

Abstract
Aging is accompanied by a decline in the healthy function of multiple organ systems, leading to increased incidence and mortality from diseases such as type II diabetes mellitus, neurodegenerative diseases, cancer, and cardiovascular disease. Historically, researchers have focused on investigating individual pathways in isolated organs as a strategy to identify the root cause of a disease, with hopes of designing better drugs. Studies of aging in yeast led to the discovery of a family of conserved enzymes known as the sirtuins, which affect multiple pathways that increase the life span and the overall health of organisms. Since the discovery of the first known mammalian sirtuin, SIRT1, 10 years ago, there have been major advances in our understanding of the enzymology of sirtuins, their regulation, and their ability to broadly improve mammalian physiology and health span. This review summarizes and discusses the advances of the past decade and the challenges that will confront the field in the coming years.
Keywords: chromatin, metabolism, deacetylase, cancer, cardiovascular, inflammation
INTRODUCTION
It has been exactly 10 years since the Silent Information Regulator 2 (SIR2) gene was shown to extend the life span of budding yeast by repressing genome instability (1, 2). Since then, we have learned that SIR2-like genes, known as sirtuins, are found in most organisms, including plants, bacteria, and animals, and that they play key roles in promoting an organism’s health and survival (3). Major strides have been made in understanding how sirtuins function at the molecular level to sense the amount of energy taken in, the timing of daylight, and stress from the environment and how they respond to such signals by inducing changes that promote survival during times of adversity. The discovery that sirtuins can carry out nicotinamide adenine dinucleotide (NAD+)-dependent deacetylation reactions (4) opened up a new line of investigation into the metabolic control of sirtuins and modulation of their activity by small molecules. Activation of sirtuins, either by restricting calories or by pharmacological means, extends the life span and promotes the health of a wide variety of organisms from yeast to mammals, thus demonstrating the feasibility of developing drugs that target the sirtuin and treat diseases of aging (5). This review covers the past 10 years of research into what sirtuins do at the molecular, cellular, and organismal levels to alter mammalian physiology. We also address the challenges still faced by researchers in the field.
SIRTUIN BIOLOGY
Identification of SIR2/SIRT1 as a Regulator of Aging
The Sir2 protein, from the budding yeast Saccharomyces cerevisiae, is the founding member of a family of NAD+-dependent deacetylases and ADP-ribosyltransferases, termed the sirtuins (6). The gene encoding the Sir2 enzyme, SIR2, was first identified by virtue of its role in the establishment of transcriptional silencing of mating-type loci in budding yeast. Subsequent studies have shown that SIR2 is also pivotal for silencing at yeast telomeres and in the recombinant DNA (rDNA) (7, 8), as well as in the partitioning of carbonylated proteins between mother and daughter cells (9).
The role of sirtuins in aging was initially discovered in yeast via a model of replicative life span, which measured the number of times a yeast mother cell produces a daughter cell before senescing. In 1997, a cause of yeast aging was identified as stemming from recombination at rDNA loci. The formation of an extrachromosomal rDNA circle (ERC) by homologous recombination is the starting event that leads to the amplification of ERCs in aging mother cells due to their replication and preferential segregation within mother cells (1). In 1999, addition of an extra copy of the SIR2 gene was shown to extend replicative life span by ~30% by suppressing rDNA recombination and decreasing ERC formation while deleting SIR2-increased ERCs and shortened life span (2).
Since the late 1990s, it has become clear that the yeast SIR2 gene is but one member of a large family of conserved genes found in organisms ranging from bacteria to mammals (10). The conserved functional role of SIR2 in aging has become evident from studies involving more complex model organisms, such as Caenorhabditis elegans and Drosophila. In C. elegans, life span extension by sir-2.1 requires the worm forkhead protein DAF-16 but may not require an intact insulin signaling pathway (11, 12). Instead, sir-2.1 binds to DAF-16 and activates it directly during stress (12). Moreover, sir-2.1 does not appear to respond to changes in insulin signaling; rather, it is activated by stress treatments such as heat shock and oxidative damage. Increasing the copy number of the SIR2 ortholog in Drosophila (dSIR2) also extends life span (13). Interestingly, overexpression of dSIR2 specifically in neurons is sufficient to drive the increase in the fly’s life span.
Mammalian Sirtuins: Enzymatic Activity and Regulation
The exciting discovery that increasing SIR2 gene dosage in yeast, worms, and flies extends life span has spurred studies of mammalian sirtuins. Do sirtuins extend mammalian life span? Do mammalian sirtuins promote health and protect against aging-associated diseases? Are these proteins important for mediating benefits of calorie restriction (CR)? What are the targets of sirtuins? These are some of the central questions driving the field. Surprising progress has been made in only a decade of research on mammalian sirtuins. However, we are just beginning to understand the involvement of sirtuins in mammalian aging and age-associated diseases, and our comprehension of many sirtuins remains rudimentary.
Mammals contain seven sirtuins, SIRT1–7 (Table 1), which are categorized by their highly conserved central NAD+-binding and catalytic domain, termed the sirtuin core domain (14). Although these sirtuins are relatively conserved, their N and C termini differ, and they are likely to have highly divergent biological functions owing to (a) different enzymatic activities, (b) unique binding partners and substrates, and (c) distinct subcellular localization and expression pattern (reviewed in Reference 15).
Table 1.
Summary of the mammalian sirtuins
| Sirtuin | Location | Interactions | Biology | Null phenotype |
|---|---|---|---|---|
| SIRT1 | Nucleus | FOXO, PGC-1α NF-κB, Ku70, etc. |
Metabolism, stress | Developmental defects, lethal in some backgrounds |
| SIRT2 | Cytosol | Tubulin, H4, FOXO | Cell cycle | Developmentally normal |
| SIRT3 | Mitochondria | AceCS2, GDH complex I |
Thermogenesis, ATP production |
Developmentally normal |
| SIRT4 | Mitochondria | GDH, IDE, ANT | Insulin secretion | Developmentally normal |
| SIRT5 | Mitochondria | CPS1 | Urea cycle | Developmentally normal |
| SIRT6 | Nucleus | Histone H3, NF-κB | Base excision repair, metabolism |
Premature aging |
| SIRT7 | Nucleolus | Pol I | rDNA transcription | Smaller size, short lifespan, heart defects |
Abbreviations: AceCS2, acetyl-CoA-synthetase 2; ANT, adenide nucleotide translocator; CPS1, carbamoyl phosphate synthetase 1; FOXO, forkhead box, subgroup O; GDH, glutamate dehydrogenase; IDE, insulin degrading enzyme; NF-κB, nuclear factor kappa B; PGC-1α, peroxisome proliferator-activated receptor gamma coactivator 1 alpha; Pol I, DNA polymerase I; rDNA, recombinant DNA.
Reaction Mechanisms
The first reaction characterized for a sirtuin was a ribosyltransfer reaction catalyzed by the Sir2 homolog cobB from bacteria that convert 5,6-dimethylbenzimidazole to alpha-ribose-5,6-benzimidazole (16). This finding led to the surprising discovery that sirtuins are NAD+-dependent deacetylases and ADP-ribosyltransferases. Most sirtuins catalyze NAD+-dependent deacetylation (Figure 1) (4, 17–19). SIRT4, however, possesses NAD+-dependent mono-ADP-ribosyltransferase activity (20, 21), whereas SIRT1 and SIRT6 have been shown to perform both auto-ADP-ribosyltransferase and substrate-specific deacetylase activities (22). Studies of SIRT4 and SIRT7 have not yet observed deacetylase activity, but such activity may require a specific substrate, as for SIRT6. Although yeast SIR2 is a histone deacetylase (HDAC), studies of sirtuins in other organisms have identified a host of nonhistone substrates. Indeed, sirtuins function by acting as substrate-specific protein deacetylases. Sirtuins perform deacetylation at modified lysine residues via a unique enzymatic mechanism that requires NAD+ cleavage with each reaction cycle (4, 18, 23–25). Thus, unlike other HDACs that hydrolyze acetyl-lysine residues, sirtuin activity is intimately tied to the metabolic state of the cell.
Figure 1.

The sirtuin deacetylation reaction and regulation by stress and nutrition. Unlike type I and II deacetylases, which hydrolyze the acetyl group on a substrate, sirtuin deacetylases catalyze an unprecedented two-step biological reaction that consumes nicotinamide adenine dinucleotide (NAD+) and releases nicotinamide (NAM), O-acetyl-ADP-ribose (AADPR), and the deacetylated substrate. Amide-to-ester acyltransfer is unfavorable, but hydrolysis of NAD+ can provide a favorable driving force for the overall sirtuin reaction. Evidence favors a mechanism in which electrophilic capture of the acetyl oxygen in an ADP-ribosyltransfer reaction forms an ADP-ribose peptide-imidate complex. This intermediate may last for a few seconds, enough time for NAM to enter the C-pocket and catalyze a reverse reaction. Activation of sirtuins can be facilitated by the removal of NAM and its conversion to NAD+ by PNC1 (yeast and simple metazoans) or NAMPT (mammals and alpha proteobacteria), two genes upregulated by stress and nutrient limitation.
The mechanistic basis for deacetylation by sirtuins is both elegant and novel. The deacetylation reaction begins with an amide cleavage of NAD+ and the formation of nicotinamide (NAM) and a covalent ADP-ribose (ADPR) peptide-imidate intermediate (see Figure 1). The intermediate is resolved to form O-acetyl-ADP-ribose (AADPR), and the deacetylated substrate is released (25–27). Although the amide-to-AADPR acyltransfer is unfavorable, hydrolysis of NAD+ can provide a favorable driving force for the overall sirtuin reaction (28–30). Although some details remain to be resolved, it is thought that peptide binding facilitates an allosteric change in enzyme structure that enables reaction of NAD+ with a nucleophile from the enzyme to generate the enzyme-stabilized ADPR intermediate (30).
Sirtuin Modulation by NAD+ Biosynthesis
There is increasing evidence that the biosynthetic pathways that generate NAD+ are also critical for the regulation of sirtuins in various subcellular compartments. Both the Sternglanz and the Sinclair labs (31–33) reported that sirtuins are inhibited by NAM and in a noncompetitive manner with NAD+. The surprising fact that it is noncompetitive inhibition led the authors to speculate that NAM binds to a novel site on sirtuins and is physiologically relevant, an idea that was subsequently verified. The mechanism of inhibition requires that NAM enter a highly conserved C-pocket adjacent to the NAD-binding site (31, 34) and react with the peptide-imidate intermediate of the reaction, thereby regenerating NAD+ (33). This type of reversible regulation is rare in biology and is thought to be a major mechanism of control for many of the sirtuins.
In yeast, worms, and flies, NAM is recycled back to NAD+ in four steps, in the first of which it is catalyzed by Pnc1 to produce nicotinic acid. PNC1 is upregulated in response to environmental stresses, such as heat and CR, leading to increased stress resistance and life span in S. cerevisiae and D. melanogaster (35–37). Thus, PNC1 promotes survival and life span in response to environmental stress, which supports the view that life span extension by stress and diet is the result of an ancient survival response. Another NAD+ precursor, nicotinamide riboside (NR), is found in yeast and mammalian cells and, when supplied exogenously to yeast, can also extend life span (38, 39). Mammals recycle NAD+ from NAM in two steps. First, a NAM phosphoribosyltransferase known as Nampt (also termed Visfatin or PBEF) converts NAM to nicotinamide mononucleotide (NMN) (40, 41). Second, NMN is utilized by the isozymes Nmnat1, -2, and -3 to regenerate NAD+ in the nucleus, Golgi, and mitochondria, respectively (42).
Consistent with the ability of PNC1 to regulate Sir2 in yeast, mammalian Nampt is one of the main regulators of SIRT1 activity (40, 43–45). Interestingly, the enzyme downstream of Nampt, Nmnat1, interacts directly with SIRT1 at promoters, indicating either that this enzyme hands off NAD+ to SIRT1 or that there are nanopools of NAD+ that influence SIRT1 activity (45).
The Nampt enzyme and NMN can also be found in the serum of mice and humans (where the enzyme is known as eNAMPT) (46). Imai and colleagues (47) proposed that NMN is a signaling molecule that allows stressed or nutrient-deprived cells to communicate with other parts of the body. This concept, termed the NAD world, is an area of considerable interest, especially given the possibility of using NMN or a downstream molecule such as NR as a therapeutic for type II diabetes mellitus (TDM) or other diseases of aging (47–49).
SIRT1 and Nampt form an essential part of the mammalian circadian clock feedback cycle. Nampt is under the transcriptional regulation of a CLOCK-BMAL-SIRT1 complex, which increases the conversion of NAM to NAD+. This in turn activates SIRT1, which reactivates Nampt expression—all in a 12-h cycle (50–53). NAD+ levels are dynamically regulated; thus, caution should be exercised when obtaining and comparing results from different times of the day.
In mammals, not only is NAD+ destroyed by the sirtuins (and by poly-ADP-ribose polymerase), it is continually catabolized by CD38, a glycohydrolase. CD38 was first described as a NAD cyclase on the cell surface involved in immunity, but this is a relatively minor activity. Knockdown or deletion of CD38 increases steady-state levels of NAD+, leading to speculation that inhibition of this enzyme could be an effective way to activate sirtuins (54, 55). As we describe below, one-year-old mice lacking CD38 were protected from age-onset obesity and diabetes, perhaps because of increased sirtuin activity (54, 55).
The AADPR produced by the sirtuin reaction is a novel metabolite that is less studied but that may have major physiological significance. For example, in yeast, AADPR promotes the assembly of the Sir complex, specifically Sir3 with the Sir2/Sir4 dimer, and catalyzes the spread of the SIR complex across chromatin (56). In mammals, AADPR binds and activates the transient receptor potential melastatin-related channel 2 (57). AADPR can also be metabolized by nudix hydrolases in vitro and may be metabolized in vivo to acetate by an unidentified enzyme (58). Thus, the cleavage products derived from NAD+ during sirtuin-mediated deacetylation yield metabolites that may mediate biologically relevant functions in aging and metabolism. Given how little is known at this stage, this area of sirtuin biology may yield some of the more interesting discoveries in coming years.
Mammalian Sirtuins: Subcellular Localization
Mammalian sirtuins are found in numerous compartments within the cell (Table 1). SIRT1, -6, and -7 are found predominantly in the nucleus (59); SIRT3–5 reside in mitochondria; and SIRT2 is primarily cytoplasmic. The subcellular localization of these proteins probably depends upon cell type, stress status, and molecular interactions. For instance, SIRT1 and -2 were found to localize in both the nucleus and the cytoplasm and to interact with both nuclear and cytosolic proteins (60–62).
What governs sirtuin localization? Why is SIRT1 sometimes nuclear and sometimes cytosolic? First, each sirtuin contains primary amino acid signal sequences that contribute to its intracellular localization. For example, the nuclear localization of SIRT1, -6, and -7 is largely attributed to their nuclear localization signals. In addition to possessing two nuclear localization signal regions, SIRT1 contains two nuclear export signals (61). Thus, the exposure of nuclear localization signals versus nuclear export signals may dictate the cytosolic versus nuclear localization of SIRT1.
SIRT3–5 contain N-terminal mitochondrial targeting sequences and are widely believed to localize to the matrix of mitochondria (21, 22, 63–65). However, studies have not conclusively ruled out localization for these proteins to other compartments of mitochondria. The localization of SIRT3 has been hotly debated (66). Initial reports found that SIRT3 was localized exclusively to mitochondria (22, 64). However, subsequent studies suggested that SIRT3 may translocate from the mitochondria to the nucleus during cellular stress (67, 68). Insights into the function of SIRT3 may come from a better understanding of its structure and enzymology. A recent paper identified a SIRT3 crystal structure of the apoenzyme and a reaction-intermediate structure trapped by a thioacetyl peptide (69). This study demonstrated that the substrate induces conformational changes and that the acetylated peptide is the first substrate to bind to SIRT3, preceding NAD+ (69). In sum, sirtuins are distributed among multiple compartments of the cell, and their localization may be dynamic, depending on tissue/cell type and physiologic condition.
Sirtuins in Calorie Restriction
Numerous studies have shown that a diet of reduced calories, also known as CR, promotes life span extension by up to 50% in a wide range of organisms, including yeast, worms, flies, and mice (70). However, there are some exceptions, including wild mice and houseflies (71, 72). In mammals, CR triggers physiological changes that improve glucose homeostasis: Rodents and humans on CR demonstrate decreased insulin and glucose levels and have improved insulin sensitivity. These metabolic changes are relevant to aging because decreasing insulin signaling is implicated in longevity regulation in studies of model organisms (73). The physiology of CR has been comprehensively reviewed elsewhere (70).
The discovery that Sir2 is a conserved regulator of life span in yeast, worms, and flies, coupled with the fact that sirtuin activity relies upon NAD+, has intensified interest in elucidating the role of Sir2 in CR. Does Sir2 activity mediate CR benefits? The answers depend on the CR regimen (moderate versus severe restriction) and genetic background. In multiple studies of yeast replicative aging (the number of times a mother cell divides), life span extension by CR does require SIR2. Moreover, a moderate CR diet (0.5% glucose) was shown to increase mitochondrial respiration, upregulate Sir2 activity, and suppress rDNA recombination (36, 74). The yeast homologs of Sir2, Hst1 and Hst2, have also been shown to promote replicative life span (75).
However, there is some debate about the exact role of sirtuins in yeast CR. A more severe CR regimen (0.05% glucose) also extends yeast replicative life span but does not require SIR2 or mitochondrial respiration (76, 77). Lamming and colleagues (75) showed that deleting both SIR2 and HST2 blocked CR-mediated life span extension, suggesting that there is functional redundancy within the yeast sirtuin family. SIR2 had no effect on yeast survival under starvation conditions (chronological life span) and appears rather to reduce survival of certain exceptionally long-lived mutant strains such as sch9 (78). In worm models of CR, sir-2.1 does not seem to be required for life span extension, although, given the variety of ways to perform CR in worms, sir-2.1 could be required for a diet that has not yet been tested; alternatively, the other three worm sirtuins could be involved (79).
Because the amount of yeast Sir2 protein does not increase during CR (36), researchers have sought other explanations for increased Sir2 activity. The Guarente laboratory (80) proposed that during CR, a reduction in NADH (an inhibitor of SIR2) results in increased Sir2 activity, whereas the Sinclair and Smith labs (36, 37, 81, 82) proposed that the increase in Sir2 activity is due to upregulation of PNC1 during CR, which depletes NAM and increases flux through the NAD+ salvage pathway. Interestingly, PNC1 is also upregulated by mild stresses that extend life span such as increased temperature (37°C) and nitrogen restriction. These data are seen as evidence that CR is a form of hormesis, or a mild stress that induces a beneficial defense response (83–85). The NADH and the PNC1 mechanisms are fundamentally different: The former regulates sirtuins directly, whereas the latter also involves an active genetic pathway that responds specifically to stress. Since 2003, there has been increasing evidence for both these mechanisms of Sir2/SIRT1 regulation in yeast and in mammals, suggesting that they probably act in concert (32, 86, 87), although some investigators have questioned whether NADH is potent enough to inhibit SIRT1 in vivo (26).
These findings have generated considerable interest in elucidating the role of mammalian sirtuins in CR. So far, most of these studies have involved SIRT1 and provide support for the model that SIRT1 mediates several salutary effects of CR. First, rodent studies show that CR upregulates SIRT1 expression in a variety of tissues, such as brain, kidney, liver, white adipose, and skeletal muscle (88). Nonetheless, SIRT1 induction is not observed in all CR studies, and it may be induced in a tissue-specific manner or even decreased (89). Notably, NAD+ levels are also increased in some tissues during CR. Thus, it is likely that in many tissues a combination of boosting SIRT1 protein levels and NAD+ concentration (or flux though the NAD salvage pathway) contributes to increased activity of this sirtuin during CR.
Mouse models have also tightened the link between SIRT1 and CR. The Guarente lab (90) was the first to show that SIRT1 is required for the induction of a phenotype by CR: an increase in physical activity. Transgenic mice that over-express SIRT1 also display several metabolic benefits that overlap with CR phenotypes. For example, the first study using a knockin mouse model with SIRT1 expression driven by the beta actin promoter demonstrated that SIRT1-overexpressing mice are leaner and more glucose tolerant and that they display reduced levels of blood cholesterol, adipokines, and insulin compared to wild-type controls (91). In another series of elegant studies, wild-type or mutant SIRT1 was overexpressed in mice using its own promoter from a bacterial artificial chromosome. These animals were used to demonstrate that elevated SIRT1 did not improve basal glucose tolerance but rather attenuated obesity-induced glucose intolerance (92). Another study showed that transgenic SIRT1 mice are resistant to liver steatosis (fatty liver) and insulin resistance (93). These SIRT1 phenotypes are similar to the effects of treating mice with SIRT1 activators such as resveratrol and SRT1720 (94–96). Because many of these studies were performed with whole-body transgenic animals, it will be useful to identify the tissues that drive these protective phenotypes. Moreover, it will be interesting to know whether overexpression of SIRT1 can extend mean or maximum life span or merely health span.
SIRT1 null mice have also provided evidence that SIRT1 mediates aspects of CR. As mentioned above, the first study (90) investigated the behavior of mice fed a CR diet. CR causes an increase in activity in wild-type mice, which may aid animal survival in the wild by triggering foraging when food is scarce. Whole-body SIRT1 null mice do not show this increase, suggesting that SIRT1 is required for this phenotype of CR (90, 97). Recently, life spans of SIRT1 null mice have been measured. SIRT1 null mice have a shorter life span than do their wild-type littermates, and CR does not increase the life span of these animals (98). These data provide important evidence that SIRT1 may be required for life span extension by CR in mammals. However, firm conclusions are complicated by the fact that SIRT1 null mice that survive to adulthood die prematurely and have developmental defects (99). Future life span studies that use SIRT1 tissue-specific knockout mice may ascertain which tissues drive this important phenotype. Notably, few studies have probed the role of SIRT2–7 in CR phenotypes, but these sirtuins may also play roles in regulating the physiologic responses to CR.
Posttranslational Regulation: Small Molecules, Interactions, and Modifications
Given that SIRT1 levels are responsive to environmental stimuli such as daylight, cell stress, and CR, it is not surprising that the gene is controlled by numerous transcription factors and microRNAs, including p53, FOXO3, HIC1:CtBP, E2F1, c-Myc, miR-34, and miR-199 (100–109). Almost nothing else is known about how the other sirtuins are regulated at the transcriptional level.
As mentioned above, sirtuins are also regulated at the posttranslational level. The discovery that sirtuins can also be modulated directly by small molecules and by protein interactors has opened up the possibility of mimicking the benefits of CR without having to restrict calories. Small-molecule modulators of SIRT1 include the inhibitors splitomycin, sirtinol (110), and EX-527 (110), as well as analogs of sirtinol that inhibit yeast SIR2, SIRT1, and SIRT2 (Figure 2) (111). SIRT1-activating compounds (STACs) include resveratrol, SRT1720 and SRT2183, and analogs of NAM (94, 112, 113). With the exception of NAM analogs such as methyl-NAM, which interfere with NAM inhibition of sirtuins (32), SIRT1 activators work by lowering the Michaelis constant (Km) for the substrate and for NAD+ and, to a lesser extent, by increasing enzyme velocity (Vmax) (94, 113).
