

Abstract
Background
The recent availability of genetic analyses has demonstrated the shortcomings of the current phenotypic method of corneal dystrophy classification. Abnormalities in different genes can cause a single phenotype, whereas different defects in a single gene can cause different phenotypes. Some disorders termed corneal dystrophies do not appear to have a genetic basis.
Purpose
The purpose of this study was to develop a new classification system for corneal dystrophies, integrating up-to-date information on phenotypic description, pathologic examination, and genetic analysis.
Methods
The International Committee for Classification of Corneal Dystrophies (IC3D) was created to devise a current and accurate nomenclature.
Results
This anatomic classification continues to organize dystrophies according to the level chiefly affected. Each dystrophy has a template summarizing genetic, clinical, and pathologic information. A category number from 1 through 4 is assigned, reflecting the level of evidence supporting the existence of a given dystrophy. The most defined dystrophies belong to category 1 (a well-defined corneal dystrophy in which a gene has been mapped and identified and specific mutations are known) and the least defined belong to category 4 (a suspected dystrophy where the clinical and genetic evidence is not yet convincing). The nomenclature may be updated over time as new information regarding the dystrophies becomes available.
Conclusions
The IC3D Classification of Corneal Dystrophies is a new classification system that incorporates many aspects of the traditional definitions of corneal dystrophies with new genetic, clinical, and pathologic information. Standardized templates provide key information that includes a level of evidence for there being a corneal dystrophy. The system is user-friendly and upgradeable and can be retrieved on the website www.corneasociety.org/ic3d.
Keywords: corneal dystrophy, inherited corneal disease, genetic corneal disease, corneal histopathology, gene, mutation, key reference, eponym, epithelial basement membrane dystrophy, epithelial recurrent erosion dystrophy, subepithelial mucinous corneal dystrophy, Meesmann corneal dystrophy, Lisch epithelial corneal dystrophy, gelatinous drop-like corneal dystrophy, Grayson-Wilbrandt corneal dystrophy, lattice corneal dystrophy, lattice gelsolin type dystrophy, granular corneal dystrophy 1, granular corneal dystrophy 2, Avellino corneal dystrophy, Reis-Bücklers corneal dystrophy, Thiel-Behnke corneal dystrophy, macular corneal dystrophy, Schnyder corneal dystrophy, Schnyder crystalline corneal dystrophy, congenital stromal corneal dystrophy, fleck corneal dystrophy, posterior amorphous corneal dystrophy, central cloudy dystrophy of François, pre-Descemet corneal dystrophy, Fuchs endothelial corneal dystrophy, posterior polymorphous corneal dystrophy, congenital hereditary endothelial dystrophy 1, congenital hereditary endothelial dystrophy 2, X-linked endothelial corneal dystrophy
HISTORY
The word dystrophy is derived from the Greek (dys = wrong, difficult; trophe = nourishment)1 and was introduced into the medical literature by Wilhelm Erb (1840–1921) in 1884, in describing a disease of the musculature.2 In 1890, Arthur Groenouw (1862–1945) published his classic paper describing 2 patients with “noduli corneae,” with 1 patient having granular corneal dystrophy and the other, macular corneal dystrophy.3 At the same time, Biber was also publishing his thesis on lattice corneal dystrophy.4
In that pre-slit lamp era, the extent of corneal examination was limited. But although Groenouw did not initially appreciate the differences between granular and macular dystrophy or recognize the familial predisposition, the 2 diseases subsequently became known as corneal dystrophies.5 Fuchs6 used the word dystrophy to refer to ophthalmologic disease and postulated that dystrophic tissues resulted from lack of nourishment, hormones, blood, and nerve supply. Uhthoff7 and later Yoshida8 continued to use the term in their publications.
CORNEAL DYSTROPHY DEFINITION
Although many definitions of the word “dystrophy” have appeared in the medical literature,1 the term is most commonly used to describe an inherited disorder affecting cells, tissues, or organs, alone or in combination. In ophthalmology, the term “corneal dystrophy” has been used to refer to a group of inherited corneal diseases that are typically bilateral, symmetric, slowly progressive, and without relationship to environmental or systemic factors.9 As knowledge has increased, exceptions to each of these definitions have been noted. Thus, most patients with epithelial basement membrane dystrophy do not have a hereditary pattern. Some patients with posterior polymorphous corneal dystrophy only manifest unilateral changes. In macular dystrophy, the level of antigenic serum keratan sulfate correlates with the immunophenotypes of the disease, indicating that systemic abnormalities are integral to the development of the characteristic corneal changes. Like-wise, there are a number of hereditary, bilateral diseases of the cornea, such as cornea plana, which have not been traditionally classified as corneal dystrophies and may be alternatively accommodated among the congenital anomalies affecting the cornea.
Consequently, experience has demonstrated that the separation of entities into the category called corneal dystrophies may have more historical than practical meaning. There remains no consensus as to the precise definition of corneal dystrophy, but, according to custom, we have chosen to primarily deal with entities formerly called corneal dystrophies.
CORNEAL DYSTROPHY LITERATURE
Bücklers,10 whose name was later attached to Reis–Bücklers corneal dystrophy (RBCD), published the first classification of the corneal dystrophies when he described the differences between granular, lattice, and macular corneal dystrophies. Although the dystrophies can be classified according to genetic pattern, severity, histopathologic features, or biochemical characteristics, the most commonly used classification system has been anatomically based.9 The dystrophies are typically classified by level of the cornea that is involved, which separates these entities into epithelial and subepithelial, Bowman layer, stromal, Descemet membrane, and endothelial dystrophies.11-14
SHORTCOMINGS OF CORNEAL DYSTROPHY CLASSIFICATION
Critical review of the corneal dystrophy literature reveals numerous apparent misconceptions and errors. For example, many publications emphasize the necessity of demonstrating corneal crystals to make the diagnosis of Schnyder crystalline corneal dystrophy (SCD).15,16 However, examination of large pedigrees of patients with SCD demonstrates that only 50% of affected patients actually have corneal crystals.17 Nevertheless, publications over the past decades erroneously emphasize that crystals are necessary for the diagnosis of SCD.18
The direct consequence is that in some patients with SCD who lack stromal crystals, the diagnosis may be delayed for decades.17 Once established in textbooks, it is exceedingly difficult to purge incorrect information about rare diseases. Many myths are perpetuated because very few ophthalmologists have seen a substantial number of the unusual corneal dystrophies.
Another difficulty in the literature is the tendency to place too much emphasis on a new or rare observation rather than wait for a full analysis of a new disorder. For example, some of the early papers describing the ultrastructure of RBCD had actually analyzed tissue from patients with Thiel–Behnke corneal dystrophy (TBCD).19 In a publication, the known entity of RBCD was renamed as an unusual variant of granular dystrophy.20,21 These inconsistencies in the literature have confounded our understanding of precise findings in specific corneal dystrophies.
DOES EVERY SINGLE DYSTROPHY ACTUALLY EXIST?
Before the 1970s, new corneal dystrophies were identified and characterized almost exclusively by their clinical appearance aided, in some cases, by light microscopic histopathology. In some cases, the description of a dystrophy was based on a report of a single family.22,23
In other cases, a new dystrophy could be misclassified as a variant of a previously described dystrophy. For years, the dystrophy of Waardenburg and Jonkers24 appeared in references and textbooks. In actuality, these patients had Thiel–Behnke corneal dystrophy.25,26
Often, it is impossible to either confirm or exclude every corneal dystrophy that has made its way into textbooks as an independent entity. Moreover, misunderstandings that became prevalent have often persisted long after they could be resolved. For example, what did Reis and Bücklers actually see when they described what is now called Reis–Bücklers corneal dystrophy?22,23 The original pedigree is lost to follow up, and their clinical description is sketchy concerning the specific signs and symptoms. Nevertheless, we still presume that the entity they described is probably what is now established as Reis–Bücklers dystrophy (RBCD), but the original patients could have had what is now called TBCD.
Before honeycomb-shaped corneal dystrophy was described by Thiel and Behnke in 1967,25 and even afterwards, patients affected with this dystrophy were instead reported as examples of RBCD.19 It took more than 30 years before the literature had separated these 2 dystrophies. On the other hand, Grayson and Wilbrandt 27 described a family with a Bowman layer dystrophy they initially reported as RBCD but actually provided insufficient evidence to determine definitively whether the unique findings, subsequently called Grayson–Wilbrandt corneal dystrophy (GWCD), indicated a distinct entity or a variant of a different Bowman layer dystrophy.
Although the original publication on central cloudy dystrophy of François28 described a hereditary corneal opacification, there have been only a few other publications that have described an entire family with this disease.29,30 Both articles were written before the advent of genotyping so no genetic information is available. Central cloudy dystrophy of François appears clinically indistinguishable from the degenerative condition, posterior crocodile shagreen.31 It is not possible to determine whether previous publications describing an individual patient with central cloudy dystrophy of François were actually describing patients with posterior crocodile shagreen.32 In the absence of additional affected pedigrees or genetic studies confirming inheritance, it is possible that central cloudy dystrophy of François and posterior crocodile shagreen are the same entity. Without genotypic information, it may be impossible to determine whether rare or newly described dystrophies are actually unique diseases or represent phenotypic variations of previously described entities.
GENETICS
The development of genotypic analyses has revolutionized our knowledge of the corneal dystrophies and further elucidated additional inaccuracies in the dystrophy nomenclature. The genetic characterization of corneal dystrophies revealed both genetic heterogeneity, that is, different genes (KRT3 and KRT12) causing a single dystrophy phenotype (Meesmann dystrophy), and phenotypic heterogeneity with a single gene (TGFBI) causing different allelic dystrophy phenotypes (RBCD, TBCD, granular type 1, granular type 2, and lattice type 1). Consequently, by enhancing our understanding of the dystrophies, newer genetic information has made the phenotypic classification system archaic.
CURRENT CLASSIFICATION OF THE CORNEAL DYSTROPHIES
The knowledge base has exploded since the first descriptions of granular, macular, and lattice dystrophies over a century ago. Not only has the word dystrophy lost importance but also the distinctive name of many of the individual dystrophies has become less meaningful. The basis of the nomenclature system seems to be more historic than scientific.
As the classification system of these disorders has taken on historic implications, it has been proposed that these conditions be classified “under the rubric of inherited corneal diseases,” although acknowledging that “the popular designation of corneal dystrophy will probably keep its place.33”
RECLASSIFICATION OF THE NOMENCLATURE IN OTHER MEDICAL SPECIALTIES
Ophthalmology is not the only medical field that has discovered that the nomenclature of certain diseases has become archaic. Rapid advances in genotyping have challenged the nomenclature of other diseases in other specialties. Some of these specialties have met the challenge by devising new nomenclature systems. In 2001, the European Academy of Allergy and Clinical Immunology published a position paper proposing a new nomenclature in allergy after discussion with “many pediatricians in Europe for several years.34” One of the authors wrote that he “set up a reference panel of pediatricians within different areas of pediatrics and at intervals I asked them for their opinion on the proposal.” Subsequent articles in this field have underscored the importance on nomenclature of atopy, atopic disease, and allergy on classifying the individual patient diseases and directing future therapy.35
The disconnect between the language of basic scientists and the language of clinicians has also presented challenges in the muscular dystrophy nomenclature. Dubovitz36 wrote about his concern regarding a “major problem, within the field of therapy for muscular dystrophy, that has arisen from inappropriate nomenclature,” namely that it “… has had a negative impact on the whole field.” Klein wrote that “From a historical perspective, 2 golden ages have shaped the current and evolving classification schemes: 1. the definition of clinical pathological entities in the early twentieth century; and 2. the application of molecular neurogenetics in the past 10–15 years.” He concluded that the shortcoming of the current classification systems resulted not only because of the complex nature of the disorders but also that “modern classification schemes was based on clinical, pathologic and genetic/molecular criteria … attempt to integrate all three levels” and although “genetic classifications are now widely used … expert clinical diagnosis remains an important step in correct diagnosis and classification.”37 The author proposed classification schemes based on clinical features, genetic features, and molecular mechanisms or protein functions.
THE FORMATION OF THE INTERNATIONAL COMMITTEE FOR CLASSIFICATION OF CORNEAL DYSTROPHIES (IC3D)
In April 2005 at the World Cornea Congress meeting, the session on corneal dystrophies clearly elucidated that nomenclature problems vexed not only SCD but also many other dystrophies. That evening, J.S.W. approached the other members of the board of directors of the Cornea Society to request their support for the creation of an international committee to revise the corneal dystrophy nomenclature. The goal was the recruitment of an international panel of interested world experts possessing firsthand experience with the clinical, genetic, and histopathologic findings of all the corneal dystrophies. In this way, the literature could be critically evaluated to distill the facts and recognize and then remove outdated inaccurate information. With the support of the Cornea Society President (M.W.B.), international ophthalmologic societies were contacted representing 5 continents to recruit representation of corneal specialists, ophthalmic pathologists, and geneticists for this collaborative effort.
The International Committee for Classification of Corneal Dystrophies (IC3D) held its first meeting in Chicago in October 2005 at the American Academy of Ophthalmology, followed by meetings in San Paulo in February 2006 at the World Ophthalmology Congress, in Ft. Lauderdale in May 2006 at the Association for Research in Vision and Ophthalmology, in Las Vegas in October 2006 at the American Academy of Ophthalmology, and in San Diego in April 2007 at the Association of Cataract and Refractive Surgeons. In between, thousands of e-mails provided online discussion to move the project forward.
CHARACTERISTICS OF THE NEW NOMENCLATURE
At the initial meeting, the group discussed the necessary characteristics of a new nomenclature that definitely would improve accuracy, would be more informative, and could be easy to use, so that it truly could replace the nomenclature that had been used for over a century—a gargantuan task, the successfulness of which only time will tell. The new nomenclature had to reflect current clinical, pathologic, and genetic knowledge, be easily adaptable to advances in understanding from the continued discovery of new genes and mutations and be linked to the old nomenclature for ease of use.
THE IC3D TEMPLATES
The development of a series of templates, which would assemble accurate and up-to-date information about each dystrophy and facilitate the development and maintenance of a revised nomenclature, was undertaken. Each dystrophy template was a brief summary of the current genetic, clinical, and pathologic information about the disease and included representative clinical images. This approach also offered the opportunity to correct errors in the literature and “set the record straight.” Published information was reviewed by all members of the committee, particularly those who had experience with a particular dystrophy. Although there were some dystrophies with which no member of the committee had personal experience, such dystrophies were exceedingly rare and sometimes had only 1 case report in the literature. The process, therefore, was found to be very effective.
CLASSIFICATION AND THE EVOLUTION OF A CORNEAL DYSTROPHY
The largest challenge to the committee was how to devise a classification that would be flexible enough to facilitate the expansion of knowledge from other sources, including genotyping. Evidence for the existence of a corneal dystrophy starts with the identification of a clinical phenotype and may proceed to the characterization of the causative gene mutation. When a corneal dystrophy is first described, there is usually a predictable chain of events. Initially, an entity is identified and characterized clinically. With corneal disorders that impair vision severely enough to warrant keratoplasty, tissue evaluations of the diseased cornea lead to the establishment of distinct clinicopathologic entities. Even in the absence of tissue evaluations, the next phase involves genetic linkage studies that lead to the mapping of the chromosomal locus of the disorder, especially if the condition has a simple Mendelian inheritance pattern. This task is much more tedious and time consuming when more than 1 gene is involved or if there is an interaction between genetic and environmental factors. Gene mapping is followed in due course by the identification of the relevant gene and particular mutations that are responsible for different phenotypical forms of the disorder. Eventually, identification of the gene product provides a better understanding of the mechanism of the disorder and may present some therapeutic possibilities.
To indicate the level of evidence supporting the existence of a given dystrophy, the IC3D committee developed a series of descriptive, evidential categories as follows:
Categories
Category 1: A well-defined corneal dystrophy in which the gene has been mapped and identified and specific mutations are known.
Category 2: A well-defined corneal dystrophy that has been mapped to 1 or more specific chromosomal loci, but the gene(s) remains to be identified.
Category 3: A well-defined corneal dystrophy in which the disorder has not yet been mapped to a chromosomal locus.
Category 4: This category is reserved for a suspected new, or previously documented, corneal dystrophy, although the evidence for it, being a distinct entity, is not yet convincing.
The category assigned to a specific corneal dystrophy can be expected to change over time as knowledge progressively advances. Eventually, all valid corneal dystrophies should attain the classification of category 1; macular corneal dystrophy is an example of a category 1 dystrophy. Conversely, over time and with further information, some entities that are category 4 may be shown not to be distinct entities and may be removed. For example, “Central Discoid Corneal Dystrophy”38 (CDCD), a category 4 dystrophy was found to be indistinguishable phenotypically from SCD sine crystals. Consequently, the IC3D committee further reviewed a case report of CDCD to determine whether this was a unique dystrophy or a variant of SCD. When the causative gene for SCD was found to be UBIAD1, 39,40 genetic testing of the proband with CDCD could be performed. Interestingly, the CDCD proband did demonstrate a unique mutation in the UBIAD1 gene (personal correspondence J.S.W.), which was not found in 100 control individuals. With a mutation in the UBIAD1 gene and corneal histopathology, which demonstrated stromal vacuoles consistent with dissolved lipid, it seemed that CDCD was actually SCD. Consequently, this category 4 dystrophy was removed and CDCD was re-classified as SCD. This case clearly illustrates the importance and utility of the IC3D classification system. If an entity is initially categorized as a level 4 dystrophy, new information can be used to determine whether the entity is indeed new or unique or is perhaps a variant of a previously described disease.
THE NEW CLASSIFICATION SYSTEM
Our proposed corneal dystrophy classification system is anatomically based, with dystrophies classified according to the layer chiefly affected (www.corneasociety.org/ic3d). Thus, they are epithelial and subepithelial, Bowman layer, stromal and those affecting Descemet membrane and the endothelium. The majority of the dystrophy names are identical or similar to those in the current nomenclature. However, dystrophies with a known common genetic basis, that is, TGFBI dystrophies, have been grouped together.
Each template provides the key genetic, clinical and histopathologic features that are characteristic for that dystrophy. Each is assigned a level of evidence category of 1, 2, 3, or 4, depending on the amount of clinical and genetic information available. A more detailed description of genetic mutations is included in the Appendix.
ACKNOWLEDGMENTS
The members of IC3D Committee are as follows: J. S. W., MD (Chair), M. W. B., MD (Vice-Chair). Membership—Asia: E. K. K., MD, PhD, and S. K., MD; Australia: Rasik Vajpayee, MS; Europe: C. B., MD, Tony Bron, MD, M. B., MD, T. K., MD, W.L., MD, H. U. M., MD, PhD, F. L.M., MD, B. S., MD, and G. V. R., MD; and North America: A. J.A., MD, M .W. B., MD, J. S., MD, G. K. K., MD, PhD, M. J.M., MD, and C. R., MD, and J. S. W., MD. We would like to acknowledge the generous financial support of the Cornea Society without which this work would not have been possible. We appreciate the support of the Eye Defects Research Foundation and Research to Prevent Blindness. We also like to acknowledge the photographic contributions of the members of the IC3D, Robert Feder, MD, Dienke Wittebol-Post, MD, and Jay Krachmer, MD.
Appendix
THE IC3D CLASSIFICATION (C = CATEGORY)
Epithelial and Subepithelial Dystrophies
Epithelial basement membrane dystrophy (EBMD)—majority degenerative, some C1
Epithelial recurrent erosion dystrophy (ERED) C4, (Smolandiensis variant) C3
Subepithelial mucinous corneal dystrophy (SMCD) C4
Mutation in keratin genes: Meesmann corneal dystrophy (MECD) C1
Lisch epithelial corneal dystrophy (LECD) C2
Gelatinous drop-like corneal dystrophy (GDLD) C1
Bowman Layer Dystrophies
Reis–Bücklers corneal dystrophy (RBCD)—Granular corneal dystrophy type 3 C1
Thiel–Behnke corneal dystrophy (TBCD) C1, potential variant C2
Grayson –Wilbrandt corneal dystrophy (GWCD) C4
Stromal Dystrophies
- TGFBI corneal dystrophies
- Lattice corneal dystrophy
- Lattice corneal dystrophy, TGFBI type (LCD): Classic lattice corneal dystrophy (LCD1) C1, variants (III, IIIA, I/IIIA, and IV) are C1
- Lattice corneal dystrophy, gelsolin type (LCD2) C1 (This is not a true corneal dystrophy but is included here for ease of differential diagnosis)
- Granular corneal dystrophy C1
- Granular corneal dystrophy, type 1 (classic) (GCD1) C1
- Granular corneal dystrophy, type 2 (granular-lattice) (GCD2) C1
- Granular corneal dystrophy, type 3 (RBCD) = Reis–Bücklers C1
Macular corneal dystrophy (MCD) C1
Schnyder corneal dystrophy (SCD) C1
Congenital stromal corneal dystrophy (CSCD) C1
Fleck corneal dystrophy (FCD) C1
Posterior amorphous corneal dystrophy (PACD) C3
Central cloudy dystrophy of François (CCDF) C4
Pre-Descemet corneal dystrophy (PDCD) C4
Descemet Membrane and Endothelial Dystrophies
Fuchs endothelial corneal dystrophy (FECD) C1, C2, or C3
Posterior polymorphous corneal dystrophy (PPCD) C1 or C2
Congenital hereditary endothelial dystrophy 1 (CHED1) C2
Congenital hereditary endothelial dystrophy 2 (CHED2) C1
X-linked endothelial corneal dystrophy (XECD) C2
Appendix
EPITHELIAL AND SUBEPITHELIAL DYSTROPHIES
Epithelial Basement Membrane Dystrophy (EBMD)
MIM #121820
Alternative Names, Eponyms
Map-dot-fingerprint dystrophy.
Cogan microcystic epithelial dystrophy.
Anterior basement membrane dystrophy.
Inheritance
Most cases have no inheritance documented. Many are considered to be degenerative or secondary to trauma. Familial cases have been reported.
Genetic Locus
5q31.
Gene
TGFBI in the minority of cases.
Onset
Present in adult life. Rarely seen in children.
Signs (Fig. 1)
Maps
Irregular islands of thickened, gray, hazy epithelium with scalloped, circumscribed borders, particularly affecting the central or paracentral cornea. Isolated or combined with other signs.
Dots (Cogan)
Irregular round, oval or comma-shaped, non-staining, putty-gray opacities. Clustered like an archipelago in the central cornea. Typically combined with other signs, especially with maps.
Fingerprint lines
Parallel, curvilinear lines, usually paracentral. Best seen in retro illumination. Isolated or combined with other signs, especially maps.
Bleb pattern (Bron)
A subepithelial pattern like pebbled glass, best seen by retro-illumination. Isolated or combined with other signs.
Poor adhesion of basal epithelial cells to abnormal basal laminar material is thought predisposition to recurrent erosions.
Symptoms
Asymptomatic or recurrent erosions with pain, lacrimation, and blurred vision. Except for the bleb pattern, on-axis lesions may also cause blurred vision due to irregular astigmatism.
Course
Location and degree of pathology can fluctuate with time.
Light Microscopy
Maps
Sheets of intraepithelial, multilamellar, basal laminar material.
Fingerprint lines
Rib-like intraepithelial extensions of basal laminar material.
Dots
Intraepithelial pseudocyst containing cytoplasmic debris.
Bleb pattern
Irregular, subepithelial accumulation of a fibrillogranular material.
FIGURE 1.

