

Abstract
Epidermal growth factor receptor (EGFR) kinase and the related human epidermal growth factor receptor-2 (HER2, ErbB2) are two growth factor receptors that have implications in cancer. The overexpression or activation of HER2 occurs frequently in breast, ovarian, and lung cancers, making it an important therapeutic target in the treatment of cancer. Blocking HER2-mediated signaling with antibodies or small molecules has been shown to be effective in inhibiting cell growth. After analyzing the crystal structure of the HER2-herceptin complex, several peptidomimetics (HERP5, 6 & 7) were designed to inhibit HER2-mediated signaling for cell growth. We have used an in silico screening method to investigate the chemical diversity of the designed compounds. Autodock software was used to dock the different analogs of HERP5 and HERP7 with HER2 protein extracellular domain. A total of 53 compounds were docked to HER2 protein, and their binding modes were analyzed in terms of docking energy, hydrogen bonding, and hydrophobic interactions. Compounds that exhibited low energy docked structures were chosen for chemical synthesis and biological activity. Two of the compounds (HERP5 and HERP7) exhibited antiproliferative activity, with IC50 values of 0.396 μM and 0.143 μM, respectively, against SKBR-3 cell lines (breast cancer cell lines) that overexpress HER2 protein.
Keywords: breast cancer, docking, HER2, MTT assay, SKBR-3 cell line, virtual screening
Human tumors express high levels of growth factors and their receptors. Epidermal growth factor receptors (EGFR) are the best-studied growth factor receptor family (1). This family consists of four homologous receptors, namely, epidermal growth factor receptor 1 (also called EGFR, ErbB1) or human epidermal growth factor receptor 1 (HER1), HER2 (ErbB2), HER3 (ErbB3), and HER4 (ErbB4) (2-7). In normal cells, activation of this receptor tyrosine kinase family triggers signaling pathways that control normal cell growth, differentiation, and motility. It is well established that binding of extracellular ligands such as epidermal growth factor (EGF) and transforming growth factor α (TGFα) to the extracellular ligand binding domain of EGFR results in receptor homo-heterodimerization, activation of tyrosine kinase activity, and autophosphorylation of the receptors, thus initiating a mitogenic signaling cascade (5). HER2-mediated heterodimerization has important implications in cancer (8-12). Deregulation of signaling pathways and overexpression of HER2 is known to occur in cancer cells, indicating the role of HER2 in tumorigenesis. Several lines of evidence make HER2 an important therapeutic target in the treatment of breast cancer (9-10). HER2 levels in human cancer cells with gene amplification are much higher than those in normal adult tissues. Since the expression of HER2 is high in breast cancer cells, molecules targeting it will have a high probability of binding to HER2-expressing cancer cells rather than normal cells. This may reduce the toxicity of HER2-based therapeutic agents.
Blockade of HER2-mediated multimerization results in inhibition of phosphorylation, ultimately leading to control of cell growth. Thus, blocking HER2-mediated signaling has potential therapeutic value. Monoclonal antibodies specifically directed against the extracellular domain of HER2 have been shown to be selective inhibitors of the growth of HER2-overexpressing cancer cells (13-14). The extracellular region of HER2 consists of four domains (I-IV). Domain II of HER2 is known to participate in dimerization with other HER receptors (15). Domain IV has an important cleavage site for matrix metalloproteases (MMP). The antibody herceptin (trastuzumab) binds to domain IV of HER2 and inhibits the cleavage site of MMPs. This leads to indirect inhibition of dimerization and phosphorylation (5,16). Various therapeutic agents directed against HER2 have provided promising alternatives to traditional non-specific chemotherapy (17-22). However, no small molecule specifically targeting an extracellular region of HER2 has yet been approved for clinical use. Small molecules targeting the cytoplasmic kinase domains of EGFR and HER2 have been developed as anticancer agents. However, developing a highly specific kinase domain inhibitor is challenging, as there are hundreds of kinases, most of which have highly homologous ATP binding sites where the inhibitors competitively bind and thereby modulate the signal for cancer growth. Even the few molecules that have been found to be specific for the ATP binding site of a kinase domain have faced the problem of drug resistance due to mutation in the binding site (18-23). Therefore, there is a need to develop small molecules that are targeted toward the extracellular region of HER2. Based on the crystal structure of HER2 complexed with its antibody herceptin, we have designed peptides/peptidomimetics to block HER2-mediated signaling and the interaction of HER2 with other ErbB receptors. The chemical space of peptidomimetics was investigated by a virtual screening procedure using different β-amino acids and organic functional groups (Tables 1, 2 & 3). Selected compounds from docking studies were synthesized and evaluated for their biological activity. Two compounds, HERP5 and HERP7 (Table 2), exhibited antiproliferative activity with IC50 values of 0.396 μM and 0.143 μM, respectively, against the breast cancer cell line SKBR-3. Compounds HERP5 and HERP7 are specific for HER2-overexpressing cell lines in contrast to colon cancer cell lines and breast cancer cell lines that do not express HER2.
Table 1.
Low energy docked structures of HERP5, 6, 7 and their analogs.
| Code no. | Structure | Docking energy kcal/mol |
|---|---|---|
| HERP5 |
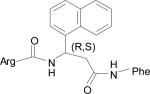
|
–13.87 |
| HERP51 |
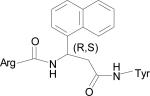
|
–14.16 |
| HERP5c3 |
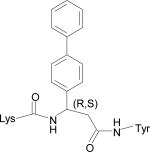
|
–12.59 |
| HERP5a |
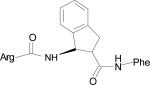
|
–12.40 |
| HERP5c |
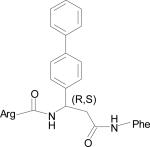
|
–12.06 |
| HERP6 |
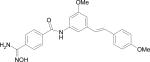
|
–10.54 |
| HERP7 |
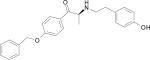
|
–10.65 |
| HERP7h |
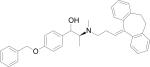
|
–11.23 |
| HERP7f |
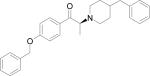
|
–10.97 |
| Arg-Tyr | Arg-Tyr | -3.91 |
Docking energy has an error of 2 kcal/mol in autodock 3.0. The chirality of amino acids was L. For β-amino acid both R & S configurations were considered. The energy difference in the docking calculations was less than 3 kcal/mol between resulting diastereoisomers of HERP5 and its analogs.
Table 2.