Figure 2.

Chemical inhibitors and activators of SIRT1. Over the past 10 years, a variety of small-molecule SIRT1-activating compounds (STACs) or inhibitors have been published. The known IC50s and EC1.5s for SIRT1 are shown in parentheses. Of all the inhibitors, only nicotinamide (NAM) is a physiological inhibitor, although analogs of NAM can activate sirtuins, apparently by occluding the C-pocket (see Figure 1). Polyphenolic activators such as resveratrol appear to bind the same site and activate via the same mechanism (i.e., primarily a lowering of the Michaelis constant effect) as that used by more potent activators such as SRT1720. The potency of inhibition is expressed as IC50 (the concentration to inhibit 50% activity), and activation is expressed as EC1.5 (the concentration to activate 1.5-fold).
During the past decade, over 3500 STACs have been synthesized and analyzed (114).With regard to resveratrol and other natural polyphenols of similar structure, such as fisetin and butein (see Figure 2), these molecules have been shown to extend life span in a wide variety of organisms from yeast to flies to obese mice (115–118). However, some labs report little or no life span extension (119, 120). The question remains: How do these molecules activate Sir2 and SIRT1? Assays used to detect activation by fluorescence or mass spectrometry have typically utilized peptide substrates that contain additional groups such as AMC (7-amino-4-methylcoumarin) or TAMRA (tetramethyl-rhodamine) (94, 113, 121), although these groups are not necessary to detect activation of SIRT1 by small molecules (122). Some studies have questioned whether this assay recapitulates SIRT1 activation on a native substrate, yet the increasing number of studies showing dependency of these molecules on SIRT1, and their ability to mimic CR and SIRT1 overexpression in vivo (95, 112, 116, 123, 124), argues that they act in large part via SIRT1 (92, 93, 96, 125). Sauve (28) speculated that the fluorophore in the assay reproduces biophysical properties of native substrates in cells, thereby providing a cognate-binding site for compounds such as resveratrol in vivo.
A number of protein modulators of SIRT1 have also been identified, including the activators AROS (active regulator of SIRT1) (126), necdin (127), and DBC1 (the inhibitor deleted in breast cancer 1) (Figure 3) (128, 129). Both DBC1 and AROS bind to the N terminus of SIRT1, the same region to which SIRT1 small-molecule activators bind, raising the possibility of a common mechanism of control. It is also clear that sirtuins can be modified directly. For example, SIRT1 has at least 13 residues that are phosphorylated in vivo, and removal of the phosphates decreases catalytic activity. Mutating two cyclin B/CDK1 sites (Thr530 and Ser540) disrupts the normal cell cycle and proliferation (130). These modulators of SIRT1 activity are described in more detail below in relation to their effects in specific disease models.
Figure 3.

SIRT1 regulation of age-related physiology. SIRT1 activity can be regulated through NAD+ (nicotinamide adenine dinucleotide) and nicotinamide (NAM) concentrations, by SIRT1 protein level, and by phosphorylation; SIRT1 can be activated by active regulator of SIRT1 (AROS) and inhibited by DBC1 (deleted in breast cancer 1). SIRT1 activation promotes survival of neurons and protects cardiomyocytes from death. In the liver, SIRT1 promotes fatty acid oxidation and gluconeogenesis during nutrient deprivation via LXR, PGC-1α, and PPARα. In white adipose tissue (WAT), SIRT1 decreases fat storage by repressing PPARγ. SIRT1 promotes insulin secretion and pancreatic beta cell survival by suppressing UCP2 and interacting with FOXO, respectively. In skeletal muscle, SIRT1 promotes mitochondrial biogenesis through the activation of PGC-1α. Abbreviations: CNS, central nervous system; FOXO, forkhead box transcription factor, subgroup O; LXR, liver X receptor; NF-κB, nuclear factor kappa B; PGC-1α, peroxisome proliferator-activated receptor gamma coactivator 1 alpha; PPARα, peroxisome proliferators-activated receptor alpha; UCP2, uncoupling protein 2. Adapted from Reference 15.
METABOLIC DISEASES
A growing area of sirtuin research involves the regulation of metabolism. Maintenance of energy homeostasis requires a coordinated regulation of energy intake, storage, and expenditure. In healthy individuals, fluctuations in any of these processes are compensated by adaptation by the other two. This elegant balance is orchestrated by the involvement of interorgan communication using endocrine and metabolic pathways. Thus, metabolic pathways are designed to sense incoming nutritional and environmental cues and to respond appropriately. Metabolic imbalance occurs when the ability to adapt to changes in energy intake, storage, or energy expenditure is lost. Abnormal metabolic homeostasis can have severe consequences and often manifests as syndromes such as obesity and TDM. Moreover, signs of metabolic imbalance, such as insulin resistance, are observed with increasing frequency during aging. The identification of central pathways that regulate metabolic homeostasis is an area of intense sirtuin research and has important implications for understanding the molecular networks that control adaptation responses and for treating metabolic diseases. This section reviews our understanding of SIRT1 and mitochondrial sirtuins in regulating adaptation to nutrients.
There has been intense scrutiny of the role of sirtuins in metabolic pathways. Again, most of the studies have focused on SIRT1. Although the scope and detail of SIRT1 functions are not yet fully elucidated, overwhelming evidence suggests that this enzyme senses nutritional availability and relays this information to proteins that govern fuel utilization and energy adaptation. Multiple SIRT1 targets and regulatory mechanisms are highlighted in Figure 3. SIRT1 binds to and deacetylates a number of important transcription factors—such as peroxisome proliferator-activated receptor gamma (PPARγ), PPARα, PPAR gamma coactivator 1 alpha (PGC-1α), and the forkhead box, subgroup O (FOXO) family of transcription factors—to drive metabolic responses such as insulin secretion, gluconeogenesis, and fatty acid oxidation. SIRT1 thus acts as a pleiotropic energy sensor to help mount an appropriate physiological response.
Mitochondrial Biogenesis and Gluconeogenesis
In vivo studies that use mouse models support the implications of biochemical and cellular data. For example, PGC-1α seems to be a predominant SIRT1 target in a number of cell types. PGC-1α was originally identified and cloned as a cold-inducible PPARγ activator in brown adipose tissue (131). Now it is clear that PGC-1α is a potent inducer of mitochondrial biogenesis as well as of the uptake and utilization of substrates for energy production in a number of cell types. A series of elegant studies (132) first described the importance of the SIRT1–PGC-1α network in hepatocytes. SIRT1 can interact with and deacetylate PGC-1α in vitro (132). The deacetylation of PGC-1α results in the upregulation of its activity.
Activated SIRT1 deacetylates PGC-1α, resulting in the SIRT1-dependent induction of gluconeogenic genes and the suppression of glycolytic genes (132). The relevance for SIRT1 in gluconeogenesis in vivo was examined through use of adenovirus to decrease the levels of SIRT1, specifically in adult mouse liver (132). In this model, knockdown of SIRT1 in liver caused mild hypoglycemia and increased glucose production. Loss of hepatic SIRT1 in these adult animals actually improved glucose clearance and increased insulin sensitivity. Similarly, knocking down SIRT1 in adult rat liver decreased hepatic glucose production in a rat model of TDM (133). In these experiments, fasting was found to increase NAD+ concentrations and SIRT1 levels in mouse liver (132). Several groups have also reported increases in hepatic NAD+ during nutrient deprivation, such as in fasting and in CR in whole-cell extracts and in mitochondria (44).
Two other studies have utilized liver-specific SIRT1 null mice and observed no change in insulin or glucose homeostasis or in gluconeogenesis (89, 134). What can account for the differences in these models? It could be that the absolute loss of SIRT1 in the null models activates compensatory pathways that restore gluconeogenesis. Although these mice appeared developmentally normal, perhaps the absence of SIRT1 during liver development activated compensation mechanisms. Another explanation is that studies of SIRT1 in the liver failed to induce the normal feedback loop provided by TORC2, which was recently shown by Montminy and colleagues to repress SIRT1-stimulated gluconeogenesis (135). Together, these data illustrate the complexity and difficulty of interpreting studies of isolated tissues in the mammalian system.
Substrate Usage: Glycolysis Versus Fatty Acid Oxidation
Later studies have shown that SIRT1 can orchestrate a coordinated shift in mitochondrial substrate usage, primarily by regulating PGC-1α activity. This switch allows the cell to preserve glucose while oxidizing fatty acids, reminiscent of the substrate utilization observed during nutrient deprivation. In skeletal muscle, SIRT1 deactylates PGC-1α to promote transcription of mitochondrial fatty acid oxidation genes, and SIRT1 is required for the switch to fatty acid oxidation under low-nutrient conditions (136–139). Additionally, activation of PGC-1α triggers the transcription of oxidative phosphorylation genes, suggesting that SIRT1 may couple fatty acid oxidation with energy production (140). Consistent with these findings, liver mitochondria isolated from whole-body SIRT1 null mice displayed lower rates of respiration and reactive oxygen species (ROS) production (97). Moreover, SIRT1 null mice had increased levels of active AMP-activated protein kinase (AMPK) after a 24-h fast, suggesting a decrease in their ATP levels (97). Thus, activation of PGC-1α may provide a direct mechanism through which SIRT1 can control mitochondrial biogenesis.
As in skeletal muscle, SIRT1 activation causes a global shift toward hepatic fatty acid oxidation during nutrient deprivation (134, 138). SIRT1 may act through PGC-1α to activate PPARα transcription to induce fatty acid catabolic genes (134). SIRT1-overexpressing transgenic mice gain weight, similar to wild-type controls, but display protection from hepatic steatosis when fed a high-fat diet (93), consistent with the effects observed for the SIRT1 activators resveratrol and SRT1720 (96, 138, 141, 142). SIRT1 null hepatocytes exhibit lower rates of fatty acid oxidation (134). Moreover, when liver-specific SIRT1 knockout mice were fed a high-fat, Western-style diet, they gained more weight than did littermate controls and exhibited higher lipids and decreased ketones after fasting (134).
Again, some evidence is conflicting. One study (143) demonstrated that similarly generated liver-specific SIRT1 null animals gained less weight when fed a high-fat diet. Another study (89) suggested that, because SIRT1 activates liver X receptor (LXR), lack of LXR activation in the SIRT1 liver-specific null animals decreased fat production in these mice, providing one mechanism for absence of weight gain on a high-fat diet. Finally, these findings suggested that SIRT1 activity is (a) increased in liver by a high-fat diet and (b) decreased in this organ by CR. Thus, it is entirely possible that sirtuin activity (or NAD+) increases in some tissues during fasting but is actually repressed in others. How can these two studies be reconciled? Both used C57BL/6 mice, so the best explanation is that differences in dietary composition may account for some discrepancies. Indeed, the latter study used considerably more carbohydrate than the other did.
Importantly, other studies have linked SIRT1 with improved glucose and insulin homeostasis and fuel utilization. For example, pharmacological activation of SIRT1 in vivo by resveratrol or by more potent SIRT1 activators (e.g., SRT1720 and SRT2183) induces changes in gene expression consistent with those induced by PGC-1α activation in liver and muscle, promotes mitochondrial respiration, and increases insulin sensitivity (94–96). Here, the lack of gluconeogenesis is probably the effect of TORC2 repressing PGC-1α in the liver (135), an effect that could be absent in liver-specific genetic alterations. Consistent with this finding, blocking SIRT1 leads to insulin resistance in cultured cells (124). The authors suggest that SIRT1 represses protein tyrosine phosphatase 1B, a negative regulator of insulin signaling, providing a potential mechanism for the effect of SIRT1 on insulin signaling in peripheral tissues (124).With regard to the starvation response, it is also notable that SIRT1 is required for full induction of autophagy (144), but follow-up in vivo studies are needed to assess the relevance of this finding to metabolism or cell survival.
AMPK-SIRT1 Axis
Exercise induces an energy deprivation state that activates similar metabolic pathways in skeletal muscle. Upon exercise, AMPK was found to boost SIRT1 activity by increasing intracellular NAD+ (87). In turn, SIRT1 deacetylates and activates transcription programs driven by PGC-1α, FOXO1, and FOXO3. These findings help to explain many of the overlapping functions of AMPK and SIRT1 activation (Figure 4). Pharmacological activation of SIRT1 by SRT1720 does not activate AMPK directly, but it does increase insulin sensitivity, improve endurance, increase respiratory quotient, and shift muscle toward oxidative pathways (138). Thus, the benefits of SIRT1 activation can occur in the absence of AMPK activation, but clearly the two pathways can act in concert to reinforce one another.
Figure 4.

The SIRT1-AMPK metabolic control network. Conditions of perceived energy deprivation, such as fasting, calorie restriction (CR), and exercise, increase the AMP/ATP ratio and activate AMP-activated protein kinase (AMPK). Energy deprivation also increases nicotinamide adenine dinucleotide (NAD+) levels and activates the NAD+-dependent deacetylase activity of SIRT1. Activated AMPK and SIRT1 converge by activating peroxisome proliferator-activated receptor gamma coactivator 1 alpha (PGC-1α) via phosphorylation and deacetylation, respectively, to induce mitochondrial biogenesis and fatty acid oxidation. Cross talk in this pathway occurs because AMPK activity increases NAD+, and SIRT1 also activates AMPK.
Insulin Secretion
In addition to modulating insulin and glucose homeostasis in liver and muscle, SIRT1 acts in pancreatic beta cells directly. SIRT1 is a positive regulator of insulin secretion, which triggers glucose uptake and utilization. Through use of a mouse model in which SIRT1 was overexpressed specifically in pancreatic beta cells (BESTO mice), it was shown that elevated levels of SIRT1 increased glucose-stimulated insulin secretion ex vivo (145). These mice consistently demonstrated improved glucose tolerance in vivo. In line with studies of BESTO mice, whole-body SIRT1 null animals demonstrated impaired insulin secretion in ex vivo studies using isolated islets (146). In both models, it was shown that SIRT1 promotes insulin secretion by suppressing uncoupling protein 2 (UCP2) expression, which could lead to a boost in cytosolic ATP/ADP ratios. Although both studies demonstrated that levels of SIRT1 protein regulate insulin secretion, elevated levels of SIRT1 protein alone were not sufficient to promote insulin secretion, as was illustrated by studies of aging BESTO mice. The increase in insulin secretion in BESTO mice declined with age but could be restored by addition of NMN. Furthermore, a subsequent study showed that a decline in NAD+ biosynthesis may be responsible for the decreased SIRT1 activity in these islets during aging (147). This finding raises the importance of investigating NAD+ biosynthesis, not sirtuin levels alone, during aging in other metabolic tissues.
Fat Tissue
SIRT1 also regulates white adipose tissue (WAT), which is significant because fat accumulation and distribution increase during aging. WAT functions both to store fats and to serve as an endocrine organ by secreting hormones, such as (a) leptin, (b) adiponectin, and (c) inflammatory agents such as tumor necrosis factor alpha (TNFα) and resistin. An increase in adiposity also elevates the risk of developing metabolic syndromes and insulin resistance, and a reduction in WAT mass is presumed to be responsible for at least part of the life span increase by CR. In cultured cells, SIRT1 has been shown to bind PPARγ and repress transcription of its target genes involved in fat storage (148). As a result, upregulation of SIRT1 in differentiated fat cells triggers lipolysis and results in decreased fat storage. Consistent with these findings, treatment of mice on a high-fat diet with resveratrol or SRT1720 reduces weight gain (94, 96). Thus, an increase in SIRT1 levels may be a part of the metabolic program to mobilize fatty acids during fasting and to reduce adiposity in WAT during CR. SIRT1 also contributes to the production of adiponectin by WAT by enhancing the interaction between FOXO1 and C/EBP(149). Because adiponectin improves insulin sensitivity, its control by SIRT1 may provide yet another mechanism for regulating metabolic homeostasis.
Mitochondrial Sirtuins in Metabolism
SIRT3–5 are the sirtuins that reside in the mitochondria—a metabolic hot spot (21, 35, 42, 55). Mitochondria are at center stage for cellular energy (ATP) production, generation of ROS, and signaling during apoptosis. These organelles consume 85%–95% of the oxygen used by cells in a series of enzymatic reactions that ultimately generate ATP from oxidative phosphorylation. When energy generation is required, mitochondrial biogenesis is often accompanied by an increase in oxidative fuel utilization to promote energy production. Mitochondria serve as a nexus for nutrient adaptation (Figure 5). Pyruvate, derived from glucose, is metabolized via the tricarboxylic acid (TCA) cycle; fatty acids and amino acids are also converted in the mitochondria by fatty acid oxidation and amino-transferase reactions, respectively. Not surprisingly, defects in mitochondrial functions have been linked to aging and can result in imbalances in metabolic homeostasis (150). For instance, impaired mitochondrial function in the pancreas could inhibit the increase in ATP/ADP needed to stimulate insulin exocytosis, and defective mitochondria in muscle may lead to insulin resistance (151).
Figure 5.

Network of the mitochondrial sirtuins (SIRT3–5). Mitochondria can metabolize fuels, such as fatty acids, amino acids, and pyruvate, derived from glucose. Electrons pass through electron transport complexes (I–IV), generating a proton gradient that is used to drive ATP synthase to generate ATP. SIRT3 binds to complex I, regulating its activity and energy levels in the cell. SIRT3 also binds and deacetylates acetyl-CoA synthetase 2 (AceCS2) and glutamate dehydrogenase (GDH), activating their enzymatic activities. SIRT4 binds and represses GDH activity via ADP-ribosylation. SIRT5 deacetylates and activates carbamoyl phosphate synthetase 1 (CPS1), the rate-limiting step of the urea cycle.
Recent studies have revealed that mitochondrial sirtuins could be a pivotal part of the mitochondrial changes driving energy adaptation. However, compared with SIRT1, little is known about the biochemical and biological functions of SIRT3–5. SIRT3 is the mitochondrial sirtuin most conserved with SIRT1, and it is the best-understood mitochondrial sirtuin. SIRT3 is expressed in all tissues, with the highest levels appearing in metabolically active tissues such as brown adipose, muscle, liver, kidney, heart, and brain (64, 152, 153).
Experiments comparing membrane-bound and -soluble mitochondrial fractions have suggested that SIRT3 is in the matrix and in the inner mitochondrial membrane. Moreover, electron microscopy studies have observed SIRT3 together with mitochondrial cristae, the site for oxidative phosphorylation. SIRT3 null mice do not have an overt developmental or metabolic phenotype (154). The mice display normal body composition and levels of mitochondrial proteins and a typical response to fasting and cold exposure, but they also display a robust biochemical phenotype—increased levels of mitochondrial acetylation (154). This observation is significant because metabolic proteins, such as TCA-cycle enzymes, fatty acid oxidation enzymes, and subunits of oxidative phosphorylation complexes, were found to be heavily acetylated in a mass spectrometry survey of acetylation. However, it remains to be determined precisely how global acetylation influences mitochondrial control of metabolism. Notably, no mitochondrial acetyltransferase has yet been identified.
SIRT3 can bind and deacetylate at least three metabolic substrates in the mitochondria: acetyl-CoA-synthetase (AceCS) (65, 155), glutamate dehydrogenase (GDH), and complex I. SIRT3-mediated deacetylation seems to have an activating effect on enzymatic activity. For example, two groups (154, 156) have shown that SIRT3 deacetylates and activates AceCS, which forms acetyl-CoA from acetate, CoA, and ATP. Mammalian cells do not require AceCS to synthesize acetyl-CoA under normal conditions. However, AceCS activity becomes critical under ketogenic conditions, such as prolonged fasting and diabetes, when energy-expending tissues such as muscle must convert acetate released by the liver into acetyl-CoA for further metabolism. SIRT3 also binds and deacetylates GDH, although an effect on its activity has not been reported (154, 156). GDH interconverts glutamate to alpha-ketoglutarate, and its regulation by SIRT3 could be a way to control amino acid flux into the TCA cycle. GDH activity also increases during fasting and CR in liver, but the role of SIRT3 in this response has not been investigated. The regulation of AceCS and GDH in vitro by SIRT3 warrants further study to evaluate whether SIRT3 regulates acetate or amino acid metabolism in vivo. These interactions suggest that under conditions of energy limitation SIRT3 may play a role in funneling carbons from alternative sources—namely ketone bodies and amino acids—into the central metabolism of the TCA cycle. Consistent with this hypothesis, SIRT3 expression in brown adipose tissue and WAT increases during CR and decreases in genetically obese mice (153).
In addition to potentially regulating central pathways of mitochondrial metabolism, SIRT3 is linked to mitochondrial respiration. SIRT3 binds to complex I and promotes NADH-driven mitochondrial respiration (157). Furthermore, mitochondria from SIRT3 null livers demonstrated decreased oxygen consumption, and heart, kidney, and liver displayed a 50% reduction in basal ATP levels (157). Overexpression of SIRT3 in brown adipocytes increases oxygen consumption, reflecting heightened electron transport activity and increased uncoupling (153). Accordingly, SIRT3 overexpression results in decreased membrane potential and ROS production in these cells (153). However, these overexpression studies are difficult to interpret because they were performed using a truncated form of SIRT3 that does not correctly localize to mitochondria (158).
Given the ties among SIRT3, metabolism, and mitochondrial function, it is especially intriguing that genetic studies have linked SIRT3 to human life span. A silent G–T transversion in the conserved sirtuin core domain is associated with survivorship in elderly males, and SIRT3 contains a variable number tandem repeat polymorphism found almost exclusively in males over the age of 90 (159, 160). Future biochemical studies to examine how SIRT3 single nucleotide polymorphisms modulate protein level or activity will be critical for a better understanding of these associations. Although these human genetic studies are limited in scale, the findings hint that SIRT3 may have a positive impact on human life span.
SIRT4 localizes to mitochondria of human and mouse cells and has been observed in the mitochondrial matrix (21, 22, 161). SIRT4 is a ubiquitously expressed gene, but in mice its protein levels are highest in the kidney, heart, brain, liver, and pancreatic beta cells (21, 22, 161). Unlike SIRT3, SIRT4 does not have a detectable NAD+-dependent deacetylase activity toward canonical sirtuin targets (21, 22, 161). It is possible that SIRT4 is more specific in its substrate specificity than SIRT3, and future surveys may identify specific substrates that are deacetylated by SIRT4. However, SIRT4 has been shown to contain a NAD+-dependent ADP-ribosyltransferase activity. One SIRT4 substrate has been identified; SIRT4 interacts with and represses GDH activity via ADP-ribosylation (21). GDH regulates the usage of amino acids into energy production. Its biological significance is evident from patients with GDH-activating mutations who exhibit hyperinsulinism/hyperammonia syndrome (162). Isolated pancreatic islets from SIRT4 null mice exhibited higher GDH activity and increased insulin secretion in response to glucose and amino acids. In a separate study, SIRT4 overexpression in insulinoma cells suppressed insulin secretion (161). SIRT4 has also been shown to interact with insulin-degrading enzyme and adenine nucleotide translocator, but the functional significance of these interactions is not known (161). It will be important for future studies to address whether SIRT4 affects fuel utilization in other tissues. Also, how SIRT4 function relays physiological changes during nutrient limitation or metabolic dysfunction remains to be evaluated.