Epithelial basement membrane dystrophy. A, Map-like changes. B, Intraepithelial dot opacities underlying map-like figures. C, Fingerprint lines viewed in retroillumination.
Transmission Electron Microscopy
Map
Thick epithelial basement membrane that extends into the epithelium as multilamellar, 2- to 6-nm-thick sheets.
Fingerprint line
Fine fibrillogranular substance in addition to basement membrane. The fibrils are about 17 nm in diameter and the granular material about 8 nm.
Dot
Intraepithelial pseudocyst contains degenerating cells with pyknotic nuclei and cytoplasmic debris.
Bleb pattern
The anterior surface of this material forms discrete mounds, which dent the overlying basal epithelial cells. May mimic cysts clinically but no cysts present histologically.
Confocal Microscopy
Map-fingerprint-dot
Intraepithelial basement membrane, which appears separated from normal basal epithelial cells. Droplet-shaped configuration in the epithelium. Ring-like structure in the basal epithelium.
Category
Most cases are sporadic and may be degenerative. Category 1 in a minority of cases.
REFERENCES
- 1.Boutboul S, Black GCM, Moore JE, et al. A subset of patients with epithelial basement membrane corneal dystrophy have mutations in TGFBI/BIGH3. Hum Mutat. 2006;27:553–557. doi: 10.1002/humu.20331. [DOI] [PubMed] [Google Scholar]
- 2.Bron AJ, Brown NA. Some superficial corneal disorders. Trans Ophthalmol Soc UK. 1971;91:13–29. [PubMed] [Google Scholar]
- 3.Bron AJ, Tripathi RC. Cystic disorders of the corneal epithelium II. Pathogenesis. Br J Ophthalmol. 1973;57:361–375. doi: 10.1136/bjo.57.6.361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cogan DG, Donaldson DD, Kuwabara T, et al. Microcystic dystrophy of the corneal epithelium. Trans Am Ophthalmol Soc. 1964;62:213–225. [PMC free article] [PubMed] [Google Scholar]
- 5.Guerry D. Fingerprint-like lines in the cornea. Am J Ophthalmol. 1950;33:724–726. doi: 10.1016/0002-9394(50)90194-2. [DOI] [PubMed] [Google Scholar]
- 6.Laibson PR, Krachmer JH. Familial occurrence of dot (microcystic), map, fingerprint dystrophy of the cornea. Invest Ophthalmol Vis Sci. 1975;14:397–399. [PubMed] [Google Scholar]
- 7.Laibson PR. Microcystic corneal dystrophy. Trans Am Ophthalmol Soc. 1976;74:488–531. [PMC free article] [PubMed] [Google Scholar]
- 8.Lisch W, Lisch C. Die epitheliale Hornhautbasalmembrandystrophie. Klin Monatsbl Augenheilkd. 1983;183:251–255. doi: 10.1055/s-2008-1054913. [DOI] [PubMed] [Google Scholar]
- 9.Munier FL, Korvatska E, Djemai A, et al. Keratoepithelin mutations in four 5q31-linked corneal dystrophies. Nat Genet. 1997;15:247–251. doi: 10.1038/ng0397-247. [DOI] [PubMed] [Google Scholar]
- 10.Rodrigues MM, Fine BS, Laibson PR, et al. Disorders of the corneal epithelium. A clinicopathologic study of dot, geographic, and fingerprint patterns. Arch Ophthalmol. 1974;92:475–482. doi: 10.1001/archopht.1974.01010010489005. [DOI] [PubMed] [Google Scholar]
- 11.Vogt A. Lehrbuch und Atlas der Spaltlampenmikroskopie des lebenden Auges (1. Teil) Springer; Berlin, Germany: 1930. pp. 119–121. [Google Scholar]
Appendix
Epithelial Recurrent Erosion Dystrophy (ERED)
MIM #122400
Alternative Names, Eponyms
Corneal erosions, recurring hereditary (Franceschetti).
Variants
Dystrophia Smolandiensis.
Inheritance
Autosomal dominant.
Genetic Locus
Unknown.
Gene
Unknown; COL8A2, TGFBI, GSN, KRT3 and KRT12 excluded in Smolandiensis variant.
Onset
First decade of life.
Signs (Fig. 2)
Recurrent corneal erosions appear typically at 4 –6 years of age but occasionally as early as 8 months of age. They are precipitated by minimal trauma or are spontaneous. The cornea may show subepithelial haze or blebs between attacks. In the Smolandiensis variant, half of the patients develop single to a few permanent central subepithelial corneal opacities, which appear at as early as 7 years of age. These vary from subepithelial fibrosis to protruding keloid-like nodules.
FIGURE 2.
Epithelial recurrent erosion corneal dystrophy (Smolandiensis variant). The right eye of a 41 -year -old female with a central keloid-like opacification found in half of the affected family members.
Symptoms
Most patients have attacks of redness, photophobia, epiphora, and ocular pain. Some experience a burning sensation and report sensitive eyes for years. Exposure to sunlight or draught, dust and smoke and lack of sleep can precipitate attacks. In the Smolandiensis variant, a quarter of patients eventually need corneal grafts at mean age of 44 years. The opacities recur within 15 months in the graft periphery, but the central graft can remain clear for many years.
Course
Attacks generally decline in frequency and intensity and cease by the age of 50 years. In the Smolandiensis variant, central subepithelial opacities will progress.
Light Microscopy
No changes consistent with either EBMD or known dystrophy of Bowman layer are reported for the Smolandiensis variant.
Transmission Electron Microscopy
Not reported.
Confocal Microscopy
Not reported.
Category
4, 3 (Smolandiensis variant).
REFERENCES
- 1.Hammar B, Björck E, Lagerstedt K, et al. A new corneal disease with recurrent erosive episodes and autosomal dominant inheritance. Acta Ophthalmol Scand. doi: 10.1111/j.1600-0420.2007.01123.x. In press. [DOI] [PubMed] [Google Scholar]
- 2.Franceschetti A. Hereditaere rezidivierende Erosion der Hornhaut. Z Augenheilk. 1928;66:309–316. [Google Scholar]
- 3.Valle O. Hereditary recurring corneal erosions: a family study, with special reference to Fuchs’ dystrophy. Acta Ophthalmol. 1967;45:829–836. doi: 10.1111/j.1755-3768.1967.tb08123.x. [DOI] [PubMed] [Google Scholar]
- 4.Wales HJ. A family history of corneal erosions. Trans Ophthalmol Soc NZ. 1956;8:77–78. [PubMed] [Google Scholar]
Appendix
Subepithelial Mucinous Corneal Dystrophy (SMCD)
MIM: None.
Alternative Names, Eponyms
None.
Inheritance
Autosomal dominant.
Genetic Locus
Unknown.
Gene
Unknown.
Onset
First decade of life.
Signs (Fig. 3)
Bilateral subepithelial opacities and haze, most dense centrally, involving the entire cornea.
Symptoms
Painful episodes of recurrent corneal erosions, which decrease during adolescence (only 1 publication of a single family).
Course
Progressive loss of vision in adolescence.
Light Microscopy
Subepithelial band of eosinophilic, periodic acid–Schiff–positive, Alcian blue–positive, hyaluronidase-sensitive material is present anterior to Bowman layer.
Transmission Electron Microscopy
Subepithelial deposits of fine fibrillar material.
Immunohistochemistry
Combination of chondroitin-4-sulfate and dermatan sulfate.
Confocal Microscopy
Not reported.
Category
4.
FIGURE 3.
Subepithelial mucinous corneal dystrophy. Subepithelial opacities and haze involving the entire cornea; these are most dense toward the center (broad oblique and slit views).
REFERENCES
- 1.Feder RS, Jay M, Yue BY, et al. Subepithelial mucinous corneal dystrophy. Clinical and pathological correlations. Arch Ophthalmol. 1993;111:1106–1114. doi: 10.1001/archopht.1993.01090080102025. [DOI] [PubMed] [Google Scholar]
Appendix
Mutations in Keratin Genes: Meesmann Corneal Dystrophy (MECD)
MIM #122100
Alternative Names, Eponyms
Juvenile hereditary epithelial dystrophy.
Variant
Stocker–Holt variant.
Inheritance
Autosomal dominant.
Genetic Loci
Locus 12q13 (KRT3).
Locus 17q12 (KRT12) Stocker–Holt variant.
Genes
Keratin K3 (KRT3).
Keratin K12 (KRT12) Stocker–Holt variant.
Onset
Early childhood.
Signs (Fig. 4)
Multiple, tiny epithelial vesicles extend to the limbus and are most numerous in the interpalpebral area with clear surrounding epithelium. Whorled and wedge-shaped epithelial patterns have been reported. The cornea may be slightly thinned and corneal sensation may be reduced.
Indirect illumination shows varying diffuse gray opacities in different patterns, which may have a distinct border. Areas of the central or peripheral cornea may be unaffected. The gray opacities appear as transparent cysts on indirect illumination. Coalescence of several cysts may result in refractile linear opacities with intervening clear cornea.
Stocker–Holt Variant
The entire cornea demonstrates fine, grayish punctate epithelial opacities that stain with fluorescein and fine linear opacities that may appear in a whorl pattern.
Course
Slowly progressive.
Symptoms
Patients are typically asymptomatic or may have mild visual reduction, although some patients complain of glare and light sensitivity. Recurrent painful punctiform epithelial erosions may occur. Rarely, blurred vision results from corneal irregularity and scarring.
FIGURE 4.

Meesmann corneal dystrophy. A, Multiple solitary microcysts that are most prominent in the interpalpebral region are seen in retroillumination. B, Diffuse gray opacity with broad oblique illumination, and multiple solitary microcysts in retroillumination.
Stocker–Holt Variant
Patients demonstrate more severe signs and symptoms with earlier onset compared with classic Meesmann corneal dystrophy.
Light Microscopy
The epithelium always demonstrates intraepithelial cysts. Cysts are filled with periodic acid–Schiff–positive cellular debris, which fluoresces. The epithelium may be thickened and disorganized. Thickened multilaminar basement membrane with projections into the basal epithelium.
Stocker–Holt Variant
Variably thickened epithelium with vacuolated cells and evidence of degeneration. Variably thickened basement membrane extending into the epithelium. Normal Bowman layer and stroma.
Transmission Electron Microscopy
Intracytoplasmic “peculiar substance” represents a focal collection of fibrogranular material surrounded by tangles of cytoplasmic filaments. Cystic round and well-delineated lesions (10–50 μm across). Some lesions with reflective points in the cytoplasm probably correspond to cell nuclei.
Stocker–Holt Variant
Not reported.
Confocal Microscopy
Hyporeflective areas in the basal epithelium ranging from 40 to 150 mm in diameter, with potential reflective spots inside.
Stocker–Holt Variant
Not reported.
Category
1, including Stocker–Holt Variant
REFERENCES
- 1.Behnke H, Thiel HJ. Über die hereditäre epitheldystrophie der Horhaut (Typ Meesman-Wilke) in Schleswig-Holstein. Klin Monatsbl Augenheilkd. 1965;147:662–672. [PubMed] [Google Scholar]
- 2.Burns RP. Meesmann’s corneal dystrophy. Trans Am Ophthalmol Soc. 1968;66:530–635. [PMC free article] [PubMed] [Google Scholar]
- 3.Fine BS, Yanoff M, Pitts E, et al. Meesmann’s epithelial dystrophy of the cornea. Am J Ophthalmol. 1977;83:633–642. doi: 10.1016/0002-9394(77)90128-3. [DOI] [PubMed] [Google Scholar]
- 4.Meesmann A. Über eine bisher nicht beschriebene dominant vererbte Dystrophia epithelialis corneae. Ber Zusammenkunft Dtsch Ophthalmol Ges. 1938;52:154–158. [Google Scholar]
- 5.Stocker FW, Holt LB. A rare form of hereditary epithelial dystrophy of the cornea: a genetic, clinical and pathologic study. Trans Am Ophthalmol Soc. 1954;52:133–144. [PMC free article] [PubMed] [Google Scholar]
- 6.Thiel HJ, Behnke H. On the extent of variation of hereditary epithelial corneal dystrophy (Meesmann-Wilke type) Ophthalmologica. 1968;155:81–86. doi: 10.1159/000305266. [DOI] [PubMed] [Google Scholar]
- 7.Tuft S, Bron AJ. Imaging the microstructural abnormalities of Meesmann corneal dystrophy by in vivo confocal microscopy. Cornea. 2006;25:868–870. doi: 10.1097/01.ico.0000251346.00439.a8. [DOI] [PubMed] [Google Scholar]
- 8.Wittebol-Post D, Van-Bijsterveld OP, Delleman JW. Meesmann’s epithelial dystrophy of the cornea. Biometrics and a hypothesis. Ophthalmologica. 1987;194:44–49. doi: 10.1159/000309733. [DOI] [PubMed] [Google Scholar]
Appendix
Lisch Epithelial Corneal Dystrophy (LECD)
MIM: None.
Genetic locus
Xp 22.3.
Gene
Unknown.
Alternative Names, Eponyms
Band-shaped and whorled microcystic dystrophy of the corneal epithelium.
Inheritance
X-chromosomal dominant.
Onset
Childhood.
Signs (Fig. 5)
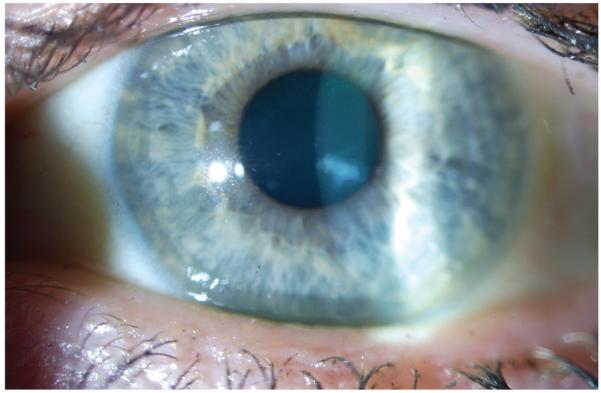
Direct illumination shows localized gray opacities in different patterns: whorl-like, radial, band shaped, flame/feathery shaped, and club shaped. Indirect illumination demonstrates multiple, densely crowded clear cysts. The surrounding epithelium is clear. Similar degree of opacities observed in men and women.
FIGURE 5.

Lisch epithelial corneal dystrophy. A, Localized, whorl-like gray opacity on direct illumination. B, Sclerotic scatter demonstrating localized whorl-like gray opacity. C, Retroillumination demonstrating crowded microcysts.
Symptoms
Asymptomatic or blurred vision if the pupillary zone is involved.
Course
Slow progression of opacities with possible deterioration in vision.
Light Microscopy
Diffuse cytoplasmic vacuolization of all cells in the affected area.
Transmission Electron Microscopy
Extensive vacuolization of the cytoplasm of the affected corneal epithelium. The vacuoles are either optically empty or contain weakly osmiophilic, partly homogenous, and partly lamellar material eventually due to collapsing and coalescing of vacuoles.
Immunohistochemistry
Scattered staining on Ki67 immunohistochemistry indicates no evidence of increased mitotic activity.
Confocal Microscopy
Many solitary dark and well-demarcated lesions (50–100 μm) with round and oval configuration. Some lesions demonstrate central reflective points, which probably correspond to the cell nuclei.
Category
2.
REFERENCES
- 1.Alvarez-Fischer M, Alvarez de Toledo J, Barraquer RI. Lisch corneal dystrophy. Cornea. 2005;24:494–495. doi: 10.1097/01.ico.0000141224.32893.c2. [DOI] [PubMed] [Google Scholar]
- 2.Butros S, Lang GK, Alvarez de Toledo J, et al. Die verschiedenen Trübungsmuster der Lisch-Hornhaut-dystrophie. Klin Monatsbl Augenheilkd. 2006;223:837–840. doi: 10.1055/s-2006-927120. [DOI] [PubMed] [Google Scholar]
- 3.Charles NC, Young JA, Kunar A, et al. Band-shaped and whorled microcystic dystrophy of the corneal epithelium. Ophthalmology. 2000;107:1761–1764. doi: 10.1016/s0161-6420(00)00228-1. [DOI] [PubMed] [Google Scholar]
- 4.Lisch W, Steuhl KP, Lisch C, et al. A new, band-shaped and whorled microcystic dystrophy of the corneal epithelium. Am J Ophthalmol. 1992;114:35–44. doi: 10.1016/s0002-9394(14)77410-0. [DOI] [PubMed] [Google Scholar]
- 5.Lisch W, Büttner A, Offner F, et al. Lisch corneal dystrophy is genetically distinct from Meesmann corneal dystrophy and maps to Xp22.3. Am J Ophthalmol. 2000;130:461–468. doi: 10.1016/s0002-9394(00)00494-3. [DOI] [PubMed] [Google Scholar]
- 6.Robin SB, Epstein RJ, Kornmehl EW. Band-shaped, whorled microcystic corneal dystrophy. Am J Ophthalmol. 1994;117:543–544. doi: 10.1016/s0002-9394(14)70024-8. [DOI] [PubMed] [Google Scholar]
- 7.Rohrbach JM, Grüb M, Szurman P. Einseitige, rezidivierende Trübung des Hornhautepithels. Ophthalmologe. 2007;104:72–74. doi: 10.1007/s00347-006-1333-8. [DOI] [PubMed] [Google Scholar]
Appendix
Gelatinous Drop-Like Corneal Dystrophy (GDLD)
MIM #204870.
Alternative Names, Eponyms
Subepithelial amyloidosis.
Primary familial amyloidosis (Grayson).
Genetic Locus
1p32.
Gene
Tumor-associated calcium signal transducer 2 (TACSTD2, previously M1S1).
Inheritance
Autosomal recessive.
Onset
First to second decade.
Signs (Fig. 6)
Initially, the subepithelial lesions may appear similar to band-shaped keratopathy or there may be groups of small multiple nodules, that is, mulberry configuration. These lesions demonstrate late staining with fluorescein, indicating extremely hyperpermeable corneal epithelium. Superficial vascularization is frequently seen. In later life, patients may also develop stromal opacification or develop larger nodular lesions, that is, kumquat-like lesions.
Symptoms
Significant decrease in vision, photophobia, irritation, redness, and tearing.
Course
Progression of protruding subepithelial deposits and stromal opacity. Almost all patients develop recurrence after superficial keratectomy, lamellar keratoplasty, or penetrating keratoplasty, typically within a few years.
Light Microscopy
Subepithelial and stromal amyloid deposits.
Transmission Electron Microscopy
Disruption of epithelial tight junctions in the superficial epithelium. Amyloid is noted in the basal epithelial layer.
Confocal Microscopy
Not reported.
FIGURE 6.

Gelatinous drop-like corneal dystrophy. A, Mulberry type. B, Band keratopathy type. C, Kumquat-like type.
Category
1.
REFERENCES
- 1.Ide T, Nishida K, Maeda N, et al. A spectrum of clinical manifestations of gelatinous drop-like corneal dystrophy in Japan. Am J Ophthalmol. 2004;137:1081–1084. doi: 10.1016/j.ajo.2004.01.048. [DOI] [PubMed] [Google Scholar]
- 2.Kinoshita S, Nishida K, Dota A, et al. Epithelial barrier function and ultrastructure of gelatinous drop-like corneal dystrophy. Cornea. 2000;19:551–555. doi: 10.1097/00003226-200007000-00029. [DOI] [PubMed] [Google Scholar]
- 3.Klintworth GK, Valnickova Z, Kielar RA, et al. Familial subepithelial corneal amyloidosis —a lactoferrin-related amyloidosis. Invest Ophthalmol Vis Sci. 1997;38:2756–2763. [PubMed] [Google Scholar]
- 4.Nakaizumi GA. A rare case of corneal dystrophy. Acta Soc Ophthalmol Jpn. 1914;18:949–950. [Google Scholar]
- 5.Ren Z, Lin PY, Klintworth GK, et al. Allelic and locus heterogeneity in autosomal recessive gelatinous drop-like corneal dystrophy. Hum Genet. 2002;110:568–577. doi: 10.1007/s00439-002-0729-z. [DOI] [PubMed] [Google Scholar]
- 6.Tsujikawa M, Kurahashi H, Tanaka T, et al. Identification of the gene responsible for gelatinous drop-like corneal dystrophy. Nat Genet. 1999;21:420–423. doi: 10.1038/7759. [DOI] [PubMed] [Google Scholar]
- 7.Yoshida S, Kumano Y, Yoshida A, et al. Two brothers with gelatinous drop-like dystrophy at different stages of the disease: role of mutational analysis. Am J Ophthalmol. 2002;133:830–832. doi: 10.1016/s0002-9394(02)01407-1. [DOI] [PubMed] [Google Scholar]
Appendix
BOWMAN LAYER DYSTROPHIES
Reis–Bücklers Corneal Dystrophy (RBCD)
MIM #608470
Alternative Names, Eponyms
Corneal Dystrophy of Bowman layer, type I (CDB I).
Geographic corneal dystrophy (Weidle).
Superficial granular corneal dystrophy.
Atypical granular corneal dystrophy.
Granular corneal dystrophy, type 3.
Anterior limiting membrane dystrophy, type I (ALMD I).
Genetic Locus
5q31.
Gene
TGFBI.
Inheritance
Autosomal dominant.
Onset
Childhood.
Signs (Fig. 7)
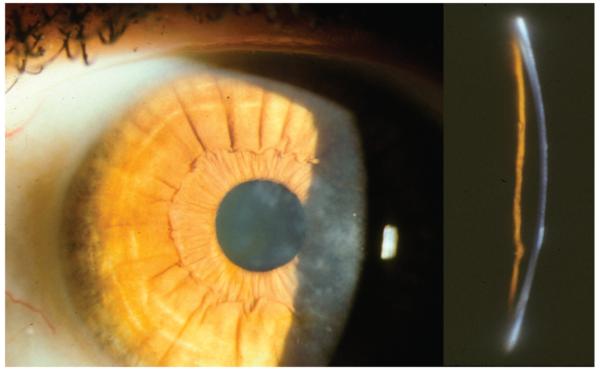
Confluent irregular and coarse geographic-like opacities with varying densities develop at the level of Bowman layer and superficial stroma, initially separated from one another. Opacities may extend to the limbus and deeper stroma with time. Can be confused with TBCD.
Symptoms
Vision is impaired from childhood. Recurrent corneal erosions cause ocular discomfort and pain in the first decade but may become less severe from the end of the second decade. Erosions are typically more frequent and severe than in TBCD.
Course
Slowly progressive deterioration of vision. Recurrent corneal erosions may resolve with time. Similar but frequently more aggressive course than TBCD but may not be able to distinguish in an individual case.
Light Microscopy
Bowman layer is replaced by a sheet-like connective tissue layer with granular Masson trichrome–red deposits, which in advanced cases can extend to subepithelial stroma.
Transmission Electron Microscopy
Subepithelial electron-dense, rod-shaped bodies identical to those in GCD1, but not the curly fibers of TBCD, are observed on electron microscopy. Electron microscopy is necessary for definitive histopathologic diagnosis to distinguish from TBCD.
Confocal Microscopy
Distinct deposits are found in the epithelium and Bowman layer. The deposits in the basal epithelial cell layer show extremely high reflectivity from small granular material without any shadows. Bowman layer is replaced by highly reflective irregular material, even more reflective than in TBCD (5q31). Fine diffuse deposits may be noted in the anterior stroma.
Immunohistochemistry
Rod-shaped bodies are immunopositive for transforming growth factor beta–induced protein (keratoepithelin).
Category
1.
FIGURE 7.

Reis–Bücklers corneal dystrophy. A, Coarse geographic opacity of the superficial cornea. B, Broad oblique illumination demonstrating dense, reticular, superficial opacity. C, Slit lamp view demonstrating irregularities in Bowman layer.
REFERENCES
- 1.Bücklers M. Über eine weitere familiäre Hornhaut-dystrophie (Reis) Klin Monatsbl Augenkeilkd. 1949;114:386–397. [Google Scholar]
- 2.Kobayashi A, Sugiyama K. In vivo laser confocal microscopy findings for Bowman’s layer dystrophies (Thiel-Behnke and Reis-Bücklers corneal dystrophies) Ophthalmology. 2007;114:69–75. doi: 10.1016/j.ophtha.2006.05.076. [DOI] [PubMed] [Google Scholar]
- 3.Konishi M, Yamada M, Nakamura Y, et al. Immunohistology of keratoepithelin in corneal stromal dystrophies associated with R124 mutations of the BIGH3 gene. Curr Eye Res. 2000;21:891–896. doi: 10.1076/ceyr.21.5.891.5536. [DOI] [PubMed] [Google Scholar]
- 4.Küchle M, Green WR, Völcker HE, et al. Reevaluation of corneal dystrophies of Bowman’s layer and the anterior stroma (Reis-Bücklers and Thiel-Behnke types): a light and electron microscopic study of eight corneas and a review of the literature. Cornea. 1995;14:333–354. doi: 10.1097/00003226-199507000-00001. [DOI] [PubMed] [Google Scholar]
- 5.Munier FL, Korvatska E, Djemai A, et al. Keratoepithelin mutations in four 5q31-linked corneal dystrophies. Nat Genet. 1997;15:247–251. doi: 10.1038/ng0397-247. [DOI] [PubMed] [Google Scholar]
- 6.Reis W. Familiäre, fleckige Hornhautentartung. Dtsch Med Wochenschr. 1917;43:575. [Google Scholar]
- 7.Ridgway AE, Akhtar S, Munier FL, et al. Ultrastructural and molecular analysis of Bowman’s layer corneal dystrophies: an epithelial origin? Invest Ophthalmol Vis Sci. 2000;41:3286–3292. [PubMed] [Google Scholar]
- 8.Small KW, Mullen L, Barletta J, et al. Mapping of Reis-Bücklers corneal dystrophy to chromosome 5q. Am J Ophthalmol. 1996;121:384–390. doi: 10.1016/s0002-9394(14)70434-9. [DOI] [PubMed] [Google Scholar]
- 9.Stone EM, Mathers WD, Rosenwasser GO, et al. Three autosomal dominant corneal dystrophies map to chromosome 5q. Nat Genet. 1994;6:47–51. doi: 10.1038/ng0194-47. [DOI] [PubMed] [Google Scholar]
- 10.Streeten BW, Qi Y, Klintworth GK, et al. Immunolocalization of beta ig-h3 protein in 5q31-linked corneal dystrophies and normal corneas. Arch Ophthalmol. 1999;117:67–75. doi: 10.1001/archopht.117.1.67. [DOI] [PubMed] [Google Scholar]
- 11.Weidle EG. Klinische und feingewebliche Abgrenzung der Reis-Bücklers‘schen Hornhaut-dystrophie. Klin Monatsbl Augenheilkd. 1989;194:217–226. doi: 10.1055/s-2008-1046362. [DOI] [PubMed] [Google Scholar]
- 12.Wittbol-Post D, Pels E. The dystrophy described by Reis and Bücklers. Ophthalmologica. 1989;199:1–9. doi: 10.1159/000310006. [DOI] [PubMed] [Google Scholar]
Appendix
Thiel–Behnke Corneal Dystrophy (TBCD)
MIM #602082
Alternative Names, Eponyms
Corneal dystrophy of Bowman layer, type II (CDB2).
Honeycomb-shaped corneal dystrophy.
Anterior limiting membrane dystrophy, type II.
Curly fibers corneal dystrophy.
Waardenburg–Jonkers corneal dystrophy.
Genetic Loci
5q31.
10q24.
Gene
5q31: TGFBI.
10q24: Unknown.
Inheritance
Autosomal dominant.
Onset
Childhood.
Signs (Fig. 8)
Symmetrical subepithelial reticular (honeycomb) opacities with peripheral cornea typically uninvolved. Variety of opacification patterns may make it impossible to distinguish from RBCD in early or individual cases. Opacities can progress to deep stromal layers and corneal periphery.
Symptoms
Recurrent corneal erosions cause ocular discomfort and pain in the first and second decade. Gradual visual impairment develops later. Erosions are less frequent, and the onset of visual impairment is later than in RBCD.
Course
Slowly progressive deterioration of vision from increasing corneal opacification. Recurrent corneal erosions may resolve with time. Similar but frequently less aggressive course than RBCD but may not be able to distinguish in an individual case.
Light Microscopy
Irregular thickening of the epithelial layer to allow for ridges and furrows of underlying stroma, with focal absences of epithelial basement membrane. Bowman layer is replaced by a fibrocellular layer between epithelium and stroma with a pathognomonic wavy saw-toothed pattern.
Transmission Electron Microscopy
Presence of curly collagen fibers with a diameter of 9–15 nm is pathognomonic and distinguishes this dystrophy from RBCD.
Confocal Microscopy
Distinct deposits are found in the epithelium and Bowman layer. The deposits in the basal epithelial cell layer show homogeneous reflectivity with round edges accompanying dark shadows. Bowman layer is replaced with reflective irregular material that is less reflective than in RBCD.
Immunohistochemistry
Curly fibers are immunopositive for transforming growth factor beta–induced protein (keratoepithelin) in TBCD (5q31).
FIGURE 8.