Antiproliferative activity of compounds studied in three cancer cell lines
| Code No. | Structure | HCT116 IC50 μM | MCF-7 IC50 μM | SKBR-3 IC50 μM |
|---|---|---|---|---|
| HERP1 |
Arg-Tyr |
>100 |
>100 |
>100 |
| HERP2 |
Arg-Pro-Tyr |
>100 |
>100 |
>100 |
| HERP3 |
Arg-Tyr-Trp-Tyr-Gly |
>100 |
>100 |
>100 |
| HERP4 |
Arg-Phe |
>100 |
>100 |
>100 |
| HERP5 |
H-Arg-[(R,S)3-amino-3-(1-napthyl)-propionic acid]-Phe-OH |
25.7 |
16.9 |
0.396 |
| HERP6 |
4-(N'-hydroxycarbamimidoyl)-N-[ 3-methoxy-5-(4-methoxystyryl) phenyl ] benzamide |
11.9 |
9.2 |
1.62 |
| HERP7 | 24.7 | 13.2 | 0.143 | |
| (S)-2-(4-hydroxyphenethylamino)-1(4-benzyloxy)phenyl)propan-1-one |
IC50 (n = 6, standard error 1.1) for HCT 116 cells; (n = 4, standard error 1.0) for MCF 7 cells; and (n = 4, standard error 0.022) for HERP5 and HERP7 for SKBR3 cells.
Table 3.
Analogs of HERP5 showing variations in R1, R2, and R3 group. A total of 36 compounds were generated. To start with, S configuration was considered at the R2 position. After selecting compounds based on low-docking energy from the first stage of docking, both R and S configurations were considered for second stage of docking.
| R1 | R2 | R3 |
|---|---|---|
| Arg |
|
Phe |
| Lys | Tyr |
Results and Discussion
Design strategy
Domains II and IV of the HER2 extracellular region play major roles in multimerization of HERs and the downstream signaling that leads to cell growth (5). The crystal structure of HER2-herceptin complex indicates that the binding site on HER2 domain IV has a pocket-like structure homologous to that of domain II of other HERs (4). This pocket accommodates binding of small peptide/peptidomimetic molecules, which can modulate HER2-mediated signaling. Analysis of 3D structures and resultant design of peptides for HER2 inhibition followed an in silico approach. The crystal structure of herceptin complexed with the HER2 extracellular region was used for the analysis (4). Herceptin (anti-HER2) binds to HER2 on the C-terminal part of domain IV. The interaction between HER2 and this antibody is mediated by three regions at the C-terminal of domain IV (Fig. 1): (a) residues 557–561, (b) residues 570–573, and (c) residues 593–603. Regions (a) and (c) form electrostatic interactions with the antibody, whereas region (b) interacts with the antibody using its hydrophobic pocket. These interactions block HER2 from proteolytic cleavage and indirectly affect the dimerization with other HERs that induce signaling pathways. Thus, blocking any interaction at these regions on HER2 can disrupt signaling pathway(s) that promote cell proliferation and cancer. The electrostatic interactions at regions (a) and (c) and the hydrophobic interaction at region (b) can be blocked by small peptide molecules or peptidomimetics that are targeted toward HER2 extracellular domain IV.
Figure 1.

Crystal structure of part of domain IV of HER2 interacting with herceptin. Three important pockets (two electrostatic and one hydrophobic) in the interaction region are shown. Residues that form the hydrogen bonding and hydrophobic interaction in the binding pocket of HER2-herceptin are shown. Residues from herceptin are shown as red sticks and residues from HER2 protein domain IV are shown in blue. Amino acid residues from herceptin are labeled with a three letter code and residues from HER2 protein are labeled with a single letter amino acid code.
We designed a template compound based on the spatial disposition of electrostatic and hydrophobic interaction sites in the HER2-herceptin complex. The hydrophobic region of the interacting surface of HER2 is dominated by residues Phe and Pro (Fig. 1). This region interacts with residues Arg50, Tyr52, Tyr33, Trp99, Tyr105, and Gly103 of the antibody. Electrostatic interaction between HER2 and herceptin is mediated by acidic and basic residues. Arg50, a basic amino acid residue from herceptin chain B, forms hydrogen bonds with Glu558 and Asp560 of HER2 (region a) (Fig. 1). The backbone of Gly103 C=O from the antibody chain B (region c) makes contact with HER2 by hydrogen bonding with Lys593. Based on these interactions, peptides with Arg-Tyr and Arg-Tyr-Trp-Tyr-Gly mimicking the HER2 binding region of antibody were designed (Table 2). These residues are in proximity in the three-dimensional structure of the antibody and are placed apart in the sequence by one or more residues. A template structure used for the design of peptides/peptidomimetics is shown in Fig. 2; it contains an Arg residue (N-terminal), a hydrophobic/aromatic residue (i.e., Phe or Tyr, Pro), and a carboxylic acid (C-terminal). These chemical groups were linked by a hydrophobic spacer with conformational constraints. The initial design was based on the amino acid residues of herceptin that interact with HER2 in the binding pocket. Peptides containing Arg, Phe, Tyr, and Trp were designed (Table 2, HERP1-4). Small peptide molecules usually acquire a flexible conformation in solution. To restrict the conformation of small peptides, constraints must be incorporated. A β-amino acid was used at the R2 position of the template (Table 3, Fig. 2). The resulting structure was HERP5 with Arg-β-amino acid-Phe-COOH (Table 1). This structure provides a suitable template to investigate how substitution at R1, R2, and R3 affects to fit into the HER2 cavity, which is the site of binding of the herceptin antibody. Synthesis of the modified structure can be readily carried out using different β-amino acids at the R2 position. Our ultimate aim is to design a compound that is conformationally constrained and stable against enzymatic degradation by peptidases and amidases. Peptidomimetics with non-peptide functional groups have been used in drug design to mimic amino acids (24). Based on the template shown in the Fig. 2, a peptidomimetic with arginine attached to stilbene was proposed with hydroxyl group on the stilbene moiety mimicking the C-terminal COOH for hydrogen bonding and spacer with double bond of stilbene and aromatic groups representing the hydrophobic groups (Fig. 3). Previous studies have shown that many naturally occurring anticancer compounds have the stilbene template (25, 26). Surrogates of amino acid arginine have been used in RGD peptidomimetics (24-26). Hence, the stilbene moiety was used with the arginine (surrogate) moiety to design new structures. The linker connecting arginine or an arginine surrogate was varied in each compound. In compound 6a (Fig. 3), direct linkage with ester group; compound 6b, proline type linker; compound 6c, amide linkage; and compound 6d, ethenyl linkage. We attempted to synthesize compounds 6a,b,c,d (after docking calculations). However, the intermediates were labile or unstable during the synthesis. Hence we modified the structure of the designed compound to arrive at HERP6 with amide linkage with arginine surrogate having amidoxime moiety. HERP6 was viable for synthesis and purification (Fig. 3). In the structure of HERP7 (Table 2), we replaced both stilbene and arginine with other functionalities. The phenyl hydroxyl group in HERP7 is a mimetic of the amino acid tyrosine. We designed several compounds based on HERP5, 6 and 7 by varying the R1, R2, and R3 groups in HERP5, and the p-hydroxyphenyl ethyl group in HERP7 (Table 1, supplementary material, bulky group like amitriptyline or 4-phenyl 2-aminobutyl, alcohol versions of these, HERP7h). Thirty-six possible analogs of HERP5 that were generated by varying R1, R2 and R3 are shown in Table 3, and analogs of HERP6 and 7 are given in the supplementary material. Synthesizing and evaluating the biological activity of a large number of compounds is expensive; hence, we sought virtual screening procedures to identify the compounds that bind with low docking energy with receptors and which are suitable for synthesis.