Recently, the first comprehensive study of SIRT5 revealed that this sirtuin regulates ammonia entry into the urea cycle. SIRT5 localizes to the mitochondrial matrix and is ubiquitously expressed. SIRT5 functions as a weak NAD+-dependent deacetylase. SIRT5 null mice have been generated and are developmentally normal without obvious metabolic defects (63, 154). SIRT5 is known to interact with at least two proteins involved in cellular metabolism: cytochrome c and carbamoyl phosphate synthetase 1 (CPS1) (63, 156). CPS1 is the rate-limiting first step of the urea cycle; its activity is required for clearing ammonia generated by amino acid metabolism. By deacetylating CPS1, SIRT5 stimulates its enzymatic activity (63). Mice lacking SIRT5 displayed elevated ammonia levels after a prolonged fast, suggesting that this sirtuin helps the liver deal with by-products of amino acid metabolism (63). It remains to be seen whether loss of SIRT5 also increases susceptibility to ammonia toxicity.
The regulation of mitochondrial sirtuins is an area of increasing interest. There are three mechanisms of regulation: First, mitochondrial NAD+ levels can rise due to increases in Nampt by up to twofold in the liver of fasted rats, with a concomitant increase in stress resistance (44, 163). Second, mitochondrial sirtuins are also regulated at the protein level: SIRT3 protein levels increase during CR, fasting, stress, and exercise. (153, 163, 164). Third, mitochondrial sirtuins are regulated by localization. For example, SIRT3 may translocate from the nucleus to the mitochondria upon cellular stress (67, 68), and SIRT5 is translocated predominantly into the mitochondrial intermembrane space or the matrix, depending on the stimulus (156).
An elegant coordination of metabolism by mitochondrial sirtuins is emerging. SIRT3–5 appear to work in concert to regulate critical aspects of mitochondrial metabolism during times of adaptation. For example, both SIRT3 and -4 regulate GDH and may stimulate ammonia production. Accordingly, SIRT5 helps to clear this ammonia by activating the urea cycle. Future studies will need to fill in the many gaps in our current understanding of how mitochondrial sirtuins regulate metabolism. SIRT4 represses GDH, whereas the effect on SIRT3 is unknown. Are SIRT3 and -4 activated to function at the same time? In sum, these sirtuins appear to function at critical junctions in mitochondrial metabolism by acting as switches to facilitate fuel entry into the TCA cycle and by regulating oxidative phosphorylation.
CANCER
An area of considerable debate is the role of sirtuins in tumorigenesis and cancer cell proliferation. One of the confusing aspects of SIRT1 is that it plays a dual role in cell survival and cell death and can be modulated in different directions by a variety of different stimuli (Figure 6). Although CR is arguably the most effective way to prevent cancer in rodents and primates, which some view as an indication that sirtuins are tumor suppressors (70), some sirtuins, such as SIRT1 and -3, have prosurvival functions, which could be a sign that they promote tumorigenesis (165, 166).
Figure 6.
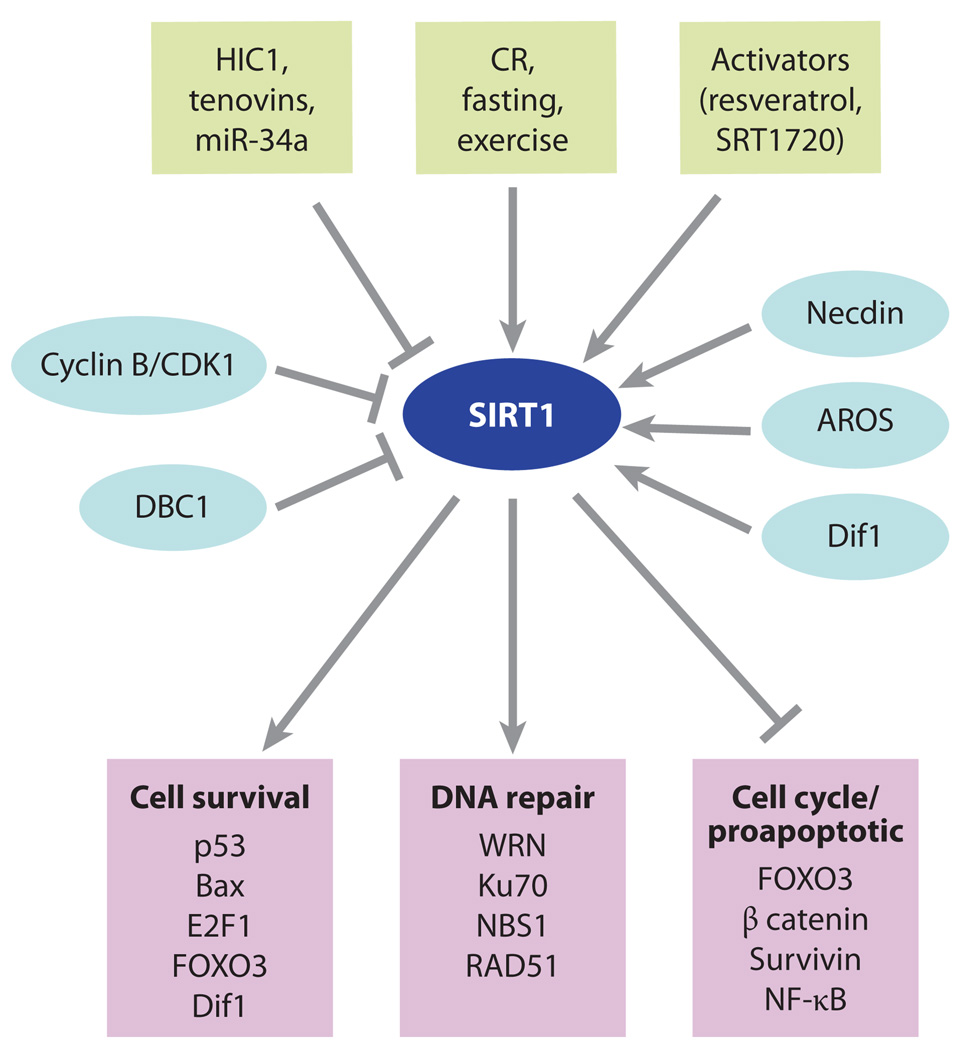
SIRT1 plays key roles in cell survival and apoptosis. The complexity of SIRT1 regulation has made it a challenge to decipher SIRT1’s role in cancer. The SIRT1 gene is under the control of both environmental stimuli such as fasting and exercise and microRNAs (miRNAs), tenovins, and the hypermethylated in cancer 1 (HIC1) transcriptional repressor. The SIRT1 enzyme can also be modulated by protein-protein interactions with deleted in breast cancer 1 (DBC1), adaptor response to oxidative stress (AROS), Dif1, and necdin. SIRT1 interacts with and deacetylates numerous proteins involved in cell survival, DNA repair, and apoptosis (Figure 3). On the basis of this information, it has been difficult to predict what SIRT1 overexpression or activation by SIRT1-activating compounds will do in vivo, but so far experiments (171) point to SIRT1 acting as a tumor suppressor in the case of the p53−/+, breast cancer 1 (BRCA1), 7,12-dimethylbenz[α]anthracene (DMBA), and APCmin+/− models of lymphoma, breast, skin, and colon cancers, respectively. Abbreviations: CR, calorie restriction; DBC1, deleted in breast cancer 1; FOXO, forkhead box, subgroup O; HIC1, hypermethylated in cancer 1; NF-κB, nuclear factor kappa B.
Initially, most of the evidence was in favor of SIRT1 being an oncogene, beginning with the identification of the first substrate of SIRT1: the tumor suppressor p53 (167, 168). Deacetylation of lysine 382 on p53 by SIRT1 reduces p53 transactivation, allowing cells to bypass p53-mediated apoptosis. These early data, combined with other studies showing that SIRT1 can suppress apoptosis (88), implied that SIRT1 might be oncogenic. Recent work, however, shows that SIRT1 plays a critical role in DNA-break repair and can prevent tumorigenesis in mouse models of cancer. Mice with additional copies of SIRT1 do not show signs of premature death or increased tumorigenesis; in fact, in models of leukemia and colon cancer, SIRT1 transgenic mice live longer (169–172). These findings have led to considerable debate as to whether inhibition or activation of SIRT1 plays a role in the development of cancer (171).
How can we resolve the apparently contradictory data on SIRT1 in cancer? It may come down to the genetics of the tumor and the stage of tumorigenesis being assessed. In assessing the potential role of sirtuins in cancer and its treatment, however, it is important to remember that most of the data so far have been generated through use of cell lines, which can be misleading when extrapolating to the in vivo situation. It is also important to recognize that some cancer models, such as the p53 lymphoma model used in SIRT1 studies, are more relevant to cancer prevention than to treatment. One reason for this finding is that SIRT1 is situated within feedback loops that allow it to promote cell survival in critical cell types without causing cancer. If and how this actually occurs are an area of major interest and will require considerably more investigation of SIRT1, not only in cell culture but also in vivo.
Altered Sirtuin Expression in Cancer
To date, the majority of information about the expression levels of sirtuins in cancer comes from studies of SIRT1 and, to a lesser extent, SIRT2. SIRT1 expression is relatively higher in many different types of cancer, including colon, skin, breast, and prostate cancers and leukemia, compared to the corresponding normal tissue (121, 173–177). But the correlation is not clear-cut. Wang et al. (178) analyzed a public database and found reduced levels of SIRT1 mRNA in prostate and bladder carcinoma, glioblastoma, and ovarian cancer compared to normal tissues. They observed significantly lower levels of SIRT1 mRNA in cancers with mutated BRCA than in cancers with wild-type BRCA1. Positive correlation between BRCA1 and SIRT1 expression was also observed through immunohistochemical staining of 45 human breast cancer samples.
A recent study of pancreatic cancer observed an increase of SIRT1 in only 1 out of 11 tumors (179). Indeed, there was a greater correlation between tumor tissue and the expression levels of the other sirtuins: SIRT2 (6 of 11), SIRT3 (4 of 11), and SIRT6 (4 of 11). The most striking result in this study was that SIRT5 transcripts were elevated in 10 of the 11 pancreatic cancers. All the patients in stages 0, IB, IIB, and IV had increased levels of SIRT5 mRNA, whereas one patient in stage III showed SIRT5 downregulation.
With regard to SIRT2, its role in regulating the cell cycle in response to stress suggests a possible role in tumorigenesis and perhaps some utility in chemotherapy. The latest evidence suggests that SIRT2 functions to release mitotic arrest in critically damaged cells, allowing them to proceed to apoptosis (17, 180, 181). Decreases in SIRT2 expression are common in gliomas and glioma-derived cell lines, and these decreases were traced to histone modifications at the promoter or deletion of the SIRT2 gene. Thus, inhibition of SIRT2 may predispose cells to uncontrolled growth (182). In agreement with this hypothesis, a recent study found that lysine 56 of histone H3, a target of SIRT1/SIRT2, is hyperacetylated in cancer tissue. Furthermore, nontumorigenic MCF10A cells show much lower levels of acetylated H3K56 than do tumorigenic MCF7 breast cancer cells (183).
Together, these data indicate that decreased SIRT1/SIRT2 activity correlates with tumorigenesis and that increasing activity of SIRT2 in certain cancers could be beneficial. Cell biology experiments also support this view. Ectopically expressed wild-type SIRT2 slows cell-cycle progression and increases the number of multinucleated cells (60, 180). Growth inhibition is dependent on the phosphorylation of serine 368 on SIRT2 by CDK1 (60, 180). SIRT2 overexpression also arrests the cell cycle prior to entry into mitosis in response to microtubule inhibitors such as nocodazole (181, 184). Conversely, SIRT2 inhibition promotes centrosome fragmentation and prolongs chronic mitotic arrest in response to nocodazole, but in so doing prevents cell death during reentry into the cell cycle (181). Interestingly, the SIRT2 catalytic mutant also increases the number of multinucleated cells (60, 180), indicating that precise levels of SIRT2 are required for mitotic fidelity and that, depending on the circumstances, an inhibitor or an activator may be useful in treating certain cancers in vivo.
Increased levels of SIRT3 and -7 mRNA have been associated with node-positive breast cancer, compared with nonmalignant breast tissue (185). A reported increase in what was thought to be SIRT8 expression in thyroid cancer turned out to be an increase in SIRT7 expression (it should be noted that SIRT8 does not exist) (186). In summary, there is no definitive correlation between a sirtuin and any particular cancer, with perhaps the exception of a lack of SIRT1 in BRCA1-negative breast cancers, and the sample sizes used to investigate the possible association of the other sirtuins with particular cancers is still too small to draw meaningful conclusions.
SIRT1 as a Potential Oncogene
In cell culture experiments, a wealth of data show that SIRT1 can prevent apoptosis and senescence (62, 128, 187, 188), suggesting that the inhibition of SIRT1 may be beneficial in treating some types of cancer in vivo (128, 165, 189). In 2001, SIRT1 was shown to interact with and target p53 for deacetylation, which reduces p53’s DNA-binding capacity (102, 168, 190). Cells with additional SIRT1 typically have lower levels of acetylated p53 lysine 382 in response to DNA damage and cell stress and are, in general, more resistant to p53-dependent cell-cycle arrest and apoptosis (167, 168). Consistent with this finding, expression of the H363Y dominant-negative allele of SIRT1 or small interfering RNA (siRNA) to SIRT1 mRNA increases p53-dependent transcriptional activity (167) and increases the sensitivity of cells to stress (171, 191). Similarly, cells derived from SIRT1 knockout mice or cells treated with siRNAs against SIRT1 show high levels of hyperacetylated p53 on lysines 382 and 320 (188, 192). The sirtuin inhibitor, sirtinol, induces senescence-like growth arrest and increases expression of plasminogen activator inhibitor 1 in human breast cancer MCF7 cells and lung cancer H1299 cells (193). Growth arrest is accompanied by impaired activation of c-Jun N-terminal kinase (JNK) and p38 mitogen-activated protein kinase in response to epidermal growth factor and insulin-like growth factor I, whereas Ras is reduced. Tyrosine phosphorylation of the receptors for epidermal growth factor and insulin-like growth factor I and activation of protein kinase B are unaltered by sirtinol treatment. The same group (194), in studies of endothelial cell function, showed that SIRT1 inhibition increases p53 acetylation and induces premature senescence-like phenotype, whereas overexpression of SIRT1 prevents senescence-like changes. Others have reported that specific inhibition of SIRT1 by siRNA knockdown can increase tumor cell death with no toxic effect on normal cells in culture (188). In the latter study, however, the mechanism of cell death remains unclear because p53 is not essential for tumor cell killing by SIRT1 depletion, although it does not rule out participation by p53 (188).
Increased SIRT1 expression in human fibroblasts has been shown to promote cellular proliferation, reduce cellular senescence, increase the growth rate, and increase the cellular life span of 2Bs human embryonic lung fibroblasts, while reducing the expression of p16INK4A and promoting phosphorylation of Rb (195). The authors (195) provided evidence that senescence was induced via the activation of ERK/S6K1, which occurred after overexpression of SIRT1 or treatment with resveratrol, a SIRT1 activator. Overexpression of SIRT1 has also been shown to repress the expression or activity of tumor-suppressor and DNA-repair genes, including FOXO1/-2/-4, WRN, Rb, p73, MLH1, and NBS1 (reviewed in References 171 and 196). A recent study of zebrafish and mouse embryos demonstrated a role for SIRT1 in promoting angiogenesis (197, 198), another indication that SIRT1 has effects that could be tumor promoting.
A study by Baylin and colleagues (172, 199) indicates that SIRT1-mediated silencing of E-cadherin may play a role in tumorigenesis. The E-cadherin CpG island is frequently silenced by hypermethlylation in epithelial cancers. If a DNA break is initiated within the CpG island, SIRT1 appears to be required for the transient recruitment of DNA methyltransferase 3B and its subsequent silencing by methylation of the DNA. This result is consistent with recent studies linking SIRT1 to the repair of broken DNA (170, 172, 178, 200), but whether SIRT1 can promote epithelial cancers via this mechanism in vivo remains to be determined. Testing the effects of SIRT1 inhibitors in such cancers may be of interest, particularly if the silencing by DNA methylation proves to be reversible.
Two tumor suppressors have been identified as negative regulators of SIRT1. A recent paper showed that hypermethylated in cancer 1 (HIC1), a zinc-finger/BTB domain protein regulated by p53, is a binding partner of SIRT1 that, in turn, represses the transcription of the SIRT1 gene (102). Inactivation of HIC1 up-regulates SIRT1 transcription, thereby inactivating p53, allowing cells to bypass apoptosis after DNA damage. Importantly, the authors found that the HIC1 promoter undergoes hypermethylation during aging, which may lead to upregulation of SIRT1 during aging and, therefore, susceptibility to cancer. Notably, inhibition of glycolysis decreases the binding of the corepressor CtBP to HIC1, which increases SIRT1 expression, linking redox status to SIRT1 expression (103).
Another tumor suppressor that negatively regulates SIRT1 is DBC1 (128, 129, 201). The DBC1 protein forms a stable interaction with the N terminus of SIRT1 that inhibits SIRT1 deacetylase activity. Knockdown of DBC1 by siRNA promotes the deacetylation of p53 and allows cells to survive genotoxic stress, an effect that depends on SIRT1. These data indicate that DBC1 may promote breast cancer in part by activating SIRT1, thereby downregulating p53 and/or other tumor-suppressor pathways.
SIRT1 as a Potential Tumor Suppressor
Although cell-based studies have indicated that SIRT1 may act as a tumor promoting gene, recent studies indicate that SIRT1 can act as a tumor suppressor. First, SIRT1 knockdown, the catalytically inactive H363Y allele, or specific inhibition of SIRT1 in cells does not affect cell viability or cell growth and is not sufficient to induce activation of endogenous p53 (110, 188, 195). In the Solomon et al. study (110), there was no increase in cell death despite DNA damage and increased p53 acetylation. Second, in cell culture studies, Mayo and colleagues (202) showed that SIRT1 stimulates TNFα-induced cell death, indicating that SIRT1 can promote apoptosis, not simply suppress it.
A recent study has highlighted a crucial feedback loop that could explain how SIRT1 could suppress oncogenesis in vivo (109). The c-Myc gene encodes a proto-oncogenic transcription factor that regulates cell proliferation, cell growth, apoptosis, and stem cell self-renewal. c-Myc binds to the SIRT1 promoter and induces SIRT1 expression, but SIRT1 then interacts with and deacetylates c-Myc, resulting in decreased c-Myc stability. This c-Myc-SIRT1 feedback loop could prevent cellular transformation and is consistent with a role for SIRT1 in tumor suppression.
In the first study to test whether SIRT1 promotes cell survival or death (i.e., oncogenic or tumor-suppressing activity) in an animal, SIRT1 was overexpressed in a mouse model of colon cancer, APCmin/+ (169). In this model, loss of the remaining wild-type copy of the min gene results in the relocalization of beta catenin to the nucleus, activating transcription of genes such as myc and cyclin D1, which in turn activate the cell cycle. CR has been shown to reduce tumor formation (203), as has resveratrol (204, 205), but it was unclear whether SIRT1 overexpression would result in more or fewer tumors. Mice overexpressing SIRT1 in the small intestine and colon showed a four-fold reduction in size and number of adenomas. Ki67 and TUNEL staining showed that the tumors in the SIRT1-overexpressing mice were growing slower and undergoing more apoptosis. Deacetylation of beta catenin by SIRT1 was the favored hypothesis for the mechanism. Together, these data indicated that SIRT1 activation may have therapeutic potential in colon cancer and other tumors driven by beta catenin signaling.
Mice that are heterozygous for p53 have been extensively used to study genomic instability in mice (206). When exposed to ionizing irradiation, p53+/− mice show accelerated tumor development due to increased loss of heterozygosity at the p53 locus (207). Given that SIRT1 downregulates p53 in cell culture experiments, SIRT1 activity was expected to exacerbate the p53+/− phenotype and shorten life span, but the opposite occurred. In one study, resveratrol-treated animals showed a 24% increase in survival and a ~45% reduction in the frequency of fatal thymic lymphomas. In accordance with protection from irradiation-induced tumorigenesis, the overall tumor spectrum was highly reminiscent of nonirradiated p53+/− mice (206, 208). Next, SIRT1 was overexpressed in thymocytes in a p53+/− model; again, after irradiation, the mean survival of the SIRT1-overexpressing mice was ~46% greater than in control animals, and the frequency of fatal thymic lymphomas was reduced by 45% in MISTO (MX-cre SIRT1-overexpressing) mice.
In a complementary study, SIRT1 null mice were crossed to the same p53+/− strain (109). On the basis of the cell culture data showing that SIRT1 downregulates p53, it was expected that the SIRT1−/− p53−/+ combination could delay the formation of cancer relative to p53+/− mice. In contrast, the SIRT1−/− mice experienced accelerated tumorigenesis, with multiple tumor types developing spontaneously from approximately 5 months of age. At 20 months of age, 76% of the SIRT1−/− p53−/+ mice had tumors compared to approximately 10%–15% of the controls. Karyotyping of primary tumors showed aneuploidy and chromosomal aberrations (178). Resveratrol, a SIRT1 activator, delayed tumorigenesis and extended life span in this strain in two independent studies (170, 178). Moreover, the relocalization of SIRT1 in response to DNA damage resulted in the absence of SIRT1 from promoters and changes in gene expression that occur during aging (170). These data support the RCM (relocalization of chromatin modifiers) hypothesis of aging, which states that the DNA damage–driven relocalization of chromatin-modifying proteins results in gene-expression changes that cause aging (170, 209). However, further work is required to validate this model (Figure 7) (196).
Figure 7.

The relocalization of chromatin modifiers (RCM) hypothesis of aging stems from S. Imai and H. Kitano, who proposed in 1998 that changes in heterochromatin underlie the aging process. The idea was based in part on observations that, in response to DNA damage and aging, yeast SIR2 is released from silent loci and relocalized to DNA breaks, where it is hypothesized to organize chromatin to facilitate repair. During relocalization, expression of silent mating-type genes (HML and HMR) cause sterility, a hallmark of aging. Recent work shows that a similar process may drive aging in mammals. In response to DNA breaks or aging, SIRT1 also relocalizes away from open reading frames (ORFs) to DNA-break sites, seemingly to alter chromatin around the break site and recruit DNA damage–repair proteins such as RAD51 and NBS1. This relocalization of SIRT1, or the epigenetic changes it induces, is proposed to alter gene-expression patterns that result in tissue dysfunction and diseases associated with aging.
SIRT1−/− mice experience embryonic lethality in inbred strain backgrounds (e.g., Reference 129). Based on the cell culture data showing that SIRT1 inactivates p53, it was initially suspected that p53 hyperactivity was responsible for the embryonic lethality of SIRT1−/− mice. Thus, deletion of p53 in the SIRT1 knockout mice was predicted to rescue the lethality of the knockout, but this was not the case (178, 210). In their paper entitled “Sirt1 fails to affect p53-mediated biological functions,” McBurney and colleagues (210) failed to find alterations in p53-downstream genes in an analysis of embryonic tissue from the SIRT1 knockouts.