Thiel–Behnke corneal dystrophy. A, Reticular honeycomb pattern of Thiel–Behnke with genetic confirmation of Arg555Gin in TGFBI. B, Superficial opacification in advanced disease.
Category
1. (TGFBI).
2. (10q24).
REFERENCES
- 1.Kobayashi A, Sugiyama K. In vivo laser confocal microscopy findings for Bowman’s layer dystrophies (Thiel-Behnke and Reis-Bücklers corneal dystrophies) Ophthalmology. 2007;114:69–75. doi: 10.1016/j.ophtha.2006.05.076. [DOI] [PubMed] [Google Scholar]
- 2.Küchle M, Green WR, Völcker HE, et al. Reevaluation of corneal dystrophies of Bowman’s layer and the anterior stroma (Reis-Bücklers and Thiel-Behnke types): a light and electron microscopic study of eight corneas and a review of the literature. Cornea. 1995;14:333–354. doi: 10.1097/00003226-199507000-00001. [DOI] [PubMed] [Google Scholar]
- 3.Lohse E, Stock EL, Jones JC, et al. Reis-Bücklers corneal dystrophy. Immunofluorescent and electron microscopic studies. Cornea. 1989;8:200–209. [PubMed] [Google Scholar]
- 4.Munier FL, Korvatska E, Djemai A, et al. Keratoepithelin mutations in four 5q31-linked corneal dystrophies. Nat Genet. 1997;15:247–251. doi: 10.1038/ng0397-247. [DOI] [PubMed] [Google Scholar]
- 5.Ridgway AE, Akhtar S, Munier FL, et al. Ultrastructural and molecular analysis of Bowman’s layer corneal dystrophies: an epithelial origin? Invest Ophthalmol Vis Sci. 2000;41:3286–3292. [PubMed] [Google Scholar]
- 6.Streeten BW, Qi Y, Klintworth GK, et al. Immunolocalization of beta ig-h3 protein in 5q31-linked corneal dystrophies and normal corneas. Arch Ophthalmol. 1999;117:67–75. doi: 10.1001/archopht.117.1.67. [DOI] [PubMed] [Google Scholar]
- 7.Thiel HJ, Behnke H. Eine bisher unbekannte subepitheliale hereditäre Hornhautdystrophie. Klin Monatsbl Augenheilkd. 1967;150:862–874. [PubMed] [Google Scholar]
- 8.Weidle EG. Die wabenförmige Hornhautdystrophie (Thiel-Behnke) Neubewertung und Abgrenzung gegenüber der Reis-Bücklerschen Hornhautdystrophie. Klin Monatsbl Augenheilkd. 1999;214:125–135. doi: 10.1055/s-2008-1034764. [DOI] [PubMed] [Google Scholar]
- 9.Wittebol-Post D, Van Schooneveld MJ. The corneal dystrophy of Waardenburg and Jonkers. Ophthalmic Paediatr Genet. 1989;10:249–255. doi: 10.3109/13816818909009879. [DOI] [PubMed] [Google Scholar]
- 10.Yee RW, Sullivan LS, Lai HT, et al. Linkage mapping of Thiel-Behnke corneal dystrophy (CDB2) to chromosome 10q23-q24. Genomics. 1997;46:152–154. doi: 10.1006/geno.1997.5028. [DOI] [PubMed] [Google Scholar]
Appendix
Grayson–Wilbrandt Corneal Dystrophy (GWCD)
MIM: None.
Alternative Names, Eponyms
None.
Genetic Locus
Unknown.
Gene
Unknown.
Inheritance
Autosomal dominant.
Onset
First to second decade.
Signs (Fig. 9)
Bowman layer demonstrates variable patterns of opacification from diffuse mottling to diffuse gray-white opacities, which extend anteriorly into the epithelium. The cornea between the deposits is clear. Refractile bodies are described in corneal stroma.
Symptoms
Decreased to normal visual acuity. Recurrent corneal erosions are less severe than in RBCD and TBCD.
Course
Progressive.
Light Microscopy
Homogeneous eosin-staining material between Bowman layer and the epithelium, which does not stain with Alcian blue or Masson trichrome stains but is positive for Periodic acid – Schiff.
Transmission Electron Microscopy
Not reported.
Confocal Microscopy
Not reported.
Category
4.
Note: There is only 1 publication describing a single family. The report does not allow definitive diagnosis or exclusion of the theory that this dystrophy may have been a dystrophy of Bowman layer or a variant of EBMD.
FIGURE 9.

Grayson–Wilbrandt corneal dystrophy. A, Irregularly shaped opacities scattered throughout the entire corneal surface best seen in diffuse illumination. B, Irregular opacities from Bowman layer extending into and involving the epithelium with prominent corneal nerves (images reprinted with permission from the American Journal of Ophthalmology 1966;61:345–349).
REFERENCES
- 1.Grayson M, Wilbrandt H. Dystrophy of the anterior limiting membrane of the cornea (Reis-Bücklers type) Am J Ophthalmol. 1966;61:345–349. doi: 10.1016/0002-9394(66)90296-0. [DOI] [PubMed] [Google Scholar]
Appendix
STROMAL DYSTROPHIES
TGFBI Dystrophies
Lattice Corneal Dystrophy, TGFBI Type (LCD): Classic Lattice Corneal Dystrophy (LCD1) and Variants
MIM #122200.
Alternative Names, Eponyms
Classic LCD.
LCD, type 1.
Biber-Haab-Dimmer.
Genetic Locus
5q31.
Gene
TGFBI
Inheritance
Autosomal dominant.
Onset
First decade.
Signs (Fig. 10)
Thin branching refractile lines and/or subepithelial, whitish, ovoid dots usually appear by the end of the first decade. The lines start centrally and more superficially, spreading centrifugally and deeply, but leaving the peripheral 1 mm, and Descemet membrane and endothelium clear. A diffuse stromal, ground-glass haze usually develops later, accompanied by recurrent erosions. The number of lattice lines may differ between the 2 eyes (unilateral cases are described), and the dystrophy may be difficult to diagnose in some younger patients.
Symptoms
Ocular discomfort, pain, and visual impairment, sometimes starting as early as in the first decade of life. Recurrent erosions are frequent. Visual impairment within the fourth decade.
Course
Progressive, often leading to keratoplasty within the fourth decade of life.
Light Microscopy
Epithelial atrophy and disruption with degeneration of basal epithelial cells; focal thinning or absence of Bowman layer, progressively increasing with age; eosinophilic layer between epithelial basement membrane and Bowman layer; and stromal deposition of amyloid substance distorts the architecture of corneal lamellae. Amyloid deposits have characteristic staining. Deposits stain positive with Congo red. Green birefringence is visible with a polarizing filter and red-green dichroism when a green filter is added with this stain. Metachromasia is noted with crystal violet and fluorescence is noted with use of thioflavin T staining.
FIGURE 10.

Lattice corneal dystrophy, TGFBI type (classic lattice). A, Early lattice corneal dystrophy (LCD1) with dots and lattice lines in retroillumination with genetic confirmation of Arg124Cys in TGFBI. B, Magnified view of lattice lines and dots in LCD1. C, Central opacification in advanced LCD1.
Transmission Electron Microscopy
Extracellular masses of fine, electron-dense, randomly aligned fibrils with a diameter of 8–10 nm. There are fewer keratocytes in the areas of amyloid deposition: Some are degenerated with cytoplasmic vacuolization, whereas others appear metabolically active. Descemet membrane and endothelium are normal.
Confocal Microscopy
Linear and branching structures in the stroma with changing reflectivity and poorly demarcated margins. Lines must be differentiated from other similar images (ie, fungi).
Category
1.
Note: Historically, multiple subtypes of lattice were created on the basis of phenotypic and genotypic variations. The LCD variants are caused by more than 2 dozen distinct heterozygous amyloidogenic mutations, nearly all of which are located in the fourth FAS1 domain of TGFBI. LCD variants (type IIIA, I/IIIA, IV, and polymorphic amyloidosis) have a delayed onset compared with classic LCD (LCD, type 1). The lattice lines may be larger, with a limbus to limbus ropy appearance (type IIIA), thinner (type I/IIIA), or even absent (polymorphic amyloidosis), although one has to keep in mind that the lattice pattern is very much dependent on age. Corneal erosions are a typical presenting sign of LCD, type IIIA and I/IIIA, but are virtually absent in LCD, type IV and polymorphic corneal amyloidosis. This erosive semiology likely reflects the anterior to posterior (type IIIA and I/IIIA) or posterior to anterior (type IV) progression of the dystrophy.
REFERENCES
- 1.Biber H. Inaugural dissertation. Zurich: 1890. Ueber einige seltene Hornhautkrankungen:die oberflächliche gittrige Keratitis. [Google Scholar]
- 2.Chiou AG, Beuermann RW, Kaufman SC, et al. Confocal microscopy in lattice corneal dystrophy. Graefes Arch Clin Exp Ophthalmol. 1999;237:7697–7701. doi: 10.1007/s004170050299. [DOI] [PubMed] [Google Scholar]
- 3.Dighiero P, Drunat S, Ellies P, et al. A new mutation (A546T) of the βig-h3 gene responsible for a French lattice corneal dystrophy type IIIA. Am J Ophthalmol. 2000;129:248–251. doi: 10.1016/s0002-9394(99)00324-4. [DOI] [PubMed] [Google Scholar]
- 4.Dighiero P, Niel F, Ellies P, et al. Histologic phenotype-genotype correlation of corneal dystrophies associated with eight distinct mutations in the TGFBI gene. Ophthalmology. 2001;108:818–823. doi: 10.1016/s0161-6420(00)00662-x. [DOI] [PubMed] [Google Scholar]
- 5.Ellies P, Renard G, Valleix S, et al. Clinical outcome of eight BIGH3-linked corneal dystrophies. Ophthalmology. 2002;109:793–797. doi: 10.1016/s0161-6420(01)01025-9. [DOI] [PubMed] [Google Scholar]
- 6.Fujiki K, Hotta Y, Nakayasu K, et al. A new L527R mutation of the betaIGH3 gene in patients with lattice corneal dystrophy with deep stromal opacities. Hum Genet. 1998;103:286–289. doi: 10.1007/s004390050818. [DOI] [PubMed] [Google Scholar]
- 7.Funayama T, Mashima Y, Kawashima M, et al. Lattice corneal dystrophy type III in patients with a homozygous L527R mutation in the TGFBI gene. Jpn J Ophthalmol. 2006;50:62–64. doi: 10.1007/s10384-005-0260-6. [DOI] [PubMed] [Google Scholar]
- 8.Hida T, Proia AD, Kigasawa K, et al. Histopathologic and immunochemical features of lattice corneal dystrophy type III. Am J Ophthalmol. 1987;104:249–254. doi: 10.1016/0002-9394(87)90412-0. [DOI] [PubMed] [Google Scholar]
- 9.Hotta Y, Fujiki K, Ono K, et al. Arg124Cys mutation of the betaig-h3 gene in a Japanese family with lattice corneal dystrophy type I. Jpn J Ophthalmol. 1998;42:450–455. doi: 10.1016/s0021-5155(98)00050-1. [DOI] [PubMed] [Google Scholar]
- 10.Kannabiran C, Klintworth GK. TGFBI gene mutations in corneal dystrophies. Hum Mut. 2006;27:615–625. doi: 10.1002/humu.20334. [DOI] [PubMed] [Google Scholar]
- 11.Kawamoto K, Morishige N, Yamada N, et al. Delayed corneal epithelial wound healing after penetrating keratoplasty in individuals with lattice corneal dystrophy. Am J Ophthalmol. 2006;142:173–174. doi: 10.1016/j.ajo.2006.01.077. [DOI] [PubMed] [Google Scholar]
- 12.Klintworth GB, Bao W, Afshari NA, et al. Two mutations in the TGFBI (BIGH3) gene associated with lattice corneal dystrophy in an extensively studied family. Invest Ophthalmol Vis Sci. 2004;45:1382–1388. doi: 10.1167/iovs.03-1228. [DOI] [PubMed] [Google Scholar]
- 13.Munier FL, Korvatska E, Djemai A, et al. Keratoepithelin mutations in four 5q31-linked corneal dystrophies. Nat Genet. 1997;15:247–251. doi: 10.1038/ng0397-247. [DOI] [PubMed] [Google Scholar]
- 14.Munier FL, Frueh BE, Othenin-Girard P, et al. BIGH3 mutation spectrum in corneal dystrophies. Invest Ophthalmol Vis Sci. 2002;43:949–954. [PubMed] [Google Scholar]
- 15.Nakagawa AS, Fujiki K, Enomoto Y, et al. Case of late onset and isolated lattice corneal dystrophy with Asn544Ser (N544S) mutation of transforming growth factor beta-induced (TGFBI, BIGH3) gene. Nippon Ganka Gakkai Zasshi. 2004;108:618–620. [PubMed] [Google Scholar]
- 16.Schmitt-Bernard CF, Guittard C, Arnaud B, et al. BIGH3 exon 14 mutations lead to intermediate type I/IIIA of lattice corneal dystrophies. Invest Ophthalmol Vis Sci. 2000;41:1302–1308. [PubMed] [Google Scholar]
- 17.Seitz B, Weidle E, Naumann GOH. Einseitige gittrige stromale Hornhaut-dystrophie Typ III (Hida) Klin Monatsbl Augenheilkd. 1993;203:279–285. doi: 10.1055/s-2008-1045681. [DOI] [PubMed] [Google Scholar]
- 18.Snead DR, Mathews BN. Differences in amyloid deposition in primary and recurrent corneal lattice dystrophy type 1. Cornea. 2002;21:308–311. doi: 10.1097/00003226-200204000-00014. [DOI] [PubMed] [Google Scholar]
- 19.Stewart H, Black GCM, Donnai D, et al. A mutation within exon 14 of the TGFBI (BIGH3) gene on chromosome 5q31 causes an asymmetric, late-onset form of lattice corneal dystrophy. Ophthalmology. 1999;106:964–970. doi: 10.1016/S0161-6420(99)00539-4. [DOI] [PubMed] [Google Scholar]
- 20.Stock EL, Feder RS, O’Grady RB, et al. Lattice corneal dystrophy type IIIA. Clinical and histopathologic correlations. Arch Ophthalmol. 1991;109:354–358. doi: 10.1001/archopht.1991.01080030056038. [DOI] [PubMed] [Google Scholar]
- 21.Stix B, Leber M, Bingemer P, et al. Hereditary lattice corneal dystrophy is associated with corneal amyloid deposits enclosing C-terminal fragments of keratoepithelin. Invest Ophthalmol Vis Sci. 2005;46:1133–1139. doi: 10.1167/iovs.04-1319. [DOI] [PubMed] [Google Scholar]
- 22.Suesskind D, Auw-Haedrich C, Schorderet DF, et al. Keratoepithelin in secondary corneal amyloidosis. Graefes Arch Clin Experiment Ophthalmol. 2006;244:725–731. doi: 10.1007/s00417-005-0153-x. [DOI] [PubMed] [Google Scholar]
- 23.Tian X, Fujiki K, Wang W, et al. Novel mutation (V505D) of the TGFBI gene found in a Chinese family with lattice corneal dystrophy, type I. Jpn J Ophthalmol. 2005;49:84–88. doi: 10.1007/s10384-004-0167-7. [DOI] [PubMed] [Google Scholar]
- 24.Tsujikawa K, Tsujikawa M, Yamamoto S, et al. Allelic homogeneity due to a founder mutation in Japanese patients with lattice corneal dystrophy type IIIA. Am J Med Genet. 2002;113:20–22. doi: 10.1002/ajmg.10709. [DOI] [PubMed] [Google Scholar]
- 25.Yamada N, Chikama TI, Morishige N, et al. Homozygous mutation (L527R) of TGFBI in an individual with lattice corneal dystrophy. Br J Ophthalmol. 2005;89:771–773. doi: 10.1136/bjo.2004.056168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yamamoto S, Okada M, Tsujikawa M, et al. A kerato-epithelin (betaig-h3) mutation in lattice corneal dystrophy type IIIA. Am J Hum Genet. 1998;62:719–722. doi: 10.1086/301765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yamamoto S, Okada M, Tsujikawa M, et al. The spectrum of beta ig-h3 gene mutations in Japanese patients with corneal dystrophy. Cornea. 2000;19:S21–S23. doi: 10.1097/00003226-200005001-00005. [DOI] [PubMed] [Google Scholar]
Appendix
Lattice Corneal Dystrophy, Gelsolin Type (LCD2) (see note below)
MIM #105120
Genetic Locus
9q34.
Gene
Gelsolin GSN (See note below).
Alternative Names, Eponyms
Part of
Familial amyloidosis, Finnish (FAF).
Meretoja syndrome.
Amyloidosis V.
Familial amyloidotic polyneuropathy IV (FAP-IV).
Inheritance
Autosomal dominant.
Onset
Third to fourth decade.
Signs (Fig. 11)
Lattice lines, more peripheral and less numerous than those of lattice dystrophy, type I, appear in the corneal stroma, spreading centripetally from the limbus. The central cornea is relatively spared. Pronounced dermatochalasis is typical and lagophthalmos common later in life. Risk of open angle glaucoma may be increased.
FIGURE 11.

Lattice corneal dystrophy, gelsolin type (Meretoja). A, Diffuse lattice lines of the stroma. B, Typical facies of the Meretoja syndrome.
Systemic signs
Cranial neuropathy, manifesting as facial paresis, bulbar palsy, and laxity of the facial skin. Gradual onset of facial drooping, causing eyebrows to fall over eyes, lagophthalmos, drooping of lower lip with drooling. Peripheral polyneuropathy affects mainly senses of vibration and touch. Carpal tunnel syndrome. Autonomic disturbance includes orthostatic hypotension, cardiac conduction abnormalities, and dysfunction of perspiration.
Symptoms
Ocular: Corneal sensitivity is reduced or absent. Visual acuity is usually normal until the sixth decade because the dystrophy progresses from the peripheral to central cornea. Dry eye symptoms are frequent, and corneal erosions may occur late in life.
Course
Slowly progressive, the majority are in good health still in the seventh decade.
Variant
In rare homozygotes, the systemic component is severe, manifesting with nephrotic syndrome and renal failure from heavy glomerular amyloid deposits.
Light Microscopy
Amyloid is deposited in the cornea in lattice lines, as a discontinuous band under Bowman layer and within the sclera. Streak-like deposits are seen between corneal lamellae, especially in the limbal cornea.
Immunohistochemistry
Deposition of mutated gelsolin is detected in the conjunctiva, in the sclera, in the stroma of the ciliary body, along the choriocapillaris, in the perineurium of ciliary nerves, in the walls of ciliary vessels, and in the optic nerve. Extraocularly, amyloid is found in arterial walls, peripheral nerves and glomeruli.
Confocal Microscopy
Prominent deposits, presumably amyloid, are seen contiguous to basal epithelial cells and stromal nerves. In severely affected corneas, sub-basal and stromal nerves are reduced or absent. Anterior stroma shows fibrosis and abnormal extracellular matrix. Thick anterior and midstromal filaments corresponding to lattice lines and thin undulating structures are visible.
Category
1.
Note: This is not a true corneal dystrophy but is listed here because it can be confused with true lattice dystrophies, which in turn may delay diagnosis of the underlying systemic amyloidosis for many years, especially in populations in which this type of familial amyloidosis is rare.
REFERENCES
- 1.Levy E, Haltia M, Fernandez-Madrid I, et al. Mutation in gelsolin gene in Finnish hereditary amyloidosis. J Exp Med. 1990;172:1865–1867. doi: 10.1084/jem.172.6.1865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kiuru S. Gelsolin-related familial amyloidosis, Finnish type (FAF), and its variants found worldwide. Amyloid. 1998;5:55–66. doi: 10.3109/13506129809007291. [DOI] [PubMed] [Google Scholar]
- 3.Kivelä T, Tarkkanen A, McLean I, et al. Immunohistochemical analysis of lattice corneal dystrophies types I and II. Br J Ophthalmol. 1993;77:799–804. doi: 10.1136/bjo.77.12.799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kivelä T, Tarkkanen A, Frangione B, et al. Ocular amyloid deposition in familial amyloidosis, Finnish: an analysis of native and variant gelsolin in Meretoja’s syndrome. Invest Ophthalmol Vis Sci. 1994;35:3759–3769. [PubMed] [Google Scholar]
- 5.Meretoja J. MD thesis. University of Helsinki; Helsinki: 1973. Inherited Systemic Amyloidosis with Lattice Corneal Dystrophy. [Google Scholar]
- 6.Rosenberg ME, Tervo TM, Gallar J, et al. Corneal morphology and sensitivity in lattice dystrophy type II (familial amyloidosis, Finnish type) Invest Ophthalmol Vis Sci. 2001;42:634–641. [PubMed] [Google Scholar]
Appendix
Granular Corneal Dystrophy, Type 1 (Classic) (GCD1)
MIM #121900.
Alternative Names, Eponyms
Corneal dystrophy Groenouw type I.
Genetic Locus
5q31.
Gene
TGFBI
FIGURE 12.

Granular corneal dystrophy, type 1. A, Discrete and confluent, axially distributed anterior stromal deposits. B, Diffuse granular opacities in an adult. C, Early subepithelial verticillate opacity in a 6-year old.
Inheritance
Autosomal dominant.
Onset
Childhood, may be seen as early as 2 years of age.
Signs (Fig. 12)
Slit lamp examination reveals well-defined granules that appear white on direct illumination. On retroillumination, these granules are composed of extremely small, translucent dots with the appearance of vacuoles, glassy splinters, or crushed bread crumbs. Opacities do not extend to the limbus. In children, there may be a vortex pattern of brownish granules superficial to Bowman layer. In later life, granules may extend into the deeper stroma down to Descemet membrane. Homozygotes have more severe manifestations.
Symptoms
Glare and photophobia are early symptoms. Visual acuity decreases as opacification progresses with age. Recurrent erosions are seen frequently. Homozygote has more severe symptoms.
Course
As the condition progresses, the opacities become more confluent in the superficial cornea, resulting in a significant reduction of visual acuity.
Light Microscopy
Multiple stromal deposits may extend from deep epithelium to Descemet membrane. The hyaline opacities stain with Masson trichrome.
Transmission Electron Microscopy
Rod-shaped bodies are found, which are similar in appearance to those in RBCD.
Immunohistochemistry
Abnormal deposits react with antibodies to transforming growth factor beta–induced protein (keratoepithelin).
Confocal Microscopy
Hyper-reflective opacities.
Category
1.
REFERENCES
- 1.Bücklers M. Die erblichen Hornhautdystrophie. Klin Monatsbl Augenheilkd. 1938;3:1–135. Beiheft. [Google Scholar]
- 2.Eiberg E, Møller HU, Berendt I, et al. Assignment of granular corneal dystrophy Groenouw type I (CDGG1) to Chromosome 5q. Eur J Hum Genet. 1994;2:132–138. doi: 10.1159/000472353. [DOI] [PubMed] [Google Scholar]
- 3.Groenouw A. Knötchenförmige Hornhauttrübungen (Noduli corneae) Arch Augenheilkd. 1890;21:281–289. [Google Scholar]
- 4.Jones ST, Zimmerman LE. Histopathologic differentiation of granular, macular and lattice dystrophies of the cornea. Am J Ophthalmol. 1961;51:394–410. doi: 10.1016/0002-9394(61)92085-2. [DOI] [PubMed] [Google Scholar]
- 5.Matsuo N, Fujiwara H, Ofuchi Y. Electron and light microscopic observations of a case of Groenouw’s nodular corneal dystrophy. Folia Ophthalmol Jpn. 1967;18:436–447. [PubMed] [Google Scholar]
- 6.Møller HU. Granular corneal dystrophy Groenouw type I. Clinical and genetic aspects. Acta Ophthalmol. 1991;69(suppl 198):1–40. [PubMed] [Google Scholar]
- 7.Munier FL, Korvatska E, Djemai A, et al. Kerato-epithelin mutations in four 5q31-linked corneal dystrophies. Nat Genet. 1997;15:247–251. doi: 10.1038/ng0397-247. [DOI] [PubMed] [Google Scholar]
- 8.Seitz B, Behrens A, Langenbucher A, et al. Morphometric analysis of deposits in granular and lattice corneal dystrophy — histopathologic implications for phototherapeutic keratectomy. Cornea. 2004;23:380–385. doi: 10.1097/00003226-200405000-00013. [DOI] [PubMed] [Google Scholar]
- 9.Stone EM, Mathers WD, Rosenwasser GOD, et al. Three autosomal dominant corneal dystrophies map to chromosome 5q. Nat Genet. 1994;6:47–51. doi: 10.1038/ng0194-47. [DOI] [PubMed] [Google Scholar]
- 10.Weidle EG, Lisch W. Die verschiedenen Trübungsformen der bröckeligen Hornhaut-dystrophie. Klin Monatsbl Augenheilkd. 1984;185:167–173. doi: 10.1055/s-2008-1054592. [DOI] [PubMed] [Google Scholar]
- 11.Wittebol-Post D, van der Want JJL, van Bijsterveld OP. Granular dystrophy of the cornea (Groenouw’s type I) Ophthalmologica. 1987;195:169–177. doi: 10.1159/000309808. [DOI] [PubMed] [Google Scholar]
Appendix
Granular Corneal Dystrophy, Type 2 (Granular-Lattice) (GCD2)
MIM #607541.
Alternative Names, Eponyms
Combined granular–lattice corneal dystrophy.
Avellino corneal dystrophy.
FIGURE 13.