Figure 2.

Template of the small molecule designed based on the structure shown in Figure 2. An Arg or Lys amino acid residue or surrogates of these amino acids can be used at the N-terminal and a hydrophobic residue can be used at the C-terminal. These can be connected by a conformationally constrained spacer group. Peptides/peptidomimetics or small organic molecules designed based on this template are proposed to fit in the cavity of HER2 domain IV.
Figure 3.

Design of analogs of HERP5 with conformational constraints and arginine surrogates with stilbene moiety. The linker connecting arginine or an arginine surrogate was varied in each compound. In compound 6a, direct linkage with ester group; compound 6b, proline-type linker; compound 6c, amide linkage; and compound 6d, ethenyl linkage. Compound HERP6 with amidoxime moiety is shown.
Docking
The automatic docking method is used to predict the binding of small flexible molecule to protein of interest. Interaction energies between ligand-receptor are calculated with a free-energy-based expression. The docked energies are listed in the increasing order of energy (27,28). From this study, the lowest docked-energy structure was analyzed in detail in an effort to identify a pharmacophoric lead compound structure on which the future analog synthesis program could be based. The low energy docked structures of HERP5, HERP7, their analogs, and HERP6 are given in Table 1.
HERP5
The five low energy docked structures of HERP5 are shown in Fig. 4A. These five low energy structures had docking energies ranging from –13.87 to –13.31 kcal/mol. Two possible modes of binding were observed for HERP5 with the docked energy within a range of 2 kcal/mol of the lowest docked energy structure. One possible binding site was near Phe573 of HER2 and the other near four prolines, Pro540, 529, 543, and 547. HERP5 formed two intermolecular hydrogen bonds near the Phe573 binding pocket, namely, the Arg side chain of HERP5 was hydrogen bonded to the carbonyl group of Cys574 and the amide of Phe of HERP5 was hydrogen bonded to the backbone carbonyl of Arg577. The naphthalene ring of HERP5 was stabilized by hydrophobic interactions with Pro571, Pro572, and Phe573 of the protein HER2. When positioned near prolines 540, 529, 543, and 547, hydrophobic/van der Waals interactions occurred between the napthelene ring of HERP5 and the proline residues. A pi-cation interaction between Arg523 of protein HER2 and Phe of the HERP5 was also observed. In addition, the Arg side chain of HERP5 was hydrogen bonded to Pro540 and Tyr532 residues. To understand the importance of functional groups of HERP5, the R1, R2, and R3 positions were varied as shown in Table 3. Only the low energy docked structures that have energies comparable to HERP5 are shown in Table 1. Among the 36 analogs of HERP5 that were docked, HERP51 and HERP5c3 showed docking energies comparable to that of HERP5. In HERP51, the Phe of HERP5 was changed to Tyr. In HERP5c3, the beta amino acid was changed to a biphenyl group. The hydrophobic groups seem to play a major role in the binding of these analogs to the HER2 protein. Changing Arg at the N-terminal of the ligand to Lys with its positively charged side chain did not significantly decrease the docking energy. For compound HERP5, both R and S configurations were considered at β-amino acid (R2) position for docking. The difference in energy of docking between the diastereoisomers for HERP5 (with R and S configuration at beta amino acid and S (L) configuration at amino acids) was less than 3kcal/mol. Thus, docking studies indicated that chirality of the peptidomimetic at R2 position may not play a major role in binding to HER2.
Figure 4.

A) Low energy docked structures of HERP5 with HER2 protein domain IV. Note the two modes of binding. The docking energies of both the clusters were within 0.5 kcal/mol. HER2 protein is shown as surface and HERP5 docked structures are shown as dark sticks.
B) Low energy docked structures of HERP6 with HER2 protein domain IV. HER2 protein is shown as surface and HERP6 docked structures are shown as dark sticks.
C) Low energy docked structures of HERP7 with HER2 protein domain IV. HER2 protein is shown as surface and HERP7 docked structures are shown as dark sticks.
HERP6
Fig. 4B shows the low energy docked structures of HERP6 with HER2 protein. The docked energies of the six structures fall in the range of –10.54 to –9.60 kcal/mol. The structures clustered around the four prolines, Pro540, 543 529 and 547 on HER2 protein. The lowest energy docked structure interacted with HER2 with two hydrogen bonds, one between the amide NH of HERP6 and the main chain C=O of Pro540 and the other between the OH of HERP6 with NH of Ala559 of the protein. The three aromatic groups of HERP6 were surrounded by prolines of HER2 protein.
HERP7
In the case of HERP7, there was only one low energy cluster of docking. The six low energy structures were clustered in the hydrophobic channel formed by three prolines, Pro529, 540, and 543 (Fig. 4C). The docking energies of the six low energy conformers were in the range of –10.65 to –9.71 kcal/mol. There were two hydrogen-bonding interactions, one with NH of HERP7 and C=O of Pro543 of HER2 and the other with the hydroxyl group of the aromatic ring to C=O of Ala566.
Since compounds HERP5, 6 and 7 are different in their characteristics we classified their analogs into three groups with HERP5, 6 and 7 as lead compounds. The energy values in each group were compared to lead compound in each group. Based on the docking energies of these structures, HERP5, 6, and 7 are good candidates for the synthesis and evaluation of antiproliferative activity.