In support of a tumor-suppressor function for SIRT1, the gene has been shown to play important roles in repairing broken DNA and maintaining genome stability. The first clue came in 1999, when SIR2 from Saccharomyces cerevisiae, the founding member of the sirtuin family, was shown by three independent laboratories to localize to broken DNA in response to DNA damage (211–213). This response required only a single DNA break and checkpoint signaling via RAD9 and MEC1 (the yeast ATR/ATM). At the time, it was unclear how SIR2, a deacetylase, was directly involved in DNA-break repair, if at all (214). The SIR2, -3, and -4 proteins were later shown to bypass G2 arrest with an unrepaired DNA break (215), and a study by Tyler and colleagues (216) showed that SIR2 binds to a double-strand-break repair site approximately 3 h after initiation of the break coincident with decreases in histone acetylation. These data were interpreted as evidence that SIR2 prepared chromatin around the break site to facilitate the repair process (216).
Four years later, a series of papers showed that mammalian SIRT1 regulates a critical repair factor in the MRE11/RAD50/NBS1 complex, NBS1 (217). Within months thereafter, it was reported that SIRT1 localizes to DNA breaks and is required for efficient DNA-break repair (170, 178). Consistent with this model, cells and mice lacking SIRT1 are more prone to DNA damage–induced aneuploidy, and the efficiency of the repair of a DNA break is reduced by approximately 50% (170). Cells lacking SIRT1 are defective in recruiting DNA-repair factors to the break in response to ATM-and H2AX-mediated signals, including NBS1 and RAD51. These data may explain why mice overexpressing SIRT1 are less prone to loss of heterozygosity at the p53 locus and subsequent tumorigenesis and why SIRT1−/− mice are more prone (171). They may also explain, in part, why SIRT1 can suppress adenomas in APCmin+/− mice that are caused by loss of heterozygosity at the min locus. However, this finding may not be the entire explanation for the reduced adenomas, because SIRT1 overexpression also increases apoptosis and reduces the number of mitoses in the tumors (169).
Resveratrol was first reported to protect mice from chemically induced skin cancers in 1997 (218). Since then, numerous papers have confirmed that resveratrol is a potent chemo-protective agent in mouse models of cancer. Interestingly, a recent paper by the McBurney laboratory (125) showed that resveratrol is significantly less protective against skin cancer in the SIRT1 null mouse. Only 20% of the resveratrol-treated wild-type mice developed tumors after 15 weeks, whereas 75% of SIRT1 null mice developed tumors. Thus, some of the protective effects of resveratrol against skin cancer in this model are mediated by SIRT1.
Given the critical role of yeast Sir2 at telomeres, many researchers have speculated that SIRT1 may also be involved in telomere biology. SIRT1 has not been detected at telomeres in mammalian cells, but hematopoietic stem cells obtained from SIRT1-deficient mice show increased growth capacity, seemingly due to increased telomerase activity (219). In these studies, the overexpression or downregulation of SIRT1 alone had no effect on the life span of human diploid .broblasts, indicating that genetic or epigenetic alterations may occur during the development of the SIRT1 knockout mouse. Growth rates, however, were lower in the SIRT1-overexpressor lines (219).
Other Sirtuins in Cancer
Another sirtuin studied in the context of cancer is SIRT2, a tubulin deacetylase that is required for normal mitotic progression (17, 180) and that controls mitotic checkpoint functions in early metaphase to prevent chromosomal instability (181, 184). SIRT2 abundance increases dramatically during mitosis, and SIRT2 is multiply phosphorylated during the G(2)-to-M transition of the cell cycle. Cells stably over-expressing the wild-type SIRT2, but not missense mutants lacking SIRT2 activity, have a marked prolongation of the mitotic phase of the cell cycle (220). In glioma and glioma-derived cell lines, SIRT2 is downregulated, indicating that increased expression of SIRT2 may be beneficial in the disease (182).
Inhibition or downregulation of SIRT2 interferes with cell-cycle progression and can promote cell-cycle arrest in vitro (180), indicating that inhibition of SIRT2 may be useful in treating some cancers. A novel SIRT2 inhibitor, AC-93253, is selective for SIRT2 over SIRT1 and has cytotoxicity for four different tumor cell lines (221). A dual inhibitor of SIRT1 and -2, cambinol, was found to inhibit Burkitt lymphoma xenografts (see Figure 2) (222), but whether this effect occurs via SIRT1 or -2 or is an off-target effect remains to be determined. One possible clue comes from a recent paper, which found that both SIRT1 and -2 deacetylate H3K56 and that this acetylation marker is increased in multiple types of cancer (183). Clearly, more work is needed to assess whether any of these compounds could be effective cancer therapies in humans.
As for the other sirtuins, SIRT3–7, there are only a few hints that they may be important in cancer biology. SIRT3 can be proapoptotic in HCT116 cells via JNK2, a pathway independent from SIRT1 (223). In other situations, such as in response to DNA damage when NAD+ levels in mitochondria fall below critical levels, SIRT3 (and SIRT4) can be antiapoptotic (44). GCIP (or CCNDBP1/DIP/HHM) is a potential tumor suppressor on chromosome 15 that is downregulated in colon, breast, and prostate cancers. GCIP specifically interacts with one of the class III HDAC proteins, SIRT6, which is important for maintaining genome stability (22, 224). This suggests a possible function of GCIP-SIRT6 in tumor suppression (225).
In summary, as researchers have tested the function of SIRT1 in mouse models, they have failed to confirm in vitro data suggesting that SIRT1 is a tumor-promoting gene. Of the various mouse strains that have been engineered to overexpress SIRT1—at least six have been reported—none has shown a predisposition for cancer, even in cancer-prone mouse models such as p53+/− and APCmin/+. The SIRT1 null mice have also failed to provide compelling evidence for SIRT1 as a tumor promoter, and in fact the mice are more prone to p53-induced cancers. Possible reasons for the discrepancy between the cell culture and in vivo data are that (a) the effects of SIRT1 are cell type specific and/or mouse model specific, (b) the in vivo role of SIRT1 is not to promote cell growth but to prevent premature senescence that leads to aging and disease, and (c) SIRT1 interacts with p53 to alter respiration or another mitochondrial function (226, 227). Only with greater numbers of studies of human tissue samples and animal models will we obtain a better sense of whether, and if so under what circumstances, SIRT1 can promote tumorigenesis and tumor growth in vivo.
CARDIOVASCULAR DISEASE
Cardiovascular disease (CVD), the leading cause of death in the world, is a progressive, multistep process that depends on complex interactions among cholesterol biosynthesis, the immune system, and vascular endothelial cell function (228). Risk factors include higher-than-normal levels of oxidized low-density lipoprotein cholesterol, vascular injury, and increased inflammation and cholesterol deposition by macrophages infiltrating smooth muscle below the vascular endothelium. For many decades, CR has been known to protect from CVD in animal (85) and human studies (229), but whether these effects could be mediated by a single genetic pathway or be susceptible to a single pharmacological intervention is unclear.
Endothelial Function
The increasing evidence that CR may be mediated, at least in part, by sirtuins (230, 231) led to speculation that sirtuins may be useful in preventing or treating CVD (5). A recent influx of data on SIRT1 in CVD has confirmed this view, making SIRT1 a promising molecular target for successful therapeutic intervention for CVD (232). SIRT1 is highly expressed in endothelial cells and controls functions critical to suppressing the development of atherosclerosis, including upregulation of endothelial nitric oxide synthase (eNOS), a reduction in cell senescence of smooth muscle cells, suppression of inflammation and ROS in arteries, and increased vascular growth (232, 233). SIRT1 has also been shown to alter cholesterol biosynthesis in the liver and macrophages (143) and to reduce serum lipid levels (138). Together, these results are consistent with SIRT1 being a major player in the prevention of CVD by CR. As yet, there is only speculation about the potential role of SIRT2–7 in CVD.
Apart from SIRT1’s link to CR in budding yeast, the first indication that this sirtuin may protect against CVD in mammals was the relationship between resveratrol and SIRT1. Since the early 1990s, resveratrol has been touted as the source of the cardioprotective benefits of red wine, and in cell culture and animal models resveratrol has been shown to be highly anti-inflammatory and protective against CVD (234). Until 2003, however, the mechanism of protection was considered to be entirely due to resveratrol’s antioxidant properties. In 2006, resveratrol was shown to decrease ROS, inflammation, and apoptosis in the aortas of elderly mice on a high-fat diet while maintaining normal endothelial function (96). Although these effects were coincident with deacetylation of a SIRT1 target, PGC-1α, this study suggested that SIRT1 was involved in the effects of resveratrol. Since then, however, evidence has mounted that SIRT1 is directly responsible for these protective effects. One of the key requirements for the assembly and repair of damage to the cardiovascular system is angiogenesis. Potente and colleagues (197) showed that mice lacking SIRT1 in the endothelium appear normal from a development standpoint but have an impaired ability to form new blood vessels in response to ischemia. Through use of endothelial cell culture and time-lapse analysis of vessel formation in developing zebrafish embryos with fluorescently labeled endothelial cells, SIRT1 was identified as a requisite factor for endothelial sprouting and vessel navigation. Whether or not the lack of a developmental defect in the adult SIRT1 knockout mice is due to redundancy of sirtuins or a difference between mice and fish remains to be seen.
Vessel Inflammation
A major function of SIRT1 that protects the cardiovascular system is its ability to suppress inflammation. Isolated macrophages and endothelial cells treated with SIRT1 activators have lower levels of inflammatory cytokines, including TNFα, intercellular adhesion molecule (ICAM)-1, interleukin (IL)-6, IL-1, and inducible NOS (iNOS) (121, 235, 236). A similar anti-inflammatory response to SIRT1 activation is seen in mice and rats exposed to a high-fat diet or to cigarette smoke (95, 237, 238). In cultured coronary arterial endothelial cells (CAECs), the protective effects of resveratrol were abolished by knockdown of SIRT1, whereas the overexpression of SIRT1 mimicked the effects of resveratrol (238).
Some of the cardiovascular benefits of CR and pharmacologic activation of SIRT1 seem to stem from SIRT1’s ability to induce nitric oxide signaling. eNOS, an enzyme that generates nitric oxide, is atheroprotective and promotes blood vessel relaxation. Resveratrol has been shown to induce the expression of eNOS when tested either alone (205, 239) or in combination with a statin (240), albeit in one rat study it failed to induce eNOS expression (241).
The first direct link between eNOS and SIRT1 was established upon the demonstration that SIRT1 physically interacts with eNOS and deacetylates lysines 496 and 506 in the calmodulin-binding domain of eNOS, leading to enhanced eNOS activity (242). Mice on a CR diet had lower levels of eNOS acetylation, consistent with the known increase in SIRT1 levels and eNOS in rodents on CR (88, 243, 244). Nampt/PBEF, a NAD+ biosynthetic gene and activator of SIRT1, endows human endothelial cells with increased replicative life span and enhanced angiogenic capacity in a high-glucose environment (245). Conversely, inhibition of SIRT1 expression decreases nitric oxide bioavailability, inhibits endothelium-dependent vasorelaxation, and induces premature senescence of endothelial cells (242). In agreement with the in vitro data, transgenic overexpression of SIRT1 in endothelial cells prevents the loss of vasorelaxation and lowers the number of atherosclerotic plaques in the apoE−/− model of atherogenesis (246), without affecting blood lipid or glucose levels (247). Interestingly, eNOS signaling is necessary for SIRT1 induction by CR (244), indicating that SIRT1-eNOS-SIRT1 signaling forms a positive feedback loop that amplifies the effects of CR and resveratrol (Figure 8).
Figure 8.

The role of SIRT1 in protection from atherosclerosis and cardiovascular disease (CVD). In the normal progression of atherosclerosis, damage and inflammation of blood vessel walls promote the infiltration of macrophages, where they take up and accumulate oxidized low-density lipoprotein, becoming foam cells that can eventually rupture and promote additional inflammation and plaque formation, which in turn occludes blood flow. Overexpression of SIRT1 in endothelial cells or treatment of mice with resveratrol SIRT1 reduces reactive oxygen species in vessel walls and slows the progression of CVD, apparently through multiple mechanisms. These include the suppression of inflammation by increasing endothelial nitric oxide synthase (eNOS) and decreasing nuclear factor kappa B (NF-κB) activity. Increased retrograde cholesterol transport in macrophages may also be a contributing factor. Abbreviations: cGMP, cyclic guanosine monophosphate; ICAM, intercellular adhesion molecule 1; IL-6, interleukin-6; iNOS, inducible NO, nitric oxide synthase; TNFα, tumor necrosis factor alpha.
Recently, Ungvari and colleagues (238) showed that resveratrol treatment attenuated the deleterious effects of cigarette smoke extract in rat arteries and CAECs. The inflammatory markers (ICAM-1, iNOS, IL-6, and TNFα) were considerably reduced, as were nuclear factor kappa B (NF-κB) activation and inflammatory gene expression. In CAECs, these protective effects, including the suppression of apoptosis, were prevented by knockdown of SIRT1, whereas the overexpression of SIRT1 mimicked the effects of resveratrol. Thus, the vasoprotective and anti-inflammatory effects of resveratrol require SIRT1, making this sirtuin a potential drug target for the treatment of CVD and vascular aging.
With regard to the other sirtuins, SIRT2–7, little is known about their potential for influencing the cardiovascular system. SIRT3 probably protects against the damage caused by myocardial infarction or chronic heart failure given that it is a stress- and exercise-responsive gene that protects cardiac myocytes from genotoxic and oxidative stress-mediated cell death. The nuclear form of SIRT3 is thought to promote cell survival by deacetylating Ku70, causing it to sequester the proapoptotic protein Bax away from mitochondria (163).
Vascularization
Another attribute of SIRT1 relevant to CVD is its ability to downregulate FOXO signaling. FOXO transcription family members are essential negative regulators of blood vessel formation, and FOXO1 is the primary mediator (248–250). SIRT1 targets FOXO factors for deacetylation, thus reducing FOXO1-dependent transcriptional activity (248). SIRT1 also interacts with a component of the Notch signaling pathway, the Hairy and Enhancer of split basic loop-helix-loop-helix repressor protein Hey2 (251). As Potente & Dimmeler (198) point out, given the role of Hey2 in vascular patterning in vertebrates, the interaction between SIRT1 and Hey2 indicates that SIRT1 may function downstream of Notch and modulate Hey2 endothelial angiogenic activity (198).
Cholesterol Metabolism
The key role of oxidized low-density lipoprotein cholesterol in the progression of atherosclerosis and the protective function of high-density lipoprotein are well known. Guarente and colleagues (143) showed that SIRT1 deacetylates and thereby activates the nuclear receptor LXR, promoting the transcription of LXR target genes involved in lipid metabolism, including the ABCA1 transporter, and in mediating the efflux of cholesterol from peripheral tissues. It is tempting to speculate that one of the long-term benefits of SIRT1 activation by CR or a SIRT1 activator could be the efflux of cholesterol from peripheral tissues to slow atherosclerotic plaque formation (198). In the liver, SIRT1 controls hepatic triglyceride synthesis by activating genes such as SREBP1. Consistent with this finding, SIRT1 knockout mice show reductions in plasma, hepatic triglyceride accumulation, and decreased high-density lipoprotein cholesterol but, surprisingly, increased glucose tolerance (143).
NEURODEGENERATION
Unlike the situation for CVD, the role of sirtuins in neuroprotection is not entirely clear. Although there are numerous papers showing that increases in NAD+ and SIRT1 are neuroprotective (252, 253), unresolved questions about SIRT1 remain. Is it always beneficial to activate SIRT1, or in some circumstances should it be inhibited? Why is the catalytic activity of SIRT1 not always required for neuroprotection? There is also debate about whether the neuroprotection provided by resveratrol and NAM are mediated via SIRT1, an alternate sirtuin, or an entirely different mechanism. One study suggested that SIRT1 is harmful to neurons (98). With regard to the other sirtuins (SIRT3–7), little is known apart from one report (254) that increased expression of either SIRT2, -3, or -6 promotes apoptosis in neurons exposed to low potassium and that SIRT6 is modestly protective against homocysteic acid toxicity in neuroblastoma cells. Two studies have reported cytoprotective functions for SIRT3 and -4 (44, 163); the latter study showing that these enzymes are required for the protection of cells by increases in Nampt and mitochondrial NAD+ (44). It will be interesting to test whether this protection can also be observed for neurons.
Wallerian Degeneration and Parkinson’s Disease
By the early 2000s, SIRT1 was already known to suppress apoptosis in a variety of cell types (31, 88, 168, 187, 255), but it was the study by Milbrandt and colleagues (252) that provided the first evidence that SIRT1 is neuroprotective. Their study showed that SIRT1 was necessary for the protection of neurons against Wallerian degeneration (WD), which is essentially the die-back of axons following a nerve crush injury (252, 256). Mutant mice that are protected from Wallerian degeneration (known as WldS mice) have greatly increased resistance to WD due to a mutation that amplifies the NAD+ biosynthetic gene Nmnat fused to a ubiquitin assembly protein, Ufd2 (257, 258). The Milbrandt group’s paper (252) proposed that increased NAD+ production by Nmnat and subsequent SIRT1 activation were necessary for neuroprotection. Although the ability of SIRT1 to provide neuroprotection in a variety of contexts is now undisputed, and although SIRT1 has been shown to require catalytic activity in human neurons, there continues to be debate about how Nmnat1 protects cells and whether SIRT1 is involved (259, 260). For example, SIRT1 has been found to be dispensable for the protective effects of NAD+, and there are conflicting data on whether Nmnat alone can protect neurons in vivo or whether catalytic activity is required (253, 259, 261–263). However, recent data in vivo support the Milbrandt model (253). A recent study showing that resveratrol abolishes the resistance of WldS mice to WD proposed that SIRT2 activation was responsible (264), although this result is confusing given that resveratrol does not activate SIRT2 in vitro (94). This work also seems to run counter to a study showing that inhibition of SIRT1 provided neuroprotection in a mouse model of Parkinson’s disease (265).
Although the role of SIRT1 and -2 in WD is unclear, SIRT1 provides neuroprotection in the context of a variety of cell stresses, both in vitro and in vivo. In 2004, Brunet et al. (187) were the first to show SIRT1-mediated neuroprotection using cerebellar granule neurons, which was traced to SIRT1’s ability to target FOXO3 for deacetylation, thereby dampening FOXO-mediated cell death. In the same year, Sinclair and colleagues (62, 88) showed that SIRT1 targets the C terminus of Ku70 for deacetylation, which promotes the binding of Bax to Ku70, thereby preventing Bax from initiating apoptosis.
Alzheimer’s Disease and Amyotrophic Lateral Sclerosis
SIRT1 and the STACs are highly neuroprotective of neurons in vitro and in vivo. Tsai and colleagues (139) reported that SIRT1 protects neurons from apoptosis resulting from overexpression of mutant SOD1-G37R, an allele linked to amyotrophic lateral sclerosis. The authors also reported protection from the p25 mutant version of CDK5, a suspected contributor to axonopathies and Alzheimer’s disease in humans. In a p25 transgenic mouse model of Alzheimer’s disease and axonopathies, resveratrol and SIRT1 provided protection from neurodegeneration and decline in cognition (139). Human SIRT1 and resveratrol were also shown by the Pasinetti and Sauve groups (86) to reduce the A beta peptide content of neurons in the Swedish Tg2576 mouse model of Alzheimer’s disease that expresses mutant human cDNA for APP. The same papers proposed that the neuroprotective effects of CR may be due to changes in the NAD+/NAM ratio and subsequent SIRT1-mediated activation of ROCK1, a Rho kinase that induces the protective, non-amyloidogenic alpha-secretase pathway (86).
Optic Neuritis and Age-Related Macular Degeneration
Optic neuritis is inflammation of the optic nerve; it causes loss of vision, usually due to swelling and destruction of the myelin sheath covering the optic nerve, and is a precursor to multiple sclerosis. Shindler and colleagues (267) found that the activation of SIRT1 by intravitreal injection of resveratrol or a precursor to NAD+, NR, significantly attenuated the loss of retinal ganglial cells and that this neuroprotective effect was blocked by sirtinol, a SIRT1 inhibitor. Another eye disease, age-related macular degeneration, is the leading cause of severe vision loss in the elderly. Polymorphisms in the complement factor H(CFH) gene that reduce activity of CFH increase the risk of age-related macular degeneration. Overexpression of SIRT1 has also been shown to attenuate FOXO3 recruitment to the regulatory region of the CFH gene and to reverse hydrogen peroxide–induced repression of CFH gene expression, raising the possibility that SIRT1 activation may ameliorate or slow the progression of age-related macular degeneration (268).
Neural Progenitors
A recent study showed that SIRT1 controls the fate of neural progenitor cells, which are thought to regenerate neurons to compensate for damage in the central nervous system. Many neurodegenerative diseases of the brain are characterized by the emergence of more astrocytes than neurons through a process known as reactive astrogliosis, which may be induced in part by the oxidative milieu caused by inflammation. A recent study found that activation of SIRT1 by resveratrol suppresses proliferation of neural progenitor cells and directs their differentiation toward the astroglial lineage rather than the neuronal lineage, mimicking the effect of oxidative conditions (269). The effect of resveratrol was blocked by siRNAs against SIRT1 mRNA. The mechanism of SIRT1 action appears to be the binding to Hes1 to deacetylate H3K9 and stabilizes the TLE1 corepressor complex that inhibits the proneuronal transcription factor Mash1. Another study concluded that SIRT1 represses the Notch1-Hes1 signaling pathway and that the transient translocation of SIRT1 into the nucleus may have a role in neural progenitor cell differentiation (270). Thus, SIRT1 may link oxidative stress, inflammation, and metabolism to the differentiation of neural progenitors (271). In this context, an inhibitor of SIRT1 may be beneficial by increasing and skewing the differentiation of neural progenitor cells into neurons. Along the same lines, Longo and colleagues (98) reported that mice lacking SIRT1 have lower levels of oxidized proteins and lipids and that knockdown of SIRT1 provided resistance to oxidative stress. It remains to be determined, however, whether increased expression of SIRT1 has the opposite effect.
INFLAMMATION AND INFECTION
SIRT1 as a Modulator of Nuclear Factor Kappa B
Within the past few years, sirtuins have been identified as novel regulators of the immune system, and numerous studies show that SIRT1 can suppress inflammation in multiple tissues (93, 121, 235, 236, 238, 276, 277). One of the master regulators of both adaptive and innate immunity is the NF-κB system, which is composed of receptors and signaling molecules whose functions have been highly conserved throughout evolution. NF-κB is a pleiotropic transcription factor that functions as a cytoplasmic sensor of danger signals, including hypoxia, oxidative stress, and genotoxic stress. NF-κB forms complexes with a number of other proteins, including Rel family members (RelA/p65, c-Rel, and RelB) and the NF-κB components p50 and p52. In the basal state, NF-κB complexes are retained in the cytoplasm via binding to the inhibitory IκB family of proteins. Upon activation, IκB proteins are phosphorylated and targeted for ubiquitination and subsequent degradation, freeing NF-κB complexes to translocate into the nucleus and trigger expression of target genes that are largely proinflammatory. Upregulation of the NF-κB-binding domain is strongly associated with aging, whereas NF-κB activity is strongly diminished by CR (273). SIRT1 has recently emerged as a potent inhibitor of the NF-κB system, providing a mechanistic link between inflammation and aging (275). After translocation to the nucleus, the p65 subunit is acetylated by p300 to enhance DNA binding. SIRT1 binds and deacetylates RelA/p65, inhibiting the transcriptional activity of NF-κB (202).
