;47:10832–10840. doi: 10.1021/ic800947z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Armarego WLF, Perrin DD. Purification of Laboratory Chemicals. 4. Butterworth-Heinemann; Boston, MA: 1996. [Google Scholar]
- 20.Szajna E, Dobrowolski PD, Fuller AL, Berreau LM. Inorg Chem. 2004;43:3988–3997. doi: 10.1021/ic040002a. [DOI] [PubMed] [Google Scholar]
- 21.Allred RA, McAlexander LH, Arif AM, Berreau LM. Inorg Chem. 2002;41:6790–6801. doi: 10.1021/ic0258072. [DOI] [PubMed] [Google Scholar]
- 22.Wolsey WC. J Chem Educ. 1973;50:A335–A337. [Google Scholar]
- 23.Plietker B. J Org Chem. 2004;69:8287–8296. doi: 10.1021/jo048822s. [DOI] [PubMed] [Google Scholar]
- 24.Allen TH, Root WS. J Biol Chem. 1955;216:309–317. [PubMed] [Google Scholar]
- 25.(a) Ming L, Guilong Z, Lirong W, Huazheng Y. Synth Comm. 2005;35:493–501. [Google Scholar]; (b) Liu Z, Chen ZC, Zheng QG. Synthesis. 2004:33–36. [Google Scholar]
- 26.Otwinowski Z, Minor M. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 27.Sheldrick GM. SHELXL-97, Program for the Refinement of Crystal Structures. University of Göttingen; Göttingen, Germany: 1997. [Google Scholar]
- 28.Wray JW, Abeles RH. J Biol Chem. 1995;270:3147–3153. doi: 10.1074/jbc.270.7.3147. [DOI] [PubMed] [Google Scholar]
- 29.Berreau LM. Comm Inorg Chem. 2007;28:123–171. [Google Scholar]
- 30.Roberts JD, Smith DR, Lee CC. J Am Chem Soc. 1951;73:618–625. [Google Scholar]
- 31.Rubin MB, Gleiter R. Chem Rev. 2000;100:1121–1164. doi: 10.1021/cr960079j. [DOI] [PubMed] [Google Scholar]
- 32.Dai Y, Pochapsky TC, Abeles RH. Biochemistry. 2001;40:6379–6387. doi: 10.1021/bi010110y. [DOI] [PubMed] [Google Scholar]
- 33.Wada A, Yamaguchi S, Jitsukawa K, Masuda H. Angew Chem Int Ed. 2005:5698–5701. doi: 10.1002/anie.200501157. [DOI] [PubMed] [Google Scholar]
- 34.Fujii T, Yamaguchi S, Hirota S, Masuda H. Dalton Trans. 2008:164–170. doi: 10.1039/b712572k. [DOI] [PubMed] [Google Scholar]
- 35.Makowska-Grzyska MM, Szajna E, Shipley C, Arif AM, Mitchell MH, Halfen JA, Berreau LM. Inorg Chem. 2003;42:7472–7488. doi: 10.1021/ic034810y. [DOI] [PubMed] [Google Scholar]
- 36.Borowski T, Bassan A, Siegbahn PEM. J Mol Struct THEOCHEM. 2006;772:89–92. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.