Synthesis of compounds
Compounds HERP1,2,3,4, and 6 were synthesized in the laboratory. A schematic diagram of the synthesis of HERP6 is shown in Fig. 5. HERP5 and 7 were custom synthesized (NeoMPS, San Diego, CA; RL Fine Chemicals, Bangalore, India). In HERP5, Arg and Phe were L amino acids. The β-amino acid could have R and S configurations, which will result in two diastereoisomers of HERP5. Compound HERP5 was synthesized as a mixture of diastereoisomers (R and S isomers at R2 position, S at amino acids at R1 and R3). Details of the synthesis are given in the experimental section. All the compounds were >90% pure and were analyzed by HPLC, MS, HR-MS and NMR.
Figure 5.

Schematic diagram of the synthesis scheme for compound HERP6 (compound 8 in the scheme).
Antiproliferative activity of the compounds using MTT assay
The synthesized compounds were screened for antiproliferative activity using the microculture tetrazolium assay, which is based on the ability of metabolically active cells to reduce the yellow tetrazolium salt to a colored formazan product (29,30). The assay was carried out on MCF-7 breast cancer cells (estrogen receptor present, express normal levels of HER2), human colon cancer cells HCT116 (express low levels of HER2) and SKBR-3 cells, a breast cancer cell line that overexpresses HER2. The peptides HERP1–HERP4 were found not to be active against the cancer cell lines used in the study. Compound HERP5 (R2 position R and S, diastereoisomers, Tables 1 &2) showed antiproliferative activity against the three cell lines used in the study with IC50 values of 25.7, 16.9 and 0.396 μM in HCT116, MCF-7, and SKBR-3 cell lines, respectively. HERP6 had IC50 values of 11.9 μM (HCT116), 9.2 μM (MCF-7), and 1.62 μM (SKBR-3), whereas HERP7 had IC50 values of 24.7 μM (HCT116), 13.2 μM (MCF-7), and 0.143 μM (SKBR-3).
In the studies described above, we obtained more than 20-fold difference in the IC50 values for the compounds HERP5, 7 between cells that overexpress HER2 protein and cells that do not overexpress HER2 protein. This major difference could be due to the specificity of the designed compound for particular cell line, presumably due to its specificity towards a surface receptor protein. MCF-7 and HCT-116 cell lines do not overexpress HER2 protein. MCF-7 cell lines are breast cancer cell lines that express estrogen positive receptors. SKBR-3 cell lines overexpress HER2 protein and do not express estrogen receptors (Breast cancer cell line database, MD Anderson cancer Center, TX). It is reported that other breast cancer cell lines such as MCF-7 express 40,000 to 100,000 HER2 protein per cell, whereas, SKBR-3 breast cancer cell lines express two million HER2 proteins per cell (31,32). Although, several factors contribute to the difference in activity of compounds in in vitro assay, the observed specificity of the compounds for SKBR-3 cell lines may be due to interaction of HERP5 with HER2 protein. Docking studies and biological activity of compounds HERP5, 6 and 7 (Tables 1 & 2) indicated that within similar series of compounds (HERP5 and its analogs, HERP6, 7 and analogs) low energy docked structures exhibited antiproliferative activity in the micromolar to nanomolar range. Arg-Tyr (Tables 1 & 2) a dipeptide showed relatively high energy of docking and did not show any antiproliferative activity against the cell lines studied. To correlate docking studies with antiproliferative activity more number of compounds have to be synthesized and their biological activity to be evaluated.
Conclusions and Future Directions
From the results obtained from these studies, it is clear that peptidimimetics designed based on the structure of the HER2-herceptin complex show antiproliferative activity against three cancer cell lines, HCT116, MCF-7, and SKBR-3. The chemical space of peptidomimetics was investigated by a virtual screening procedure using different β-amino acids. Selected compounds from docking studies were synthesized and evaluated for their biological activity. Compounds HERP5 and HERP7 seem to have specificity for HER2-overexpressing breast cancer cell lines rather than for colon cancer and breast cancer cell lines that do not overexpress HER2. Establishment of the structure-activity relationships of these compounds requires the study of additional compounds with varying anticancer activity against SKBR-3 cell lines. Synthesis of several analogs of HERP5, diastereoisomers of HERP5, and 7 and evaluation of biological activity in cancer cell lines are in progress.
Experimental
Chemistry
1H NMR spectra were recorded on Bruker DRX 300 MHz and 500 MHz NMR spectrometers. Chemical shifts were reported in δ (ppm), relative to tetramethylsilane as internal standard. For compounds in water, 2,2-Dimethyl-2-silapentane-5-sulfonic acid (DSS) was used as reference. Thin layer chromatography (TLC) was carried out on pre-coated plates (silica gel 60 F254, Merck) and visualized with ultraviolet light. Flash column chromatography was performed with silica gel 60 (70–230 mesh) using hexane/ethyl acetate as eluting solvent. Microwave experiments were carried out on Biotage Initator™. Reagents used for synthesis were of synthetic or analytical grade.
Synthesis of HERP6 (compound 8)
A schematic diagram of the synthesis of HERP6 (compound 8) is shown in Fig. 5.
4-methoxy benzyltriphenyl phosphonium chloride (1)
Triphenylphosphine (27.7 g) and 4-methoxybenzyl chloride (16.5 g) were dissolved in 100 ml of toluene and refluxed under nitrogen protection for 24 hours. Upon cooling, the solid was collected by suction and dried under reduced pressure to give 1 as a white powder in 95% yield (39.8 g). It was used without further purification for subsequent reactions. 1H NMR (CDCl3): δ 7.7 (15H, m) 7.0 (2H, d, J = 8.70), 6.6 (2H, d, J = 8.70), 5.7 (2H, s,), 3.7 (3H, s, OCH3).
3-(tert-Butyldimethylsilyloxy)-5-hydroxybenzaldehyde (2)
3,5-Dihydroxybenzaldehyde (10.0 g, 72.4 mmol) and DIPEA (17.7 ml) were dissolved in 80 ml of N,N-dimethylforamide (DMF) at 0°C. An equivalent amount of tert-butyldimethylsilyl chloride dissolved in 50 ml DMF was added drop wise over an hour. The mixture was stirred overnight at 28°C. After workup, 8.4 g (46%) of white solid 2 was obtained through column chromatography and was used without further purification for the next step of the reaction.