Granular corneal dystrophy, type 2 (granular–lattice). A, Icicle and star-shaped stromal opacities among disk-shaped opacities in a heterozygote with histopathologic confirmation of granular corneal dystrophy, type 2 (GCD2), and genetic confirmation of R124H mutation. B, Finger-like stars and disks in diffuse and retroillumination. C, Seventeen-year old with few white dots and a family history of GCD2. D, Homozygote with denser and confluent opacities.
For close to 100 years, this entity was considered a mild variety of granular corneal dystrophy (Groenouw type I). Bücklers, as early as 1938, described a large family with illustrative pictures of this phenotype. Fifty years later, Weidle published the same patients and subdivided granular dystrophy according to subtle differences of clinical appearance. In 1988, Folberg et al described the histopathology of deposition of both amyloid and hyaline deposits in these patients. In 1992, the clinical findings of these patients were published. The combined granular corneal dystrophy–LCD was now called Avellino dystrophy. Avellino, which is the Italian district of the progenitor of the pedigree, became the popular name that appears in most modern textbooks to describe the granular-lattice findings.
Genetic Locus
5q31.
Gene
TGFBI
Inheritance
Autosomal dominant.
Onset
Homozygous patients have earlier onset with dystrophy diagnosed, as early as 3 years of age, compared with heterozygote patients, who may be diagnosed as early as the age of 8 years. Most often, GCD2 is diagnosed during teens or during early adulthood.
Signs (Fig. 13)
Initial slit lamp signs are subtle superficial stromal tiny whitish dots. In the next stage, rings or stellate-shaped snowflake stromal opacities appear between the superficial stroma and the mid stroma. Some patients may also demonstrate lattice lines in deeper cornea. Typically, these lines are located deeper than the snowflake stromal opacity. In the final stage, there is a more superficial, translucent flattened breadcrumb opacity, which may coalesce in the anterior stroma. Some patients only manifest multiple white dots. Patients with GCD2 have fewer opacities than those with GCD1. Homozygote patients initially demonstrate numerous small dots in the superficial cornea in early childhood. By adulthood, there are larger, very dense subepithelial irregularly shaped opacities, which may become deeper with time.
Symptoms
Vision decreases with age as the central visual axis becomes affected. Pain may accompany mild corneal erosions.
Course
Slowly progressive. Homozygotes demonstrate more rapid progression.
Light Microscopy
Corneal opacities extend from the basal epithelium to the deep stroma. Although there is deposition of both typical GCD1 deposits and amyloid; individual opacities stain with either Masson trichrome or Congo red. Homozygotes demonstrate more severe findings.
Transmission Electron Microscopy
Anterior stromal rod-shaped, very electron-dense deposits are similar to the deposits noted in GCD1. On higher magnification, the rod-shaped deposit is composed of extracellular masses of fine, electron-dense, highly aligned fibrils.
An extremely common ultrastructural finding is the presence of randomly aligned fibrils of amyloid (see LCD1 template).
Homozygotes demonstrate more severe findings.
Confocal Microscopy
Findings are a combination of GCD1 and LCD. Reflective, breadcrumb-like round deposits with well-delineated borders or highly reflective, irregular trapezoidal deposits are present in the anterior stroma (similar to GCD1). Linear and branching deposits with changing reflectivity are observed (similar to LCD).
Category
1.
Note: Injury to the central cornea results in exacerbation of the corneal dystrophy with increased opacification. LASIK is contraindicated in this dystrophy.
REFERENCES
- 1.Bücklers M. Die erblichen Hornhautdystrophie. Klin Monatsbl Augenheilkd. 1938;3:1–135. Beiheft. [Google Scholar]
- 2.Folberg R, Alfonso E, Croxatto JO, et al. Clinically atypical granular corneal dystrophy with pathologic features of lattice-like amyloid deposits: a study of three families. Ophthalmology. 1988;95:46–51. doi: 10.1016/s0161-6420(88)33226-4. [DOI] [PubMed] [Google Scholar]
- 3.Holland EJ, Daya SM, Stone EM, et al. Avellino corneal dystrophy. Clinical manifestations and natural history. Ophthalmology. 1992;99:1564–1568. doi: 10.1016/s0161-6420(92)31766-x. [DOI] [PubMed] [Google Scholar]
- 4.Jones ST, Zimmerman LE. Histopathologic differentiation of granular, macular and lattice dystrophies of the cornea. Am J Ophthalmol. 1961;51:394–410. doi: 10.1016/0002-9394(61)92085-2. [DOI] [PubMed] [Google Scholar]
- 5.Jun RM, Tchah H, Kim TI, et al. Avellino corneal dystrophy after LASIK. Ophthalmology. 2004;111:463–468. doi: 10.1016/j.ophtha.2003.06.026. [DOI] [PubMed] [Google Scholar]
- 6.Lee JH, Chung SH, Stulting RD, et al. Effects of corneal neovascularization on the manifestations of Avellino corneal dystrophy (granular corneal dystrophy type II) Cornea. 2006;25:914–918. doi: 10.1097/01.ico.0000224645.89342.55. [DOI] [PubMed] [Google Scholar]
- 7.Matsuo N, Fujiwara H, Ofuchi Y. Electron and light microscopic observations of a case of Groenouw’s nodular corneal dystrophy. Folia Ophthalmol Jpn. 1967;18:436–447. [PubMed] [Google Scholar]
- 8.Moon JW, Kim SW, Kim T, et al. Homozygous granular corneal dystrophy type II (Avellino corneal dystrophy): natural history and progression after treatment. Cornea. 2007;26:1095–1100. doi: 10.1097/ICO.0b013e3181484013. [DOI] [PubMed] [Google Scholar]
- 9.Munier FL, Korvatska E, Djemai A, et al. Kerato-epithelin mutations in four 5q31-linked corneal dystrophies. Nat Genet. 1997;15:247–251. doi: 10.1038/ng0397-247. [DOI] [PubMed] [Google Scholar]
- 10.Roh MI, Chung SH, Stulting RD, et al. Preserved peripheral corneal clarity after surgical trauma in patients with Avellino corneal dystrophy. Cornea. 2006;25:497–498. doi: 10.1097/01.ico.0000214236.59356.43. [DOI] [PubMed] [Google Scholar]
- 11.Roh MI, Grossniklaus HE, Chung SH, et al. Avellino corneal dystrophy exacerbated after LASIK: scanning electron microscopic findings. Cornea. 2006;25:306–311. doi: 10.1097/01.ico.0000183536.07275.9a. [DOI] [PubMed] [Google Scholar]
- 12.Weidle EG. Granular corneal dystrophy: two variants. In: Ferraz de Olivera LN, editor. Ophthalmology Today. Elsevier; Amsterdam, The Netherlands: 1988. pp. 617–619. [Google Scholar]
Appendix
Macular Corneal Dystrophy (MCD)
MIM #217800.
Alternative Names, Eponyms
Groenouw corneal dystrophy type II.
Fehr spotted dystrophy.
Genetic Locus
16q22.
Gene
Carbohydrate sulfotransferase 6 gene—CHST6.
Inheritance
Autosomal recessive.
Onset
Childhood.
Signs (Fig. 14)
Initially, diffuse stromal haze extending to the limbus; later, superficial, central, elevated, irregular whitish opacities (macules) develop and give the condition its name. Unlike granular dystrophy, there are no clear areas between corneal opacities. There are also more posterior peripheral white lesions. The cornea is thinner than normal in early disease. In the advanced stage, the corneal endothelium is affected and Descemet membrane develops guttate excrescenses. In addition, the stroma thickens from the inbibition of water from endothelial decompensation.
Symptoms
Severe visual impairment occurs between 10 and 30 years of age. Reduction of corneal sensitivity. Photophobia. Painful attacks can sometimes occur due to recurrent erosions.
Course
Slowly progressive.
Light Microscopy
Glycosaminoglycans (GAGs) accumulate intracellularly and extracellularly in the corneal stroma, corneal endothelium, and Descemet membrane (stain positively with Hale colloidal iron or Alcian blue). Guttae are commonly present on Descemet membrane.
FIGURE 14.

Macular corneal dystrophy. A, Early macular corneal dystrophy with few central opacities. B, Slit-lamp photograph of advanced macular dystrophy with stromal opacities at multiple levels and diffuse stromal haze. C, More advanced macular dystrophy at higher magnification revealing more numerous and diffuse corneal opacities and stromal haze.
Transmission Electron Microscopy
Keratocytes and endothelial cells stain positively for GAGs and contain vacuoles and lamellar bodies. The extracellular matrix contains clumps of fibrillogranular material that stain positively for GAGs.
Confocal Microscopy
Blurred limited accumulations of light reflective material are located in the anterior part of the corneal stroma.
Additional Findings
There are 3 variants of macular corneal dystrophy, which are based on the immunoreactivity of the macular deposits. These variants are indistinguishable from each other clinically.
The immunophenotype of macular corneal dystrophy determines the reactivity of the abnormal deposits with an antibody that is specific for the sulfated epitopes on antigenic keratan sulfate (AgKS).
The serum AgKS correlates with the immunophenotypes in the corneal tissue.
Macular corneal dystrophy type I: No AgKS reactivity in the cornea or in the serum.
Macular corneal dystrophy type IA: Keratocytes manifest AgKS reactivity but the extracellular material does not. Serum lacks AgKS.
Macular corneal dystrophy type II: All the abnormal accumulations react positively with AgKS and the serum has normal or lower levels of AgKS.
Category
1.
REFERENCES
- 1.Akama TO, Nishida K, Nakayama J, et al. Macular corneal dystrophy type I and type II are caused by distinct mutations in a new sulphotransferase gene. Nat Genet. 2000;26:237–241. doi: 10.1038/79987. [DOI] [PubMed] [Google Scholar]
- 2.Groenouw A. Knötchenförmige Hornhauttrübungen (Noduli corneae) Arch Augenheilkd. 1890;21:281–289. [Google Scholar]
- 3.Klintworth GK, Smith CF, Bowling BL. CHST6 mutations in North American subjects with macular corneal dystrophy: a comprehensive molecular genetic review. Mol Vis. 2006;12:159–176. [PubMed] [Google Scholar]
- 4.Klintworth GK, Vogel FS. Macular corneal dystrophy: an inherited acid mucopolysaccharide storage disease of the corneal fibroblast. Am J Pathol. 1964;45:565–586. [PMC free article] [PubMed] [Google Scholar]
- 5.Szentmáry N, Seitz B, Langenbucher A, et al. Histologic and ultrastructural changes in corneas with granular and macular dystrophy after excimer laser phototherapeutic keratectomy. Cornea. 2006;25:257–263. doi: 10.1097/01.ico.0000176603.87376.d8. [DOI] [PubMed] [Google Scholar]
- 6.Vance JM, Jonasson F, Lennon F, et al. Linkage of a gene for macular corneal dystrophy to chromosome 16. Am J Hum Genet. 1996;58:757–762. [PMC free article] [PubMed] [Google Scholar]
Appendix
Schnyder Corneal Dystrophy (SCD)
MIM #21800.
Alternative Names, Eponyms
Schnyder crystalline corneal dystrophy (SCCD).
Schnyder crystalline dystrophy sine crystals.
Hereditary crystalline stromal dystrophy of Schnyder.
Crystalline stromal dystrophy.
Central stromal crystalline corneal dystrophy.
Corneal crystalline dystrophy of Schnyder.
Schnyder corneal crystalline dystrophy.
Genetic Locus
1p36.
Gene
UbiA prenyltransferase domain containing 1—UBIAD1.
Inheritance
Autosomal dominant.
Onset
Maybe as early as childhood, but diagnosis usually made by the second or third decade. Diagnosis may be further delayed in patients who have the acrystalline form of the disease.
Signs (Fig. 15)
Corneal changes are predictable on the basis of age. Patients aged 23 years or younger have central corneal haze and/or subepithelial crystals. Between 23 and 38 years of age, arcus lipoides is noted. After age 38, mid-peripheral panstromal haze develops causing the entire cornea to appear hazy. Despite the name, only 50% of patients demonstrate corneal crystals. Crystals may be unilateral, may rarely regress, and can occur late in the disease.
FIGURE 15.

Schnyder corneal dystrophy (SCD). A, Central stromal opacity in early SCD without crystals. B, Central subepithelial crystals in early SCD with crystals. C, Central ring-like opacity, prominent peripheral arcus lipoides, and moderate mid-peripheral haze in a middle-aged individual with non crystalline Schnyder. D, Central dense opacity, peripheral arcus lipoides, and prominent mid-peripheral haze. E, Advanced SCD with dense corneal opacification, subepithelial crystals, and peripheral arcus lipoides.
Symptoms
Visual acuity decreases with age. Complaints of glare increase with age. Although scotopic vision may be remarkably good (considering the slit lamp appearance), photopic vision may be disproportionately decreased. Corneal sensation decreases with age. Both affected and unaffected members of the pedigrees may have hyperlipoproteinemia (type IIa, III, or IV).
Course
Slowly progressive, although majority of patients older than 50 years may require keratoplasty for decreased photopic vision.
Light Microscopy
Abnormal deposition of intra- and extracellular esterified and unesterified phospholipids and cholesterol in basal epithelial cells, Bowman layer, and stroma. Organic solvents and resins can dissolve lipids. Consequently, to process the corneal specimen to allow special lipid stains such as oil red O or Sudan black to be performed, the ophthalmologist should inform the pathologist before placing corneal specimen in fixative that lipid stains are requested.
Transmission Electron Microscopy
Abnormal accumulation of intracellular and extracellular esterified and unesterified phospholipids and cholesterol are deposited in epithelium, in Bowman layer, and throughout the stroma. Endothelial lipid has rarely been reported.
Confocal Microscopy
Intracellular and extracellular highly reflective deposits may lead to eventual disruption of the basal epithelial/subepithelial nerve plexus.
Category
1.
Note: Although Schnyder crystalline corneal dystrophy has been the more commonly used name for this entity, this name has led to confusion in diagnosis because only 50% of the patients have crystals. Consequently, the name Schnyder corneal dystrophy should be the preferred name. If the ophthalmologist does not suspect Schnyder corneal dystrophy when performing penetrating keratoplasty, the opportunity to perform lipid stains may be lost if the corneal specimen is not preserved correctly and lipid is dissolved. In addition, there has been 1 published report of positive Congo red staining, suggesting amyloid deposition in a corneal specimen from a patient with Schnyder corneal dystrophy. More recently, a patient with an entity previously called central discoid corneal dystrophy was found to have a mutation in the UBIAD1 gene, which causes Schnyder corneal dystrophy. Although the corneal pathology demonstrated GAGs, the phenotype was identical to Schnyder corneal dystrophy sine crystals and the genotype demonstrated the UBIAD1 mutation, and there was autosomal dominant inheritance. The entity called central discoid corneal dystrophy is actually Schnyder corneal dystrophy, although GAGs were found on histopathology.
REFERENCES
- 1.Aldave AJ, Edward DP, Park AJ, et al. Central discoid corneal dystrophy. Cornea. 2002;21:739–744. doi: 10.1097/00003226-200211000-00001. [DOI] [PubMed] [Google Scholar]
- 2.Delleman JW, Winkelman JE. Degeneratio corneae cristallinea hereditaria. A clinical, genetical and histological study. Ophthalmologica. 1968;155:409–426. doi: 10.1159/000305324. [DOI] [PubMed] [Google Scholar]
- 3.Eiferman RA, Rodrigues MM, Laibson PR, et al. Schnyder crystalline dystrophy associated with amyloid deposition. Metab Pediatr Syst Ophthalmol. 1979;3:15. [Google Scholar]
- 4.Gaynor PM, Zhang WY, Weiss JS, et al. Accumulation of HDL apolipoproteins accompanies abnormal cholesterol accumulation in Schnyder’s corneal dystrophy. Arterioscler Thromb Vasc Biol. 1996;16:993–999. doi: 10.1161/01.atv.16.8.992. [DOI] [PubMed] [Google Scholar]
- 5.Lisch W, Weidle EG, Lisch C, et al. Schnyder’s dystrophy. Progression and metabolism. Ophthalmic Paediatr Genet. 1986;7:45–56. doi: 10.3109/13816818609058041. [DOI] [PubMed] [Google Scholar]
- 6.Orr A, Sube MP, Marcadier, et al. Mutations in the UBIAD1 gene encoding a potential prenyltransferase are causal for Schnyder crystalline corneal dystrophy. PLoS ONE. 2007;2:e685. doi: 10.1371/journal.pone.0000685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pameijer JK. Über eine fremdartige familiäre oberflächliche Hornhautveränderung. Klin Monatsbl Augenheilkd. 1935;95:516–517. [Google Scholar]
- 8.Schnyder WF. Mitteilung über einen neuen Typus von familiärer Hornhauterkrankung. Schweiz Med Wschr. 1929;10:559–571. [Google Scholar]
- 9.Schnyder WF. Scheibenförmige Kristalleinlagerungen in der Hornhautmitte als Erbleiben. Klin Monatsbl Augenheilkd. 1939;103:494–502. [Google Scholar]
- 10.Vesaluoma MH, Linna TU, Sankila EM, et al. In vivo confocal microscopy of a family with Schnyder crystalline corneal dystrophy. Ophthalmology. 1999;106:944–951. doi: 10.1016/S0161-6420(99)00514-X. [DOI] [PubMed] [Google Scholar]
- 11.van Went JM, Wibaut F. En zeldzame erfelijke hoornvliessandoening. Niederl Tijdschr Geneesks. 1924;68:2996–2997. [Google Scholar]
- 12.Weiss JS, Rodrigues MM, Kruth HS, et al. Panstromal Schnyder’s corneal dystrophy. Ultrastructural and histochemical studies. Ophthalmology. 1992;99:1072–1081. doi: 10.1016/s0161-6420(92)31848-2. [DOI] [PubMed] [Google Scholar]
- 13.Weiss JS. Schnyder’s dystrophy of the cornea. A Swede-Finn connection. Cornea. 1992;11:93–101. doi: 10.1097/00003226-199203000-00001. [DOI] [PubMed] [Google Scholar]
- 14.Weiss JS. Schnyder crystalline dystrophy sine crystals. Recommendation for a revision of nomenclature. Ophthalmology. 1996;103:465–473. doi: 10.1016/s0161-6420(96)30670-2. [DOI] [PubMed] [Google Scholar]
- 15.Weiss JS. Visual morbidity in thirty-four families with Schnyder’s corneal dystrophy. Trans Am Soc Ophthamol. 2007;105:1–33. [PMC free article] [PubMed] [Google Scholar]
- 16.Weiss JS, Kruth HS, Kuivaniemi H, et al. Mutations in the UBIAD1 gene on chromosome short arm 1, region 36 cause Schnyder crystalline corneal dystrophy. Invest Ophthalmol Vis Sci. 2007;48:5007–5012. doi: 10.1167/iovs.07-0845. [DOI] [PubMed] [Google Scholar]
Appendix
Congenital Stromal Corneal Dystrophy (CSCD)
MIM #610048.
Alternative Names, Eponyms
Congenital hereditary stromal dystrophy.
Congenital stromal dystrophy of the cornea.
Genetic Locus
12q21.33.
Gene
Decorin—DCN.
Inheritance
Autosomal dominant.
Onset
Congenital.
Signs (Fig. 16)
Diffuse, bilateral, corneal clouding with flake-like, whitish stromal opacities throughout the stroma. The changes are equally pronounced in all areas of the cornea. There are no signs of vascularization or staining with fluorescein. Pachymetry demonstrates increased thickness.
FIGURE 16.
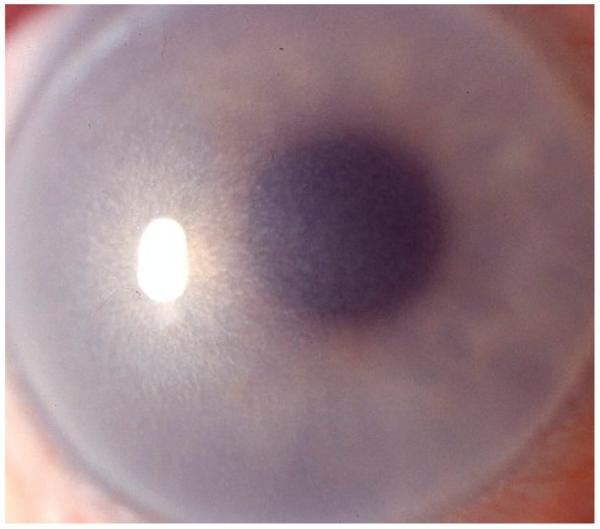
Congenital stromal corneal dystrophy: diffuse bilateral clouding with flake-like opacities throughout the stroma.
Symptoms
Moderate to severe visual loss.
Course
Nonprogressive or slowly progressive.
Light Microscopy
The stromal lamellae are separated from each other in a regular manner, may have areas of deposition of amorphous material.
Transmission Electron Microscopy
Abnormal lamellar layers consisting of thin filaments randomly arranged in an electron-lucent ground substance separate lamellae of normal appearance. The changes can be seen at all levels of the stroma. The collagen fibril diameter in all lamellae is roughly half that of normal collagen fibrils. The abnormal layers are broader in the posterior stroma. The keratocytes and endothelium are normal, although absence of the anterior banded zone of Descemet membrane has been reported.
Confocal Microscopy
Epithelial cells appear normal. Increased reflectivity from the anterior stroma prevents further studies.
Category
1.
REFERENCES
- 1.Bredrup C, Knappskog PM, Majewski J, et al. Congenital stromal dystrophy of the cornea caused by a mutation in the decorin gene. Invest Ophthalmol Vis Sci. 2005;46:420–426. doi: 10.1167/iovs.04-0804. [DOI] [PubMed] [Google Scholar]
- 2.Desvignes P, Vigo A propos d’un cas de dystrophie cornéenne parenchymateuse familiale à hérédité dominante. Bull Soc Ophtalmol Fr. 1955;4:220–225. [PubMed] [Google Scholar]
- 3.Odland M. Dystrophia corneae parenchymatosa congenita. A clinical, morphological and histochemical examination. Acta Ophthalmol (Copenh) 1968;46:477–485. doi: 10.1111/j.1755-3768.1968.tb02832.x. [DOI] [PubMed] [Google Scholar]
- 4.Pouliquen Y, Lacombe E, Schreinzer C, et al. La dystrophie congénitale héréditaire du stroma cornéen de Turpin. J Fr Ophtalmol. 1979;2:115–125. [PubMed] [Google Scholar]
- 5.Turpin R, Tisserand M, Sérane J. Opacités cornéennes héréditaires et congénitales réparties sur trois générations et atteignant deux jumelles monozygotes. Arch Ophtalmol. 1939;3:109–111. [Google Scholar]
- 6.Van Ginderdeuren R, De Vos R, Casteels I, et al. Report of a new family with dominant congenital hereditary stromal dystrophy of the cornea. Cornea. 2002;21:118–120. doi: 10.1097/00003226-200201000-00025. [DOI] [PubMed] [Google Scholar]
- 7.Witschel H, Fine BS, Grützner P, et al. Congenital hereditary stromal dystrophy of the cornea. Arch Ophthalmol. 1978;96:1043–1051. doi: 10.1001/archopht.1978.03910050563015. [DOI] [PubMed] [Google Scholar]
Appendix
Fleck Corneal Dystrophy (FCD)
MIM #121850.
Alternative Names, Eponyms
François-Neetens speckled corneal dystrophy.
Gene Locus
2q35.
Gene
Phosphatidylinositol-3-phosphate/phosphatidylinositol 5-Kinase type III—PIP5K3.
Inheritance
Autosomal dominant.
FIGURE 17.

Fleck corneal dystrophy: Dandruff-like opacities seen in 2 different patients throughout the stroma using (A) broad oblique illumination and indirect illumination, and (B) at varying depths in the slit-lamp photograph.
Onset
Congenital.
Signs (Fig. 17)
Distinctive appearance, with “small, translucent, discoid opacities” or “discrete, flat, gray-white, dandruff-like (sometimes ring-shaped)” opacities scattered sparsely throughout any level of the otherwise clear stroma. Flecks may be present up to the limbus. Epithelium, Bowman layer, Descemet membrane, and the endothelium are not involved. There may be asymmetric or unilateral corneal involvement.
Symptoms
Asymptomatic.
Course
Nonprogressive.
Light Microscopy
Swollen, vacuolated keratocytes, which contain GAG and complex lipids (excess GAG stains with Alcian blue and colloidal iron; lipids are demonstrated by Sudan black and oil red O).
Transmission Electron Microscopy
Some keratocytes show membrane-based inclusions with delicate granular material.
Confocal Microscopy
Accumulation of pathologic material in stromal cells and inclusions in the basal nerves.
Category
1.
REFERENCES
- 1.Assi A, Ebenezer N, Ficker L. Corneal fleck dystrophy in an English family. Br J Ophthalmol. 1999;83:1407–1408. doi: 10.1136/bjo.83.12.1403g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gillespie F, Covelli B. Fleck (mouchetée) dystrophy of the cornea. Report of a family. South Med J. 1963;56:1265–1267. doi: 10.1097/00007611-196311000-00014. [DOI] [PubMed] [Google Scholar]
- 3.Holopainen JM, Moilanen JA, Tervo TM. In vivo confocal microscopy of Fleck dystrophy and pre-Descemet’s membrane corneal dystrophy. Cornea. 2003;22:160–163. doi: 10.1097/00003226-200303000-00016. [DOI] [PubMed] [Google Scholar]
- 4.Jiao X, Munier FL, Schorderet DF, et al. Genetic linkage of François-Neetens fleck (mouchetee) corneal dystrophy to chromosome 2q35. Hum Genet. 2003;112:593–599. doi: 10.1007/s00439-002-0905-1. [DOI] [PubMed] [Google Scholar]
- 5.Li S, Tiab L, Jiao X, et al. Mutations in PIP5K3 are associated with François-Neetens mouchetee fleck corneal dystrophy. Am J Hum Genet. 2005;77:54–63. doi: 10.1086/431346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Patten JT, Hyndiuk RA, Donaldson DD, et al. Fleck (Mouchetee) dystrophy of the cornea. Ann Ophthalmol. 1976;8:25–32. [PubMed] [Google Scholar]
- 7.Purcell JJ, Jr, Krachmer JH, Weingeist TA. Fleck corneal dystrophy. Arch Ophthalmol. 1977;95:440–444. doi: 10.1001/archopht.1977.04450030082009. [DOI] [PubMed] [Google Scholar]
Appendix
Posterior Amorphous Corneal Dystrophy (PACD)
MIM: None.
Alternative Names, Eponyms
Posterior amorphous stromal dystrophy.
Gene
Unknown.
Inheritance
Autosomal dominant.
Onset
Often occurs in the first decade of life; it has been noted as early as 16 weeks, suggesting a congenital nature.
Signs (Fig. 18)
PACD presents as diffuse gray-white, sheet-like opacities that can involve any layer of the stroma but are most prominent posteriorly. The lesions can be centroperipheral, extending to the limbus, or peripheral, the latter with less pronounced findings and symptoms. There are often transparent stromal breaks in the opacification. Corneal thinning to as low as 380 μm, a flattened corneal topography (<41.00 D) and hyperopia are present particularly in the centroperipheral form. Descemet membrane and endothelium may be indented by the opacities and focal endothelial abnormalities have been observed. Prominent Schwalbe line, fine iris processes, pupillary remnants, iridocorneal adhesions, corectopia, pseudopolycoria, and anterior stromal tags have been reported, particularly in patients with a centroperipheral pattern. No association with glaucoma is noted.
Symptoms
The visual acuity is mildly affected, usually better than 20/40.
FIGURE 18.
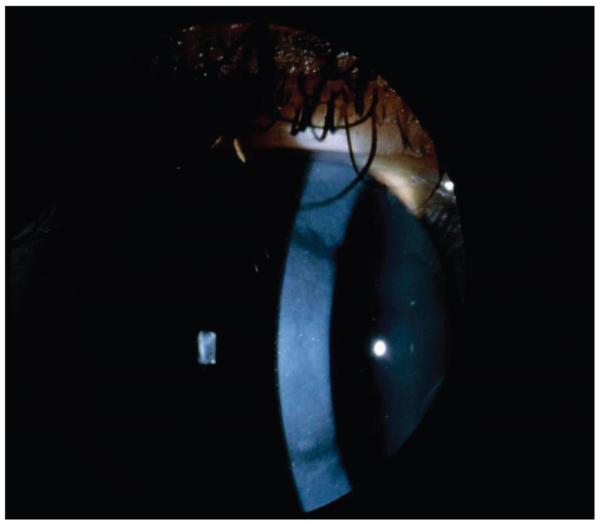
Posterior amorphous corneal dystrophy: central deep stromal/pre-Descemet opacity with some degree of peripheral extension interrupted by a clear ring in the mid-peripheral cornea.
Course
None or slowly progressive. Usually no treatment is needed, although sometimes penetrating keratoplasty is required.
Light Microscopy
Irregular stromal architecture just anterior to a thin Descemet membrane and focal attenuation of endothelial cells.
Transmission Electron Microscopy
There are abnormally oriented collagen fibers and abnormal keratocytes with disorganization of the posterior stromal lamellae. A fibrillar layer resembling stromal collagen fibers interrupts Descemet membrane. These findings are not pathognomonic of this dystrophy and may be found in other abnormalities. In a patient with more pronounced changes, additional subepithelial deposits and a thick collagenous layer posterior to Descemet membrane were present.
Confocal Microscopy
Microfolds and a hyper-reflective layer in the posterior stroma are present.
Category
3.
Note: The possible congenital onset, lack of progression, and association with iris abnormalities have raised the question whether this may in fact be a mesodermal dysgenesis rather than a corneal dystrophy.
REFERENCES
- 1.Carpel EF, Sigelman RJ, Doughman DJ. Posterior amorphous corneal dystrophy. Am J Ophthalmol. 1977;83:629–632. doi: 10.1016/0002-9394(77)90127-1. [DOI] [PubMed] [Google Scholar]
- 2.Dunn SP, Krachmer JH, Ching SS. New findings in posterior amorphous corneal dystrophy. Arch Ophthalmol. 1984;102:236–239. doi: 10.1001/archopht.1984.01040030186023. [DOI] [PubMed] [Google Scholar]
- 3.Erdem U, Muftuoglu O, Hurmeric V. In vivo confocal microscopy findings in a patient with posterior amorphous corneal dystrophy. Clin Exp Ophthalmol. 2007;35:99–102. doi: 10.1111/j.1442-9071.2007.01426.x. [DOI] [PubMed] [Google Scholar]
- 4.Johnson AT, Folberg R, Vrabec MP, et al. The pathology of posterior amorphous corneal dystrophy. Ophthalmology. 1990;97:104–109. doi: 10.1016/s0161-6420(90)32638-6. [DOI] [PubMed] [Google Scholar]
- 5.Moshegov CN, Hoe WK, Wiffen SJ, et al. Posterior amorphous corneal dystrophy. A new pedigree with phenotypic variation. Ophthalmology. 1996;103:474–478. doi: 10.1016/s0161-6420(96)30669-6. [DOI] [PubMed] [Google Scholar]
Appendix
Central Cloudy Dystrophy of François (CCDF)
MIM #217600.
Alternative Names, Eponyms
None.
Gene/Genetic Locus
None.
Inheritance
Unknown. Autosomal dominant inheritance is reported in a few articles describing the entity. This entity may be phenotypically indistinguishable from posterior crocodile shagreen, which is a corneal degeneration.
Onset
First decade (youngest affected patient was 8 years old).
Signs (Fig. 19)
Fortuitous finding of cloudy central polygonal or rounded stromal opacities that fade anteriorly and peripherally and are surrounded by clear tissue. The changes are very similar to Vogt posterior crocodile shagreen.
Symptoms
Mostly asymptomatic.
Course
Nonprogressive.
Light Microscopy
No description in familial cases. Faint undulating appearance of the deep stroma and positive staining for GAGs.
Transmission Electron Microscopy
No description in familial cases. One publication described an elderly patient with no familial history. Corneal pathology revealed extracellular vacuoles, some of which contained fibrillogranular material and electron-dense deposits. Endothelial vacuoles with fibrillogranular material. A saw-toothed lamellar pattern has been reported.
FIGURE 19.