Sirtuins may also activate NF-κB signaling by regulating other factors, such as FOXO proteins, which converge on insulin signaling and stress pathways and suppress NF-κB signaling. FOXO3a overexpression can inhibit the TNFα-induced activation of NF-κB, and FOXO3a can promote apoptosis by suppressing NF-κB activity (reviewed in Reference 283). Interestingly, several studies have reported that SIRT1 interacts with and deacetylates FOXO proteins in worms and in mammalian systems (118, 187, 285), indicating that there may be an additional layer of NF-κB signaling control.
Metabolic disorders are associated with a state of chronic inflammation and the release from adipocytes and macrophages of the pro-inflammatory cytokines TNF-alpha and IL-6, which appear to impair insulin action in peripheral tissues. Several animal studies indicate that the beneficial effect of SIRT1 on metabolic disorders is mediated in part by its ability to suppress inflammation in adipocytes and macrophages. For example, SIRT1-overexpressing and resveratrol treated mice have lower levels of lipid-induced hepatic inflammation when challenged by a high-fat diet (93, 95). These animals have decreased NF-κB activity, which results in correlates with decreased expression of proinflammatory cytokines, such as TNFα and IL-6. Similarly, SIRT1 expression in macrophages is inversely correlated to inflammatory gene expression (252). Modulation of SIRT1 activity also affects lipid accumulation in adipocytes, which has an impact on the etiology of metabolic diseases such as obesity and TDM. Consistent with this evidence, liver-specific SIRT1 null mice show increased signs of inflammation and NF-κB signaling when fed a high-fat diet (134). In contrast to these findings, a recent study that used SIRT1 whole-body null mice demonstrated that these animals accumulate immunoglobin complexes in the kidney and liver but do not show alterations in their immune responses (278).
Smoking-Induced Inflammation
Interaction between SIRT1 and RelA/p65 is diminished by cigarette smoke, which causes increased acetylation and activation of NF-κB proinflammatory responses in macrophages (279). As discussed above, resveratrol treatment attenuates the deleterious effects of cigarette smoke extract in rat arteries and CAECs, effects that are prevented by knockdown of SIRT1 (238). Interestingly, CAECs cultured in the presence of serum from normal-fed mice exhibited NF-κB activation and inflammatory gene expression. In contrast, treatment of CAECs with serum from CR mice showed far less ROS generation, NF-κB activation, and induction of inflammatory genes, changes that were diminished by siRNA knockdown of SIRT1. Thus, the antioxidant and anti-inflammatory effects of CR on the vasculature may be mediated by circulating factors via a SIRT1-dependent mechanism (272).
Along similar lines, the lungs of smokers and patients with chronic obstructive pulmonary disease (COPD) have decreased levels of nuclear SIRT1, as compared with nonsmokers (280, 281). Intranasal treatment with a selective STAC, SRT2172, prevented the pulmonary neutrophilia and reduction in exercise tolerance observed in a mouse smoking model of COPD, ostensibly by blocking an increase of matrix metalloproteinase 9 (281). This work suggests that SIRT1 activation may be a useful therapeutic approach to treating chronic inflammatory diseases such as COPD.
Inflammatory Uveitis
Uveitis, or inflammation of the middle layer of the eye, is estimated to be responsible for approximately 10% of the blindness in the United States. Resveratrol pretreatment has been shown to provide significant and dose-dependent suppression of leukocyte adhesion to retinal vessels at sites of ocular inflammation in endotoxin-induced uveitis mice (282). Levels of MCP-1, ICAM-1, 8-OhdG, and NF-κB are significantly reduced by resveratrol administration, coincident with increased SIRT1 activity in retinal pigment epithelium chondroidal cells (282).
Viral Infections
There is some evidence that human immunodeficiency virus (HIV)-1 suppresses SIRT1 during viral infection. NF-κB signaling has been implicated in the hyperactive immune response that contributes to HIV-1 infectivity and replication (286). The Tat protein of HIV-1 is required for the transcriptional activation of HIV. SIRT1 has been directly linked to this pathway on several levels. First, Tat itself is a target for SIRT1 (287, 288); SIRT1 and Tat form a complex and synergistically activate the HIV promoter. Deacetylation by SIRT1 recycles Tat to its un-acetylated form, allowing it to bind transactivation responsive RNA and transcriptional elongation factors (288). Next, Tat also seems to regulate SIRT1 activity. TAT directly binds to SIRT1, blocking its ability to deacetylate p65 (288). In doing so, Tat hyperactivates NF-κB target gene expression in wild-type cells, but not in SIRT1 null cells. Thus, SIRT1, Tat, and NF-κB act in a circuit, in which SIRT1 promotes Tat and Tat neutralizes the negative effect of SIRT1 on NF-κB signaling.
SIRT6 and the Immune Response
The only other sirtuin linked to the regulation of immune responses is SIRT6, which may be a prolongevity protein. SIRT6 null mice have severe developmental defects, display hypoglycemia, and have a premature aging phenotype (224). Recently, it was found that SIRT6 interacts with the RelA/p65 component of the NF-κB complex and is recruited to some promoters of NF-κB target genes (289). SIRT6 deacetylates histone H3 lysine 9 at these promoters and represses the expression of NF-κB targets. Consistently, the expression of several NF-κB target genes was elevated in SIRT6 null mice. An independent study found that SIRT6 regulates TNF production (290). Strikingly, hyperactive NF-κB signaling may be responsible for the short life span and degenerative phenotype of SIRT6 null mice, as haploinsufficiency of RelA rescues the premature aging phenotype of SIRT6 knockout animals.
PERSPECTIVE
It was only 10 years ago that yeast SIR2 was shown to extend life span. Since then, it has become clear that SIR2 is only one example of a large and highly conserved family of enzymes that control key cellular processes, such as fuel adaptation to low-nutrient conditions and mitochondrial biogenesis and function, in settings that include DNA repair, neuronal survival, and the maintenance of a youthful pattern of gene expression, among others.
However, much remains to be elucidated about the biology of sirtuins. For example, differences in mouse data reveal a complicated scenario that brings to light the fact that there is still much unknown regarding SIRT1 function, even though many of this sirtuin’s substrates have been identified. Some of the confusion in interpreting these studies comes from conflicting data about when SIRT1 is active. For example, is SIRT1 induced in liver by fasting or by CR? Are earlier studies on SIRT1 now more difficult to interpret given that we know SIRT1 and its regulator, Nampt, are key components of the circadian clock that vary over a 12-h cycle (51, 53, 291, 292)? NAD+ is an important physiological regulator of sirtuins, even in mitochondria (44), yet it is still not clear how NAD+ is synthesized in various cellular compartments. Is it generated from NAM or NR, or is it transported as NMN through the membranes?
Despite the rapid advances in sirtuin biology, there are still no tools for accurately measuring SIRT1 activity on a native substrate in vitro or for assessing sirtuin activity in vivo. Currently, investigators use sirtuin protein level, NAD+ concentration, and the acetylation state of sirtuin substrates in vivo as readouts. When used in combination, these methods can generate a rough snapshot. However, this snapshot may not be entirely accurate because sirtuins have overlapping targets, as in the case of SIRT1–3 and FOXO proteins. Sirtuins’ activities may also be regulated by posttranslational modification or by inhibitory proteins, as in the case of DBC1 for SIRT1 (27, 62). Thus, the above considerations highlight a need for assays that can provide a quantitative readout for the specific activities of SIRT1–7.
A major outstanding question is whether SIRT1 activation is safe enough to be used as a therapy for human diseases such as diabetes or CVD. Clearly CR has some drawbacks, including decreased fertility and osteoporosis, but these negative effects have not yet been reported for SIRT1-overexpressing mice or pharmacological STACs given to mice or humans (94, 96, 114, 293). Now that STACs are in human clinical trials for multiple diseases, we should know the answers to some of these questions within the next few years.
Although there are significant hurdles to overcome, in contrast to 10 years ago when the mammalian sirtuins were first investigated, there is little doubt that studies of these enzymes have greatly increased our knowledge about the elegant mechanisms that coordinate energy intake with organismal health and survival. In the coming decades, we should know if it is possible to safely target the sirtuins to treat diseases of aging.
ACKNOWLEDGMENTS
M.C.H. was supported by a Brookdale Leadership in Aging Fellowship and by the Paul F. Glenn Foundation. D.A.S. was supported by the National Institutes of Health, the Paul F. Glenn Foundation, and the Ellison Medical Foundation.
Footnotes
DISCLOSURE STATEMENT
D.A.S. is a consultant to Sirtris, a GSK company aiming to develop drugs that target the sirtuins to treat diseases of aging.
Contributor Information
Marcia C. Haigis, Email: Marcia_Haigis@hms.harvard.edu.
David A. Sinclair, Email: David_Sinclair@hms.harvard.edu.
LITERATURE CITED
- 1.Sinclair DA, Guarente L. Extrachromosomal rDNA circles—a cause of aging in yeast. Cell. 1997;91:1033–1042. doi: 10.1016/s0092-8674(00)80493-6. [DOI] [PubMed] [Google Scholar]
- 2.Kaeberlein M, McVey M, Guarente L. The SIR2/3/4 complex and SIR2 alone promote longevity in Saccharomyces cerevisiae by two different mechanisms. Genes Dev. 1999;13:2570–2580. doi: 10.1101/gad.13.19.2570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sinclair DA, Guarente L. Unlocking the secrets of longevity genes. Sci. Am. 2006;294:48–57. doi: 10.1038/scientificamerican0306-48. [DOI] [PubMed] [Google Scholar]
- 4.Imai S, Armstrong CM, Kaeberlein M, Guarente L. Transcriptional silencing and longevity protein Sir2 is an NAD-dependent histone deacetylase. Nature. 2000;403:795–800. doi: 10.1038/35001622. [DOI] [PubMed] [Google Scholar]
- 5.Westphal CH, Dipp MA, Guarente L. A therapeutic role for sirtuins in diseases of aging? Trends Biochem. Sci. 2007;32:555–560. doi: 10.1016/j.tibs.2007.09.008. [DOI] [PubMed] [Google Scholar]
- 6.Frye RA. Characterization of five human cDNAs with homology to the yeast SIR2 gene: Sir2-like proteins (sirtuins) metabolize NAD and may have protein ADP-ribosyltransferase activity. Biochem. Biophys. Res. Commun. 1999;260:273–279. doi: 10.1006/bbrc.1999.0897. [DOI] [PubMed] [Google Scholar]
- 7.Smith JS, Boeke JD. An unusual form of transcriptional silencing in yeast ribosomal DNA. Genes Dev. 1997;11:241–254. doi: 10.1101/gad.11.2.241. [DOI] [PubMed] [Google Scholar]
- 8.Bryk M, Banerjee M, Murphy M, Knudsen KE, Garfinkel DJ, Curcio MJ. Transcriptional silencing of Ty1 elements in the RDN1 locus of yeast. Genes Dev. 1997;11:255–269. doi: 10.1101/gad.11.2.255. [DOI] [PubMed] [Google Scholar]
- 9.Aguilaniu H, Gustafsson L, Rigoulet M, Nystrom T. Asymmetric inheritance of oxidatively damaged proteins during cytokinesis. Science. 2003;299:1751–1753. doi: 10.1126/science.1080418. [DOI] [PubMed] [Google Scholar]
- 10.North BJ, Verdin E. Sirtuins: Sir2-related NAD-dependent protein deacetylases. Genome Biol. 2004;5:224. doi: 10.1186/gb-2004-5-5-224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tissenbaum HA, Guarente L. Increased dosage of a sir-2 gene extends lifespan in Caenorhabditis elegans. Nature. 2001;410:227–230. doi: 10.1038/35065638. [DOI] [PubMed] [Google Scholar]
- 12.Wang Y, Tissenbaum HA. Overlapping and distinct functions for a Caenorhabditis elegans SIR2 and DAF-16/FOXO. Mech. Ageing Dev. 2006;127:48–56. doi: 10.1016/j.mad.2005.09.005. [DOI] [PubMed] [Google Scholar]
- 13.Rogina B, Helfand SL. Sir2 mediates longevity in the fly through a pathway related to calorie restriction. Proc. Natl. Acad. Sci. USA. 2004;101:15998–16003. doi: 10.1073/pnas.0404184101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Frye RA. Phylogenetic classification of prokaryotic and eukaryotic Sir2-like proteins. Biochem. Biophys. Res. Commun. 2000;273:793–798. doi: 10.1006/bbrc.2000.3000. [DOI] [PubMed] [Google Scholar]
- 15.Haigis MC, Guarente LP. Mammalian sirtuins—emerging roles in physiology, aging, and calorie restriction. Genes Dev. 2006;20:2913–2921. doi: 10.1101/gad.1467506. [DOI] [PubMed] [Google Scholar]
- 16.Tsang AW, Escalante-Semerena JC. CobB, a new member of the SIR2 family of eucaryotic regulatory proteins, is required to compensate for the lack of nicotinate mononucleotide:5,6-dimethylbenzimidazole phosphoribosyltransferase activity in cobT mutants during cobalamin biosynthesis in Salmonella typhimurium LT2. J. Biol. Chem. 1998;273:31788–31794. doi: 10.1074/jbc.273.48.31788. [DOI] [PubMed] [Google Scholar]
- 17.North BJ, Marshall BL, Borra MT, Denu JM, Verdin E. The human Sir2 ortholog, SIRT2, is an NAD+-dependent tubulin deacetylase. Mol. Cell. 2003;11:437–444. doi: 10.1016/s1097-2765(03)00038-8. [DOI] [PubMed] [Google Scholar]
- 18.Tanner KG, Landry J, Sternglanz R, Denu JM. Silent information regulator 2 family of NAD-dependent histone/protein deacetylases generates a unique product, 1-O-acetyl-ADP-ribose. Proc. Natl. Acad. Sci. USA. 2000;97:14178–14182. doi: 10.1073/pnas.250422697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Landry J, Slama JT, Sternglanz R. Role of NAD+ in the deacetylase activity of the SIR2-like proteins. Biochem. Biophys. Res. Commun. 2000;278:685–690. doi: 10.1006/bbrc.2000.3854. [DOI] [PubMed] [Google Scholar]
- 20.Liszt G, Ford E, Kurtev M, Guarente L. Mouse Sir2 homolog SIRT6 is a nuclear ADP-ribosyltransferase. J. Biol. Chem. 2005;280:21313–21320. doi: 10.1074/jbc.M413296200. [DOI] [PubMed] [Google Scholar]
- 21.Haigis MC, Mostoslavsky R, Haigis KM, Fahie K, Christodoulou DC, et al. SIRT4 inhibits glutamate dehydrogenase and opposes the effects of calorie restriction in pancreatic β cells. Cell. 2006;126:941–954. doi: 10.1016/j.cell.2006.06.057. [DOI] [PubMed] [Google Scholar]
- 22.Michishita E, McCord RA, Berber E, Kioi M, Padilla-Nash H, et al. SIRT6 is a histone H3 lysine 9 deacetylase that modulates telomeric chromatin. Nature. 2008;452:492–496. doi: 10.1038/nature06736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tanny JC, Dowd GJ, Huang J, Hilz H, Moazed D. An enzymatic activity in the yeast Sir2 protein that is essential for gene silencing. Cell. 1999;99:735–745. doi: 10.1016/s0092-8674(00)81671-2. [DOI] [PubMed] [Google Scholar]
- 24.Moazed D. Enzymatic activities of Sir2 and chromatin silencing. Curr. Opin. Cell Biol. 2001;13:232–238. doi: 10.1016/s0955-0674(00)00202-7. [DOI] [PubMed] [Google Scholar]
- 25.Sauve AA, Celic I, Avalos J, Deng H, Boeke JD, Schramm VL. Chemistry of gene silencing: the mechanism of NAD+-dependent deacetylation reactions. Biochemistry. 2001;40:15456–15463. doi: 10.1021/bi011858j. [DOI] [PubMed] [Google Scholar]
- 26.Schmidt MT, Smith BC, Jackson MD, Denu JM. Coenzyme specificity of Sir2 protein deacetylases: implications for physiological regulation. J. Biol. Chem. 2004;279:40122–40129. doi: 10.1074/jbc.M407484200. [DOI] [PubMed] [Google Scholar]
- 27.Borra MT, Langer MR, Slama JT, Denu JM. Substrate specificity and kinetic mechanism of the Sir2 family of NAD+-dependent histone/protein deacetylases. Biochemistry. 2004;43:9877–9887. doi: 10.1021/bi049592e. [DOI] [PubMed] [Google Scholar]
- 28.Sauve AA. Pharmaceutical strategies for activating sirtuins. Curr. Pharm. Des. 2009;15:45–56. doi: 10.2174/138161209787185797. [DOI] [PubMed] [Google Scholar]
- 29.Smith BC, Hallows WC, Denu JM. Mechanisms and molecular probes of sirtuins. Chem. Biol. 2008;15:1002–1013. doi: 10.1016/j.chembiol.2008.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sauve AA, Wolberger C, Schramm VL, Boeke JD. The biochemistry of sirtuins. Annu. Rev. Biochem. 2006;75:435–465. doi: 10.1146/annurev.biochem.74.082803.133500. [DOI] [PubMed] [Google Scholar]
- 31.Bitterman KJ, Anderson RM, Cohen HY, Latorre-Esteves M, Sinclair DA. Inhibition of silencing and accelerated aging by nicotinamide, a putative negative regulator of yeast sir2 and human SIRT1. J. Biol. Chem. 2002;277:45099–45107. doi: 10.1074/jbc.M205670200. [DOI] [PubMed] [Google Scholar]
- 32.Sauve AA, Moir RD, Schramm VL, Willis IM. Chemical activation of Sir2-dependent silencing by relief of nicotinamide inhibition. Mol. Cell. 2005;17:595–601. doi: 10.1016/j.molcel.2004.12.032. [DOI] [PubMed] [Google Scholar]
- 33.Sauve AA, Schramm VL. Sir2 regulation by nicotinamide results from switching between base exchange and deacetylation chemistry. Biochemistry. 2003;42:9249–9256. doi: 10.1021/bi034959l. [DOI] [PubMed] [Google Scholar]
- 34.Avalos JL, Boeke JD, Wolberger C. Structural basis for the mechanism and regulation of Sir2 enzymes. Mol. Cell. 2004;13:639–648. doi: 10.1016/s1097-2765(04)00082-6. [DOI] [PubMed] [Google Scholar]
- 35.Balan V, Miller GS, Kaplun L, Balan K, Chong ZZ, et al. Life span extension and neuronal cell protection by Drosophila nicotinamidase. J. Biol. Chem. 2008;283:27810–27819. doi: 10.1074/jbc.M804681200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Anderson RM, Bitterman KJ, Wood JG, Medvedik O, Sinclair DA. Nicotinamide and Pnc1 govern lifespan extension by calorie restriction in S. cerevisiae. Nature. 2003;423:181–185. doi: 10.1038/nature01578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gallo CM, Smith DL, Jr, Smith JS. Nicotinamide clearance by Pnc1 directly regulates Sir2-mediated silencing and longevity. Mol. Cell Biol. 2004;24:1301–1312. doi: 10.1128/MCB.24.3.1301-1312.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bieganowski P, Brenner C. Discoveries of nicotinamide riboside as a nutrient and conserved NRK genes establish a Preiss-Handler independent route to NAD+ in fungi and humans. Cell. 2004;117:495–502. doi: 10.1016/s0092-8674(04)00416-7. [DOI] [PubMed] [Google Scholar]
- 39.Belenky P, Racette FG, Bogan KL, McClure JM, Smith JS, Brenner C. Nicotinamide riboside promotes Sir2 silencing and extends lifespan via Nrk and Urh1/Pnp1/Meu1 pathways to NAD+ Cell. 2007;129:473–484. doi: 10.1016/j.cell.2007.03.024. [DOI] [PubMed] [Google Scholar]
- 40.Revollo JR, Grimm AA, Imai S. The NAD biosynthesis pathway mediated by nicotinamide phosphoribosyltransferase regulates Sir2 activity in mammalian cells. J. Biol. Chem. 2004;279:50754–50763. doi: 10.1074/jbc.M408388200. [DOI] [PubMed] [Google Scholar]
- 41.Rongvaux A, Shea RJ, Mulks MH, Gigot D, Urbain J, et al. Pre-B-cell colony-enhancing factor, whose expression is up-regulated in activated lymphocytes, is a nicotinamide phosphoribosyltransferase, a cytosolic enzyme involved in NAD biosynthesis. Eur. J. Immunol. 2002;32:3225–3234. doi: 10.1002/1521-4141(200211)32:11<3225::AID-IMMU3225>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 42.Berger F, Lau C, Dahlmann M, Ziegler M. Subcellular compartmentation and differential catalytic properties of the three human nicotinamide mononucleotide adenylyltransferase isoforms. J. Biol. Chem. 2005;280:36334–36341. doi: 10.1074/jbc.M508660200. [DOI] [PubMed] [Google Scholar]
- 43.Yang H, Lavu S, Sinclair DA. Nampt/PBEF/Visfatin: a regulator of mammalian health and longevity? Exp. Gerontol. 2006;41:718–726. doi: 10.1016/j.exger.2006.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yang H, Yang T, Baur JA, Perez E, Matsui T, et al. Nutrient-sensitive mitochondrial NAD+ levels dictate cell survival. Cell. 2007;130:1095–1107. doi: 10.1016/j.cell.2007.07.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhang T, Berrocal JG, Frizzell KM, Gamble MJ, Dumond ME, et al. Enzymes in the NAD+ salvage pathway regulate SIRT1 activity at target gene promoters. J. Biol. Chem. 2009;284:20408–20417. doi: 10.1074/jbc.M109.016469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fukuhara A, Matsuda M, Nishizawa M, Segawa K, Tanaka M, et al. Visfatin: a protein secreted by visceral fat that mimics the effects of insulin. Science. 2005;307:426–430. doi: 10.1126/science.1097243. [DOI] [PubMed] [Google Scholar]