3-(tert-Butyldimethylsilyloxy)-5-methoxybenzaldehyde (3)
To a solution of 2 (0.7 g, 2.8 mmol) in 10 ml dichloromethane was added molecular sieves (0.8 g), proton sponge, and an equivalent amount of trimethyloxonium tetrafluoroborate. The reaction mixture was stirred overnight, filtered through celite 545, and rinsed with dichloromethane. The solvent was removed under reduced pressure and the residue was purified by column chromatography to give 3 as an oil (0.4 g, 54%). 1H NMR (CDCl3) δ 9.88 (1H, s), 7.02–7.03 (1H, dd), 6.93 (1H, dd), 6.64–6.66 (1H, t), 3.84 (3H, s), 0.99 (9H, s), 0.23 (6H, s).
(E)-tert-Butyl(3-methoxy-5-(4-methoxystyryl)phenoxy)dimethylsilane (4)
13.72 g of 1 was suspended in 70 ml of dry tetrahydrofuran and cooled to –78 °C. 20.5 ml of lithium butyl was added dropwise over 30 minutes. After an hour, the bath was removed and the reaction mixture was stirred for another 2 h. 3-(tert-Butyldimethylsilyloxy)-5-methoxybenzaldehyde 3 (8.7 g) in 30 ml of tetrahydrofuran was added dropwise, stirred for 3 hours at –78 °C, and subsequently warmed to room temperature overnight. After workup, an oil (10.4g, 86%) made up of the E and Z isomers was obtained. The E isomer 4 was obtained by treating the mixture (10 g) dissolved in 100 ml dichloromethane with bis (acetonitrile) dichloropalladium (0.35 g) and stirring for 14 hours. Purification by column chromatography gave 8.5 g of the E isomer 4. 1H-NMR (CDCl3) δ: 0.23 (6H, S), 1.00 (9H, s), 3.81 (3H, s), 3.83 (3H, s), 6.30–6.31 (1H, t, J = 1.89, 2.25 Hz), 6.58 (1H, s), 6.66 (1H, s), 6.84–6.91 (3H, m), 6.98–7.03 (1H, d, J = 16.17), 7.43–7.46 (2H, d, J = 8.66 Hz).
3-Hydroxy-4',5 -dimethoxystilbene (5)
To a solution of 4 (8.46 g) in dry tetrahydrofuran (130 ml) was added tetrabutylammonium fluoride (1N in tetrahydrofuran, 22.9 ml) at 0°C. The reaction mixture was stirred at room temperature (28°C) for 45 minutes, then poured into ice water (200 ml) and extracted with dichloromethane. The organic layer was dried over anhydrous MgSO4 and removed under reduced pressure. The crude product was purified by column chromatography to give 5 (5.4 g, 92%) as a white solid. 1H-NMR (CDCl3) δ: 3.82 (3H, s), 3.83 (3H, s), 4.79 (1H, s), 6.31–6.32 (1H, t, J = 1.89, 2.25 Hz), 6.58 (1H, s), 6.63 (1H, s), 6.84–6.91 (3H, m), 6.99–7.05 (1H, d, J = 16.2), 7.42–7.45 (2H, d, J = 9.06 Hz).
(E)-3-Methoxy-5-(4-methoxystyryl)phenyl trifluoromethanesulfonate (6)
2.29 g of 5 was dissolved in 22.5 ml of pyridine and 22.5 ml of dichloromethane, and the mixture was cooled to –10 °C. Trifluoromethanesulfonic anhydride (5.05 g, 17.90 mmol) was added. After stirring for 3 hours at 0°C, the mixture was poured into 60 ml of water and extracted with ether (4x). The combined organic phase was washed with water, dilute hydrochloric acid and water, successively, then dried over anhydrous MgSO4. Solvent was removed under reduced pressure and the residue was purified by column chromatography. 6 was obtained as a white solid (3.4 g) in 98% yield. 1H-NMR (CDCl3) δ: 3.84 (3H, s), 3.86 (3H, s), 6.62–6.68 (1H, t, J = 1.86, 1.89 Hz), 6.86–6.91 (1H, d, J = 16.2 Hz), 6.90–6.93 (2H, d, J = 8.64 Hz), 6.98–6.99 (1H, d, J = 1.89 Hz), 6.99–7.00 (1H, d, J = 1.89 Hz), 7.04–7.09 (1H, d, J = 16.17 Hz), 7.44–7.47 (2H, d, J = 8.67 Hz).
3-[(4-Cyanobenzoyl)-amido]-4',5-dimethoxystilbene (7)
Triflate 6 (1.52g), 4-cyanobenzamide (0.68 g), cerium carbonate (1.78g), palladium acetate (43.8mg), 4,5-bis(diphenylphosphino)-9,9-dimethylxanthene (0.17 g) and tetrahydrofuran (5 ml) were placed into a microwave vial. The mixture was heated to 110°C with microwave irradiation for 8 hours. After workup, 1.2 g of 7 was obtained in 80% yield. 1H-NMR (CDCl3) δ: 3.84 (3H, s), 3.87 (3H, s), 6.87–6.94 (4H, m), 7.05–7.11 (1H, d, J = 16.2 Hz), 7.22 (1H, s), 7.32 (1H, s), 7.43–7.46 (2H, d, J = 8.67 Hz), 7.78 (1H, br), 7.80–7.82 (2H, d, J = 8.28 Hz), 7.97–8.00 (2H, d, J = 8.28 Hz).
4-(N'-Hydroxycarbamimidoyl)-N-[ 3-methoxy-5-(4-methoxystyryl) phenyl]benzamide (8)
7 (130 mg) and sodium bicarbonate (28 mg) were placed in a microwave vial, followed by the addition of THF (1 ml) and 50% v/v hydroxylamine (50 μl). The reaction mixture was irradiated by MW for 30 minutes at 110°C. On cooling, the mixture was filtered and the filtrate was concentrated under reduced pressure. The residue was dissolved in dichloromethane; the organic layer was washed with brine (3x) and dried over anhydrous MgSO4. The solvent was evaporated to give the crude product, which was recrystallized from methanol to give 8 (80 mg, 57%) as a white solid.
1H-NMR (acetone-d6): δ 9.16 (1H, S (br)) 8.07 (1H, S (br), NH-CO), 8.02 (2H, d, J 7.8 Hz), 7.87 (2H, d, J 7.8 Hz), 7.54 (2H, d, J 8.1 Hz), 7.48, 1H, S), 7.65 (1H, S), 7.19 (1H, d, J 16.5 Hz), 7.05 (1H, d, J 16.5 Hz), 6.94 (2H, d, J 8.7 Hz), 6.93 (1H, S), 5.60 (2H, S (br), NH2), 3.82 (6H, S). 13C-NMR (acetone-d6): δ 165.5 (C=ONH-), 160.9 (C-OMe), 160.2 (C-OMe), 151.1 (C=N-OH), 141.2, 139.8, 137.0, 136.1, 130.4, 129.2, 128.3, 127.9, 126.8, 125.9, 114.6, 111.0, 107.6, 105.6, 55.2.