Central cloudy dystrophy of Francois. A, Axially distributed, polygonal gray-white stromal opacities separated by linear areas of clear cornea. B, Broad beam slit lamp photograph demonstrating central stromal opacities with linear clear areas and “cracked ice” appearance.
Confocal Microscopy
No description in familial cases. In 2 unrelated patients, there were small highly refractile granules and deposits in the anterior stroma. Multiple dark striae in the extracellular matrix with increased intensity in the posterior stroma, which is adjacent to the corneal endothelial layer.
Category
4.
Note: Many of the publications referenced did not provide documentation that the corneal disease was familial. Consequently, it is entirely possible that these cases of CCDF were actually posterior crocodile shagreen.
REFERENCES
- 1.Bramsen T, Ehlers N, Baggesen LH. Central cloudy corneal dystrophy of François. Acta Ophthalmol (Copenh) 1976;54:221–226. doi: 10.1111/j.1755-3768.1976.tb00435.x. [DOI] [PubMed] [Google Scholar]
- 2.François J. Une nouvelle dystrophie hérédo-familiale de la cornée. J Genet Hum. 1956;5:189–196. [PubMed] [Google Scholar]
- 3.Karp CL, Scott IU, Green WR, et al. Central cloudy corneal dystrophy of François. A clinicopathologic study. Arch Ophthalmol. 1997;115:1058–1062. doi: 10.1001/archopht.1997.01100160228013. [DOI] [PubMed] [Google Scholar]
- 4.Kobayashi A, Sugiyama K, Huang AJ. In vivo confocal microscopy in patients with central cloudy dystrophy of François. Arch Ophthalmol. 2004;122:1676–1679. doi: 10.1001/archopht.122.11.1676. [DOI] [PubMed] [Google Scholar]
- 5.Meyer JC, Quantock AJ, Thonar EJ, et al. Characterization of a central corneal cloudiness sharing features of posterior crocodile shagreen and central cloudy dystrophy of François. Cornea. 1996;15:347–354. doi: 10.1097/00003226-199607000-00003. [DOI] [PubMed] [Google Scholar]
- 6.Strachan IM. Cloudy central corneal dystrophy of François. Five cases in the same family. Br J Ophthalmol. 1969;53:192–194. doi: 10.1136/bjo.53.3.192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zaidi A, McLeod SD. Laser in situ keratomileusis in a patient with presumed central cloudy corneal dystrophy of François. Am J Ophthalmol. 2005;139:376–377. doi: 10.1016/j.ajo.2004.08.007. [DOI] [PubMed] [Google Scholar]
Appendix
Pre-Descemet Corneal Dystrophy (PDCD)
MIM: None.
Alternative Names, Eponyms
None.
Gene
Unknown.
Inheritance
Pre-Descemet dystrophy is not a well-defined entity. Although there is no definite pattern of inheritance, it has been described in families over 2–4 generations. The subtype punctiform and polychromatic pre-Descemet dystrophy reported to be autosomal dominant in 1 pedigree may represent a specific dystrophy.
Onset
Usually after 30 years of age but has been found in children as young as 3 years (punctiform and polychromatic pre-Descemet dystrophy).
Signs (Fig. 20)
Pre-Descemet dystrophy has several subgroups; many of these may represent sporadic, age-related degenerative, and secondary changes. There are focal, fine, gray opacities in the deep stroma immediately anterior to Descemet membrane with a variety of shapes. Larger lesions occur. The opacities may be central, annular, or diffuse. In the subtype, punctiform and polychromatic pre-Descemet dystrophy, the changes are more uniform and polychromatic. The rest of the cornea is normal. Similar opacities have been noted in association with other ocular and systemic diseases, such as pseudoxanthoma elasticum, X-linked and recessive ichthyosis, keratoconus, PPCD, EBMD, and CCDF.
Symptoms
The vision is usually unaffected and the patients are asymptomatic.
Course
Punctiform and polychromatic pre-Descemet dystrophy is nonprogressive. Other forms show progression.
Light Microscopy
Histopathologic studies are not consistent. Normal cornea except enlarged keratocytes in the posterior stroma with vacuoles and intracytoplasmic inclusions containing lipid-like material has been described.
Transmission Electron Microscopy
Membrane-bound intracellular vacuoles containing electron-dense material suggestive of secondary lysosomes and inclusions consistent with lipofuscin-like lipoprotein suggesting a degenerative process. No extracellular deposits noted.
FIGURE 20.

Pre-Descemet corneal dystrophy: punctate opacities anterior to Descemet membrane demonstrated with indirect illumination and slit lamp beam.
Confocal Microscopy
Hyper-reflective dots located anterior to Descemet membrane; in 1 case reported to be present throughout the stroma.
Category
4.
Note: Similar deep corneal opacities are frequently seen in patients with ichthyosis and in carriers of X-linked ichthyosis (MIM #308100). It is unclear whether pre-Descemet dystrophy is a hereditary or a degenerative disorder.
REFERENCES
- 1.Curran RE, Kenyon KR, Green WR. Pre-Descemet’s membrane corneal dystrophy. Am J Ophthalmol. 1974;77:7110–7116. doi: 10.1016/0002-9394(74)90536-4. [DOI] [PubMed] [Google Scholar]
- 2.Fernandez-Sasso D, Acosta JE, Malbran E. Punctiform and polychromatic pre-Descemet’s dominant corneal dystrophy. Br J Ophthalmol. 1979;63:336–338. doi: 10.1136/bjo.63.5.336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Grayson M, Wilbrandt M. Pre-Descemet dystrophy. Am J Ophthalmol. 1967;64:227. doi: 10.1016/0002-9394(67)92524-x. [DOI] [PubMed] [Google Scholar]
- 4.Kempster RC, Hirst LW, de la Cruz Z, et al. Clinicopathologic study of the cornea in X-linked ichthyosis. Arch Ophthalmol. 1997;115:409–415. doi: 10.1001/archopht.1997.01100150411017. [DOI] [PubMed] [Google Scholar]
- 5.Lisch W, Weidle EG. Die posteriore kristalline Hornhautdystrophie. Klin Monatsbl Augenheilkd. 1984;185:128–131. doi: 10.1055/s-2008-1054585. [DOI] [PubMed] [Google Scholar]
- 6.Yassa NH, Font RL, Fine BH. Corneal immunoglobulin deposition in the posterior stroma. A case report including immunohistochemical and ultrastructural observations. Arch Ophthalmol. 1987;105:99–103. doi: 10.1001/archopht.1987.01060010105040. [DOI] [PubMed] [Google Scholar]
- 7.Ye YF, Yao YF, Zhou P, et al. In vivo confocal microscopy of pre-Descemet’s membrane corneal dystrophy. Clin Exp Ophthalmol. 2006;34:614–616. doi: 10.1111/j.1442-9071.2006.01288.x. [DOI] [PubMed] [Google Scholar]
Appendix
DESCEMET MEMBRANE AND ENDOTHELIAL DYSTROPHIES
Fuchs Endothelial Corneal Dystrophy (FECD)
MIM #136800.
Alternative Names, Eponyms
Endoepithelial corneal dystrophy.
Inheritance
Cases without known inheritance are most common.
Some cases with autosomal dominant inheritance reported.
Genetic Locus
Fuchs endothelial corneal dystrophy 13pTel –13q12.13, 15q, 18q21.2 –q21.32.
Early-onset variant Fuchs endothelial corneal dystrophy 1p34.3 – p32
Gene
None.
Early-onset variant collagen type VIII, Alpha 2—COL8A2.
Onset
Cases without known heredity as early as fifth decade. Fuchs endothelial corneal dystrophy fourth decade and later.
Early-onset variant FECD first decade. Most cases begin in the fourth decade or later but the early variant starts in the first decade.
Signs (Fig. 21)
Cornea guttata accompanied by stromal edema: central beaten metal-like endothelial changes with or without pigment dusting. Corneal guttae in adult-onset Fuchs endothelial corneal dystrophy are larger than those seen in early-onset Fuchs endothelial corneal dystrophy. Stromal edema due to endothelial decompensation. Intra- and interepithelial edema (epithelial bullae); bullous keratopathy. Subepithelial fibrous scarring and peripheral superficial vascularization may occur in longstanding cases from chronic edema.
Symptoms
Intermittent reduced vision from epithelial/stromal edema. Visual acuity worse in the morning due to increased epithelial/stromal edema. Pain, photophobia, and epiphora due to epithelial erosions resulting from burst epithelial bullae. Progressive visual loss.
Course
Progressive.
FIGURE 21.

Fuchs endothelial corneal dystrophy. A, Central guttae viewed in retroillumination and in the slit beam. B, Cornea guttae as seen in specular reflection. C, Advanced stromal edema. D, Advanced endothelial decompensation with epithelial microcystic and bullous edema.
Light Microscopy
Diffuse thickening and lamination of Descemet membrane. Sparse and atrophic endothelial cells, hyaline excrescences on thickened Descemet membrane (guttae). Guttae become buried or confluent or may be absent. Degeneration, thinning, and reduction of endothelial cells. Increasing waviness of the stromal collagen lamellae. Thickening of Descemet membrane is noted.
Transmission Electron Microscopy
Multiple layers of basement membrane–like material on the posterior part of Descemet membrane. Degeneration of endothelial cells. Stromal thickening with severe disorganization and disruption of the lamellar pattern.
Confocal Microscopy
Polymegathism and pleomorphism of the endothelial cells. Early-onset variant Fuchs endothelial corneal dystrophy has smaller guttae than typical Fuchs endothelial corneal dystrophy.
Category
3 Fuchs endothelial corneal dystrophy in patients with no known inheritance.
2 Fuchs endothelial corneal dystrophy with known genetic loci but gene not yet localized.
1 Early-onset Fuchs endothelial corneal dystrophy.
REFERENCES
- 1.Fuchs E. Dystrophia epithelialis corneae. Albrecht von Graefes Arch Clin Exp Ophthalmol. 1910;76:478–508. [Google Scholar]
- 2.Fuchs E. Erkrankung der Hornhaut durch Schädigung von hinten. Albrecht von Graefes Arch Clin Exp Ophthalmol. 1917;92:145–236. [Google Scholar]
- 3.Gottsch JD, Zhang C, Sundin OH, et al. Fuchs corneal dystrophy: aberrant collagen distribution in an L450W mutant of the COL8A2 gene. Invest Ophthalmol Vis Sci. 2005;46:4504–4511. doi: 10.1167/iovs.05-0497. [DOI] [PubMed] [Google Scholar]
- 4.Hogan MJ, Wood J, Fine M. Fuchs’ endothelial dystrophy of the cornea. Am J Ophthalmol. 1974;78:363–383. doi: 10.1016/0002-9394(74)90224-4. [DOI] [PubMed] [Google Scholar]
- 5.Krachmer JH, Purcell JJ, Jr, Joung CW, et al. Corneal endothelial dystrophy. A study of 64 families. Arch Ophthalmol. 1978;96:2036–2039. doi: 10.1001/archopht.1978.03910060424004. [DOI] [PubMed] [Google Scholar]
- 6.Sundin OH, Jun A, Broman KW, et al. Linkage of late-onset Fuchs corneal dystrophy to a novel locus at 13ptel-13q12.13. Invest Ophthalmol Vis Sci. 2006;47:140–145. doi: 10.1167/iovs.05-0578. [DOI] [PubMed] [Google Scholar]
- 7.Sundin OH, Broman KW, Chang HH, et al. A common locus for late-onset Fuchs corneal dystrophy maps to 18q21.2-q21.32. Invest Ophthalmol Vis Sci. 2006;47:3912–3920. [Google Scholar]
- 8.Waring GO, III, Rodrigues MM, Laibson PR. Corneal dystrophies. II. Endothelial dystrophies. Surv Ophthalmol. 1978;23:147–168. doi: 10.1016/0039-6257(78)90151-0. [DOI] [PubMed] [Google Scholar]
- 9.Zhang C, Bell WR, Sundin OH, et al. Immunohistochemistry and electron microscopy of early-onset Fuchs corneal dystrophy in three cases with the same L450W COL8A2 mutation. Trans Am Ophthalmol Soc. 2006;104:85–97. [PMC free article] [PubMed] [Google Scholar]
Appendix
Posterior Polymorphous Corneal Dystrophy (PPCD)
MIM PPCD1 #122000, PPCD2 #609140, PPCD3 #609141.
Alternative Names, Eponyms
Posterior polymorphous dystrophy (PPMD).
Schlichting dystrophy.
Inheritance
Autosomal dominant.
Isolated unilateral cases, with similar phenotype but no heredity.
Genetic Locus
PPCD 1—20p11.2–q11.2.
PPCD 2—1p34.3–p32.3.
PPCD 3—10p11.2.
Gene
PPCD 1—unknown.
PPCD 2—collagen type VIII alpha 2, COL8A2
PPCD 3—two-handed zinc-finger homeodomain transcription factor 8—ZEB1.
Onset
Early childhood.
FIGURE 22.

Posterior polymorphous corneal dystrophy. A, Endothelial plaque-like lesions. B, Irregular crater-like figures on Descemet membrane viewed with specular reflection. C, Railroad track opacities as seen in broad oblique illumination and retroillumination.
Signs (Fig. 22)
Often asymmetric. Deep corneal lesions of various shapes including nodular, vesicular (isolated, in clusters, or confluent) and blister-like lesions. “Railroad tracks” appearance (multiple and isolated). Varying gray tissue at the level of Descemet membrane. Rarely stromal and epithelial edema ranging to ground-glass, milky appearance due to endothelial decompensation. Peripheral iridocorneal adhesions in about 25% of cases. In about 15% of cases, intraocular pressure (IOP) was elevated. Rarely, secondary subepithelial band keratopathy.
Symptoms
Endothelial alterations often asymptomatic. Rarely extensive and progressive visual impairment due to stromal clouding.
Course
Rarely congenital corneal clouding. Endothelial changes often unchanged over years. Possible slow progression of polymorphic vesicles and greater thickness of Descemet membrane over years occasionally causing endothelial decompensation.
Light Microscopy
Descemet membrane with multiple layers of collagen on its posterior surface manifesting focal fusiform or nodular excrescences.
Transmission Electron Microscopy
Extreme thinning or absence of the posterior nonbanded layer of Descemet membrane. Two types of collagenous tissue posterior to Descemet membrane form layers up to 25 nm thick. Multilayered epithelial-like cells with microcilia and desmosomes.
Confocal Microscopy
Vesicular lesions: Rounded dark areas with some cell detail apparent in the middle giving a doughnut-like appearance. Multilayered nests of cells. Railroad track: band-like dark area with irregular edges enclosing some smaller lighter cells resembling epithelium-like cells. Polymegathism of the endothelium.
Immunohistochemistry
PPCD 1: Positive with anti-CK7 antibodies.
Category
PPCD 1—2.
PPCD 2—1.
PPCD 3—1.
REFERENCES
- 1.Aldave AJ, Yellore VS, Principe AH, et al. Candidate gene screening for posterior polymorphous dystrophy. Cornea. 2005;24:151–155. doi: 10.1097/01.ico.0000141235.26096.1d. [DOI] [PubMed] [Google Scholar]
- 2.Cibis GW, Krachmer JA, Phelps CD, et al. The clinical spectrum of posterior polymorphous dystrophy. Arch Ophthalmol. 1977;95:1529–1537. doi: 10.1001/archopht.1977.04450090051002. [DOI] [PubMed] [Google Scholar]
- 3.Heon E, Mathers WD, Alward WL, et al. Linkage of posterior polymorphous corneal dystrophy to 20q11. Hum Mol Genet. 1995;4:485–488. doi: 10.1093/hmg/4.3.485. [DOI] [PubMed] [Google Scholar]
- 4.Koeppe L. Klinische Beobachtungen mit der Nernstspaltlampe und dem Hornhautmikroskop. Albr Graefe Arch Klin Exp Ophthalmol. 1916;91:363–379. [Google Scholar]
- 5.Krafchak CM, Pawar H, Moroi SE, et al. Mutations in TCF8 cause posterior polymorphous corneal dystrophy and ectopic expression of COL4A3 by corneal endothelial cells. Am J Hum Genet. 2005;77:694–708. doi: 10.1086/497348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Patel DV, Grupcheva CN, McGhee CNJ. In vivo microscopy of posterior polymorphous dystrophy. Cornea. 2005;24:550–554. doi: 10.1097/01.ico.0000153557.59407.20. [DOI] [PubMed] [Google Scholar]
- 7.Rodrigues MM, Waring GO, Laibson PR, et al. Endothelial alterations in congenital corneal dystrophies. Am J Ophthalmol. 1975;80:678–689. doi: 10.1016/0002-9394(75)90400-6. [DOI] [PubMed] [Google Scholar]
- 8.Shimizu S, Krafchak C, Fuse N, et al. A locus for posterior polymorphous corneal dystrophy (PPCD3) maps to chromosome 10. Am J Med Genet. 2004;130:372–377. doi: 10.1002/ajmg.a.30267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schlichting H. Blasen-und dellenförmige Endotheldystrophie der Hornhaut. Klin Monatsbl Augenkeilkd. 1941;107:425–435. [Google Scholar]
Appendix
Congenital Hereditary Endothelial Dystrophy 1 (CHED1)
MIM #121700.
Alternative Names, Eponyms
None.
Genetic Locus
20p 11.2–q11.2 (pericentromeric region).
Gene
Unknown.
Inheritance
Autosomal dominant.
Onset
First or second year, occasionally congenital.
Signs (Fig. 23A)
Often asymmetric. Corneal clouding ranging from a diffuse haze to a ground-glass, milky appearance with occasional focal gray spots. Thickening of the cornea (can be 2–3 times normal thickness). Rarely subepithelial band keratopathy. Asymptomatic patients have only endothelial changes in form of moon crater–like appearance and peau d’orange texture. Rarely elevated IOP.
FIGURE 23.

Congenital hereditary endothelial dystrophy. A, CHED1—Milky appearance of cornea with diffuse illumination. B, CHED2—Slit beam photograph demonstrating diffuse stromal thickening in a homozygote individual with SLC4A11 mutations.
Symptoms
Corneal clouding with blurred vision, photophobia, and tearing. Worsening of vision in the morning. Exclusively peau d’orange–like endothelial alterations with no or little objective reduction of vision.
Course
Progression of corneal clouding over 1–10 years. Slow progression of endothelial alterations with the possibility of endothelial decompensation over a prolonged period.
Light Microscopy
Diffuse thickening and lamination of Descemet membrane. Sparse and atrophic endothelial cells. Parts of the endothelium are replaced by keratin containing stratified squamous epithelium.
Transmission Electron Microscopy
Multiple layers of basement membrane–like material on the posterior part of Descemet membrane. Degeneration of endothelial cells with many vacuoles. Stromal thickening with severe disorganization and disruption of the lamellar pattern.
Confocal Microscopy
Not reported.
Category
2.
REFERENCES
- 1.Cibis GW, Krachmer JA, Phelps CD, et al. The clinical spectrum of posterior polymorphous dystrophy. Arch Ophthalmol. 1977;95:1529–1537. doi: 10.1001/archopht.1977.04450090051002. [DOI] [PubMed] [Google Scholar]
- 2.Judisch GF, Maumenee IH. Clinical differences of recessive congenital hereditary endothelial dystrophy and dominant hereditary endothelial dystrophy. Am J Ophthalmol. 1978;85:606–612. doi: 10.1016/s0002-9394(14)77091-6. [DOI] [PubMed] [Google Scholar]
- 3.Levenson JE, Chandler JW, Kaufman HE. Affected asymptomatic relatives in congenital hereditary endothelial dystrophy. Am J Ophthalmol. 1973;76:967–971. doi: 10.1016/0002-9394(73)90090-1. [DOI] [PubMed] [Google Scholar]
- 4.McCartney AC, Kirkness CM. Comparison between posterior polymorphous dystrophy and congenital hereditary endothelial dystrophy of the cornea. Eye. 1988;2:63–70. doi: 10.1038/eye.1988.14. [DOI] [PubMed] [Google Scholar]
- 5.Pearce WG, Tripathi RC, Morgan G. Congenital endothelial corneal dystrophy. Clinical, pathological, and genetic study. Br J Ophthalmol. 1969;53:577–591. doi: 10.1136/bjo.53.9.577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Toma NMG, Ebenezer ND, Inglehearn CF, et al. Linkage of congenital hereditary endothelial dystrophy to chromosome 20. Hum Mol Genet. 1995;4:2395–2398. doi: 10.1093/hmg/4.12.2395. [DOI] [PubMed] [Google Scholar]
Appendix
Congenital Hereditary Endothelial Dystrophy 2 (CHED2)
MIM #217700.
Alternative Names, Eponyms
Maumenee corneal dystrophy.
Genetic Locus
20p13 (telomeric portion).
Gene
Solute carrier family 4, sodium borate transporter, member 11—SLC4A11.
Inheritance
Autosomal recessive.
Onset
Congenital.
Signs (Fig. 23B)
Often asymmetric. More common and severe than CHED1. Corneal clouding ranging from a diffuse haze to ground-glass, milky appearance with occasional focal gray spots. Thickening of the cornea (can be 2–3 times normal thickness). Rarely secondary subepithelial band keratopathy. Rarely elevated IOP.
Symptoms
Corneal clouding with blurred vision often accompanied by nystagmus. Minimal to no tearing or photophobia.
Course
Relatively stationary.
Light Microscopy
Diffuse thickening and lamination of Descemet membrane. Sparse and atrophic endothelial cells.
Transmission Electron Microscopy
Multiple layers of basement membrane–like material on the posterior part of Descemet membrane. Degeneration of endothelial cells with many vacuoles. Stromal thickening with severe disorganization and disruption of the lamellar pattern.
Confocal Microscopy
Not reported.
Immunohistochemistry
Distribution of collagen types I and III–V, and laminin within the posterior collagenous layer of Descemet membrane. SLC4A11 encodes Bicarbonate transporter-related protein-1 (BTR1). BTR1 mutants remain in cytoplasm, whereas wild-type BTR1 localizes mostly to the plasma membrane.
Category
1.
REFERENCES
- 1.Callaghan M, Hand CK, Kennedy SM, et al. Homozygosity mapping and linkage analysis demonstrate that autosomal recessive congenital hereditary endothelial dystrophy (CHED) and autosomal dominant CHED are genetically distinct. Br J Ophthalmol. 1999;83:115–119. doi: 10.1136/bjo.83.1.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jiao X, Sultana A, Garg P, et al. Autosomal recessive corneal endothelial dystrophy (CHED2) is associated with mutations in SLC4A11. J Med Genet. 2007;44:64–68. doi: 10.1136/jmg.2006.044644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kenyon KR, Antine B. The pathogenesis of congenital hereditary endothelial dystrophy of the cornea. Am J Ophthalmol. 1971;72:787–795. doi: 10.1016/0002-9394(71)90019-5. [DOI] [PubMed] [Google Scholar]
- 4.Kirkness CM, McCartney A, Rice NS, et al. Congenital hereditary corneal edema of Maumenee: its clinical features, management, and pathology. Br J Ophthalmol. 1987;71:130–144. doi: 10.1136/bjo.71.2.130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Maumenee AE. Congenital hereditary corneal dystrophy. Am J Ophthalmol. 1960;50:1114–1124. doi: 10.1016/0002-9394(60)90998-3. [DOI] [PubMed] [Google Scholar]
- 6.Sekundo W, Marshall GE, Lee WR, et al. Immuno-electron labelling of matrix components in congenital hereditary endothelial dystrophy. Graefes Arch Clin Exp Ophthalmol. 1994;232:337–346. doi: 10.1007/BF00175985. [DOI] [PubMed] [Google Scholar]
- 7.Vithana EN, Morgan P, Sundavesan P, et al. Mutations in sodium-borate cotransporter SLC4A11 cause recessive congenital hereditary endothelial dystrophy (CHED 2) Nat Genet. 2006;38:755–757. doi: 10.1038/ng1824. [DOI] [PubMed] [Google Scholar]
Appendix
X-linked Endothelial Corneal Dystrophy (XECD)
MIM: None.
Alternative Names, Eponyms
None.
Genetic Locus
Xq25.
Gene
Unknown.
Inheritance
X-chromosomal dominant.
Onset
Congenital.
FIGURE 24.