- 47.Imai S. The NAD world: a new systemic regulatory network for metabolism and aging—Sirt1, systemic NAD biosynthesis, and their importance. Cell Biochem. Biophys. 2009;53:65–74. doi: 10.1007/s12013-008-9041-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bogan KL, Brenner C. Nicotinic acid, nicotinamide, and nicotinamide riboside: a molecular evaluation of NAD+ precursor vitamins in human nutrition. Annu. Rev. Nutr. 2008;28:115–130. doi: 10.1146/annurev.nutr.28.061807.155443. [DOI] [PubMed] [Google Scholar]
- 49.Belenky P, Christensen KC, Gazzaniga F, Pletnev AA, Brenner C. Nicotinamide riboside and nicotinic acid riboside salvage in fungi and mammals. Quantitative basis for Urh1 and purine nucleoside phosphorylase function in NAD+ metabolism. J. Biol. Chem. 2009;284:158–164. doi: 10.1074/jbc.M807976200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rutter J, Reick M, Wu LC, McKnight SL. Regulation of clock and NPAS2 DNA binding by the redox state of NAD cofactors. Science. 2001;293:510–514. doi: 10.1126/science.1060698. [DOI] [PubMed] [Google Scholar]
- 51.Ramsey KM, Yoshino J, Brace CS, Abrassart D, Kobayashi Y, et al. Circadian clock feedback cycle through NAMPT-mediated NAD+ biosynthesis. Science. 2009;324:651–654. doi: 10.1126/science.1171641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Jung-Hynes B, Ahmad N. SIRT1 controls circadian clock circuitry and promotes cell survival: a connection with age-related neoplasms. FASEB J. 2009;23:2803–2809. doi: 10.1096/fj.09-129148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Nakahata Y, Sahar S, Astarita G, Kaluzova M, Sassone-Corsi P. Circadian control of the NAD+ salvage pathway by CLOCK-SIRT1. Science. 2009;324:654–657. doi: 10.1126/science.1170803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Aksoy P, Escande C, White TA, Thompson M, Soares S, et al. Regulation of SIRT 1 mediated NAD dependent deacetylation: a novel role for the multifunctional enzyme CD38. Biochem. Biophys. Res. Commun. 2006;349:353–359. doi: 10.1016/j.bbrc.2006.08.066. [DOI] [PubMed] [Google Scholar]
- 55.Lund FE. Signaling properties of CD38 in the mouse immune system: enzyme-dependent and -independent roles in immunity. Mol. Med. 2006;12:328–333. doi: 10.2119/2006-00099.Lund. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Liou GG, Tanny JC, Kruger RG, Walz T, Moazed D. Assembly of the SIR complex and its regulation by O-acetyl-ADP-ribose, a product of NAD-dependent histone deacetylation. Cell. 2005;121:515–527. doi: 10.1016/j.cell.2005.03.035. [DOI] [PubMed] [Google Scholar]
- 57.Grubisha O, Rafty LA, Takanishi CL, Xu X, Tong L, et al. Metabolite of SIR2 reaction modulates TRPM2 ion channel. J. Biol. Chem. 2006;281:14057–14065. doi: 10.1074/jbc.M513741200. [DOI] [PubMed] [Google Scholar]
- 58.Rafty LA, Schmidt MT, Perraud AL, Scharenberg AM, Denu JM. Analysis of O-acetyl-ADP-ribose as a target for nudix ADP-ribose hydrolases. J. Biol. Chem. 2002;277:47114–47122. doi: 10.1074/jbc.M208997200. [DOI] [PubMed] [Google Scholar]
- 59.Michan S, Sinclair D. Sirtuins in mammals: insights into their biological function. Biochem. J. 2007;404:1–13. doi: 10.1042/BJ20070140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.North BJ, Verdin E. Interphase nucleo-cytoplasmic shuttling and localization of SIRT2 during mitosis. PLoS ONE. 2007;2:e784. doi: 10.1371/journal.pone.0000784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tanno M, Sakamoto J, Miura T, Shimamoto K, Horio Y. Nucleocytoplasmic shuttling of the NAD+-dependent histone deacetylase SIRT1. J. Biol. Chem. 2007;282:6823–6832. doi: 10.1074/jbc.M609554200. [DOI] [PubMed] [Google Scholar]
- 62.Cohen HY, Lavu S, Bitterman KJ, Hekking B, Imahiyerobo TA, et al. Acetylation of the C terminus of Ku70 by CBP and PCAF controls bax-mediated apoptosis. Mol. Cell. 2004;13:627–638. doi: 10.1016/s1097-2765(04)00094-2. [DOI] [PubMed] [Google Scholar]
- 63.Nakagawa T, Lomb DJ, Haigis MC, Guarente L. SIRT5 deacetylates carbamoyl phosphate synthetase 1 and regulates the urea cycle. Cell. 2009;137:560–570. doi: 10.1016/j.cell.2009.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Schwer B, North BJ, Frye RA, Ott M, Verdin E. The human silent information regulator (Sir)2 homologue hSIRT3 is a mitochondrial nicotinamide adenine dinucleotide-dependent deacetylase. J. Cell Biol. 2002;158:647–657. doi: 10.1083/jcb.200205057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Schwer B, Bunkenborg J, Verdin RO, Andersen JS, Verdin E. Reversible lysine acetylation controls the activity of the mitochondrial enzyme acetyl-CoA synthetase 2. Proc. Natl. Acad. Sci. USA. 2006;103:10224–10229. doi: 10.1073/pnas.0603968103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hallows WC, Albaugh BN, Denu JM. Where in the cell is SIRT3? Functional localization of an NAD+-dependent protein deacetylase. Biochem. J. 2008;411:e11–e13. doi: 10.1042/BJ20080336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Nakamura Y, Ogura M, Tanaka D, Inagaki N. Localization of mouse mitochondrial SIRT proteins: shift of SIRT3 to nucleus by co-expression with SIRT5. Biochem. Biophys. Res. Commun. 2008;366:174–179. doi: 10.1016/j.bbrc.2007.11.122. [DOI] [PubMed] [Google Scholar]
- 68.Scher MB, Vaquero A, Reinberg D. SirT3 is a nuclear NAD+-dependent histone deacetylase that translocates to the mitochondria upon cellular stress. Genes Dev. 2007;21:920–928. doi: 10.1101/gad.1527307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Jin L, Wei W, Jiang Y, Peng H, Cai J, et al. Crystal structures of human SIRT3 displaying substrate-induced conformational changes. J. Biol. Chem. 2009;284:24394–24405. doi: 10.1074/jbc.M109.014928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sinclair DA. Toward a unified theory of caloric restriction and longevity regulation. Mech. Ageing Dev. 2005;126:987–1002. doi: 10.1016/j.mad.2005.03.019. [DOI] [PubMed] [Google Scholar]
- 71.Harper JM, Leathers CW, Austad SN. Does caloric restriction extend life in wild mice? Aging Cell. 2006;5:441–449. doi: 10.1111/j.1474-9726.2006.00236.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Cooper TM, Mockett RJ, Sohal BH, Sohal RS, Orr WC. Effect of caloric restriction on life span of the housefly, Musca domestica. FASEB J. 2004;18:1591–1593. doi: 10.1096/fj.03-1464fje. [DOI] [PubMed] [Google Scholar]
- 73.Guarente L, Kenyon C. Genetic pathways that regulate ageing in model organisms. Nature. 2000;408:255–262. doi: 10.1038/35041700. [DOI] [PubMed] [Google Scholar]
- 74.Lin SJ, Kaeberlein M, Andalis AA, Sturtz LA, Defossez PA, et al. Calorie restriction extends Saccharomyces cerevisiae lifespan by increasing respiration. Nature. 2002;418:344–348. doi: 10.1038/nature00829. [DOI] [PubMed] [Google Scholar]
- 75.Lamming DW, Latorre-Esteves M, Medvedik O, Wong SN, Tsang FA, et al. HST2 mediates SIR2-independent life-span extension by calorie restriction. Science. 2005;309:1861–1864. doi: 10.1126/science.1113611. [DOI] [PubMed] [Google Scholar]
- 76.Kaeberlein M, Hu D, Kerr EO, Tsuchiya M, Westman EA, et al. Increased life span due to calorie restriction in respiratory-deficient yeast. PLoS Genet. 2005;1:e69. doi: 10.1371/journal.pgen.0010069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kaeberlein M, Kirkland KT, Fields S, Kennedy BK. Sir2-independent life span extension by calorie restriction in yeast. PLoS Biol. 2004;2:E296. doi: 10.1371/journal.pbio.0020296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Fabrizio P, Gattazzo C, Battistella L, Wei M, Cheng C, et al. Sir2 blocks extreme life-span extension. Cell. 2005;123:655–667. doi: 10.1016/j.cell.2005.08.042. [DOI] [PubMed] [Google Scholar]
- 79.Bishop NA, Guarente L. Two neurons mediate diet-restriction-induced longevity in C. elegans. Nature. 2007;447:545–549. doi: 10.1038/nature05904. [DOI] [PubMed] [Google Scholar]
- 80.Lin SJ, Ford E, Haigis M, Liszt G, Guarente L. Calorie restriction extends yeast life span by lowering the level of NADH. Genes Dev. 2004;18:12–16. doi: 10.1101/gad.1164804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Medvedik O, Lamming DW, Kim KD, Sinclair DA. MSN2 and MSN4 link calorie restriction and TOR to sirtuin-mediated lifespan extension in Saccharomyces cerevisiae. PLoS Biol. 2007;5:e261. doi: 10.1371/journal.pbio.0050261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Anderson RM, Bitterman KJ, Wood JG, Medvedik O, Cohen H, et al. Manipulation of a nuclear NAD+ salvage pathway delays aging without altering steady-state NAD+ levels. J. Biol. Chem. 2002;277:18881–18890. doi: 10.1074/jbc.M111773200. [DOI] [PubMed] [Google Scholar]
- 83.Masoro EJ. Caloric restriction and aging: an update. Exp. Gerontol. 2000;35:299–305. doi: 10.1016/s0531-5565(00)00084-x. [DOI] [PubMed] [Google Scholar]
- 84.Rattan SI. Aging, anti-aging, and hormesis. Mech. Ageing Dev. 2004;125:285–289. doi: 10.1016/j.mad.2004.01.006. [DOI] [PubMed] [Google Scholar]
- 85.Sinclair DA, Howitz KT. Dietary Restriction, Hormesis, and Small Molecule Mimetics. Amsterdam/Boston: Elsevier; 2006. 660 pp. [Google Scholar]
- 86.Qin W, Yang T, Ho L, Zhao Z, Wang J, et al. Neuronal SIRT1 activation as a novel mechanism underlying the prevention of Alzheimer disease amyloid neuropathology by calorie restriction. J. Biol. Chem. 2006;281:21745–21754. doi: 10.1074/jbc.M602909200. [DOI] [PubMed] [Google Scholar]
- 87.Canto C, Gerhart-Hines Z, Feige JN, Lagouge M, Noriega L, et al. AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature. 2009;458:1056–1060. doi: 10.1038/nature07813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Cohen HY, Miller C, Bitterman KJ, Wall NR, Hekking B, et al. Calorie restriction promotes mammalian cell survival by inducing the SIRT1 deacetylase. Science. 2004;305:390–392. doi: 10.1126/science.1099196. [DOI] [PubMed] [Google Scholar]
- 89.Chen D, Bruno J, Easlon E, Lin SJ, Cheng HL, et al. Tissue-specific regulation of SIRT1 by calorie restriction. Genes Dev. 2008;22:1753–1757. doi: 10.1101/gad.1650608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Chen D, Steele AD, Lindquist S, Guarente L. Increase in activity during calorie restriction requires Sirt1. Science. 2005;310:1641. doi: 10.1126/science.1118357. [DOI] [PubMed] [Google Scholar]
- 91.Bordone L, Cohen D, Robinson A, Motta MC, van Veen E, et al. SIRT1 transgenic mice show phenotypes resembling calorie restriction. Aging Cell. 2007;6:759–767. doi: 10.1111/j.1474-9726.2007.00335.x. [DOI] [PubMed] [Google Scholar]
- 92.Banks AS, Kon N, Knight C, Matsumoto M, Gutierrez-Juarez R, et al. SirT1 gain of function increases energy efficiency and prevents diabetes in mice. Cell Metab. 2008;8:333–341. doi: 10.1016/j.cmet.2008.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Pfluger PT, Herranz D, Velasco-Miguel S, Serrano M, Tschop MH. Sirt1 protects against high-fat diet-induced metabolic damage. Proc. Natl. Acad. Sci. USA. 2008;105:9793–9798. doi: 10.1073/pnas.0802917105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Milne JC, Lambert PD, Schenk S, Carney DP, Smith JJ, et al. Small molecule activators of SIRT1 as therapeutics for the treatment of type 2 diabetes. Nature. 2007;450:712–716. doi: 10.1038/nature06261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Baur JA, Pearson KJ, Price NL, Jamieson HA, Lerin C, et al. Resveratrol improves health and survival of mice on a high-calorie diet. Nature. 2006;444:337–342. doi: 10.1038/nature05354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Lagouge M, Argmann C, Gerhart-Hines Z, Meziane H, Lerin C, et al. Resveratrol improves mitochondrial function and protects against metabolic disease by activating SIRT1 and PGC-1α. Cell. 2006;127:1109–1122. doi: 10.1016/j.cell.2006.11.013. [DOI] [PubMed] [Google Scholar]
- 97.Boily G, Seifert EL, Bevilacqua L, He XH, Sabourin G, et al. SirT1 regulates energy metabolism and response to caloric restriction in mice. PLoS One. 2008;3:e1759. doi: 10.1371/journal.pone.0001759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Li Y, Xu W, McBurney MW, Longo VD. SirT1 inhibition reduces IGF-I/IRS-2/Ras/ERK1/2 signaling and protects neurons. Cell Metab. 2008;8:38–48. doi: 10.1016/j.cmet.2008.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.McBurney MW, Yang X, Jardine K, Hixon M, Boekelheide K, et al. The mammalian SIR2α protein has a role in embryogenesis and gametogenesis. Mol. Cell Biol. 2003;23:38–54. doi: 10.1128/MCB.23.1.38-54.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Wang C, Chen L, Hou X, Li Z, Kabra N, et al. Interactions between E2F1 and SirT1 regulate apoptotic response to DNA damage. Nat. Cell Biol. 2006;8:1025–1031. doi: 10.1038/ncb1468. [DOI] [PubMed] [Google Scholar]
- 101.Nemoto S, Fergusson MM, Finkel T. Nutrient availability regulates SIRT1 through a forkhead-dependent pathway. Science. 2004;306:2105–2108. doi: 10.1126/science.1101731. [DOI] [PubMed] [Google Scholar]
- 102.Chen WY, Wang DH, Yen RC, Luo J, Gu W, Baylin SB. Tumor suppressor HIC1 directly regulates SIRT1 to modulate p53-dependent DNA-damage responses. Cell. 2005;123:437–448. doi: 10.1016/j.cell.2005.08.011. [DOI] [PubMed] [Google Scholar]
- 103.Zhang Q, Wang SY, Fleuriel C, Leprince D, Rocheleau JV, et al. Metabolic regulation of SIRT1 transcription via a HIC1:CtBP corepressor complex. Proc. Natl. Acad. Sci. USA. 2007;104:829–833. doi: 10.1073/pnas.0610590104. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 104.Rane S, He M, Sayed D, Vashistha H, Malhotra A, et al. Downregulation of miR-199a derepresses hypoxia-inducible factor-1α and sirtuin 1 and recapitulates hypoxia preconditioning in cardiac myocytes. Circ. Res. 2009;104:879–886. doi: 10.1161/CIRCRESAHA.108.193102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Yamakuchi M, Lowenstein CJ. MiR-34, SIRT1 and p53: the feedback loop. Cell Cycle. 2009;8:712–715. doi: 10.4161/cc.8.5.7753. [DOI] [PubMed] [Google Scholar]
- 106.Pogribny IP, Muskhelishvili L, Tryndyak VP, Beland FA. The tumor-promoting activity of 2-acetylaminofluorene is associated with disruption of the p53 signaling pathway and the balance between apoptosis and cell proliferation. Toxicol. Appl. Pharmacol. 2009;235:305–311. doi: 10.1016/j.taap.2008.12.021. [DOI] [PubMed] [Google Scholar]
- 107.Fujita Y, Kojima K, Hamada N, Ohhashi R, Akao Y, et al. Effects of miR-34a on cell growth and chemoresistance in prostate cancer PC3 cells. Biochem. Biophys. Res. Commun. 2008;377:114–119. doi: 10.1016/j.bbrc.2008.09.086. [DOI] [PubMed] [Google Scholar]
- 108.Yamakuchi M, Ferlito M, Lowenstein CJ. miR-34a repression of SIRT1 regulates apoptosis. Proc. Natl. Acad. Sci. USA. 2008;105:13421–13426. doi: 10.1073/pnas.0801613105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Yuan J, Minter-Dykhouse K, Lou Z. A c-Myc-SIRT1 feedback loop regulates cell growth and transformation. J. Cell Biol. 2009;185:203–211. doi: 10.1083/jcb.200809167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Solomon JM, Pasupuleti R, Xu L, McDonagh T, Curtis R, et al. Inhibition of SIRT1 catalytic activity increases p53 acetylation but does not alter cell survival following DNA damage. Mol. Cell Biol. 2006;26:28–38. doi: 10.1128/MCB.26.1.28-38.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Mai A, Massa S, Lavu S, Pezzi R, Simeoni S, et al. Design, synthesis, and biological evaluation of sirtinol analogues as class III histone/protein deacetylase (sirtuin) inhibitors. J. Med. Chem. 2005;48:7789–7795. doi: 10.1021/jm050100l. [DOI] [PubMed] [Google Scholar]
- 112.Smith JJ, Kenney RD, Gagne DJ, Frushour BP, Ladd W, et al. Small molecule activators of SIRT1 replicate signaling pathways triggered by calorie restriction in vivo. BMC Syst. Biol. 2009;3:31. doi: 10.1186/1752-0509-3-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Howitz KT, Bitterman KJ, Cohen HY, Lamming DW, Lavu S, et al. Small molecule activators of sirtuins extend Saccharomyces cerevisiae lifespan. Nature. 2003;425:191–196. doi: 10.1038/nature01960. [DOI] [PubMed] [Google Scholar]
- 114.Lavu S, Boss O, Elliott PJ, Lambert PD. Sirtuins—novel therapeutic targets to treat age-associated diseases. Nat. Rev. Drug Discov. 2008;7:841–853. doi: 10.1038/nrd2665. [DOI] [PubMed] [Google Scholar]
- 115.Bauer JH, Goupil S, Garber GB, Helfand SL. An accelerated assay for the identification of lifespan-extending interventions in Drosophila melanogaster. Proc. Natl. Acad. Sci. USA. 2004;101:12980–12985. doi: 10.1073/pnas.0403493101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Wood JG, Rogina B, Lavu S, Howitz K, Helfand SL, et al. Sirtuin activators mimic caloric restriction and delay ageing in metazoans. Nature. 2004;430:686–689. doi: 10.1038/nature02789. [DOI] [PubMed] [Google Scholar]
- 117.Jarolim S, Millen J, Heeren G, Laun P, Goldfarb DS, Breitenbach M. A novel assay for replicative lifespan in Saccharomyces cerevisiae. FEMS Yeast Res. 2004;5:169–177. doi: 10.1016/j.femsyr.2004.06.015. [DOI] [PubMed] [Google Scholar]
- 118.Viswanathan M, Kim SK, Berdichevsky A, Guarente L. A role for SIR-2.1 regulation of ER stress response genes in determining C. elegans life span. Dev. Cell. 2005;9:605–615. doi: 10.1016/j.devcel.2005.09.017. [DOI] [PubMed] [Google Scholar]
- 119.Bass TM, Weinkove D, Houthoofd K, Gems D, Partridge L. Effects of resveratrol on lifespan in Drosophila melanogaster and Caenorhabditis elegans. Mech. Ageing Dev. 2007;128:546–552. doi: 10.1016/j.mad.2007.07.007. [DOI] [PubMed] [Google Scholar]
- 120.Kaeberlein M, McDonagh T, Heltweg B, Hixon J, Westman EA, et al. Substrate-specific activation of sirtuins by resveratrol. J. Biol. Chem. 2005;280:17038–17045. doi: 10.1074/jbc.M500655200. [DOI] [PubMed] [Google Scholar]
- 121.Nayagam VM, Wang X, Tan YC, Poulsen A, Goh KC, et al. SIRT1 modulating compounds from high-throughput screening as anti-inflammatory and insulin-sensitizing agents. J. Biomol. Screen. 2006;11:959–967. doi: 10.1177/1087057106294710. [DOI] [PubMed] [Google Scholar]
- 122.Galonek H, Miller C, Israelian K, Ribish S, Lynch AV, et al. Enzymatic and cellular activity of small molecule activators of SIRT1; Presented at NAD Metab. Signal., FASEB J. Conf.; Carefree, Ariz.. 2009. [Google Scholar]
- 123.Wang RH, Zheng Y, Kim HS, Xu X, Cao L, et al. Interplay among BRCA1, SIRT1, and Survivin during BRCA1-associated tumorigenesis. Mol. Cell. 2008;32:11–20. doi: 10.1016/j.molcel.2008.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Sun C, Zhang F, Ge X, Yan T, Chen X, et al. SIRT1 improves insulin sensitivity under insulin-resistant conditions by repressing PTP1B. Cell Metab. 2007;6:307–319. doi: 10.1016/j.cmet.2007.08.014. [DOI] [PubMed] [Google Scholar]
- 125.Boily G, He XH, Pearce B, Jardine K, McBurney MW. SirT1-null mice develop tumors at normal rates but are poorly protected by resveratrol. Oncogene. 2009;28:2882–2893. doi: 10.1038/onc.2009.147. [DOI] [PubMed] [Google Scholar]
- 126.Kim EJ, Kho JH, Kang MR, Um SJ. Active regulator of SIRT1 cooperates with SIRT1 and facilitates suppression of p53 activity. Mol. Cell. 2007;28:277–290. doi: 10.1016/j.molcel.2007.08.030. [DOI] [PubMed] [Google Scholar]
- 127.Hasegawa K, Yoshikawa K. Necdin regulates p53 acetylation via Sirtuin1 to modulate DNA damage response in cortical neurons. J. Neurosci. 2008;28:8772–8784. doi: 10.1523/JNEUROSCI.3052-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Zhao W, Kruse JP, Tang Y, Jung SY, Qin J, Gu W. Negative regulation of the deacetylase SIRT1 by DBC1. Nature. 2008;451:587–590. doi: 10.1038/nature06515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Kim JE, Chen J, Lou Z. DBC1 is a negative regulator of SIRT1. Nature. 2008;451:583–586. doi: 10.1038/nature06500. [DOI] [PubMed] [Google Scholar]
- 130.Sasaki T, Maier B, Koclega KD, Chruszcz M, Gluba W, et al. Phosphorylation regulates SIRT1 function. PLoS One. 2008;3:e4020. doi: 10.1371/journal.pone.0004020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Puigserver P, Wu Z, Park CW, Graves R, Wright M, Spiegelman BM. A cold-inducible coactivator of nuclear receptors linked to adaptive thermogenesis. Cell. 1998;92:829–839. doi: 10.1016/s0092-8674(00)81410-5. [DOI] [PubMed] [Google Scholar]