Peptides HERP1–4 were synthesized in the laboratory by solid phase peptide synthesis using standard Fmoc chemistry. PAL resin (5-(4-N-Fmoc-aminomethyl-3,5-dimethoxyphenoxy)valeryl) was used as a solid support for the linear peptides (33,34). First, the Fmoc protecting group on the resin was removed by treatment with 20% piperidine/dimethylformamide (DMF). The Nα-Fmoc-amino acids were preactivated by mixing with the coupling reagent 2-(7-aza-1H-benzotriazole-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate/N,N'-diisopropylethylamine (HATU/DIPEA) (1:1); the activated amino acid was then added to the resin and mixed at room temperature. Cycles of deprotection of Fmoc and coupling with the subsequent amino acids were repeated until the desired peptide-bound resin was completed. The resin was washed manually with DMF, dichloromethane (DCM), and methanol successively to remove the excess solvents and dried in vacuo over KOH overnight before cleavage and deprotection. The dried peptidyl-resin was mixed with the cleavage cocktail (90% TFA, 5% thioanisole and 5% 1,2-ethanedithiol, 5 ml per gram of resin) and precipitated with cold ether. For maximum recovery, the ether-peptide mixture was kept at –20°C overnight. Then the precipitated material was collected by centrifugation and washed three times with cold ether to remove any residual scavengers. After evaporation of the ether, the peptide was dissolved in water with a few drops of acetic acid and lyophilized.
The lyophilized peptide was dissolved in water/acetonitrile. The peptide was purified by preparative HPLC (Waters 600 HPLC system), on a reversed-phase C18 column (Inertsil, 10 × 250 mm, 5 μm, 300 Å) with a linear gradient of solvent A (0.1% TFA/H2O) and solvent B (0.1% TFA/acetonitrile). The peptides were detected by UV at λ = 215 nm and 280 nm. The purity of each peptide was verified by an analytical HPLC (Shimadzu LC-10AT VP) using a reversed-phase C18 column (Lichrosorb RP18, 4.6 × 200 mm, 10 μm) with the same solvent system as in the preparative HPLC. The molecular weight of the peptides was confirmed by using electrospray ionization mass-spectrometry (ESI-MS, Finnigon MAT). The HPLC chromatogram showed that the purity of the peptides was more than 90%, while ESI-MS showed the correct molecular ion for the peptide.
Compound HERP5 and its analogs were custom synthesized and purchased from NeoMPS (San Diego, CA). Compound HERP7 was purchased from Fine Chemicals (Bangalore, India).
Docking
The crystal structure of HER2 complexed with herceptin was downloaded from the protein data bank (PDB ID 1N8Z). The antibody part of the molecule, solvent and counter ions were removed from the crystal structure using Insight II software (Accelrys, Inc. San Diego, CA) (35). A grid box with dimension of 128 Å3 was created around the binding site of herceptin on HER2 protein using autodock tools. The center of the box was set at Phe573 of HER2 protein (Fig. 1) and grid energy calculations were carried out. The three-dimensional structures of ligand HERP5 and its analogs were built using Insight II software, and molecules were saved as mol2 and pdb files. A total of 36 compounds were generated. The three-dimensional structures of HERP6, HERP7 and its analogs (17 compounds) were generated using PROGRD2, a web based software. A tripeptide, Ala-Ala-Ala, was used as a control. Docking of different ligands to protein was performed using autodock. Docking calculations were carried out in two stages. In the first stage, 2.5 million energy evaluations were performed for each of the HERP5 and HERP7 analogs. Docked ligand conformations were analyzed in terms of energy, hydrogen bonding, and hydrophobic interaction between ligand and receptor protein HER2. Low energy docked structures were chosen for chemical synthesis. In the second stage, a smaller grid box (40 × 40 × 40 Å3) with a ligand binding site on HER2 was created and docking calculations were repeated for ligands that were chosen from the first stage. The chirality of amino acids Arg and Phe was considered to be L; for beta-amino acids both R and S configurations were considered during docking. Twenty-five million energy evaluations were performed for each molecule. Detailed analyses of the ligand-receptor interactions were carried out, and final coordinates of the ligand and receptor were saved as pdb files. For display of the receptor with the ligand binding site, PyMol software was used. Only three compounds (in addition to peptides HERP1 and HERP4) with low energies of docking were selected for evaluation of biological activity. The results are shown in Table 2.
MTT Assay for determination of antiproliferative activity
The growth inhibitory activity of target compounds was determined on cell lines HCT 116 (human colon cancer cell line), MCF-7 (human breast cancer cell line), and SKBR-3 using a modified version of the microculture tetrazolium assay (29,30). HCT116 cells were cultured in McCoy's 5A modified medium with 1.5 mM L-glutamine (ATCC) supplemented with 10% FBS and 1% antibiotics and cells of passages 19–22 were used. MCF-7 cells were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% FBS and 1% antibiotics; cells of passages 3–8 were used, and SKBR-3 cell lines were maintained according to the supplier's guidelines (ATCC, Manassas, VA).
Once the cells reached 90% confluency, a cell suspension was prepared by trypsinization of monolayer cultures (other cell lines). Cell counts were performed, and the suspensions were diluted accordingly to give 1 × 105 cells/ml with the appropriate medium. Aliquots (100 μl) of the cell suspension were added to each well in a 96-well microtitre plate. The cells were incubated for 24 hours (37°C, 5% CO2) for incubation. Stock solutions of the test compound were prepared in DMSO, and serial dilutions were made with medium to over a hundred-fold concentration range. Not more than 1% DMSO (final concentration) was present in each well. The test sample was incubated with the cells for 72 hours. Wells without cells and those with cells in culture medium/DMSO were examined in parallel. 0.1% SDS and dextran were included as positive and negative controls, respectively. At the end of the incubation period, the medium was decanted and replaced with 100 μl MTT solution (0.5 mg /ml in 1x phosphate buffer saline solution (PBS). The cells were incubated for another 3 hours, after which the medium was removed from each well by pipetting and the cells were carefully washed with PBS (100 μl). DMSO (150 μl) was added to each well to lyse the cells and dissolve the purple formazan crystals. The absorbance of the formazan product was measured within 30 minutes at 590 nm on a microtitre plate reader. The absorbance values obtained at each concentration (triplicates for each run and 3-6 independent runs were carried out) were averaged, adjusted by subtraction of blank values (wells without cells), and expressed as a percentage of the average absorbance obtained from control wells (in the absence of test compound). IC50 values were determined from logarithmic plots of the % absorbance versus concentration generated using GraphPad Prism (San Diego, CA, USA).