X-linked endothelial corneal dystrophy. Seven-year-old boy with milk glass appearance of the cornea.
Signs (Fig. 24)
Males
Congenital clouding ranging from a diffuse haze to a ground-glass, milky appearance. Possible nystagmus.
Only moon crater–like endothelial changes.
Secondary subepithelial band keratopathy combined with moon crater–like endothelial changes.
Females
Only moon crater–like endothelial changes.
Symptoms
Males: Often blurred vision.
Females: Asymptomatic.
Course
Males: Progressive. Females: Nonprogressive.
Light Microscopy
Moon crater endothelial changes and subepithelial band keratopathy. Irregular thinning of the epithelium and Bowman lamella. Anterior stroma with irregularly arranged collagen lamellae. Irregular thickening of Descemet membrane with small excavations and pits. Loss of endothelial cells or atypical appearance.
Transmission Electron Microscopy
Moon crater endothelial changes and subepithelial band keratopathy. Subepithelial accumulations of an amorphous granular material. Irregular thinning of Bowman layer (up to 0.5 mm) with many interruptions and gaps. Thickening of Descemet membrane (20 -35 mm) consisting of an abnormal anterior and posterior banded zone. Complete absence of the posterior nonbanded zone. Discontinuous endothelial layer with partly normal and partly degenerative appearing cells. No evidence of desmosome-like adherent junctions between the cells or tonofilament bundles within the cytoplasm.
Confocal Microscopy
Not reported.
Category
2.
REFERENCES
- 1.Lisch W. Primäre bandförmige Hornhautdegeneration und ihre Assoziation mit anderen erblichen Hornhautveränderungen. Klin Monatsbl Augenheilk. 1976;169:717–727. [PubMed] [Google Scholar]
- 2.Schmid E, Lisch W, Philipp W, et al. A new, X-linked endothelial corneal dystrophy. Am J Ophthalmol. 2006;141:478–487. doi: 10.1016/j.ajo.2005.10.020. [DOI] [PubMed] [Google Scholar]
Appendix
RECOMMENDATIONS OF THE IC3D-THE ESTABLISHMENT OF ACCEPTED CRITERIA FOR PUBLICATION OF POTENTIAL NEW OR VARIANT CORNEAL DYSTROPHIES
We have attempted to present a revision of corneal dystrophy nomenclature that is accurate, is easy to use, and can be updated with new discoveries. For over a century, the corneal dystrophy nomenclature has been confounded by the reports of new corneal dystrophies or corneal dystrophy variants with inadequate factual substantiation. Sometimes, these “new” diseases have been variants of previously described dystrophies. However, the advent of genetic testing has provided the opportunity to obtain genetic information to accurately substantiate whether or not a corneal dystrophy is actually new. Ophthalmologists should adopt a more scientific approach to the field of genetic corneal disease by both detailed characterization of phenotypic changes and obtaining genetic testing when indicated. We hope the IC3D nomenclature classification will effectively endorse a more scientific and objective criteria for determining whether a “new” corneal dystrophy or dystrophy variant has indeed been discovered. We urge authors and reviewers alike that more stringent criteria must be met before publication of these entities.
APPENDIX
Table of Genes and Mutations Associated with the Corneal Dystrophies
The tables are grouped into four categories:
Epithelial and subepithelial dystrophies
Bowman layer dystrophies
Stromal dystrophies
Descemet membrane and Endothelial dystrophies
Each table is organized into columns:
Gene (locus) —The abbreviation and chromosomal location for each gene are provided.
RefSeq ( reference sequence)—The reference sequence used to determine the nucleotide and amino acid position of each mutation is listed.
Exon—The exon in which each mutation is located is given.
Nucleotide change—Each mutation is described at the nucleotide level. All nucleotide changes are numbered according to the Human Genome Variation Society (HGVS) mutation nomenclature system, in which nucleotide 1 is the A of the ATG-translation initiation codon.
AA change (amino acid change) —Each mutation is given at the amino acid level. The HGVS mutation nomenclature system is used, employing the three letter amino acid abbreviations, with the translation initiator methionine numbered as +1.
Original—Each mutation, as originally reported, is listed to allow the reader to correlate the mutations listed in the nucleotide and amino acid change columns with the nomenclature utilized by the original authors.
Reference—References are provided for each reported mutation.
Epithelial Dystrophies
TABLE 1.
The IC3D Classification—Abbreviations and MIM Number
| MIM Abbreviation |
IC3D Abbreviation |
MIM # | |
|---|---|---|---|
| Epithelial basement membrane dystrophy |
EBMD | EBMD | 121820 |
| Epithelial recurrent erosion dystrophy |
None | ERED | 122400 |
| Subepithelial mucinous CD | None | SMCD | None |
| Meesmann CD | None | MECD | 122100 |
| Lisch epithelial CD | None | LECD | None |
| Gelatinous drop-like CD | GDLD, CDGDL | GDLD | 204870 |
| Reis–Bücklers CD | CDB1, CDRB, RBCD | RBCD | 608470 |
| Thiel–Behnke CD | CDB2, CDTB | TBCD | 602082 |
| Grayson –Wilbrandt CD | None | GWCD | None |
| Classic Lattice CD | CDL1 | LCD1 | 122200 |
| Lattice CD, Meretoja type | None | LCD2 | 105120 |
| Granular CD, type 1 | CGDD1 | GCD1 | 121900 |
| Granular CD, type 2 (granular–lattice) |
CDA, ACD | GCD2 | 607541 |
| Macular CD | MCDC1 | MCD | 217800 |
| Schnyder CD | None | SCD | 121800 |
| Congenital stromal CD | CSCD | CSCD | 610048 |
| Fleck CD | None | FCD | 121850 |
| Posterior amorphous CD | None | PACD | None |
| Central cloudy dystrophy of François |
None | CCDF | 217600 |
| Pre-Descemet CD | None | PDCD | None |
| Fuchs endothelial CD | FECD1 | FECD | 136800 |
| Posterior polymorphous CD | PPCD1 | PPCD | 122000 |
| Congenital hereditary endothelial dystrophy 1 |
CHED1 | CHED1 | 121700 |
| Congenital hereditary endothelial dystrophy 2 |
CHED2 | CHED2 | 217700 |
| X-linked endothelial CD | None | XECD | None |
Online MIM (McKusick VA et al. http://www.ncbi.nlm.nih.gov/sites/entrez)
CD, corneal dystrophy; MIM, Mendelian Inheritance in Man.
| Meesmann Corneal Dystrophy (MECD) | ||||||
|---|---|---|---|---|---|---|
| Gene (Locus) | Refseq | Exon | Nucleotide Change | AA Change | Original | Reference |
| KRT3 (12q13) | NM_057088 | 7 | c.1508G>C | p.Arg503Pro | R503P | 2 |
| c.1525G>A | p.Glu509Lys | E509K | 3 | |||
| KRT12 (17q12) | NM_000223 | 1 | c.386T>C | p.Met129Thr | M129T | 4, 5 |
| c.389A>C | p.Gln130Pro | Q130P | 6 | |||
| c.394C>G | p.Leu132Val | p.Leu132Val | 7 | |||
| c.399T>G | p.Asn133Lys | N133K | 8 | |||
| c.403A>G | p.Arg135Gly | Arg135Gly | 9 | |||
| c.404G>T | p.Arg135Ile | Arg135Ile | 9 | |||
| c.404G>C | p.Arg135Thr | R135T | 3, 4 | |||
| c.405A>C | p.Arg135Ser | Arg135Ser | 10 | |||
| c.409G>C | p.Ala137Pro | Ala137Pro | 11 | |||
| c.419T>G | p.Leu140Arg | Leu140Arg | 9 | |||
| c.427G>C | p.Val143Leu | V143L | 3 | |||
| 6 | c.1171_1197dup | p.Lle391_Leu399dup | 1222ins27 | 10 | ||
| c.1276A>G | p. Ile 426Val | I426V | 12 | |||
| c.1277T>G | p. Ile 426Ser | I426S | 5 | |||
| c.1285T>G* | p.Tyr429Asp | Tyr429Asp | 9 | |||
| c.1286A>G | p.Tyr429Cys | Y429C | 2 | |||
Reported as c.4046T>G ( GenBank accession number AF137286).
| Gelatinous Drop-Like Corneal Dystrophy (GDLD) | ||||||
|---|---|---|---|---|---|---|
| Gene (Locus) | Refseq | Exon | Nucleotide Change | AA Change | Original | Reference |
| TACSTD2 (M1S1) (1p32) | NM_002353 | 1 | c.2T>G | p.Met1Arg | M1R | 13 |
| c.198C>A | p.Cys66X | C66X | 14 | |||
| c.250A>T | p.Lys84X | K84X | 15 | |||
| c.322T>C | p.Cys108Arg | C108R | 15 | |||
| c.341T>G | p.Phe114Cys | F114C | 14 | |||
| c.352C>T | p.Gln118X | Q118X | 16-22 | |||
| c.352C>G | p.Gln118Glu | Q118E | 13 | |||
| c.355T>A | p.Cys119Ser | C119S | 13 | |||
| c.493_494ins CCACCGCC |
p.Gly165AlafsX15 | 8-bp ins | 13 | |||
| c.509C>A | p.Ser170X | S170X | 22 | |||
| c.519dupC | p.Ala174ArgfsX43 | 520insC | 23 | |||
| c.551A>G | p.Tyr184Cys | Y184C | 16 | |||
| c.557T>C | p.Leu186Pro | L186P | 14, 24 | |||
| c.564delC | p.Lys189SerfsX82 | 870delC | 13 | |||
| c.581T>A | p.Val194Glu | V194E | 13 | |||
| c.619C>T | p.Gln207X | Q207X | 22 | |||
| c. 632delA | p.Gln211ArgfsX60 | 632delA | 22 | |||
| c.653delA | p.Asp218ValfsX53 | c.653delA | 25 | |||
| c.679G>A | p.Glu227Lys | E227K | 14 | |||
| c.772_783del ATCTATTACCTGinsT |
p.Lle258X | 772 to 783del (ATCTATTACCTG) + 772insT |
26 | |||
| c.811delA | p.Lys271SerfsX26 | 1117delA | 13 | |||
Bowman Layer Dystrophies
Reis–Bücklers Corneal Dystrophy (RBCD) = Granular Corneal Dystrophy, type 3 (see TGFBI corneal dystrophies) Thiel-Behnke Corneal Dystrophy (TBCD) (see TGFBI corneal dystrophies)
Stromal Dystrophies
| Lattice Corneal Dystrophy, Gelsolin Type (LCD2) | ||||||
|---|---|---|---|---|---|---|
| Gene (Locus) | Refseq | Exon | Nucleotide Change | AA Change | Original | Reference |
| GSN (9q34) | NM_000177 | 4 | c.654G>A* | p.Asp187Asn* | G654→A654 | 27-32 |
| c.654G>T* | p.Asp187Tyr* | G654→T654 | 33-36 | |||
Nucleotide and codon numbering system used by the authors who first reported mutations in gelsolin gene,28 with the amino acid numbering starting at the 28th translated residue Ala, preceded by a 27-residue signal peptide. If the initiation Met is designated codon +1, the mutations would be documented as c.640G>A (p.Asp214Asn) and c.640G>T (p.Asp214Pyr).
| TGFBI Corneal Dystrophies | |||||||
|---|---|---|---|---|---|---|---|
| Dystrophy | Gene (Locus) | Refseq | Exon | Nucleotide Change |
AA Change | Original | Reference |
| Reis–Bücklers = granular corneal dystrophy type 3 (RBCD) | TGFBI(5q31) | NM_000358 | 4 | c.371G>T | p.Arg124Leu | p.Arg124Leu | 37-40 |
| Thiel–Behnke corneal dystrophy (TBCD) | 12 | c.1664G>A | p.Arg555Gln | p.Arg555Gln | 37, 38 | ||
| Classic Lattice corneal dystrophy (LCD1) | 4 | c.370C>T | p.Arg124Cys | p.Arg124Cys | 37, 41 | ||
| Granular corneal dystrophy, type 1 (classic) (GCD1) | 12 | c.1663C>T | p.Arg555Trp | p.Arg555Trp | 37 | ||
| Granular corneal dystrophy, type 2 (granular-lattice) (GCD2) | 4 | c.371G>A | p.Arg124His | p.Arg124His | 37, 40 | ||
| Granular Corneal Dystrophy—Variants | |||||||
|---|---|---|---|---|---|---|---|
| Classification | Gene (Locus) | Refseq | Exon | Nucleotide Change | AA Change | Original | Reference |
| Variant GCD | TGFBI(5q31) | NM_000358 | 4 | c.337G>A | p.Val113Ile | Val113Ile | 42 |
| Variant GCD | c.367G>C | p.Asp123His | D123H | 43, 44 | |||
| Variant GCD | c.370C>A | p.Arg124Ser | R124S | 45, 46 | |||
| Variant GCD | c.371G>T & c.373_ 378delACGGAG |
p.Arg124Leu p.Thr125_Glu126del |
R124L and DeltaT125–DeltaE126 |
47, 48 | |||
| Lattice Corneal Dystrophy—Variants | |||||||
|---|---|---|---|---|---|---|---|
| Classification | Gene (Locus) | Refseq | Exon | Nucleotide Change | AA Change | Original | Reference |
| Variant LCD | TGFBI (5q31) | NM_000358 | 11 | c.1501C>A | p.Pro501Thr | Pro501Thr | 19, 20, 49-52 |
| Variant LCD | c.1514T>A | p.Val505Asp | V501D | 53 | |||
| Variant LCD | 12 | c.1553T>C | p.Leu518Pro | p.Leu518Pro | 20, 54-57 | ||
| Variant LCD | c.1580T>G | p.Leu527Arg | L527R | 20, 54, 55, 58-63 | |||
| Variant LCD | c.1612A>C | p.Thr538Pro | Thr538Pro | 64 | |||
| Variant LCD | c.1613C>G | p.Thr538Arg | T538R | 45 | |||
| Variant LCD | c.1616T>A | p.Val539Asp | Val539Asp | 65 | |||
| Variant LCD | c.1618_1620delTTT | p.Phe540del | DF540 | 45, 66 | |||
| Variant LCD | c.1619T>C | Phe540Ser | Phe540Ser | 67 | |||
| Variant LCD | c.1631A>G | p.Asn544Ser | N544S | 49, 61, 68 | |||
| Variant LCD | c.1636G>A | p.Ala546Thr | A546T | 47, 69, 70 | |||
| Variant LCD | c.1637C>A | p.Ala546Asp | A546D | 71-75 | |||
| Variant LCD | c.1640T>C | p.Phe547Ser | F547S | 76 | |||
| Variant LCD | c.1652C>A | p.Pro551Gln | P551Q | 71-73 | |||
| Variant LCD | 13 | c.1706T>G | p.Leu569Arg | Leu569Arg | 77 | ||
| Variant LCD | c.1714_1716delCAC | p.His572del | His572del | 78 | |||
| Variant LCD | c.1715A>G | p.His572Arg | H572R | 79 | |||
| Variant LCD | c.1781G>T | p.Gly594Val | Gly594Val | 65 | |||
| Variant LCD | 14 | c.1903T>A | p.Met619Ly | Met619Lys | 80 | ||
| Variant LCD | c.1864A>C | p.Asn622His | A→C transition at nucleotide 1911 |
81 | |||
| Variant LCD | c.1866T>A | p.Asn622Lys | N622K(A) | 45 | |||
| Variant LCD | c.1866T>G | p.Asn622Lys | N622K(G) | 45 | |||
| Variant CBD I & Variant LCD |
c.1868G>A | p.Gly623Asp | G623D | 27, 45, 82 | |||
| Variant LCD | c.1870_1875del GTGGTC |
p.Val624_Val625del | Val624-Val625del | 65 | |||
| Variant LCD | c.1874T>A | p.Val625Asp | V625D | 83 | |||
| Variant LCD | c.1877A>G | p.His626Arg | H626R | 45-47, 65, 84 | |||
| Variant LCD | c.1877A>C | p.His626Pro | H626P | 45 | |||
| Variant LCD | c.1879delG | p.Val627SerfsX44 | V627S | 45 | |||
| Variant LCD | c.1886_1894dup | p.Thr629_Asn630insAsnValPro | NVP629-630ins | 85 | |||
| Variant LCD | c.1892T>A | p.Val631Asp | V631D | 45 | |||
Descemet membrane and Endothelial Dystrophies
| Macular Corneal Dystrophy (MCD) | ||||||
|---|---|---|---|---|---|---|
| Gene (Locus) |
Refseq | Exon | Nucleotide Change |
AA Change |
Original | Reference |
|
CHST6 (16q22) |
NM_021615 | 1 | c.1A>T | p.M1? | p.M1? | 86 |
| c.6G>A | p.Trp2X | Trp2Ter | 87 | |||
| c.7C>A | p.Leu3Met | Leu3Met | 87 | |||
| c.15_16delCG | p.Val6LeufsX102 | delCG707-708 | 88 | |||
| c.16_40del25 | p.Val6_Leu14. SerfsX56 |
c.16_40del, Val6fs, R5fs; c.708-732del, R5fs |
87, 89 | |||
| c.15_16ins ATGCTGTGCG |
p.Val6MetfsX106 | c.15_16ins ATGCTGTGCG, V6fs |
90 | |||
| c.44T>C | p.Leu15Pro | 736T>C, L15P | 91 | |||
| c.51delG | p.Gln18ArgfsX52 | c.51delG, Gln18fs | 92 | |||
| c.52C>T | p.Gln18X | c.744C>T, Q18X | 89 | |||
| c.65T>G | p.Leu22Arg | Leu22Arg | 88 | |||
| c.91C>T | p.Pro31Ser | 783C>T, P31S | 93 | |||
| c.94_100del TCGTCCC |
p.Ser32GlnfsX36 | c.786-792del, P31fs | 89 | |||
| c.124C>T | p.His42Tyr | His42Tyr | 88 | |||
| c.137T>C | p.Leu46Pro | c.137T>C, Leu46Pro | 92 | |||
| c.148C>A* | p.Arg50Cys | Arg50Cys | 94 | |||
| c.148C>T | p.Arg50Cys | C840T, Arg50Cys | 88 | |||
| c.149G>T | p.Arg50Leu | Arg50Leu | 88 | |||
| c.152C>T | p.Ser51Leu | C844T, S51L; Ser51Leu | 95 | |||
| c.155G>A | p.Gly52Asp | c.847G>A, G52D | 89 | |||
| c.158C>T | p.Ser53Leu | Ser53Leu | 88, 89 | |||
| c.161C>T | p.Ser54Phe | Ser54Phe | 87 | |||
| c.166_167 delGTinsAG |
p.Val56Arg | Val56Arg | 87 | |||
| c.172C>T | p.Gln58X | 864C>T, Q58X | 91 | |||
| c.176T>C | p.Leu59Pro | T868C, L59P | 96 | |||
| c.180delC | p.Phe60LeufsX10 | c.180delC, Phe60fs; c.872delC, F60fs |
87, 89 | |||
| c.182A>C | p.Asn61Thr | 874A>C, N61T | 91 | |||
| c.189C>G | p.His63Gln | c.189C>G, His63Gln | 92 | |||
| c.196G>T | p.Val66Phe | Val66Phe | 97 | |||
| c.196G>C | p.Val66Leu | G888C, V66L | 96 | |||
| c.198delC | p.Phe67SerfsX3 | delC890; c.890delC, V66fs | 88, 89 | |||
| c.202T>C | p.Tyr68His | 894T>C, Y68H | 91 | |||
| c.209T>A | p.Met70Leu | 891T>A, M70L | 91 | |||
| C214C>T | p.Pro72Ser | 906C>T, P72S, Pro72Ser | 93, 95 | |||
| c.217G>A | p.Ala73Thr | Ala73Thr | 87 | |||
| c.217G>C | p.Ala73Pro | c.217G>C, Ala73Pro | 92 | |||
| c.226G>A | p.Val76Met | G918A, V76M | 96 | |||
| c.231G>C | p.Trp77Cys | c.923G>C | ||||
| c.231G>A | p.Trp77X | c.231G>A, Trp77X | 92 | |||
| c.244C>T | p.Gln82X | 936C>T, Q82X, Gln82Stop | 91, 98 | |||
| c.271_273 delGCTinsA |
p.Ala91SerfsX17 | 962_965delGCTinsA | 91 | |||
| c.274G>C | p.Val92Leu | c.274G>C, Val92Leu | 92 | |||
| c.277C>A | p.Arg93Ser | c.969C>A | ||||
| c.278G>A | p.Arg93His | Arg93His | 88 | |||
| c.290G>C | p.Arg97Pro | Arg97Pro | 88 | |||
| c.293_294 delCCinsGG |
p.Ser98Trp | c.985C>G, c.986C>G, S98W | 89 | |||
| c.293_294 delCCinsTG |
p.Ser98Leu | Ser98Leu | 87 | |||
| c.304T>G | p.Cys102Gly | 996T>G, Cys102Gly | 91, 95 | |||
| c.305G>A | p.Cys102Tyr | Cys102Tyr | 88 | |||
| c.310A>G | p.Met104Val | Met104Val | 95 | |||
| c.320T>C | p.Phe107Ser | c.1012T>C, F107S | 89 | |||
| c.329A>G | p.Tyr110Cys | Tyr110Cys | 95 | |||
| c.340C>T | p.Arg114Cys | c.340C>T, Arg114Cys | 92 | |||
| c.363C>G | p.Phe121Leu | c.1055C>G, F121L | 89 | |||
| c.364dupC | p.Gln122ProfsX100 | 1055-1056insC | 91 | |||
| c.365A>C | p.Gln122Pro | Gln122Pro | 95 | |||
| c.369G>A | p.Trp123X | c.369G>A, Trp123X; c.1061G>A, W123X |
87, 89 | |||
| c.369_375 dupGGCCGTG |
p.Ser126GlyfsX98 | 1067-1068ins(GGCCGTG) | 98 | |||
| c.379C>T | p.Arg127Cys | Arg127Cys | 88 | |||
| c.383C>T | p.Ala128Val | p.A128V | 90 | |||
| c.391T>C | p.Ser131Pro | c.391T>C, Ser131Pro; 1083T>C, S131P |
87, 91 | |||
| c.392C>T | p.Ser131Leu | c.392C>T, Ser131Leu | 92 | |||
| c.413_414 dupTT |
p.Pro139PhefsX243 | 2T insertion after 1106T, frameshift after 137A |
94 | |||
| c.418C>T | p.Arg140X | C1110T, R140X, Arg140end | 99, 100 | |||
| c.455T>C | p.Leu152Pro | 1147T>C, L152P | 91 | |||
| c.459C>A | p.Cys153X | c.459C>A, Cys153X; c.1151C>A, C153X |
87, 89 | |||
| c.484C>G | p.Arg162Gly | p.Arg162Gly | 92 | |||
| c.494G>A | p.Cys165Tyr | p.C165Y | 86 | |||
| c.494_495delGCinsCT | p.Cys165Ser | c.494G>C, c.495C>T, Cys165Ser |
87 | |||
| c.495C>G | p.Cys165Trp | Cys165Trp | 87 | |||
| c.497G>C | p.Arg166Pro | 1189G>C, R166P | 91 | |||
| c.500C>T | p.Ser167Phe | Ser167Phe | 87 | |||
| c.518T > C | p.Leu173Pro | p.Leu173Pro | 101 | |||
| c.521A>G | p.Lys174Arg | A1213G, K174R | 94 | |||
| c.529C>T | p.Arg177Cys | c.529C>T, Arg177Cys | 92 | |||
| c.530G>A | p.Arg177His | R177H | 102 | |||
| c.533T>G | p.Phe178Cys | Phe178Cys | 87 | |||
| c.545delA | p.Gln182ArgfsX199 | c.545delA, Gln182fs;delA1237 | 87, 88 | |||
| c.573dupC | p.Ala192ArgfsX30 | c.573_574insC, Ala192fs | 92 | |||
| c.578T>C | p.Leu193Pro | Leu193Pro | 87 | |||
| c.581_586 delACCTACinsGGT |
p.Asn194_Arg196 delinsArgCys |
ACCTAC 1273 GGT | 88 | |||
| c.585_587 dupACG |
p.Arg196_Lle197insArg | c.1279insACG, R195-196ins | 89 | |||
| c.593T>A | p.Val198Glu | c.1285T>A, V198E | 103 | |||
| c.599T>G | p.Leu200Arg | 1291T>G, L200R;Leu200Arg; T1291G, L200R |
91, 99 | |||
| c.604C>A | p.Arg202Ser | c.1296C>A, R202S | 89, 91 | |||
| c.607G>A | p.Asp203Asn | c.607G>A, Asp203Asn | 92 | |||
| c.609C>A | p.Asp203Glu | C1301A, D203E | 94 | |||
| c.611C>A | p.Pro204Gln | P204Q;c.1303C>A, P204G; 1303C>A, P204Q |
89, 91, 102 | |||
| c.611C>G | p.Pro204Arg | Pro204Arg | 87 | |||
| c.612_614 delGCGinsAT |
p.Arg205TrpfsX176 | GCG 1304 AT | 88 | |||
| c.614G>A | p.Arg205Gln | Arg205Gln | 88 | |||
| c.614G>T | p.Arg205Leu | R205L | 102 | |||
| c.616G>A | p.Ala206Thr | Ala206Thr | 88 | |||
| c.617C>T | p.Ala206Val | 1309C>T, A206V | 93 | |||
| c.629C>T | p.Ser210Phe | c.1321C>T, S210F | 89 | |||
| c.631C>T | p.Arg211Trp | C1323T, 1323C>T, R211W | 94, 102 | |||
| c.632G>A | p.Arg211Gln | Arg211Gln | 98 | |||
| c.649G>A | p.Ala217Thr | A217T | 102 | |||
| c.656_657insCTG | p.Ala219_Arg220insTrp | c.656_657insCTG, Ala219_Arg220insTrp; c.1348insCTG, W219-220ins |
87, 89 | |||
| c.661G>T | p.Asp221Tyr | c.661G>T, Asp221Tyr;D221Y | 87, 89 | |||
| c.663C>G | p.Asp221Glu | c.663C>G, Asp221Glu; c.1355C>G, D221E |
87, 89 | |||
| c.668G>A | p.Gly223Asp | Gly223Asp | 100 | |||
| c.682_683 delACinsGA |
p.Thr228Asp | c.682A>G, 683C>A, Thr228Asp |
92 | |||
| c.696G>A | p.Trp232X | G1388A, W232X | 96 | |||
| c.738C>G | p.Cys246Trp | c.738C>G, Cys246Trp | 92 | |||
| c.740delG | p.Arg247LeufsX134 | c.740delG, Arg247fs | 92 | |||
| c.744C>G | p.Ser248Arg | c.744C>G, Ser248Arg | 92 | |||
| c.746A>C | p.His249Pro | His249Pro | 88 | |||
| c.803A>G | p.Tyr268Cys | A1495G, Y268C | 96 | |||
| c.814C>A | p.Arg272Ser | Arg272Ser | 87 | |||
| c.815G>A | p.Arg272His | c.815G>A, Arg272His | 92 | |||
| c.820G>A | p.Glu274Lys | Glu274Lys | 88, 94 | |||
| c.827T>C | p.Leu276Pro | Leu276Pro, c.1519T>C; L276P, T1519C |
87, 99 | |||
| c.925G>T | p.Gly309X | c.1617G>T, G309X | 89 | |||
| c.985G>C | p.Val329Leu | c.985G>C, p.V329L | 90 | |||
| c.991C>T | p.Gln331X | Gln331X | 95 | |||
| c.993G>T | p.Gln331His | Gln331His | 100 | |||
| c.1000C>T | p.Arg334Cys | Arg334Cys | 87 | |||
| c.1001G>A | p.Arg334His | c.1693G>A, Arg334Cys | 97 | |||
| c.1002_1012delinsTTG | p.His335CysfsX27 | His335fs | 87 | |||
| c.1039G>T | p.Glu347X | c.1731G>T, E347X | 89 | |||
| c.1046G>A | p.Cys349Tyr | c.1046G>A, Cys349Tyr | 92 | |||
| c.1047C>G | p.Cys349Trp | c.1047C>G, Cys349Trp | 92 | |||
| c.1052_1059 dupCTGCGCTG |
p.Gln354ValfsX30 | c.1744_1751 dupGTGCGCTG |
95 | |||
| c.1056_1078del23 | p.Ala352AlafsX5; p.Leu353CysfsX4 |
del1748-1770 | 88 | |||
| c.1072T>G | p.Tyr358Asp | T1764G, Y358D | 99 | |||
| delORF | Absent Protein | delORF | 88 | |||
c.148C>A translates to p.Arg50Ser. Authors reported Arg50Cys as amino acid change, which would mean that nucleotide change is actually c.148C>T.
| Schnyder Corneal Dystrophy (SCD) | ||||||
|---|---|---|---|---|---|---|
| Gene (Locus) | Refseq | Exon | Nucleotide Change | AA Change | Original | Reference |
| UBIAD1 (1p36) | NM_013319 | 1 | c.305A>G | p.Asn102Ser | p.Asn102Ser | 104-107 |
| c.335A>G | p.Asp112Gly | p.Asp112Gly | 104 | |||
| c.353A>G | p.Asp118Gly | p.Asp118Gly | 107 | |||
| c.355A>G | p. Arg119Gly | p. Arg119Gly | 104, 106 | |||
| c.361C>G | p.Leu121Val | p.Leu121Val | 106, 107 | |||
| c.511T>C | p.Ser171Pro | p.Ser171Pro | 107 | |||
| c.524C>T | p.Thr175Ile | p.Thr175Ile | 104, 107 | |||
| c.529G>A | p.Gly177Arg | p.Gly177Arg | 105, 107 | |||
| c.556G>A | p.Gly186Arg | p.Gly186Arg | 107 | |||