- 132.Rodgers JT, Lerin C, Haas W, Gygi SP, Spiegelman BM, Puigserver P. Nutrient control of glucose homeostasis through a complex of PGC-1α and SIRT1. Nature. 2005;434:113–118. doi: 10.1038/nature03354. [DOI] [PubMed] [Google Scholar]
- 133.Rodgers JT, Lerin C, Gerhart-Hines Z, Puigserver P. Metabolic adaptations through the PGC-1α and SIRT1 pathways. FEBS Lett. 2008;582:46–53. doi: 10.1016/j.febslet.2007.11.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Purushotham A, Schug TT, Xu Q, Surapureddi S, Guo X, Li X. Hepatocyte-specific deletion of SIRT1 alters fatty acid metabolism and results in hepatic steatosis and inflammation. Cell Metab. 2009;9:327–328. doi: 10.1016/j.cmet.2009.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Liu Y, Dentin R, Chen D, Hedrick S, Ravnskjaer K, et al. A fasting inducible switch modulates gluconeogenesis via activator/coactivator exchange. Nature. 2008;456:269–273. doi: 10.1038/nature07349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Canto C, Auwerx J. PGC-1α, SIRT1 and AMPK, an energy sensing network that controls energy expenditure. Curr. Opin. Lipidol. 2009;20:98–105. doi: 10.1097/MOL.0b013e328328d0a4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Canto C, Auwerx J. Glucose restriction: longevity SIRTainly, but without building muscle? Dev. Cell. 2008;14:642–644. doi: 10.1016/j.devcel.2008.04.012. [DOI] [PubMed] [Google Scholar]
- 138.Feige JN, Lagouge M, Canto C, Strehle A, Houten SM, et al. Specific SIRT1 activation mimics low energy levels and protects against diet-induced metabolic disorders by enhancing fat oxidation. Cell Metab. 2008;8:347–358. doi: 10.1016/j.cmet.2008.08.017. [DOI] [PubMed] [Google Scholar]
- 139.Kim D, Nguyen MD, Dobbin MM, Fischer A, Sananbenesi F, et al. SIRT1 deacetylase protects against neurodegeneration in models for Alzheimer’s disease and amyotrophic lateral sclerosis. EMBO J. 2007;26:3169–3179. doi: 10.1038/sj.emboj.7601758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Gerhart-Hines Z, Rodgers JT, Bare O, Lerin C, Kim SH, et al. Metabolic control of muscle mitochondrial function and fatty acid oxidation through SIRT1/PGC-1α. EMBO J. 2007;26:1913–1923. doi: 10.1038/sj.emboj.7601633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Ajmo JM, Liang X, Rogers CQ, Pennock B, You M. Resveratrol alleviates alcoholic fatty liver in mice. Am. J. Physiol. Gastrointest. Liver Physiol. 2008;295:G833–G842. doi: 10.1152/ajpgi.90358.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.You M, Liang X, Ajmo JM, Ness GC. Involvement of mammalian sirtuin 1 in the action of ethanol in the liver. Am. J. Physiol. Gastrointest. Liver Physiol. 2008;294:G892–G898. doi: 10.1152/ajpgi.00575.2007. [DOI] [PubMed] [Google Scholar]
- 143.Li X, Zhang S, Blander G, Tse JG, Krieger M, Guarente L. SIRT1 deacetylates and positively regulates the nuclear receptor LXR. Mol. Cell. 2007;28:91–106. doi: 10.1016/j.molcel.2007.07.032. [DOI] [PubMed] [Google Scholar]
- 144.Lee IH, Cao L, Mostoslavsky R, Lombard DB, Liu J, et al. A role for the NAD-dependent deacetylase Sirt1 in the regulation of autophagy. Proc. Natl. Acad. Sci. USA. 2008;105:3374–3379. doi: 10.1073/pnas.0712145105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Moynihan KA, Grimm AA, Plueger MM, Bernal-Mizrachi E, Ford E, et al. Increased dosage of mammalian Sir2 in pancreatic β cells enhances glucose-stimulated insulin secretion in mice. Cell Metab. 2005;2:105–117. doi: 10.1016/j.cmet.2005.07.001. [DOI] [PubMed] [Google Scholar]
- 146.Bordone L, Motta MC, Picard F, Robinson A, Jhala US, et al. Sirt1 regulates insulin secretion by repressing UCP2 in pancreatic β cells. PLoS Biol. 2006;4:e31. doi: 10.1371/journal.pbio.0040031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Ramsey KM, Mills KF, Satoh A, Imai S. Age-associated loss of Sirt1-mediated enhancement of glucose-stimulated insulin secretion in β cell-specific Sirt1-overexpressing (BESTO) mice. Aging Cell. 2008;7:78–88. doi: 10.1111/j.1474-9726.2007.00355.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Picard F, Kurtev M, Chung N, Topark-Ngarm A, Senawong T, et al. Sirt1 promotes fat mobilization in white adipocytes by repressing PPAR-γ. Nature. 2004;429:771–776. doi: 10.1038/nature02583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Qiao L, Shao J. SIRT1 regulates adiponectin gene expression through Foxo1-C/enhancer-binding protein α transcriptional complex. J. Biol. Chem. 2006;281:39915–39924. doi: 10.1074/jbc.M607215200. [DOI] [PubMed] [Google Scholar]
- 150.Wallace DC. A mitochondrial paradigm of metabolic and degenerative diseases, aging, and cancer: a dawn for evolutionary medicine. Annu. Rev. Genet. 2005;39:359–407. doi: 10.1146/annurev.genet.39.110304.095751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Lowell BB, Shulman GI. Mitochondrial dysfunction and type 2 diabetes. Science. 2005;307:384–387. doi: 10.1126/science.1104343. [DOI] [PubMed] [Google Scholar]
- 152.Onyango P, Celic I, McCaffery JM, Boeke JD, Feinberg AP. SIRT3, a human SIR2 homologue, is an NAD-dependent deacetylase localized to mitochondria. Proc. Natl. Acad. Sci. USA. 2002;99:13653–13658. doi: 10.1073/pnas.222538099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Shi T, Wang F, Stieren E, Tong Q. SIRT3, a mitochondrial sirtuin deacetylase, regulates mitochondrial function and thermogenesis in brown adipocytes. J. Biol. Chem. 2005;280:13560–13567. doi: 10.1074/jbc.M414670200. [DOI] [PubMed] [Google Scholar]
- 154.Lombard DB, Alt FW, Cheng HL, Bunkenborg J, Streeper RS, et al. Mammalian Sir2 homolog SIRT3 regulates global mitochondrial lysine acetylation. Mol. Cell Biol. 2007;27:8807–8814. doi: 10.1128/MCB.01636-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Hallows WC, Lee S, Denu JM. Sirtuins deacetylate and activate mammalian acetyl-CoA synthetases. Proc. Natl. Acad. Sci. USA. 2006;103:10230–10235. doi: 10.1073/pnas.0604392103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Schlicker C, Gertz M, Papatheodorou P, Kachholz B, Becker CF, Steegborn C. Substrates and regulation mechanisms for the human mitochondrial sirtuins Sirt3 and Sirt5. J. Mol. Biol. 2008;382:790–801. doi: 10.1016/j.jmb.2008.07.048. [DOI] [PubMed] [Google Scholar]
- 157.Ahn BH, Kim HS, Song S, Lee IH, Liu J, et al. A role for the mitochondrial deacetylase Sirt3 in regulating energy homeostasis. Proc. Natl. Acad. Sci. USA. 2008;105:14447–14452. doi: 10.1073/pnas.0803790105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Jin L, Galonek H, Israelian K, Choy W, Morrison M, et al. Biochemical characterization, localization, and tissue distribution of the longer form of mouse SIRT3. Protein Sci. 2009;18:514–525. doi: 10.1002/pro.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Bellizzi D, Dato S, Cavalcante P, Covello G, Di Cianni F, et al. Characterization of a bidirectional promoter shared between two human genes related to aging: SIRT3 and PSMD13. Genomics. 2007;89:143–150. doi: 10.1016/j.ygeno.2006.09.004. [DOI] [PubMed] [Google Scholar]
- 160.Bellizzi D, Rose G, Cavalcante P, Covello G, Dato S, et al. A novel VNTR enhancer within the SIRT3 gene, a human homologue of SIR2, is associated with survival at oldest ages. Genomics. 2005;85:258–263. doi: 10.1016/j.ygeno.2004.11.003. [DOI] [PubMed] [Google Scholar]
- 161.Ahuja N, Schwer B, Carobbio S, Waltregny D, North BJ, et al. Regulation of insulin secretion by SIRT4, a mitochondrial ADP-ribosyltransferase. J. Biol. Chem. 2007;282:33583–33592. doi: 10.1074/jbc.M705488200. [DOI] [PubMed] [Google Scholar]
- 162.Stanley CA. Hyperinsulinism/hyperammonemia syndrome: insights into the regulatory role of glutamate dehydrogenase in ammonia metabolism. Mol. Genet. Metab. 2004;81 Suppl. 1:S45–S51. doi: 10.1016/j.ymgme.2003.10.013. [DOI] [PubMed] [Google Scholar]
- 163.Sundaresan NR, Samant SA, Pillai VB, Rajamohan SB, Gupta MP. SIRT3 is a stress-responsive deacetylase in cardiomyocytes that protects cells from stress-mediated cell death by deacetylation of Ku70. Mol. Cell Biol. 2008;28:6384–6401. doi: 10.1128/MCB.00426-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Palacios OM, Carmona JJ, Michan S, Chen K, Manabe Y, et al. Diet and exercise signals regulate SIRT3 and activate AMPK and PGC-1α in skeletal muscle. Aging. 2009 doi: 10.18632/aging.100075. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Brooks CL, Gu W. p53 Activation: a case against Sir. Cancer Cell. 2008;13:377–378. doi: 10.1016/j.ccr.2008.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Brooks CL, Gu W. How does SIRT1 affect metabolism, senescence and cancer? Nat. Rev. Cancer. 2009;9:123–128. doi: 10.1038/nrc2562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Luo J, Nikolaev AY, Imai S, Chen D, Su F, et al. Negative control of p53 by Sir2α promotes cell survival under stress. Cell. 2001;107:137–148. doi: 10.1016/s0092-8674(01)00524-4. [DOI] [PubMed] [Google Scholar]
- 168.Vaziri H, Dessain SK, Eaton EN, Imai SI, Frye RA, et al. hSIR2(SIRT1) functions as an NAD-dependent p53 deacetylase. Cell. 2001;107:149–159. doi: 10.1016/s0092-8674(01)00527-x. [DOI] [PubMed] [Google Scholar]
- 169.Firestein R, Blander G, Michan S, Oberdoerffer P, Ogino S, et al. The SIRT1 deacetylase suppresses intestinal tumorigenesis and colon cancer growth. PLoS ONE. 2008;3:e2020. doi: 10.1371/journal.pone.0002020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Oberdoerffer P, Michan S, McVay M, Mostoslavsky R, Vann J, et al. SIRT1 redistribution on chromatin promotes genomic stability but alters gene expression during aging. Cell. 2008;135:907–918. doi: 10.1016/j.cell.2008.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Deng CX. SIRT1, is it a tumor promoter or tumor suppressor? Int. J. Biol. Sci. 2009;5:147–152. doi: 10.7150/ijbs.5.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.O’Hagan HM, Mohammad HP, Baylin SB. Double strand breaks can initiate gene silencing and SIRT1-dependent onset of DNA methylation in an exogenous promoter CpG island. PLoS Genet. 2008;4:e1000155. doi: 10.1371/journal.pgen.1000155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Zhang Y, Zhang M, Dong H, Yong S, Li X, et al. Deacetylation of cortactin by SIRT1 promotes cell migration. Oncogene. 2009;28:445–460. doi: 10.1038/onc.2008.388. [DOI] [PubMed] [Google Scholar]
- 174.Huffman DM, Grizzle WE, Bamman MM, Kim JS, Eltoum IA, et al. SIRT1 is significantly elevated in mouse and human prostate cancer. Cancer Res. 2007;67:6612–6618. doi: 10.1158/0008-5472.CAN-07-0085. [DOI] [PubMed] [Google Scholar]
- 175.Bradbury CA, Khanim FL, Hayden R, Bunce CM, White DA, et al. Histone deacetylases in acute myeloid leukaemia show a distinctive pattern of expression that changes selectively in response to deacetylase inhibitors. Leukemia. 2005;19:1751–1759. doi: 10.1038/sj.leu.2403910. [DOI] [PubMed] [Google Scholar]
- 176.Stunkel W, Peh BK, Tan YC, Nayagam VM, Wang X, et al. Function of the SIRT1 protein deacetylase in cancer. Biotechnol. J. 2007;2:1360–1368. doi: 10.1002/biot.200700087. [DOI] [PubMed] [Google Scholar]
- 177.Hida Y, Kubo Y, Murao K, Arase S. Strong expression of a longevity-related protein, SIRT1, in Bowen’s disease. Arch. Dermatol. Res. 2007;299:103–106. doi: 10.1007/s00403-006-0725-6. [DOI] [PubMed] [Google Scholar]
- 178.Wang RH, Sengupta K, Li C, Kim HS, Cao L, et al. Impaired DNA damage response, genome instability, and tumorigenesis in SIRT1 mutant mice. Cancer Cell. 2008;14:312–323. doi: 10.1016/j.ccr.2008.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Ouaissi M, Sielezneff I, Silvestre R, Sastre B, Bernard JP, et al. High histone deacetylase 7 (HDAC7) expression is significantly associated with adenocarcinomas of the pancreas. Ann. Surg. Oncol. 2008;15:2318–2328. doi: 10.1245/s10434-008-9940-z. [DOI] [PubMed] [Google Scholar]
- 180.North BJ, Verdin E. Mitotic regulation of SIRT2 by cyclin-dependent kinase 1-dependent phosphorylation. J. Biol. Chem. 2007;282:19546–19555. doi: 10.1074/jbc.M702990200. [DOI] [PubMed] [Google Scholar]
- 181.Inoue T, Nakayama Y, Yamada H, Li YC, Yamaguchi S, et al. SIRT2 downregulation confers resistance to microtubule inhibitors by prolonging chronic mitotic arrest. Cell Cycle. 2009;8:1279–1291. doi: 10.4161/cc.8.8.8245. [DOI] [PubMed] [Google Scholar]
- 182.Hiratsuka M, Inoue T, Toda T, Kimura N, Shirayoshi Y, et al. Proteomics-based identification of differentially expressed genes in human gliomas: down-regulation of SIRT2 gene. Biochem. Biophys. Res. Commun. 2003;309:558–566. doi: 10.1016/j.bbrc.2003.08.029. [DOI] [PubMed] [Google Scholar]
- 183.Das C, Lucia MS, Hansen KC, Tyler JK. CBP/p300-mediated acetylation of histone H3 on lysine 56. Nature. 2009;459:113–117. doi: 10.1038/nature07861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Inoue T, Hiratsuka M, Osaki M, Yamada H, Kishimoto I, et al. SIRT2, a tubulin deacetylase, acts to block the entry to chromosome condensation in response to mitotic stress. Oncogene. 2007;26:945–957. doi: 10.1038/sj.onc.1209857. [DOI] [PubMed] [Google Scholar]
- 185.Ashraf N, Zino S, Macintyre A, Kingsmore D, Payne AP, et al. Altered sirtuin expression is associated with node-positive breast cancer. Br. J. Cancer. 2006;95:1056–1061. doi: 10.1038/sj.bjc.6603384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Frye R. “SIRT8” expressed in thyroid cancer is actually SIRT7. Br. J. Cancer. 2002;87:1479. doi: 10.1038/sj.bjc.6600635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Brunet A, Sweeney LB, Sturgill JF, Chua KF, Greer PL, et al. Stress-dependent regulation of FOXO transcription factors by the SIRT1 deacetylase. Science. 2004;303:2011–2015. doi: 10.1126/science.1094637. [DOI] [PubMed] [Google Scholar]
- 188.Ford J, Jiang M, Milner J. Cancer-specific functions of SIRT1 enable human epithelial cancer cell growth and survival. Cancer Res. 2005;65:10457–10463. doi: 10.1158/0008-5472.CAN-05-1923. [DOI] [PubMed] [Google Scholar]
- 189.Lara E, Mai A, Calvanese V, Altucci L, Lopez-Nieva P, et al. Salermide, a Sirtuin inhibitor with a strong cancer-specific proapoptotic effect. Oncogene. 2009;28:781–791. doi: 10.1038/onc.2008.436. [DOI] [PubMed] [Google Scholar]
- 190.Luo J, Nikolaev AY, Imai S, Chen D, Su F, et al. Negative control of p53 by Sir2α promotes cell survival under stress. Cell. 2001;107:137–148. doi: 10.1016/s0092-8674(01)00524-4. [DOI] [PubMed] [Google Scholar]
- 191.Lain S, Hollick JJ, Campbell J, Staples OD, Higgins M, et al. Discovery, in vivo activity, and mechanism of action of a small-molecule p53 activator. Cancer Cell. 2008;13:454–463. doi: 10.1016/j.ccr.2008.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Cheng HL, Mostoslavsky R, Saito S, Manis JP, Gu Y, et al. Developmental defects and p53 hyperacetylation in Sir2 homolog (SIRT1)-deficient mice. Proc. Natl. Acad. Sci. USA. 2003;100:10794–10799. doi: 10.1073/pnas.1934713100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Ota H, Tokunaga E, Chang K, Hikasa M, Iijima K, et al. Sirt1 inhibitor, Sirtinol, induces senescence-like growth arrest with attenuated Ras-MAPK signaling in human cancer cells. Oncogene. 2006;25:176–185. doi: 10.1038/sj.onc.1209049. [DOI] [PubMed] [Google Scholar]
- 194.Ota H, Akishita M, Eto M, Iijima K, Kaneki M, Ouchi Y. Sirt1 modulates premature senescence-like phenotype in human endothelial cells. J. Mol. Cell Cardiol. 2007;43:571–579. doi: 10.1016/j.yjmcc.2007.08.008. [DOI] [PubMed] [Google Scholar]
- 195.Huang J, Gan Q, Han L, Li J, Zhang H, et al. SIRT1 overexpression antagonizes cellular senescence with activated ERK/S6k1 signaling in human diploid fibroblasts. PLoS ONE. 2008;3:e1710. doi: 10.1371/journal.pone.0001710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Vijg J, Maslov AY, Suh Y. Aging: a sirtuin shake-up? Cell. 2008;135:797–798. doi: 10.1016/j.cell.2008.11.008. [DOI] [PubMed] [Google Scholar]
- 197.Potente M, Ghaeni L, Baldessari D, Mostoslavsky R, Rossig L, et al. SIRT1 controls endothelial angiogenic functions during vascular growth. Genes Dev. 2007;21:2644–2658. doi: 10.1101/gad.435107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Potente M, Dimmeler S. Emerging roles of SIRT1 in vascular endothelial homeostasis. Cell Cycle. 2008;7:2117–2122. doi: 10.4161/cc.7.14.6267. [DOI] [PubMed] [Google Scholar]
- 199.Pruitt K, Zinn RL, Ohm JE, McGarvey KM, Kang SH, et al. Inhibition of SIRT1 reactivates silenced cancer genes without loss of promoter DNA hypermethylation. PLoS Genet. 2006;2:e40. doi: 10.1371/journal.pgen.0020040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Yuan Z, Seto E. A functional link between SIRT1 deacetylase and NBS1 in DNA damage response. Cell Cycle. 2007;6:2869–2871. doi: 10.4161/cc.6.23.5026. [DOI] [PubMed] [Google Scholar]
- 201.Anantharaman V, Aravind L. Analysis of DBC1 and its homologs suggests a potential mechanism for regulation of sirtuin domain deacetylases by NAD metabolites. Cell Cycle. 2008;7:1467–1472. doi: 10.4161/cc.7.10.5883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Yeung F, Hoberg JE, Ramsey CS, Keller MD, Jones DR, et al. Modulation of NF-κB-dependent transcription and cell survival by the SIRT1 deacetylase. EMBO J. 2004;23:2369–2380. doi: 10.1038/sj.emboj.7600244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Masoro EJ. Retardation of aging processes by food restriction: an experimental tool. Am. J. Clin. Nutr. 1992;55:1250S–1252S. doi: 10.1093/ajcn/55.6.1250S. [DOI] [PubMed] [Google Scholar]
- 204.Jang M, Cai L, Udeani GO, Slowing KV, Thomas CF, et al. Cancer chemopreventive activity of resveratrol, a natural product derived from grapes. Science. 1997;275:218–220. doi: 10.1126/science.275.5297.218. [DOI] [PubMed] [Google Scholar]
- 205.Baur JA, Sinclair DA. Therapeutic potential of resveratrol: the in vivo evidence. Nat. Rev. Drug Discov. 2006;5:493–506. doi: 10.1038/nrd2060. [DOI] [PubMed] [Google Scholar]
- 206.Jacks T, Remington L, Williams BO, Schmitt EM, Halachmi S, et al. Tumor spectrum analysis in p53-mutant mice. Curr. Biol. 1994;4:1–7. doi: 10.1016/s0960-9822(00)00002-6. [DOI] [PubMed] [Google Scholar]
- 207.Kemp CJ, Wheldon T, Balmain A. p53-deficient mice are extremely susceptible to radiation-induced tumorigenesis. Nat. Genet. 1994;8:66–69. doi: 10.1038/ng0994-66. [DOI] [PubMed] [Google Scholar]
- 208.Donehower LA, Harvey M, Slagle BL, McArthur MJ, Montgomery CA, Jr, et al. Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature. 1992;356:215–221. doi: 10.1038/356215a0. [DOI] [PubMed] [Google Scholar]
- 209.Imai S, Kitano H. Heterochromatin islands and their dynamic reorganization: a hypothesis for three distinctive features of cellular aging. Exp. Gerontol. 1998;33:555–570. doi: 10.1016/s0531-5565(98)00037-0. [DOI] [PubMed] [Google Scholar]
- 210.Kamel C, Abrol M, Jardine K, He X, McBurney MW. Sirt1 fails to affect p53-mediated biological functions. Aging Cell. 2006;5:81–88. doi: 10.1111/j.1474-9726.2006.00191.x. [DOI] [PubMed] [Google Scholar]
- 211.Mills KD, Sinclair DA, Guarente L. MEC1-dependent redistribution of the Sir3 silencing protein from telomeres to DNA double-strand breaks. Cell. 1999;97:609–620. doi: 10.1016/s0092-8674(00)80772-2. [DOI] [PubMed] [Google Scholar]
- 212.McAinsh AD, Scott-Drew S, Murray JA, Jackson SP. DNA damage triggers disruption of telomeric silencing and Mec1p-dependent relocation of Sir3p. Curr. Biol. 1999;9:963–966. doi: 10.1016/s0960-9822(99)80424-2. [DOI] [PubMed] [Google Scholar]
- 213.Martin SG, Laroche T, Suka N, Grunstein M, Gasser SM. Relocalization of telomeric Ku and SIR proteins in response to DNA strand breaks in yeast. Cell. 1999;97:621–633. doi: 10.1016/s0092-8674(00)80773-4. [DOI] [PubMed] [Google Scholar]
- 214.Lee SE, Paques F, Sylvan J, Haber JE. Role of yeast SIR genes and mating type in directing DNA double-strand breaks to homologous and non-homologous repair paths. Curr. Biol. 1999;9:767–770. doi: 10.1016/s0960-9822(99)80339-x. [DOI] [PubMed] [Google Scholar]