Supplementary Material
Acknowledgments
The project described was supported by Grant Number P20RR016456 from the National Center for Research Resources. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Center for Research Resources or the National Institutes of Health. Dr. Jianchao carried out the synthesis of the compound HERP6 and was supported by the National University of Singapore, Singapore. Part of the docking calculations were performed using Dell Clusters, Louisiana Optical Network Initiative (LONI) computer resources. Authors would like to thank Mass Spectrometry Facility, Department of Chemistry, Louisiana State University Baton Rouge for high resolution mass spectra of compounds and Nancy Harmony for editing the manuscript.
Footnotes
Supplementary material
The following supplementary material is available for this article.
HPLC, mass spectra, NMR data for compound HERP5 and HR-MS data for HERP5, 6 and 7.
References
- 1.Hynes NE, Lane HA. ERBB receptor and cancer: the complexity of targeted inhibitors. Nat Rev Cancer. 2005;5:341–354. doi: 10.1038/nrc1609. [DOI] [PubMed] [Google Scholar]
- 2.Woodburn JR. The epidermal growth factor receptor and its inhibition in cancer therapy. Pharmacol Ther. 1999;82:241–250. doi: 10.1016/s0163-7258(98)00045-x. [DOI] [PubMed] [Google Scholar]
- 3.Hobbs S, Cameron EM, Hammer RP, Le ATD, Gallo RM, Blommel EN, Coffing SL, Chang H, David J, Riese DJ., II Five carboxyl-terminal residues of neuregulin2 are critical for stimulation of signaling by the ErbB4 receptor tyrosine kinase. Oncogene. 2004;23:883–893. doi: 10.1038/sj.onc.1207250. [DOI] [PubMed] [Google Scholar]
- 4.Cho HS, Mason K, Ramyar KX, Stanley AM, Gabelll SB, Denney DW, Leahy DJ. Structure of the extracellular region of HER2 alone and in complex with herceptin Fab. Nature. 2003;421:756–760. doi: 10.1038/nature01392. [DOI] [PubMed] [Google Scholar]
- 5.Burgess AW, Cho HS, Eigenbrot C, Ferguson KM, Garrett TP, Leahy DJ, Lemmon MA, Silwkowski MX, Ward CW, Yokoyama S. An open-and-shut case? Recent insights into the activation of EGF/ErbB receptors. Mol Cell. 2003;12:541–552. doi: 10.1016/s1097-2765(03)00350-2. [DOI] [PubMed] [Google Scholar]
- 6.Yarden Y, Baselga J, Miles D. Molecular approach to breast cancer treatment. Semin Oncol. 2004;31:6–13. doi: 10.1053/j.seminoncol.2004.07.016. [DOI] [PubMed] [Google Scholar]
- 7.Cristofanilli M, Hortobagyi GN. Molecular targets in breast cancer: current status and future directions. Endocr Relat Cancer. 2002;9:249–266. doi: 10.1677/erc.0.0090249. [DOI] [PubMed] [Google Scholar]
- 8.Mendelsohn J, Baselga J. The EGF receptor family as targets for cancer therapy. Oncogene. 2000;19:6550–6565. doi: 10.1038/sj.onc.1204082. [DOI] [PubMed] [Google Scholar]
- 9.Nahta R, Esteva FJ. HER2 targeted therapy: lessions learned and future directions. Clin Cancer Res. 2003;9:5078–5084. [PubMed] [Google Scholar]
- 10.Menard S, Pupa SM, Campiglio M, Tagliabue E. Biologic and therapeutic role of HER2 in cancer. Oncogene. 2003;22:6570–6578. doi: 10.1038/sj.onc.1206779. [DOI] [PubMed] [Google Scholar]
- 11.Perez-Soler R. HER1/EGFR targeting: refining the strategy. Oncologist. 2004;9:58–67. doi: 10.1634/theoncologist.9-1-58. [DOI] [PubMed] [Google Scholar]
- 12.Slamon DJ, Leyland-Jones B, Shak S, Fuchs H, Paton V, Bajmonde A, Fleming T, Eiermann W, Wolter J, Pegram M, Baselga J, Norton LN. Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. Engl J Med. 2001;344:783–792. doi: 10.1056/NEJM200103153441101. [DOI] [PubMed] [Google Scholar]
- 13.Cobleigh MA, Vogel CL, Tripathy D, Robert NJ, Scholl S, Fehrenbacher L, Wolter JM, Paton V, Shak S, Lieberman G, Slamon DJ. Multinational study of the efficacy and safety of humanized anti-HER2 monoclonal antibody in women who have HER2-overexpressing metastatic breast cancer that has progressed after chemotherapy for metastatic disease. J Clin Oncol. 1999;17:2639–2648. doi: 10.1200/JCO.1999.17.9.2639. [DOI] [PubMed] [Google Scholar]
- 14.Leonard DS, Hill ADK, Kelly L, Dijkstra B, McDermott E, O'Higgins NJ. Anti-epidermal growth factor receptor 2 monoclonal antibody therapy for breast cancer. British J Surgery. 2002;89:262–271. doi: 10.1046/j.0007-1323.2001.02022.x. [DOI] [PubMed] [Google Scholar]
- 15.Agus DB, Akita RW, Fox WD, Lewis GD, Higgins B, Pisacane PI, Lofgren JA, Tindell C, Evans DP, Maiese K, Scher HI, Sliwkowski MX. Targeting ligand-activated ErbB2 signaling inhibits breast and prostate tumor growth. Cancer Cell. 2002;2:127–137. doi: 10.1016/s1535-6108(02)00097-1. [DOI] [PubMed] [Google Scholar]
- 16.Molina MA, Codony-Servat J, Albanell J, Rojo F, Arribas J, Baselga J. Trastuzumab (herceptin), a humanized anti-HER2 receptor monoclonal antibody, inhibits basal and activated HEr2 ectodomain cleavage in breast cancer cells. Cancer Res. 2001;61:4744–4749. [PubMed] [Google Scholar]
- 17.Franklin MC, Carey KD, Vajdos FF, Leahy DJ, De Vos AM, Sliwkowski MX. Insights into ErbB signaling from the structure of the ErbB2-pertuzumab complex. Cancer Cell. 2004;5:317–328. doi: 10.1016/s1535-6108(04)00083-2. [DOI] [PubMed] [Google Scholar]