| 2 | c.695A>G | p.Asn232Ser | p.Asn232Ser | 104 | ||
| c.708C>G | p.Asp236Glu | p.Asp236Glu | 107 | |||
| Fleck Corneal Dystrophy (FCD) | ||||||
|---|---|---|---|---|---|---|
| Gene (Locus) | Refseq | Exon | Nucleotide Change | AA Change | Original | Reference |
| PIP5K3 (2q35) | NM_015040 | 17 | c.2098delA | p.Asn701ThrfsX7 | 2256delA | 110 |
| 17 | c.2116_2117delCT | p.Leu706ValfsX6 | 2274delCT | 110 | ||
| Intron 20 | c.3619 -1G>C | p.Val1207AlafsX11 | IVS19-1G→C, intron 19 |
110 | ||
| 20 | c.2551C>T | p.Arg851X | R851X | 110 | ||
| 20 | c.2962C>T | p.Gln988X | Q988X | 110 | ||
| 20 | c.3088G>T | p.Glu1030X | E1030X | 110 | ||
| 20 | c.3112C>T | p.Arg1038X | R1038X | 110 | ||
| 20 | c.3308A>G | p.Lys1103Arg | K1103R | 110 | ||
| Posterior Polymorphous Corneal Dystrophy 3 (PPCD3) | ||||||
|---|---|---|---|---|---|---|
| Gene (Locus) | Refseq | Exon | Nucleotide Change | AA Change | Original | Reference |
| TCF8(10p11–10q11) | NM_030751 | 1 | c.2T>G | p.Met1Arg | Met1Arg | 113 |
| c.34C>T | p.Gln12X | Gln12X | 113 | |||
| 5 | c.640C>T | p.Gln214X | Gln214X | 113 | ||
| 7 | c.929dupA | p.Cys311ValfsX25 | c.953_954insA | 113 | ||
| c.973C>T | p.Arg325X | Arg325X | 113 | |||
| c.1124delT | p.Phe375SerfsX31 | p.F375fs | 114 | |||
| c.1332_1335delCAAT | p. Ile444MetfsX48 | c.1332_1335delCAAT | 115 | |||
| c.1348C>T | p.Gln450X | c.1350C→T | 115 | |||
| c.1387_1390delCCTT | p.Pro463_Leu464 >TrpfsX29 |
p.P463fs | 114 | |||
| c.1482dupA | p.Glu495ArgfsX10 | c.1506dupA | 113 | |||
| c.1568delA | p.Val526X | c.1592delA | 113 | |||
| c.1576dupG | p.Val526GlyfsX3 | c.1578_1579insG | 115 | |||
| c.2157C>G | p.Tyr719X | p.Y719X | 114 | |||
| c.2182G>T | p.Glu728X | c.2184G→T | 115 | |||
| c.2324dupA | p.Glu776GlyfsX44 | c.2324_2325dupA | 114 | |||
| 9 | c.2916_2917delTG | p.Gly973ValfsX14 | c.2916_2917delTG | 115 | ||
| c.2988_2989delAG | p.Glu997AlafsX7 | c.3012_3013delAG | 113 | |||
| Congenital Hereditary Endothelial Dystrophy 2 (CHED2) | ||||||
|---|---|---|---|---|---|---|
| Gene (Locus) | Refseq | Exon | Nucleotide Change | AA Change | Original | Reference |
|
SLC4A11 (20p11.2-20q11.2) |
NM_032034 | 2 | c.140delA | p.Tyr47SerfsX69 | Tyr47SerfsX69 | 116 |
| c.246_247delTTinsA | p.Phe84LeufsX32 | p.Arg82ArgfsX33 | 117 | |||
| 3 | c.306delC | p.Gly103ValfsX13 | c.[306delC]+[?] | 116 | ||
| c.334C>T | p.Arg112X | Arg112X | 116 | |||
| 4 | c.353_356delAGAA | p.Lys118ThrfsX12 | 353_356delAGAA | 118 | ||
| c.473_480 delGCTTCGCC |
p.Arg158ProfsX4 | p.Arg158ProfsX4; Arg158GlnfsX4 |
116, 119 | |||
| 5 | c.618_619delAG | p.Val208AlafsX38 | Val208AlafsX38 | 116 | ||
| c.625C>T | p.Arg209Trp | Arg209Trp | 116 | |||
| c.637T>C | p.Ser213Pro | p.Ser213Pro | 119 | |||
| c.638C>T | p.Ser213Leu | Ser213Leu | 116 | |||
| 6 | c.695G>A | p.Ser232Asn | p.Ser232Asn | 120 | ||
| c.697C>T | p.Arg233Cys | Arg233Cys | 116 | |||
| 7 | c.859_862 delGAGAinsCCT |
p.Glu287ProfsX21 | E287fsX21 | 121 | ||
| c.878_889del12 | p.Glu293_Glu296del | Glu293_Glu296del | 116 | |||
| c.985A>T | p.Arg329X | p.Arg329X | 120 | |||
| IVS-7 | c.996 + 26C_+44Cdel19 | Unknown | Unknown | 116 | ||
| IVS-8 | c.1091-1G>C | Unknown | Unknown | 116 | ||
| 9 | c.1202C>A | Thr401Lys | Thr401Lys | 116 | ||
| 10 | c.1253G>A | p.Gly418Asp | Gly418Asp | 116 | ||
| c.1317_1322del6ins8 | p.Leu440ValfsX6 | Leu440ValfsX6 | 116 | |||
| 11 | c.1378_1381 delTACGinsA |
p.Tyr460_Ala461 delinsThr |
p.Tyr460_Ala461 delinsThr |
119 | ||
| c.1391G>A | p.Gly464Asp | G464D | 118 | |||
| c.1418T>G | p.Leu473Arg | Leu473Arg | 116 | |||
| c.1463G>A | p.Arg488Lys | p.Arg488Lys | 119 | |||
| 12 | c.1466C>T | p.Ser489Leu | S489L | 116, 118 | ||
| 13 | c.1704_1705delCT | p.Ser569ArgfsX177 | p.His568HisfsX177 | 117 | ||
| c.1751C>A | p.Thr584Lys | Thr584Lys | 116 | |||
| 14 | c.1813C>T | p.Arg605X | p.Arg605X | 116-118 | ||
| c.1894G>T | p.Glu632X | p.Glu632X | 116, 117 | |||
| 15 | c.2014_2016delTTC or c.2017_2019delTTC |
p.Phe672del or p.Phe673del |
F672del or F673del | 121 | ||
| IVS 15 | c.2067 -6_-16 delins GGCCGGCCGG |
Inactivation of splice acceptor site |
IVS15 –6_-16delins GGCCGGCCGG |
118 | ||
| 16 | c.2233_2240 dupTATGACAC |
p.Ile748MetfsX5 | p.Thr747ThrfsX6 | 119 | ||
| 17 | c.2263C>T | p.Arg755Trp | Arg755Trp | 116 | ||
| c.2264G>A (g.9044G>A) | p.Arg755Gln | p.Arg755Gln | 117, 118 | |||
| c.2318C>T | p.Pro773Leu | Pro773Leu | 116 | |||
| c.2389_2391delGAT | p.Asp797del | Asp797del | 116 | |||
| c.2407C>T | p.Gln803X | Gln803X | 116 | |||
| c.2411G>A | p.Arg804His | p.Arg804His | 117 | |||
| c.2420delTinsGG | p.Leu807ArgfsX71 | p.Leu807ArgfsX71 | 117 | |||
| c.2423_2454del | p.Leu808ArgfsX110 | p.Leu808ArgfsX110 | 119 | |||
| 18 | c.2470G>A | p.Val824Met | p.Val824Met | 116, 119 | ||
| c.2498C>T | p.Thr833Met | p.Thr833Met | 117 | |||
| c.2528T>C | p.Leu843Pro | p.Leu843Pro | 119 | |||
| c.2566A>G | p.Met856Val | p.Met856Val | 119 | |||
| c.2606G>A | p.Arg869His | p.Arg869His | 117 | |||
| c.2605C>T | p.Arg869Cys | R869C | 116, 118 | |||
| 19 | c.2623C>T | p.Arg875X | Arg875X | 116 | ||
REFERENCES
- 1.Boutboul S, Black GC, Moore JE, et al. A subset of patients with epithelial basement membrane corneal dystrophy have mutations in TGFBI/BIGH3. Hum Mutat. 2006;27:553–557. doi: 10.1002/humu.20331. [DOI] [PubMed] [Google Scholar]
- 2.Chen YT, Tseng SH, Chao SC. Novel mutations in the helix termination motif of keratin 3 and keratin 12 in 2 Taiwanese families with Meesmann corneal dystrophy. Cornea. 2005;24:928–932. doi: 10.1097/01.ico.0000159732.29930.26. [DOI] [PubMed] [Google Scholar]
- 3.Irvine AD, Corden LD, Swensson O, et al. Mutations in cornea-specific keratin K3 or K12 genes cause Meesmann’s corneal dystrophy. Nat Genet. 1997;16:184–187. doi: 10.1038/ng0697-184. [DOI] [PubMed] [Google Scholar]
- 4.Corden LD, Swensson O, Swensson B, et al. Molecular genetics of Meesmann’s corneal dystrophy: ancestral and novel mutations in keratin 12 (K12) and complete sequence of the human KRT12 gene. Exp Eye Res. 2000;70:41–49. doi: 10.1006/exer.1999.0769. [DOI] [PubMed] [Google Scholar]
- 5.Nichini O, Manzi V, Munier FL, et al. Meesmann corneal dystrophy (MECD): report of 2 families and a novel mutation in the cornea specific keratin 12 (KRT12) gene. Ophthalmic Genet. 2005;26:169–173. doi: 10.1080/13816810500374391. [DOI] [PubMed] [Google Scholar]
- 6.Corden LD, Swensson O, Swensson B, et al. A novel keratin 12 mutation in a German kindred with Meesmann’s corneal dystrophy. Br J Ophthalmol. 2000;84:527–530. doi: 10.1136/bjo.84.5.527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Aldave AJ. The clinical utility of genetic analysis in the diagnosis and management of inherited corneal disorders. Contemp Ophthalmol. 2005;4:1–10. [Google Scholar]
- 8.Irvine AD, Coleman CM, Moore JE, et al. A novel mutation in KRT12 associated with Meesmann’s epithelial corneal dystrophy. Br J Ophthalmol. 2002;86:729–732. doi: 10.1136/bjo.86.7.729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nishida K, Honma Y, Dota A, et al. Isolation and chromosomal localization of a cornea-specific human keratin 12 gene and detection of four mutations in Meesmann corneal epithelial dystrophy. Am J Hum Genet. 1997;61:1268–1275. doi: 10.1086/301650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yoon MK, Warren JF, Holsclaw DS, et al. A novel arginine substitution mutation in 1A domain and a novel 27 bp insertion mutation in 2B domain of keratin 12 gene associated with Meesmann’s corneal dystrophy. Br J Ophthalmol. 2004;88:752–756. doi: 10.1136/bjo.2003.032870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Takahashi K, Murakami A, Okisaka S, et al. Heterozygous Ala137Pro mutation in keratin 12 gene found in Japanese with Meesmann’s corneal dystrophy. Jpn J Ophthalmol. 2002;46:673–674. doi: 10.1016/s0021-5155(02)00563-4. [DOI] [PubMed] [Google Scholar]
- 12.Coleman CM, Hannush S, Covello SP, et al. A novel mutation in the helix termination motif of keratin K12 in a US family with Meesmann corneal dystrophy. Am J Ophthalmol. 1999;128:687–691. doi: 10.1016/s0002-9394(99)00317-7. [DOI] [PubMed] [Google Scholar]
- 13.Ren Z, Lin PY, Klintworth GK, et al. Allelic and locus heterogeneity in autosomal recessive gelatinous drop-like corneal dystrophy. Hum Genet. 2002;110:568–577. doi: 10.1007/s00439-002-0729-z. [DOI] [PubMed] [Google Scholar]
- 14.Alavi A, Elahi E, Tehrani MH, et al. Four mutations (three novel, one founder) in TACSTD2 among Iranian GDLD patients. Invest Ophthalmol Vis Sci. 2007;48:4490–4497. doi: 10.1167/iovs.07-0264. [DOI] [PubMed] [Google Scholar]
- 15.Murakami A, Kimura S, Fujiki K, et al. Mutations in the membrane component, chromosome 1, surface marker 1 (M1S1) gene in gelatinous drop-like corneal dystrophy. Jpn J Ophthalmol. 2004;48:317–320. doi: 10.1007/s10384-003-0064-5. [DOI] [PubMed] [Google Scholar]
- 16.Tian X, Fujiki K, Li Q, et al. Compound heterozygous mutations of M1S1 gene in gelatinous droplike corneal dystrophy. Am J Ophthalmol. 2004;137:567–569. doi: 10.1016/j.ajo.2003.08.008. [DOI] [PubMed] [Google Scholar]
- 17.Yoshida S, Kumano Y, Yoshida A, et al. Two brothers with gelatinous drop-like dystrophy at different stages of the disease: role of mutational analysis. Am J Ophthalmol. 2002;133:830–832. doi: 10.1016/s0002-9394(02)01407-1. [DOI] [PubMed] [Google Scholar]
- 18.Yajima T, Fujiki K, Murakami A, et al. Gelatinous drop-like corneal dystrophy: mutation analysis of membrane component, chromosome 1, surface marker 1. Nippon Ganka Gakkai Zasshi. 2002;106:265–272. [PubMed] [Google Scholar]
- 19.Ha NT, Fujiki K, Hotta Y, et al. Q118X mutation of M1S1 gene caused gelatinous drop-like corneal dystrophy: the P501T of BIGH3 gene found in a family with gelatinous drop-like corneal dystrophy. Am J Ophthalmol. 2000;130:119–120. doi: 10.1016/s0002-9394(00)00596-1. [DOI] [PubMed] [Google Scholar]
- 20.Fujiki K, Nakayasu K, Kanai A. Corneal dystrophies in Japan. J Hum Genet. 2001;46:431–435. doi: 10.1007/s100380170041. [DOI] [PubMed] [Google Scholar]
- 21.Tsujikawa M, Tsujikawa K, Maeda N, et al. Rapid detection of M1S1 mutations by the protein truncation test. Invest Ophthalmol Vis Sci. 2000;41:2466–2468. [PubMed] [Google Scholar]
- 22.Tsujikawa M, Kurahashi H, Tanaka T, et al. Identification of the gene responsible for gelatinous drop-like corneal dystrophy. Nat Genet. 1999;21:420–423. doi: 10.1038/7759. [DOI] [PubMed] [Google Scholar]
- 23.Tasa G, Kals J, Muru K, et al. A novel mutation in the M1S1 gene responsible for gelatinous droplike corneal dystrophy. Invest Ophthalmol Vis Sci. 2001;42:2762–2764. [PubMed] [Google Scholar]
- 24.Taniguchi Y, Tsujikawa M, Hibino S, et al. A novel missense mutation in a Japanese patient with gelatinous droplike corneal dystrophy. Am J Ophthalmol. 2005;139:186–188. doi: 10.1016/j.ajo.2004.06.090. [DOI] [PubMed] [Google Scholar]
- 25.Markoff A, Bogdanova N, Uhlig CE, et al. A novel TACSTD2 gene mutation in a Turkish family with a gelatinous drop-like corneal dystrophy. Mol Vis. 2006;12:1473–1476. [PubMed] [Google Scholar]
- 26.Ha NT, Chau HM, Cung le X, et al. A novel mutation of M1S1 gene found in a Vietnamese patient with gelatinous droplike corneal dystrophy. Am J Ophthalmol. 2003;135:390–393. doi: 10.1016/s0002-9394(02)01952-9. [DOI] [PubMed] [Google Scholar]
- 27.Afshari NA, Mullally JE, Afshari MA, et al. Survey of patients with granular, lattice, avellino, and Reis-Bucklers corneal dystrophies for mutations in the BIGH3 and gelsolin genes. Arch Ophthalmol. 2001;119:16–22. [PubMed] [Google Scholar]
- 28.Kwiatkowski DJ, Stossel TP, Orkin SH, et al. Plasma and cytoplasmic gelsolins are encoded by a single gene and contain a duplicated actinbinding domain. Nature. 1986;323:455–458. doi: 10.1038/323455a0. [DOI] [PubMed] [Google Scholar]
- 29.Levy E, Haltia M, Fernandez-Madrid I, et al. Mutation in gelsolin gene in Finnish hereditary amyloidosis. J Exp Med. 1990;172:1865–1867. doi: 10.1084/jem.172.6.1865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Maury CP, Kere J, Tolvanen R, et al. Finnish hereditary amyloidosis is caused by a single nucleotide substitution in the gelsolin gene. FEBS Lett. 1990;276:75–77. doi: 10.1016/0014-5793(90)80510-p. [DOI] [PubMed] [Google Scholar]
- 31.Paunio T, Sunada Y, Kiuru S, et al. Haplotype analysis in gelsolin-related amyloidosis reveals independent origin of identical mutation (G654A) of gelsolin in Finland and Japan. Hum Mutat. 1995;6:60–65. doi: 10.1002/humu.1380060112. [DOI] [PubMed] [Google Scholar]
- 32.Stewart HS, Parveen R, Ridgway AE, et al. Late onset lattice corneal dystrophy with systemic familial amyloidosis, amyloidosis V, in an English family. Br J Ophthalmol. 2000;84:390–394. doi: 10.1136/bjo.84.4.390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Boysen G, Galassi G, Kamieniecka Z, et al. Familial amyloidosis with cranial neuropathy and corneal lattice dystrophy. J Neurol Neurosurg Psychiatry. 1979;42:1020–1030. doi: 10.1136/jnnp.42.11.1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chastan N, Baert-Desurmont S, Saugier-Veber P, et al. Cardiac conduction alterations in a French family with amyloidosis of the Finnish type with the p. Asp187Tyr mutation in the GSN gene. Muscle Nerve. 2006;33:113–119. doi: 10.1002/mus.20448. [DOI] [PubMed] [Google Scholar]
- 35.de la Chapelle A, Tolvanen R, Boysen G, et al. Gelsolin-derived familial amyloidosis caused by asparagine or tyrosine substitution for aspartic acid at residue 187. Nat Genet. 1992;2:157–160. doi: 10.1038/ng1092-157. [DOI] [PubMed] [Google Scholar]
- 36.Maury CP, Liljestrom M, Boysen G, et al. Danish type gelsolin related amyloidosis: 654G-T mutation is associated with a disease pathogenetically and clinically similar to that caused by the 654G-A mutation (familial amyloidosis of the Finnish type) J Clin Pathol. 2000;53:95–99. doi: 10.1136/jcp.53.2.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Munier FL, Korvatska E, Djemai A, et al. Kerato-epithelin mutations in four 5q31-linked corneal dystrophies. Nat Genet. 1997;15:247–251. doi: 10.1038/ng0397-247. [DOI] [PubMed] [Google Scholar]
- 38.Ridgway AE, Akhtar S, Munier FL, et al. Ultrastructural and molecular analysis of Bowman’s layer corneal dystrophies: an epithelial origin? Invest Ophthalmol Vis Sci. 2000;41:3286–3292. [PubMed] [Google Scholar]
- 39.Mashima Y, Nakamura Y, Noda K, et al. A novel mutation at codon 124 (R124L) in the BIGH3 gene is associated with a superficial variant of granular corneal dystrophy. Arch Ophthalmol. 1999;117:90–93. doi: 10.1001/archopht.117.1.90. [DOI] [PubMed] [Google Scholar]
- 40.Konishi M, Yamada M, Nakamura Y, et al. Immunohistology of keratoepithelin in corneal stromal dystrophies associated with R124 mutations of the BIGH3 gene. Curr Eye Res. 2000;21:891–896. doi: 10.1076/ceyr.21.5.891.5536. [DOI] [PubMed] [Google Scholar]
- 41.Hotta Y, Fujiki K, Ono K, et al. Arg124Cys mutation of the betaig-h3 bene in a Japanese family with lattice corneal dystrophy type I. Jpn J Ophthalmol. 1998;42:450–455. doi: 10.1016/s0021-5155(98)00050-1. [DOI] [PubMed] [Google Scholar]
- 42.Zenteno JC, Ramirez-Miranda A, Santacruz-Valdes C, et al. Expanding the mutational spectrum in TGFBI-linked corneal dystrophies: identification of a novel and unusual mutation (Val113Ile) in a family with granular dystrophy. Mol Vis. 2006;12:331–335. [PubMed] [Google Scholar]
- 43.Ha NT, Cung le X, Chau HM, et al. A novel mutation of the TGFBI gene found in a Vietnamese family with atypical granular corneal dystrophy. Jpn J Ophthalmol. 2003;47:246–248. doi: 10.1016/s0021-5155(03)00019-4. [DOI] [PubMed] [Google Scholar]
- 44.Cung le X, Ha NT, Chau HM, et al. Mutation analysis of the TGFBI gene in Vietnamese with granular and Avellino corneal dystrophy. Jpn J Ophthalmol. 2004;48:12–16. doi: 10.1007/s10384-003-0009-z. [DOI] [PubMed] [Google Scholar]
- 45.Munier FL, Frueh BE, Othenin-Girard P, et al. BIGH3 mutation spectrum in corneal dystrophies. Invest Ophthalmol Vis Sci. 2002;43:949–954. [PubMed] [Google Scholar]
- 46.Stewart HS, Ridgway AE, Dixon MJ, et al. Heterogeneity in granular corneal dystrophy: identification of three causative mutations in the TGFBI (BIGH3) gene-lessons for corneal amyloidogenesis. Hum Mutat. 1999;14:126–132. doi: 10.1002/(SICI)1098-1004(1999)14:2<126::AID-HUMU4>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- 47.Dighiero P, Niel F, Ellies P, et al. Histologic phenotype-genotype correlation of corneal dystrophies associated with eight distinct mutations in the TGFBI gene. Ophthalmology. 2001;108:818–823. doi: 10.1016/s0161-6420(00)00662-x. [DOI] [PubMed] [Google Scholar]
- 48.Dighiero P, Drunat S, D’Hermies F, et al. A novel variant of granular corneal dystrophy caused by association of 2 mutations in the TGFBI gene-R124L and DeltaT125-DeltaE126. Arch Ophthalmol. 2000;118:814–818. doi: 10.1001/archopht.118.6.814. [DOI] [PubMed] [Google Scholar]
- 49.Mashima Y, Yamamoto S, Inoue Y, et al. Association of autosomal dominantly inherited corneal dystrophies with BIGH3 gene mutations in Japan. Am J Ophthalmol. 2000;130:516–517. doi: 10.1016/s0002-9394(00)00571-7. [DOI] [PubMed] [Google Scholar]
- 50.Kawasaki S, Nishida K, Quantock AJ, et al. Amyloid and Pro501 Thrmutated (beta)ig-h3 gene product colocalize in lattice corneal dystrophy type IIIA. Am J Ophthalmol. 1999;127:456–458. doi: 10.1016/s0002-9394(98)00360-2. [DOI] [PubMed] [Google Scholar]
- 51.Tsujikawa K, Tsujikawa M, Yamamoto S, et al. Allelic homogeneity due to a founder mutation in Japanese patients with lattice corneal dystrophy type IIIA. Am J Med Genet. 2002;113:20–22. doi: 10.1002/ajmg.10709. [DOI] [PubMed] [Google Scholar]
- 52.Yamamoto S, Okada M, Tsujikawa M, et al. A kerato-epithelin (betaig-h3) mutation in lattice corneal dystrophy type IIIA. Am J Hum Genet. 1998;62:719–722. doi: 10.1086/301765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tian X, Fujiki K, Wang W, et al. Novel mutation (V505D) of the TGFBI gene found in a Chinese family with lattice corneal dystrophy, type I. Jpn J Ophthalmol. 2005;49:84–88. doi: 10.1007/s10384-004-0167-7. [DOI] [PubMed] [Google Scholar]
- 54.Fujiki K, Hotta Y, Nakayasu K, et al. Six different mutations of TGFBI (betaig-h3, keratoepithelin) gene found in Japanese corneal dystrophies. Cornea. 2000;19:842–845. doi: 10.1097/00003226-200011000-00015. [DOI] [PubMed] [Google Scholar]
- 55.Hirano K, Nakamura M, Yamamoto N, et al. Geographical feature of lattice corneal dystrophy patients in Aichi Prefecture: an analysis of the TGFBI gene. Nippon Ganka Gakkai Zasshi. 2002;106:352–359. [PubMed] [Google Scholar]
- 56.Endo S, Nguyen TH, Fujiki K, et al. Leu518Pro mutation of the beta ig-h3 gene causes lattice corneal dystrophy type I. Am J Ophthalmol. 1999;128:104–106. doi: 10.1016/s0002-9394(99)00053-7. [DOI] [PubMed] [Google Scholar]
- 57.Hirano K, Hotta Y, Fujiki K, et al. Corneal amyloidosis caused by Leu518Pro mutation of betaig-h3 gene. Br J Ophthalmol. 2000;84:583–585. doi: 10.1136/bjo.84.6.583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hirano K, Hotta Y, Nakamura M, et al. Late-onset form of lattice corneal dystrophy caused by leu527Arg mutation of the TGFBI gene. Cornea. 2001;20:525–529. doi: 10.1097/00003226-200107000-00017. [DOI] [PubMed] [Google Scholar]
- 59.Fujiki K, Hotta Y, Nakayasu K, et al. A new L527R mutation of the betaIGH3 gene in patients with lattice corneal dystrophy with deep stromal opacities. Hum Genet. 1998;103:286–289. doi: 10.1007/s004390050818. [DOI] [PubMed] [Google Scholar]
- 60.Funayama T, Mashima Y, Kawashima M, et al. Lattice corneal dystrophy type III in patients with a homozygous L527R mutation in the TGFBI gene. Jpn J Ophthalmol. 2006;50:62–64. doi: 10.1007/s10384-005-0260-6. [DOI] [PubMed] [Google Scholar]
- 61.Kawashima M, Yamada M, Funayama T, et al. Six cases of late-onset lattice corneal dystrophy associated with gene mutations induced by the transforming growth factor-beta. Nippon Ganka Gakkai Zasshi. 2005;109:93–100. [PubMed] [Google Scholar]
- 62.Nakagawa E, Sakimoto T, Inada N, et al. Histopathological study of lattice corneal dystrophy with L 527 R mutation of transforming growth factor-beta induced gene. Nippon Ganka Gakkai Zasshi. 2004;108:118–123. [PubMed] [Google Scholar]
- 63.Yamada N, Chikama TI, Morishige N, et al. Homozygous mutation (L527R) of TGFBI in an individual with lattice corneal dystrophy. Br J Ophthalmol. 2005;89:771–773. doi: 10.1136/bjo.2004.056168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Yu P, Gu Y, Yang Y, et al. A clinical and molecular-genetic analysis of Chinese patients with lattice corneal dystrophy and novel Thr538Pro mutation in the TGFBI (BIGH3) gene. J Genet. 2006;85:73–76. doi: 10.1007/BF02728974. [DOI] [PubMed] [Google Scholar]
- 65.Chakravarthi SV, Kannabiran C, Sridhar MS, et al. TGFBI gene mutations causing lattice and granular corneal dystrophies in Indian patients. Invest Ophthalmol Vis Sci. 2005;46:121–125. doi: 10.1167/iovs.04-0440. [DOI] [PubMed] [Google Scholar]
- 66.Rozzo C, Fossarello M, Galleri G, et al. A common beta ig-h3 gene mutation (delta f540) in a large cohort of Sardinian Reis Bucklers corneal dystrophy patients. Hum Mutat. 1998;12:215–216. Mutations in brief no. 180 [Online] [PubMed] [Google Scholar]