- 215.Bennett CB, Snipe JR, Westmoreland JW, Resnick MA. SIR functions are required for the toleration of an unrepaired double-strand break in a dispensable yeast chromosome. Mol. Cell Biol. 2001;21:5359–5373. doi: 10.1128/MCB.21.16.5359-5373.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Tamburini BA, Tyler JK. Localized histone acetylation and deacetylation triggered by the homologous recombination pathway of double-strand DNA repair. Mol. Cell Biol. 2005;25:4903–4913. doi: 10.1128/MCB.25.12.4903-4913.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Yuan Z, Zhang X, Sengupta N, Lane WS, Seto E. SIRT1 regulates the function of the Nijmegen breakage syndrome protein. Mol. Cell. 2007;27:149–162. doi: 10.1016/j.molcel.2007.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Jang M, Cai L, Udeani GO, Slowing KV, Thomas CF, et al. Cancer chemopreventive activity of resveratrol, a natural product derived from grapes. Science. 1997;175:218–220. doi: 10.1126/science.275.5297.218. [DOI] [PubMed] [Google Scholar]
- 219.Narala SR, Allsopp RC, Wells TB, Zhang G, Prasad P, et al. SIRT1 acts as a nutrient-sensitive growth suppressor and its loss is associated with increased AMPK and telomerase activity. Mol. Biol. Cell. 2008;19:1210–1219. doi: 10.1091/mbc.E07-09-0965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Dryden SC, Nahhas FA, Nowak JE, Goustin AS, Tainsky MA. Role for human SIRT2 NAD-dependent deacetylase activity in control of mitotic exit in the cell cycle. Mol. Cell Biol. 2003;23:3173–3185. doi: 10.1128/MCB.23.9.3173-3185.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Zhang Y, Au Q, Zhang M, Barber JR, Ng SC, Zhang B. Identification of a small-molecule SIRT2 inhibitor with selective tumor cytotoxicity. Biochem. Biophys. Res. Commun. 2009;386:729–733. doi: 10.1016/j.bbrc.2009.06.113. [DOI] [PubMed] [Google Scholar]
- 222.Heltweg B, Gatbonton T, Schuler AD, Posakony J, Li H, et al. Antitumor activity of a small-molecule inhibitor of human silent information regulator 2 enzymes. Cancer Res. 2006;66:4368–4377. doi: 10.1158/0008-5472.CAN-05-3617. [DOI] [PubMed] [Google Scholar]
- 223.Allison SJ, Milner J. SIRT3 is pro-apoptotic and participates in distinct basal apoptotic pathways. Cell Cycle. 2007;6:2669–2677. doi: 10.4161/cc.6.21.4866. [DOI] [PubMed] [Google Scholar]
- 224.Mostoslavsky R, Chua KF, Lombard DB, Pang WW, Fischer MR, et al. Genomic instability and aging-like phenotype in the absence of mammalian SIRT6. Cell. 2006;124:315–329. doi: 10.1016/j.cell.2005.11.044. [DOI] [PubMed] [Google Scholar]
- 225.Ma W, Stafford LJ, Li D, Luo J, Li X, et al. GCIP/CCNDBP1, a helix-loop-helix protein, suppresses tumorigenesis. J. Cell Biochem. 2007;100:1376–1386. doi: 10.1002/jcb.21140. [DOI] [PubMed] [Google Scholar]
- 226.Matoba S, Kang JG, Patino WD, Wragg A, Boehm M, et al. p53 regulates mitochondrial respiration. Science. 2006;312:1650–1653. doi: 10.1126/science.1126863. [DOI] [PubMed] [Google Scholar]
- 227.Bakhanashvili M, Grinberg S, Bonda E, Simon AJ, Moshitch-Moshkovitz S, Rahav G. p53 in mitochondria enhances the accuracy of DNA synthesis. Cell Death Differ. 2008;15:1865–1874. doi: 10.1038/cdd.2008.122. [DOI] [PubMed] [Google Scholar]
- 228.Malik S, Wong ND, Franklin SS, Kamath TV, L’Italien GJ, et al. Impact of the metabolic syndrome on mortality from coronary heart disease, cardiovascular disease, and all causes in United States adults. Circulation. 2004;110:1245–1250. doi: 10.1161/01.CIR.0000140677.20606.0E. [DOI] [PubMed] [Google Scholar]
- 229.Heilbronn LK, de Jonge L, Frisard MI, DeLany JP, Larson-Meyer DE, et al. Effect of 6-month calorie restriction on biomarkers of longevity, metabolic adaptation, and oxidative stress in overweight individuals: a randomized controlled trial. JAMA. 2006;295:1539–1548. doi: 10.1001/jama.295.13.1539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Lin SJ, Defossez PA, Guarente L. Requirement of NAD and SIR2 for life-span extension by calorie restriction in Saccharomyces cerevisiae. Science. 2000;289:2126–2128. doi: 10.1126/science.289.5487.2126. [DOI] [PubMed] [Google Scholar]
- 231.Bitterman KJ, Medvedik O, Sinclair DA. Longevity regulation in Saccharomyces cerevisiae: linking metabolism, genome stability, and heterochromatin. Microbiol. Mol. Biol. Rev. 2003;67:376–399. doi: 10.1128/MMBR.67.3.376-399.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Borradaile NM, Pickering JG. NAD+, sirtuins, and cardiovascular disease. Curr. Pharm. Des. 2009;15:110–117. doi: 10.2174/138161209787185742. [DOI] [PubMed] [Google Scholar]
- 233.Brandes RP. Activating SIRT1: a new strategy to prevent atherosclerosis? Cardiovasc. Res. 2008;80:163–164. doi: 10.1093/cvr/cvn245. [DOI] [PubMed] [Google Scholar]
- 234.Opie LH, Lecour S. The red wine hypothesis: from concepts to protective signalling molecules. Eur. Heart J. 2007;28:1683–1693. doi: 10.1093/eurheartj/ehm149. [DOI] [PubMed] [Google Scholar]
- 235.Shen Z, Ajmo JM, Rogers CQ, Liang X, Le L, et al. Role of SIRT1 in regulation of LPS- or two ethanol metabolites-induced TNFα production in cultured macrophage cell lines. Am. J. Physiol . Gastrointest. Liver Physiol. 2009;296:G1047–G1053. doi: 10.1152/ajpgi.00016.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Yoshizaki T, Milne JC, Imamura T, Schenk S, Sonoda N, et al. SIRT1 exerts anti-inflammatory effects and improves insulin sensitivity in adipocytes. Mol. Cell Biol. 2009;29:1363–1374. doi: 10.1128/MCB.00705-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Pearson KJ, Baur JA, Lewis KN, Peshkin L, Price NL, et al. Resveratrol delays age-related deterioration and mimics transcriptional aspects of dietary restriction without extending life span. Cell Metab. 2008;8:157–168. doi: 10.1016/j.cmet.2008.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Csiszar A, Labinskyy N, Podlutsky A, Kaminski PM, Wolin MS, et al. Vasoprotective effects of resveratrol and SIRT1: attenuation of cigarette smoke-induced oxidative stress and proinflammatory phenotypic alterations. Am. J. Physiol. Heart Circ. Physiol. 2008;294:H2721–H2735. doi: 10.1152/ajpheart.00235.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Wallerath T, Deckert G, Ternes T, Anderson H, Li H, et al. Resveratrol, a polyphenolic phytoalexin present in red wine, enhances expression and activity of endothelial nitric oxide synthase. Circulation. 2002;106:1652–1658. doi: 10.1161/01.cir.0000029925.18593.5c. [DOI] [PubMed] [Google Scholar]
- 240.Penumathsa SV, Thirunavukkarasu M, Koneru S, Juhasz B, Zhan L, et al. Statin and resveratrol in combination induces cardioprotection against myocardial infarction in hypercholesterolemic rat. J. Mol. Cell Cardiol. 2007;42:508–516. doi: 10.1016/j.yjmcc.2006.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Rush JW, Quadrilatero J, Levy AS, Ford RJ. Chronic resveratrol enhances endothelium-dependent relaxation but does not alter eNOS levels in aorta of spontaneously hypertensive rats. Exp. Biol. Med. 2007;232:814–822. [PubMed] [Google Scholar]
- 242.Mattagajasingh I, Kim CS, Naqvi A, Yamamori T, Hoffman TA, et al. SIRT1 promotes endothelium-dependent vascular relaxation by activating endothelial nitric oxide synthase. Proc. Natl. Acad. Sci. USA. 2007;104:14855–14860. doi: 10.1073/pnas.0704329104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Civitarese AE, Carling S, Heilbronn LK, Hulver MH, Ukropcova B, et al. Calorie restriction increases muscle mitochondrial biogenesis in healthy humans. PLoS Med. 2007;4:e76. doi: 10.1371/journal.pmed.0040076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Nisoli E, Tonello C, Cardile A, Cozzi V, Bracale R, et al. Calorie restriction promotes mitochondrial biogenesis by inducing the expression of eNOS. Science. 2005;310:314–317. doi: 10.1126/science.1117728. [DOI] [PubMed] [Google Scholar]
- 245.Borradaile NM, Pickering JG. Nicotinamide phosphoribosyltransferase imparts human endothelial cells with extended replicative lifespan and enhanced angiogenic capacity in a high glucose environment. Aging Cell. 2009;8:100–112. doi: 10.1111/j.1474-9726.2009.00453.x. [DOI] [PubMed] [Google Scholar]
- 246.Zhang QJ, Wang Z, Chen HZ, Zhou S, Zheng W, et al. Endothelium-specific overexpression of class III deacetylase SIRT1 decreases atherosclerosis in apolipoprotein E-deficient mice. Cardiovasc. Res. 2008;80:191–199. doi: 10.1093/cvr/cvn224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Liang CP, Han S, Senokuchi T, Tall AR. The macrophage at the crossroads of insulin resistance and atherosclerosis. Circ. Res. 2007;100:1546–1555. doi: 10.1161/CIRCRESAHA.107.152165. [DOI] [PubMed] [Google Scholar]
- 248.Potente M, Urbich C, Sasaki K, Hofmann WK, Heeschen C, et al. Involvement of Foxo transcription factors in angiogenesis and postnatal neovascularization. J. Clin. Investig. 2005;115:2382–2392. doi: 10.1172/JCI23126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Paik JH. FOXOs in the maintenance of vascular homoeostasis. Biochem. Soc. Trans. 2006;34:731–734. doi: 10.1042/BST0340731. [DOI] [PubMed] [Google Scholar]
- 250.Paik JH, Kollipara R, Chu G, Ji H, Xiao Y, et al. FoxOs are lineage-restricted redundant tumor suppressors and regulate endothelial cell homeostasis. Cell. 2007;128:309–323. doi: 10.1016/j.cell.2006.12.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Takata T, Ishikawa F. Human Sir2-related protein SIRT1 associates with the bHLH repressors HES1 and HEY2 and is involved in HES1- and HEY2-mediated transcriptional repression. Biochem. Biophys. Res. Commun. 2003;301:250–257. doi: 10.1016/s0006-291x(02)03020-6. [DOI] [PubMed] [Google Scholar]
- 252.Araki T, Sasaki Y, Milbrandt J. Increased nuclear NAD biosynthesis and SIRT1 activation prevent axonal degeneration. Science. 2004;305:1010–1013. doi: 10.1126/science.1098014. [DOI] [PubMed] [Google Scholar]
- 253.Sasaki Y, Vohra BP, Baloh RH, Milbrandt J. Transgenic mice expressing the Nmnat1 protein manifest robust delay in axonal degeneration in vivo. J. Neurosci. 2009;29:6526–6534. doi: 10.1523/JNEUROSCI.1429-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Pfister JA, Ma C, Morrison BE, D’Mello SR. Opposing effects of sirtuins on neuronal survival: SIRT1-mediated neuroprotection is independent of its deacetylase activity. PLoS ONE. 2008;3:e4090. doi: 10.1371/journal.pone.0004090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Giannakou ME, Partridge L. The interaction between FOXO and SIRT1: tipping the balance towards survival. Trends Cell Biol. 2004;14:408–412. doi: 10.1016/j.tcb.2004.07.006. [DOI] [PubMed] [Google Scholar]
- 256.Raff MC, Whitmore AV, Finn JT. Axonal self-destruction and neurodegeneration. Science. 2002;296:868–871. doi: 10.1126/science.1068613. [DOI] [PubMed] [Google Scholar]
- 257.Coleman MP, Perry VH. Axon pathology in neurological disease: a neglected therapeutic target. Trends Neurosci. 2002;25:532–537. doi: 10.1016/s0166-2236(02)02255-5. [DOI] [PubMed] [Google Scholar]
- 258.Conforti L, Tarlton A, Mack TG, Mi W, Buckmaster EA, et al. A Ufd2/D4Cole1e chimeric protein and overexpression of Rbp7 in the slow Wallerian degeneration (WldS) mouse. Proc. Natl. Acad. Sci. USA. 2000;97:11377–11382. doi: 10.1073/pnas.97.21.11377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Conforti L, Fang G, Beirowski B, Wang MS, Sorci L, et al. NAD+ and axon degeneration revisited: Nmnat1 cannot substitute for WldS to delay Wallerian degeneration. Cell Death Differ. 2007;14:116–127. doi: 10.1038/sj.cdd.4401944. [DOI] [PubMed] [Google Scholar]
- 260.Zhai RG, Cao Y, Hiesinger PR, Zhou Y, Mehta SQ, et al. Drosophila NMNAT maintains neural integrity independent of its NAD synthesis activity. PLoS Biol. 2006;4:e416. doi: 10.1371/journal.pbio.0040416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Yahata N, Yuasa S, Araki T. Nicotinamide mononucleotide adenylyltransferase expression in mitochondrial matrix delays Wallerian degeneration. J. Neurosci. 2009;29:6276–6284. doi: 10.1523/JNEUROSCI.4304-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Watanabe M, Tsukiyama T, Hatakeyama S. Protection of vincristine-induced neuropathy by WldS expression and the independence of the activity of Nmnat1. Neurosci. Lett. 2007;411:228–232. doi: 10.1016/j.neulet.2006.09.068. [DOI] [PubMed] [Google Scholar]
- 263.Avery MA, Sheehan AE, Kerr KS, Wang J, Freeman MR. WldS requires Nmnat1 enzymatic activity and N16-VCP interactions to suppress Wallerian degeneration. J. Cell Biol. 2009;184:501–513. doi: 10.1083/jcb.200808042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Suzuki K, Koike T. Resveratrol abolishes resistance to axonal degeneration in slow Wallerian degeneration (WldS) mice: activation of SIRT2, an NAD-dependent tubulin deacetylase. Biochem. Biophys. Res. Commun. 2007;359:665–671. doi: 10.1016/j.bbrc.2007.05.164. [DOI] [PubMed] [Google Scholar]
- 265.Outeiro TF, Kontopoulos E, Altmann SM, Kufareva I, Strathearn KE, et al. Sirtuin 2 inhibitors rescue α-synuclein-mediated toxicity in models of Parkinson’s disease. Science. 2007;317:516–519. doi: 10.1126/science.1143780. [DOI] [PubMed] [Google Scholar]
- 266.Qin W, Zhao W, Ho L, Wang J, Walsh K, et al. Regulation of forkhead transcription factor FoxO3a contributes to calorie restriction-induced prevention of Alzheimer’s disease-type amyloid neuropathology and spatial memory deterioration. Ann. N.Y. Acad. Sci. 2008;1147:335–347. doi: 10.1196/annals.1427.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Shindler KS, Ventura E, Rex TS, Elliott P, Rostami A. SIRT1 activation confers neuroprotection in experimental optic neuritis. Investig. Ophthalmol. Vis. Sci. 2007;48:3602–3609. doi: 10.1167/iovs.07-0131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.Wu Z, Lauer TW, Sick A, Hackett SF, Campochiaro PA. Oxidative stress modulates complement factor H expression in retinal pigmented epithelial cells by acetylation of FOXO3. J. Biol. Chem. 2007;282:22414–22425. doi: 10.1074/jbc.M702321200. [DOI] [PubMed] [Google Scholar]
- 269.Prozorovski T, Schulze-Topphoff U, Glumm R, Baumgart J, Schroter F, et al. Sirt1 contributes critically to the redox-dependent fate of neural progenitors. Nat. Cell Biol. 2008;10:385–394. doi: 10.1038/ncb1700. [DOI] [PubMed] [Google Scholar]
- 270.Hisahara S, Chiba S, Matsumoto H, Tanno M, Yagi H, et al. Histone deacetylase SIRT1 modulates neuronal differentiation by its nuclear translocation. Proc. Natl. Acad. Sci. USA. 2008;105:15599–15604. doi: 10.1073/pnas.0800612105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271.Libert S, Cohen D, Guarente L. Neurogenesis directed by Sirt1. Nat. Cell Biol. 2008;10:373–374. doi: 10.1038/ncb0408-373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272.Csiszar A, Labinskyy N, Jimenez R, Pinto JT, Ballabh P, et al. Anti-oxidative and anti-inflammatory vasoprotective effects of caloric restriction in aging: role of circulating factors and SIRT1. Mech. Ageing Dev. 2009;130:518–527. doi: 10.1016/j.mad.2009.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.Jung KJ, Lee EK, Kim JY, Zou Y, Sung B, et al. Effect of short term calorie restriction on pro-inflammatory NF-κB and AP-1 in aged rat kidney. Inflamm. Res. 2009;58:143–150. doi: 10.1007/s00011-008-7227-2. [DOI] [PubMed] [Google Scholar]
- 274.Phillips T, Leeuwenburgh C. Muscle fiber specific apoptosis and TNF-α signaling in sarcopenia are attenuated by life-long calorie restriction. FASEB J. 2005;19:668–670. doi: 10.1096/fj.04-2870fje. [DOI] [PubMed] [Google Scholar]
- 275.Salminen A, Huuskonen J, Ojala J, Kauppinen A, Kaarniranta K, Suuronen T. Activation of innate immunity system during aging: NF-κB signaling is the molecular culprit of inflamm-aging. Ageing Res. Rev. 2008;7:83–105. doi: 10.1016/j.arr.2007.09.002. [DOI] [PubMed] [Google Scholar]
- 276.Yang SR, Wright J, Bauter M, Seweryniak K, Kode A, Rahman I. Sirtuin regulates cigarette smoke-induced proinflammatory mediator release via RelA/p65 NF-κB in macrophages in vitro and in rat lungs in vivo: implications for chronic inflammation and aging. Am. J. Physiol. Lung Cell Mol. Physiol. 2007;292:L567–L576. doi: 10.1152/ajplung.00308.2006. [DOI] [PubMed] [Google Scholar]
- 277.Csiszar A, Smith K, Labinskyy N, Orosz Z, Rivera A, Ungvari Z. Resveratrol attenuates TNF-α-induced activation of coronary arterial endothelial cells: role of NF-κB inhibition. Am. J. Physiol. Heart Circ. Physiol. 2006;291:H1694–H1699. doi: 10.1152/ajpheart.00340.2006. [DOI] [PubMed] [Google Scholar]
- 278.Sequeira J, Boily G, Bazinet S, Saliba S, He X, et al. Sirt1-null mice develop an autoimmune-like condition. Exp. Cell Res. 2008;314:3069–3074. doi: 10.1016/j.yexcr.2008.07.011. [DOI] [PubMed] [Google Scholar]
- 279.Yang SR, Wright J, Bauter M, Seweryniak K, Kode A, Rahman I. Sirtuin regulates cigarette smoke induced proinflammatory mediators release via RelA/p65 NF-κB in macrophages in vitro and in rat lungs in vivo. Am. J. Physiol. Lung Cell Mol. Physiol. 2006;292:L567–L576. doi: 10.1152/ajplung.00308.2006. [DOI] [PubMed] [Google Scholar]
- 280.Rajendrasozhan S, Yang SR, Kinnula VL, Rahman I. SIRT1, an antiinflammatory and antiaging protein, is decreased in lungs of patients with chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 2008;177:861–870. doi: 10.1164/rccm.200708-1269OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281.Nakamaru Y, Vuppusetty C, Wada H, Milne JC, Ito M, et al. A protein deacetylase SIRT1 is a negative regulator of metalloproteinase-9. FASEB J. 2009;23:2810–2819. doi: 10.1096/fj.08-125468. [DOI] [PubMed] [Google Scholar]
- 282.Kubota S, Kurihara T, Mochimaru H, Satofuka S, Noda K, et al. Prevention of ocular inflammation in endotoxin-induced uveitis with resveratrol by inhibiting oxidative damage and nuclear factor-κB activation. Investig. Ophthalmol. Vis. Sci. 2009;50:3512–3519. doi: 10.1167/iovs.08-2666. [DOI] [PubMed] [Google Scholar]
- 283.Peng SL. Immune regulation by Foxo transcription factors. Autoimmunity. 2007;40:462–469. doi: 10.1080/08916930701464913. [DOI] [PubMed] [Google Scholar]
- 284.Lin L, Hron JD, Peng SL. Regulation of NF-κB, Th activation, and autoinflammation by the forkhead transcription factor Foxo3a. Immunity. 2004;21:203–213. doi: 10.1016/j.immuni.2004.06.016. [DOI] [PubMed] [Google Scholar]
- 285.Motta MC, Divecha N, Lemieux M, Kamel C, Chen D, et al. Mammalian SIRT1 represses forkhead transcription factors. Cell. 2004;116:551–563. doi: 10.1016/s0092-8674(04)00126-6. [DOI] [PubMed] [Google Scholar]
- 286.Blazek D, Peterlin BM. Tat-SIRT1 tango. Mol. Cell. 2008;29:539–540. doi: 10.1016/j.molcel.2008.02.007. [DOI] [PubMed] [Google Scholar]
- 287.Pagans S, Pedal A, North BJ, Kaehlcke K, Marshall BL, et al. SIRT1 regulates HIV transcription via Tat deacetylation. PLoS Biol. 2005;3:e41. doi: 10.1371/journal.pbio.0030041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288.Kwon HS, Brent MM, Getachew R, Jayakumar P, Chen LF, et al. Human immunodeficiency virus type 1 Tat protein inhibits the SIRT1 deacetylase and induces T cell hyperactivation. Cell Host Microbe. 2008;3:158–167. doi: 10.1016/j.chom.2008.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289.Kawahara TL, Michishita E, Adler AS, Damian M, Berber E, et al. SIRT6 links histone H3 lysine 9 deacetylation to NF-κB-dependent gene expression and organismal life span. Cell. 2009;136:62–74. doi: 10.1016/j.cell.2008.10.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290.Van Gool F, Galli M, Gueydan C, Kruys V, Prevot PP, et al. Intracellular NAD levels regulate tumor necrosis factor protein synthesis in a sirtuin-dependent manner. Nat. Med. 2009;15:206–210. doi: 10.1038/nm.1906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291.Nakahata Y, Kaluzova M, Grimaldi B, Sahar S, Hirayama J, et al. The NAD+-dependent deacetylase SIRT1 modulates CLOCK-mediated chromatin remodeling and circadian control. Cell. 2008;134:329–340. doi: 10.1016/j.cell.2008.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292.Asher G, Gatfield D, Stratmann M, Reinke H, Dibner C, et al. SIRT1 regulates circadian clock gene expression through PER2 deacetylation. Cell. 2008;134:317–328. doi: 10.1016/j.cell.2008.06.050. [DOI] [PubMed] [Google Scholar]
- 293.Elliott PJ, Jirousek M. Sirtuins: novel targets for metabolic disease. Curr. Opin. Investig. Drugs. 2008;9:371–378. [PubMed] [Google Scholar]