- 18.Kamath S, Buolamwini JK. Receptor-guided alignment based comparative 3D-QSAR studies of benzylidene malonitrile tryphostins as EGFR and HER2 kinase inhibitor. J Med Chem. 2003;46:4657–4668. doi: 10.1021/jm030065n. [DOI] [PubMed] [Google Scholar]
- 19.Rabindran SK, Discafani CM, Rosfjord EC, Baxter M, Floyd MB, Golas J, Hallett WA, Johnson BD, Nilakantan R, Overbeek E, Reich MF, Shen R, Shi X, Tsou HR, Wang YF, Wissner A. Antitumor activity of HKI-272, an orally active, irreversible inhibitor of the HER2 tyrosine kinase. Cancer Res. 2004;64:3958–3965. doi: 10.1158/0008-5472.CAN-03-2868. [DOI] [PubMed] [Google Scholar]
- 20.Fink BE, Vite GD, Mastalerz H, Kadow JF, Kim SH, Leavitt KJ, Du K, Crews D, Mitt T, Wong TW, Hunt JT, Vyas DM, Tokarski JS. New dual inhibitors of EGFR and HER2 protein tyrosine kinases. Bioorg Med Chem Lett. 2005;15:4774–4779. doi: 10.1016/j.bmcl.2005.07.027. [DOI] [PubMed] [Google Scholar]
- 21.Brezov A, Chen J, Liu Q, Zhang HT, Greene MI, Murali R. Disabling receptor ensembles with rationally designed interface peptidomimetics. J Biol Chem. 2002;277:28330–28339. doi: 10.1074/jbc.M202880200. [DOI] [PubMed] [Google Scholar]
- 22.Wissner A, Overbeek E, Reich MF, Floyd MB, Johnson BD, Mamuya N, Rosfjord EC, Discafani C, Davis R, Shi X, Rabindran SK, Gruber BC, Ye F, Hallet WA, Nilakantan R, Shen R, Wang YF, Greenberger LM, Tsou HR. Synthesis and structure-activity relationship of 6,7-disubstituted 4-anilinoquinoline-3-carbonotrile. The design of an orally active, irreversible inhibitor of the tyrosine kinase activity of the epidermal growth factor receptor (EGFR) and the human epidermal growth factor receptor-2 (HER2). J Med Chem. 2003;46:49–63. doi: 10.1021/jm020241c. [DOI] [PubMed] [Google Scholar]
- 23.Moulder SL, Yakes FM, Muthuswamy SK, Bianco R, Simpson JF, Arteaga CL. Epidermal growth factor receptor (HER1) tyrosine kinase inhibitor ZD1839 (Iressa) inhibits HER2/neu (erbB2)-overexpressing breast cancer cells in vitro and in vivo. Cancer Res. 2001;61:8887–8895. [PubMed] [Google Scholar]
- 24.Fisher MJ, Gunn B, Harms CS, Kline AD, Mullaney JT, Nunes A, Scarborough RM, Arfsten AE, Skelton MA, Um SL, Utterback BG, Jakubowski JA. Non-peptide RGD surrogates which mimic a Gly-Asp beta-turn: potent antagonists of platelet glycoprotein IIb-IIIa. J Med Chem. 1997;40:2085–2101. doi: 10.1021/jm9701076. [DOI] [PubMed] [Google Scholar]
- 25.Castello L, Tessitore L. Resveratrol inhibits cell cycle progression in U937 cells. Oncol Rep. 2005;13:133–137. [PubMed] [Google Scholar]
- 26.Kundu JK, Surh YJ. Cancer chemopreventive and therapeutic potential of resveratrol: mechanistic perspectives. Cancer Lett. 2008;269:243–261. doi: 10.1016/j.canlet.2008.03.057. [DOI] [PubMed] [Google Scholar]
- 27.Morris GM, Goodsell DS, Halliday RS, Huey R, Hart WE, Belew RK, Olson AJ. Automated docking using a Lamarckian genetic algorithm and empirical binding free energy function. J Comput Chem. 1998;19:1639–1662. [Google Scholar]
- 28.Huey R, Morris GA, Olson AJ, Goodsell DS. A semiempirical free energy force field with charge based desolvation. A semiempirical free energy force field with charge based desolvation. J Compu Chem. 2006;28:1145–1152. doi: 10.1002/jcc.20634. [DOI] [PubMed] [Google Scholar]
- 29.Mosmann T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Methods. 1983;5:55–63. doi: 10.1016/0022-1759(83)90303-4. [DOI] [PubMed] [Google Scholar]
- 30.Alley MC, Scudiero DA, Monks A, Huresy ML, Abbott BJ, Mayo JG, Shoemaker RH, Boyd MR. Feasibility of drug screening with panels of human tumor cell lines a microculture tetrazolium assay. Bioorg Med Chem. 2003;11:729–2738. [PubMed] [Google Scholar]
- 31.Dakappagari NK, Lunte KD, Rawale S, Steele JT, Allen SD, Phillips G, Reilly RT, Kaumaya TP. Conformational Her-2/neu B-cell epitope vaccine designed to incorporate two native disulfide bonds enhances tumor cell binding and antitumor activities. J. Biol. Chem. 2005;280:54–63. doi: 10.1074/jbc.M411020200. [DOI] [PubMed] [Google Scholar]
- 32.Allen S, Garrett JT, Rawale SV, Jones AL, Philips G, Forni G, Morris JC, Oshima RG, Kaumaya TP. peptide vaccine of the HER-2/neu dimerization loop are effective in inhibiting mammary tumor growth in vivo. J. Immunol. 2007;179:472–482. doi: 10.4049/jimmunol.179.1.472. [DOI] [PubMed] [Google Scholar]
- 33.Handbook of Combinatorial and Solid Phase Organic Chemistry: a Guide to Principles, Products and Protocols. Advanced Chemtech; Louisville, KY: 1998. pp. 329–372. [Google Scholar]
- 34.Lloyd-Williams P, Albericio F, Giralt E. Chemical Approaches to the Synthesis of Peptides and Proteins. CRC; Boca Raton, Florida: 1997. pp. 19–82. Chapter 2. [Google Scholar]
- 35.Insight II, Accelrys, Inc. San Diego CA. Insight II is a commercial, licensed molecular modeling software. Obtained from Accelrys, Inc. http://www.accelrys.com/products/insight/
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.