- 67.Stix B, Leber M, Bingemer P, et al. Hereditary lattice corneal dystrophy is associated with corneal amyloid deposits enclosing C-terminal fragments of keratoepithelin. Invest Ophthalmol Vis Sci. 2005;46:1133–1139. doi: 10.1167/iovs.04-1319. [DOI] [PubMed] [Google Scholar]
- 68.Nakagawa Asahina S, Fujiki K, Enomoto Y, et al. Case of late onset and isolated lattice corneal dystrophy with Asn544Ser (N544S) mutation of transforming growth factor beta-induced (TGFBI, BIGH3) gene. Nippon Ganka Gakkai Zasshi. 2004;108:618–620. [PubMed] [Google Scholar]
- 69.Solari HP, Ventura MP, Perez AB, et al. TGFBI gene mutations in Brazilian patients with corneal dystrophy. Eye. 2007;21:587–590. doi: 10.1038/sj.eye.6702264. [DOI] [PubMed] [Google Scholar]
- 70.Dighiero P, Drunat S, Ellies P, et al. A new mutation (A546T) of the betaig-h3 gene responsible for a French lattice corneal dystrophy type IIIA. Am J Ophthalmol. 2000;129:248–251. doi: 10.1016/s0002-9394(99)00324-4. [DOI] [PubMed] [Google Scholar]
- 71.Klintworth GK, Bao W, Afshari NA. Two mutations in the TGFBI (BIGH3) gene associated with lattice corneal dystrophy in an extensively studied family. Invest Ophthalmol Vis Sci. 2004;45:1382–1388. doi: 10.1167/iovs.03-1228. [DOI] [PubMed] [Google Scholar]
- 72.Aldave AJ, Gutmark JG, Yellore VS, et al. Lattice corneal dystrophy associated with the Ala546Asp and Pro551Gln missense changes in the TGFBI gene. Am J Ophthalmol. 2004;138:772–781. doi: 10.1016/j.ajo.2004.06.021. [DOI] [PubMed] [Google Scholar]
- 73.Aldave AJ, Yellore VS, Self CA, et al. The usefulness of buccal swabs for mutation screening in patients with suspected corneal dystrophies. Ophthalmology. 2004;111:1407–1409. doi: 10.1016/j.ophtha.2003.10.033. [DOI] [PubMed] [Google Scholar]
- 74.Eifrig DE, Jr, Afshari NA, Buchanan HW, 4th, et al. Polymorphic corneal amyloidosis: a disorder due to a novel mutation in the transforming growth factor beta-induced (BIGH3) gene. Ophthalmology. 2004;111:1108–1114. doi: 10.1016/j.ophtha.2003.09.043. [DOI] [PubMed] [Google Scholar]
- 75.Correa-Gomez V, Villalvazo-Cordero L, Zenteno JC. The TGFBI A546D mutation causes an atypical type of lattice corneal dystrophy. Mol Vis. 2007;13:1695–1700. [PubMed] [Google Scholar]
- 76.Takacs L, Losonczy G, Matesz K, et al. TGFBI (BIGH3) gene mutations in Hungary—report of the novel F547S mutation associated with polymorphic corneal amyloidosis. Mol Vis. 2007;13:1976–1983. [PubMed] [Google Scholar]
- 77.Warren JF, Abbott RL, Yoon MK, et al. A new mutation (Leu569Arg) within exon 13 of the TGFBI (BIGH3) gene causes lattice corneal dystrophy type I. Am J Ophthalmol. 2003;136:872–878. doi: 10.1016/s0002-9394(03)00541-5. [DOI] [PubMed] [Google Scholar]
- 78.Aldave AJ, Rayner SA, Kim BT, et al. Unilateral lattice corneal dystrophy associated with the novel His572del mutation in the TGFBI gene. Mol Vis. 2006;12:142–146. [PubMed] [Google Scholar]
- 79.Atchaneeyasakul LO, Appukuttan B, Pingsuthiwong S, et al. A novel H572R mutation in the transforming growth factor-beta-induced gene in a Thai family with lattice corneal dystrophy type I. Jpn J Ophthalmol. 2006;50:403–408. doi: 10.1007/s10384-006-0357-6. [DOI] [PubMed] [Google Scholar]
- 80.Aldave AJ, Yellore VS, Sonmez B, et al. A novel variant of combined granular-lattice corneal dystrophy associated with the Met619Lys mutation in the TGFBI gene. Arch Ophthalmol. doi: 10.1001/archopht.126.3.371. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Stewart H, Black GC, Donnai D, et al. A mutation within exon 14 of the TGFBI (BIGH3) gene on chromosome 5q31 causes an asymmetric, late-onset form of lattice corneal dystrophy. Ophthalmology. 1999;106:964–970. doi: 10.1016/S0161-6420(99)00539-4. [DOI] [PubMed] [Google Scholar]
- 82.Aldave AJ, Rayner SA, King JA, et al. A unique corneal dystrophy of Bowman’s layer and stroma associated with the Gly623Asp mutation in the transforming growth factor beta-induced (TGFBI) gene. Ophthalmology. 2005;112:1017–1022. doi: 10.1016/j.ophtha.2004.12.044. [DOI] [PubMed] [Google Scholar]
- 83.Tian X, Fujiki K, Zhang Y, et al. A novel variant lattice corneal dystrophy caused by association of mutation (V625D) in TGFBI gene. Am J Ophthalmol. 2007;144:473–475. doi: 10.1016/j.ajo.2007.04.015. [DOI] [PubMed] [Google Scholar]
- 84.Chau HM, Ha NT, Cung LX, et al. H626R and R124C mutations of the TGFBI (BIGH3) gene caused lattice corneal dystrophy in Vietnamese people. Br J Ophthalmol. 2003;87:686–689. doi: 10.1136/bjo.87.6.686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Schmitt-Bernard CF, Guittard C, Arnaud B, et al. BIGH3 exon 14 mutations lead to intermediate type I/IIIA of lattice corneal dystrophies. Invest Ophthalmol Vis Sci. 2000;41:1302–1308. [PubMed] [Google Scholar]
- 86.Liskova P, Veraitch B, Jirsova K, et al. Sequencing of the CHST6 gene in Czech macular corneal dystrophy patients supports the evidence of a founder mutation. Br J Ophthalmol. 2007 doi: 10.1136/bjo.2007.125252. [DOI] [PubMed] [Google Scholar]
- 87.Sultana A, Sridhar MS, Klintworth GK, et al. Allelic heterogeneity of the carbohydrate sulfotransferase-6 gene in patients with macular corneal dystrophy. Clin Genet. 2005;68:454–460. doi: 10.1111/j.1399-0004.2005.00517.x. [DOI] [PubMed] [Google Scholar]
- 88.Warren JF, Aldave AJ, Srinivasan M, et al. Novel mutations in the CHST6 gene associated with macular corneal dystrophy in southern India. Arch Ophthalmol. 2003;121:1608–1612. doi: 10.1001/archopht.121.11.1608. [DOI] [PubMed] [Google Scholar]
- 89.Sultana A, Sridhar MS, Jagannathan A, et al. Novel mutations of the carbohydrate sulfotransferase-6 (CHST6) gene causing macular corneal dystrophy in India. Mol Vis. 2003;9:730–734. [PubMed] [Google Scholar]
- 90.Liu NP, Smith CF, Bowling BL, et al. Macular corneal dystrophy types I and II are caused by distinct mutations in the CHST6 gene in Iceland. Mol Vis. 2006;12:1148–1152. [PubMed] [Google Scholar]
- 91.Niel F, Ellies P, Dighiero P, et al. Truncating mutations in the carbohydrate sulfotransferase 6 gene (CHST6) result in macular corneal dystrophy. Invest Ophthalmol Vis Sci. 2003;44:2949–2953. doi: 10.1167/iovs.02-0740. [DOI] [PubMed] [Google Scholar]
- 92.Klintworth GK, Smith CF, Bowling BL. CHST6 mutations in North American subjects with macular corneal dystrophy: a comprehensive molecular genetic review. Mol Vis. 2006;12:159–176. [PubMed] [Google Scholar]
- 93.El-Ashry MF, Abd El-Aziz MM, Wilkins S, et al. Identification of novel mutations in the carbohydrate sulfotransferase gene (CHST6) causing macular corneal dystrophy. Invest Ophthalmol Vis Sci. 2002;43:377–382. [PubMed] [Google Scholar]
- 94.Akama TO, Nishida K, Nakayama J, et al. Macular corneal dystrophy type I and type II are caused by distinct mutations in a new sulphotransferase gene. Nat Genet. 2000;26:237–241. doi: 10.1038/79987. [DOI] [PubMed] [Google Scholar]
- 95.Aldave AJ, Yellore VS, Thonar EJ, et al. Novel mutations in the carbohydrate sulfotransferase gene (CHST6) in American patients with macular corneal dystrophy. Am J Ophthalmol. 2004;137:465–473. doi: 10.1016/j.ajo.2003.09.036. [DOI] [PubMed] [Google Scholar]
- 96.Ha NT, Chau HM, Cung le X, et al. Mutation analysis of the carbohydrate sulfotransferase gene in Vietnamese with macular corneal dystrophy. Invest Ophthalmol Vis Sci. 2003;44:3310–3316. doi: 10.1167/iovs.03-0031. [DOI] [PubMed] [Google Scholar]
- 97.Gulias-Canizo R, Castaneda-Diez R, Gomez-Leal A, et al. Corneal macular dystrophy: clinical, histopathologic and ultrastructural features. Arch Soc Esp Oftalmol. 2006;81:315–320. doi: 10.4321/s0365-66912006000600004. [DOI] [PubMed] [Google Scholar]
- 98.Ha NT, Chau HM, Cung le X, et al. Identification of novel mutations of the CHST6 gene in Vietnamese families affected with macular corneal dystrophy in two generations. Cornea. 2003;22:508–511. doi: 10.1097/00003226-200308000-00004. [DOI] [PubMed] [Google Scholar]
- 99.El-Ashry MF, Abd El-Aziz MM, Shalaby O, et al. Novel CHST6 nonsense and missense mutations responsible for macular corneal dystrophy. Am J Ophthalmol. 2005;139:192–193. doi: 10.1016/j.ajo.2004.07.001. [DOI] [PubMed] [Google Scholar]
- 100.Liu NP, Bao W, Smith CF, et al. Different mutations in carbohydrate sulfotransferase 6 (CHST6) gene cause macular corneal dystrophy types I and II in a single sibship. Am J Ophthalmol. 2005;139:1118–1120. doi: 10.1016/j.ajo.2004.11.054. [DOI] [PubMed] [Google Scholar]
- 101.Yellore VS, Sonmez B, Chen MC, et al. An unusual presentation of macular corneal dystrophy associated with uniparental isodisomy and a novel Leu173Pro mutation. Ophthalmic Genet. 2007;28:169–174. doi: 10.1080/13816810701407925. [DOI] [PubMed] [Google Scholar]
- 102.Iida-Hasegawa N, Furuhata A, Hayatsu H, et al. Mutations in the CHST6 gene in patients with macular corneal dystrophy: immunohistochemical evidence of heterogeneity. Invest Ophthalmol Vis Sci. 2003;44:3272–3277. doi: 10.1167/iovs.02-0910. [DOI] [PubMed] [Google Scholar]
- 103.Abbruzzese C, Kuhn U, Molina F, et al. Novel mutations in the CHST6 gene causing macular corneal dystrophy. Clin Genet. 2004;65:120–125. doi: 10.1111/j.0009-9163.2004.00191.x. [DOI] [PubMed] [Google Scholar]
- 104.Orr A, Dube MP, Marcadier J, et al. Mutations in the UBIAD1 gene, encoding a potential prenyltransferase, are causal for Schnyder crystalline corneal dystrophy. PLoS ONE. 2007;2:e685. doi: 10.1371/journal.pone.0000685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Weiss JS, Kruth HS, Kuivaniemi H, et al. Mutations in the UBIAD1 gene on chromosome short arm 1, region 36, cause Schnyder crystalline corneal dystrophy. Invest Ophthalmol Vis Sci. 2007;48:5007–5012. doi: 10.1167/iovs.07-0845. [DOI] [PubMed] [Google Scholar]
- 106.Yellore VS, Khan MA, Bourla N, et al. Identification of mutations in UBIAD1 following exclusion of coding mutations in the chromosome 1p36 locus for Schnyder crystalline corneal dystrophy. Mol Vis. 2007;13:1777–1782. [PubMed] [Google Scholar]
- 107.Weiss JW, Kruth HS, Kuivaniemi H, et al. Genetic analysis of 14 families with Schnyder crystalline corneal dystrophy reveals clues to UBIAD1 protein function. Am J Med Genet. 2008;146:271–283. doi: 10.1002/ajmg.a.32201. [DOI] [PubMed] [Google Scholar]
- 108.Rodahl E, Van Ginderdeuren R, Knappskog PM, et al. A second decorin frame shift mutation in a family with congenital stromal corneal dystrophy. Am J Ophthalmol. 2006;142:520–521. doi: 10.1016/j.ajo.2006.03.064. [DOI] [PubMed] [Google Scholar]
- 109.Bredrup C, Knappskog PM, Majewski J, et al. Congenital stromal dystrophy of the cornea caused by a mutation in the decorin gene. Invest Ophthalmol Vis Sci. 2005;46:420–426. doi: 10.1167/iovs.04-0804. [DOI] [PubMed] [Google Scholar]
- 110.Li S, Tiab L, Jiao X, et al. Mutations in PIP5K3 are associated with Francois-Neetens mouchetee fleck corneal dystrophy. Am J Hum Genet. 2005;77:54–63. doi: 10.1086/431346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Gottsch JD, Sundin OH, Liu SH, et al. Inheritance of a novel COL8A2 mutation defines a distinct early-onset subtype of Fuchs corneal dystrophy. Invest Ophthalmol Vis Sci. 2005;46:1934–1939. doi: 10.1167/iovs.04-0937. [DOI] [PubMed] [Google Scholar]
- 112.Biswas S, Munier FL, Yardley J, et al. Missense mutations in COL8A2, the gene encoding the alpha2 chain of type VIII collagen, cause two forms of corneal endothelial dystrophy. Hum Mol Genet. 2001;10:2415–2423. doi: 10.1093/hmg/10.21.2415. [DOI] [PubMed] [Google Scholar]
- 113.Aldave AJ, Yellore VS, Yu F, et al. Posterior polymorphous corneal dystrophy is associated with TCF8 gene mutations and abdominal hernia. Am J Med Genet A. 2007;143:2549–2556. doi: 10.1002/ajmg.a.31978. [DOI] [PubMed] [Google Scholar]
- 114.Liskova P, Tuft SJ, Gwilliam R, et al. Novel mutations in the ZEB1 gene identified in Czech and British patients with posterior polymorphous corneal dystrophy. Hum Mutat. 2007;28:638. doi: 10.1002/humu.9495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Krafchak CM, Pawar H, Moroi SE, et al. Mutations in TCF8 cause posterior polymorphous corneal dystrophy and ectopic expression of COL4A3 by corneal endothelial cells. Am J Hum Genet. 2005;77:694–708. doi: 10.1086/497348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Sultana A, Garg P, Ramamurthy B, et al. Mutational spectrum of the SLC4A11 gene in autosomal recessive congenital hereditary endothelial dystrophy. Mol Vis. 2007;13:1327–1332. [PubMed] [Google Scholar]
- 117.Jiao X, Sultana A, Garg P, et al. Autosomal recessive corneal endothelial dystrophy (CHED2) is associated with mutations in SLC4A11. J Med Genet. 2007;44:64–68. doi: 10.1136/jmg.2006.044644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Vithana EN, Morgan P, Sundaresan P, et al. Mutations in sodium-borate cotransporter SLC4A11 cause recessive congenital hereditary endothelial dystrophy (CHED2) Nat Genet. 2006;38:755–757. doi: 10.1038/ng1824. [DOI] [PubMed] [Google Scholar]
- 119.Desir J, Moya G, Reish O, et al. Borate transporter SLC4A11 mutations cause both Harboyan syndrome and non-syndromic corneal endothelial dystrophy. J Med Genet. 2007;44:322–326. doi: 10.1136/jmg.2006.046904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Aldave AJ, Yellore VS, Bourla N, et al. Autosomal recessive CHED associated with novel compound heterozygous mutations in SLC4A11. Cornea. 2007;26:896–900. doi: 10.1097/ICO.0b013e318074bb01. [DOI] [PubMed] [Google Scholar]
- 121.Kumar A, Bhattacharjee S, Prakash DR, et al. Genetic analysis of two Indian families affected with congenital hereditary endothelial dystrophy: two novel mutations in SLC4A11. Mol Vis. 2007;13:39–46. [PMC free article] [PubMed] [Google Scholar]
REFERENCES
- 1.Warburg M, Møller HU. Dystrophy: a revised definition. J Med Genet. 1989;26:769–771. doi: 10.1136/jmg.26.12.769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Erb W. Ueber die “juvenile Form” der progressiven Muskelatrophie und ihre Beziehungen der sogenannten Pseudohypertrophie der Muskeln. Dtsch Arch Klin Med. 1884;34:467–519. [Google Scholar]
- 3.Groenouw A. Knötchenförmige Hornhauttrübungen (Noduli corneae) Arch Augenheilkd. 1890;21:281–289. [Google Scholar]
- 4.Biber H. Ueber einige seltene Hornhautekrankungen:die oberflächliche gittrige Keratitis. In: Diggelmann Zürich A, editor. Inaugural dissertation. 1890. [Google Scholar]
- 5.Møller HU. Granular corneal dystrophy Groenouw type I. Clinical and genetic aspects. Acta Ophthalmol (Copenh) 1991;69(suppl 198):1–40. [PubMed] [Google Scholar]
- 6.Fuchs E. Dystrophia epithelialis corneae. Albrecht Von Graefes Arch Clin Exp Ophthalmol. 1910;76:478–508. [Google Scholar]
- 7.Uhthoff W. Ein Fall von doppelseitiger zentraler, punktförmiger, supepithelialer knötchenförmiger Keratitis, Groenouw mit anatomischem Befunde. Klin Monatsbl Augenheilkd. 1915;54:377–383. [Google Scholar]
- 8.Yoshida Y. Über eine neue Art der Dystrophia corneae mit histologischem Befunde. Albrecht Von Graefes Arch Clin Exp Ophthalmol. 1924;114:91–100. [Google Scholar]
- 9.American Academy of Ophthalmology . External diseases and cornea. In: Sutphin JE, editor. Basic and Clinical Sciences Course 2007–2008. American Academy of Ophthalmology; San Francisco, CA: 2007. pp. 305–329. [Google Scholar]
- 10.Bücklers M. Die erblichen Hornhautdystrophie. Klin Monatsbl Augenheilkd. 1938:1–135. Beiheft 3. [Google Scholar]
- 11.Duke-Elder S, Leigh AG. Corneal dystrophies. In: Duke-Elder S, editor. System of Ophthalmology. Part 2. Vol. 8. Kimpton; London, England: 1965. pp. 864–867. [Google Scholar]
- 12.Waring GO, Rodrigues MM, Laibson PR. Corneal dystrophies. I. Dystrophies of the epithelium, Bowman’s layer and stroma. Surv Ophthalmol. 1978;23:71–122. doi: 10.1016/0039-6257(78)90090-5. [DOI] [PubMed] [Google Scholar]
- 13.Klein D, Franceschetti A. Heredo-familiäre Hornhautdystrophie. In: Becker PD, editor. Humangenetik. Vol. 4. Georg Thieme; Stuttgart, Germany: 1964. pp. 80–81. [Google Scholar]
- 14.Klintworth GK. The molecular genetics of the corneal dystrophies—current status. Front Biosci. 2003;8:687–713. doi: 10.2741/1018. [DOI] [PubMed] [Google Scholar]
- 15.Weiss JS. Schnyder’s dystrophy of the cornea. A Swede-Finn connection. Cornea. 1992;11:93–100. doi: 10.1097/00003226-199203000-00001. [DOI] [PubMed] [Google Scholar]
- 16.Weiss JS. Schnyder crystalline dystrophy sine crystals. Recommendation for a revision of nomenclature. Ophthalmology. 1996;103:465–473. doi: 10.1016/s0161-6420(96)30670-2. [DOI] [PubMed] [Google Scholar]
- 17.Weiss JS. Visual morbidity in 34 families with Schnyder’s crystalline corneal dystrophy. Trans Am Ophthamol Soc. 2007;105:616–648. [PMC free article] [PubMed] [Google Scholar]
- 18.McCarthy MM, Innis S, Dubord P, et al. Panstromal Schnyder corneal dystrophy. A clinical pathologic report with quantitative analysis of corneal lipid composition. Ophthalmology. 1994;101:895–901. [PubMed] [Google Scholar]
- 19.Kanai A, Kaufman HE, Polack FM. Electron microscopic study of Reis-Bücklers dystrophy. Ann Ophthalmol. 1973;5:953–962. [PubMed] [Google Scholar]
- 20.Haddad R, Font RL, Fine BS. Unusual superficial variant of granular dystrophy of the cornea. Am J Ophthalmol. 1977;83:213–218. doi: 10.1016/0002-9394(77)90619-5. [DOI] [PubMed] [Google Scholar]
- 21.Møller HU. Interfamilial variability and intra-familial similarities of granular corneal dystrophy Groenouw type I with respect to biomicroscopical appearance and symptomatology. Acta Ophthalmol. 1989;67:669–677. doi: 10.1111/j.1755-3768.1989.tb04400.x. [DOI] [PubMed] [Google Scholar]
- 22.Reis W. Familiäre, fleckige Hornhautentartung. Dtsch Med Wochenschr. 1917;43:575. [Google Scholar]
- 23.Bücklers M. Über eine weitere familiäre Hornhautdystrophie (Reis) Klin Monatsbl Augenkeilkd. 1949;114:386–397. [Google Scholar]
- 24.Waardenburg PJ, Jonkers GH. A specific type of dominant progressive dystrophy of the cornea, developing after birth. Acta Ophthalmol (Copenh) 1961;39:919–923. doi: 10.1111/j.1755-3768.1961.tb07756.x. [DOI] [PubMed] [Google Scholar]
- 25.Thiel H-J, Behnke H. Eine bisher unbekannte subepitheliale hereditäre Hornhautdystrophie. Klin Monatsbl Augenheilkd. 1967;150:862–874. [PubMed] [Google Scholar]
- 26.Wittebol-Post D, Van Schooneveld MJ, Pels E. The corneal dystrophy of Waardenburg and Jonkers. Ophthalmic Paediatr Genet. 1989;10:249–255. doi: 10.3109/13816818909009879. [DOI] [PubMed] [Google Scholar]
- 27.Grayson M, Wilbrandt H. Dystrophy of the anterior limiting membrane of the cornea (Reis-Bücklers type) Am J Ophthalmol. 1966;61:345–349. doi: 10.1016/0002-9394(66)90296-0. [DOI] [PubMed] [Google Scholar]
- 28.François J. Une nouvelle dystrophie hérédo-familiale de la cornée. J Genet Hum. 1956;5:189–196. [PubMed] [Google Scholar]
- 29.Strachan IM. Cloudy central corneal dystrophy of François. Five cases in the same family. Br J Ophthalmol. 1969;53:192–194. doi: 10.1136/bjo.53.3.192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bramsen T, Ehlers N, Baggesen LH. Central cloudy corneal dystrophy of François. Acta Ophthalmol (Copenh) 1976;54:221–226. doi: 10.1111/j.1755-3768.1976.tb00435.x. [DOI] [PubMed] [Google Scholar]
- 31.Meyer JC, Quantock AJ, Thonar EJ, et al. Characterization of a central corneal cloudiness sharing features of posterior crocodile shagreen and central cloud dystrophy of François. Cornea. 1996;15:347–354. doi: 10.1097/00003226-199607000-00003. [DOI] [PubMed] [Google Scholar]
- 32.Karp CL, Scott IU, Green WR, et al. Central cloudy corneal dystrophy of François. A clinicopathologic study. Arch Ophthalmol. 1997;115:1058–1062. doi: 10.1001/archopht.1997.01100160228013. [DOI] [PubMed] [Google Scholar]
- 33.Klintworth GK. Genetic disorders of the cornea: from research to practical diagnostic testing. Graefes Arch Clin Exp Ophthalmol. 2005;33:231–232. doi: 10.1111/j.1442-9071.2005.01024.x. [DOI] [PubMed] [Google Scholar]
- 34.Johansson G, Hourihane JO, Bousquet J, et al. A revised nomenclature for allergy. An EAACI position statement from the EAACI nomenclature task force. Allergy. 2001;56:813–824. doi: 10.1034/j.1398-9995.2001.t01-1-00001.x. [DOI] [PubMed] [Google Scholar]
- 35.Dreborg S. The implications of nomenclature. Ann Allergy Asthma Immunol. 2002;89:S83–S85. doi: 10.1016/s1081-1206(10)62129-1. [DOI] [PubMed] [Google Scholar]
- 36.Dubovitz V. Current and future therapy in muscular dystrophy; need for a common language between basic scientists and clinicians. Acta Myol. 2004;23:V–IX. [PubMed] [Google Scholar]
- 37.Klein C. Movement disorders: classification. J Inherit Metab Dis. 2005;28:425–439. doi: 10.1007/s10545-005-7495-8. [DOI] [PubMed] [Google Scholar]
- 38.Aldave AJ, Edward DP, Park AJ, et al. Central discoid corneal dystrophy. Cornea. 2002;21:739–744. doi: 10.1097/00003226-200211000-00001. [DOI] [PubMed] [Google Scholar]
- 39.Weiss JS, Kruth HS, Kuivaniemi H, et al. Mutations in the UBIAD1 gene on chromosome short arm 1, region 36 cause Schnyder crystalline corneal dystrophy. Invest Ophthalmol Vis Sci. 2007;48:5007–5012. doi: 10.1167/iovs.07-0845. [DOI] [PubMed] [Google Scholar]
- 40.Orr A, Sube MP, Marcadier, et al. Mutations in the UBIAD1 gene encoding a potential prenyltransferase are causal for Schnyder crystalline corneal dystrophy. PLoS ONE. 2007;2:e685. doi: 10.1371/journal.pone.0000685. [DOI] [PMC free article] [PubMed] [Google Scholar]